

Abstract
During behavioral quiescence, the neocortex generates spontaneous slow oscillations, which may consist of up-states and down-states. Up-states are short epochs of persistent activity that resemble the activated neocortex during arousal and cognition. Neural activity in neocortical pathways can trigger up-states, but the variables that control their occurrence are poorly understood. We used thalamocortical slices from adult mice to explore the role of thalamocortical and intracortical synaptic cooperativity (the number of coincident afferents) in driving up-states. Cooperativity was adjusted by varying the intensity of electrical or blue-light stimuli in pathways that express channelrhodopsin-2. We found that optogenetics greatly improves the study of thalamocortical pathways in slices because it produces thalamocortical responses that resemble those observed in vivo. The results indicate that more synaptic cooperativity, caused by either thalamocortical or intracortical fast AMPA-receptor excitation, leads to more robust inhibition of up-states because it drives stronger feedforward inhibition. Conversely, during strong synaptic cooperativity that suppresses up-states, blocking fast excitation, and as a result the feedforward inhibition it drives, unmasks up-states entirely mediated by slow NMDA-receptor excitation. Regardless of the pathway's origin, cooperativity mediated by fast excitation is inversely related to the ability of excitatory synaptic pathways to trigger up-states in neocortex.
Introduction
Synchronous stimulation of neocortex pathways in slices trigger a stereotypical response characterized by short-latency (<10 ms) and long-latency (>10 ms) components. The short-latency component consists of an EPSP that is rapidly curtailed by a lasting IPSP (Agmon and Connors, 1992; Gibson et al., 1999; Gabernet et al., 2005; Sun et al., 2006; Cruikshank et al., 2010). During the long-latency component, up-states can occur (Rigas and Castro-Alamancos, 2007, 2009; Favero et al., 2012). Thalamocortical and intracortical synaptic pathways of neocortex have distinct abilities to trigger up-states in slices (Rigas and Castro-Alamancos, 2007). Electrical or chemical stimulation of the thalamus (thalamocortical) triggers cortical up-states very effectively, while electrical stimulation of the cortex (intracortical) triggers up-states only at very low intensities. However, as the intensity increases, intracortical electrical stimulation has a suppressive effect on up-states. Thus, increasing the stimulus intensity suppresses up-states in the intracortical but not in the thalamocortical pathway.
One possible reason for this difference in the ability to trigger up-states, which we noted previously, is reduced synaptic cooperativity (number of coincident afferent inputs) recruited in the thalamocortical pathway in the slice because many thalamocortical synapses have their axons severed and are not activated by thalamic stimulation (Rigas and Castro-Alamancos, 2007, 2009; Castro-Alamancos, 2009). The limitation of the slice preparation is reflected in weak short-latency thalamocortical (population) field potential (FP) responses, and corresponding EPSPs in single cells that rarely reach firing threshold during down-states even at the highest stimulus intensities (Rigas and Castro-Alamancos, 2009). This contrasts with the situation in vivo where relatively strong thalamic electrical stimulation drive much more robust short-latency thalamocortical FP responses and corresponding EPSPs that effectively reach firing threshold and spread activity through the somatosensory cortex (Castro-Alamancos and Connors, 1996a; Castro-Alamancos and Oldford, 2002; Hirata and Castro-Alamancos, 2006, 2008). Indeed, thalamocortical convergence is well known to drive cortical spikes in single cells in vivo (Bruno and Simons, 2002; Alonso and Swadlow, 2005; Bruno and Sakmann, 2006; Hirata and Castro-Alamancos, 2008), but the role of cooperativity in triggering persistent network activity (up-states) is not well understood.
Here we explore the role of thalamocortical and intracortical synaptic cooperativity in driving cortical up-states in thalamocortical slices of adult mice. Cooperativity was adjusted by varying the intensity of electrical or blue-light stimuli in pathways that express channelrhodopsin-2 (ChR2) (Zhang et al., 2006, 2007; Cruikshank et al., 2010). Fast and slow glutamatergic excitation was manipulated by using AMPA-receptor and NMDA-receptor antagonists, respectively. Compared with electrical stimulation of the thalamus in thalamocortical slices, we found that optogenetic stimulation of thalamocortical synapses in neocortex greatly improves the study thalamocortical pathways in slices because it produces thalamocortical responses that closely resemble those observed in vivo. Moreover, this led to the finding that cooperativity determines the ability of neocortical synaptic pathways to trigger up-states regardless of their origin; more cooperativity inhibits up-states by driving stronger feedforward inhibition.
Materials and Methods
All procedures were reviewed and approved by the Animal Care Committee of Drexel University. Slices were prepared as previously described (Rigas and Castro-Alamancos, 2007, 2009; Favero et al., 2012) from adult (>8 weeks) CD-1 (male) or Thy1-COP4/EYFP (line18) (of either sex) mice. Some CD-1 mice were anesthetized with ketamine-xylazine (100–5 mg/kg) and injected with AAV2/9-hSyn-ChR2(H134R)-eYFP (AAV, UPenn Vector core) into the somatosensory thalamus (0.2–0.4 μl) using the following coordinates (from bregma): posterior, 1.5 mm; lateral, 2.0 mm; ventral, 3.0 mm. Slices were prepared from these animals 1–2 weeks after the injection.
For slice preparation, mice were deeply anesthetized with an overdose of ketamine. Upon losing all responsiveness to a strong tail pinch, the animal was decapitated and the brain was rapidly extracted. Slices (400 μm thick) were cut in the thalamocortical plane (Agmon and Connors, 1991) using a vibratome. Slices were transferred to an interface chamber where they were bathed constantly (1–1.5 ml/min) with artificial CSF (ACSF) at 32.5°C. The ACSF contained the following (in mm): 126 NaCl, 3.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgSO4 7H2O, 10 dextrose, 1 CaCl2 2H2O. This buffer resulted in the generation of persistent network activity resembling slow oscillations in cortical slices (McCormick et al., 2003; Rigas and Castro-Alamancos, 2007, 2009; Favero et al., 2012). FP recordings were made using low-impedance (∼0.5 MΩ) glass pipettes filled with ACSF. Blind whole-cell recordings were obtained from layers V, IV, and III cells of somatosensory cortex using patch electrodes of 4–12 MΩ impedance. For current-clamp recordings, the electrodes were filled with internal solution containing the following (in mm): 135 K-gluconate, 4 KCl, 2 NaCl, 0.2 EGTA, 10 Tris-phosphocreatine, 0.3 TrisGTP, 10 HEPES, 4 MgATP (290 mOsm). Under our conditions, the Nernst equilibrium potential for Cl− is −81 mV and for K+ is −96.7 mV. In most cases, the internal solution contained neurobiotin (0.2%) to label the recorded cells.
After each experiment, the slices were fixated in 4% paraformaldehye with 1% glutaraldehyde, later cryoprotected with sucrose (30%) and resectioned on a cryostat (80 μm). Nonfluorescent sections were incubated in 3% hydrogen peroxide, followed by 0.2% Triton X-100 and by incubation in 2% goat serum. Incubation with ABC reagent (Vectorlabs) occurred overnight. The following day, diaminobenzidine was applied to the sections. After color development, sections were mounted and cleared in xylene. Fluorescent sections (from animals injected with AAV or line18) were mounted, coverslipped with DAPI mounting media, and photographed using a fluorescent microscope. Fluorescent sections that contained neurobiotin-filled cells were incubated in 0.2% Triton X-100 and 2% goat serum followed by DyLight594-streptavidin. Sections were then mounted, coverslipped with DAPI mounting media, and photographed using a fluorescent microscope. The enhanced yellow fluorescent protein (eYFP) from the ChR2 expression appears greenish, the neurobiotin-filled cell appears redish, and DAPI appears blueish. All labeled cells were faithfully reconstructed using Neurolucida (Microbrightfield).
Concentric bipolar stimulating electrodes (diameter, 125 μm) were used to electrically stimulate the thalamus (thalamocortical) or cortex (intracortical). A 200-μm-core-diameter multimode optic fiber was used to apply pulses of blue light in the cortex (around the recording site in layer IV) or thalamus (around the electrical stimulating electrode). The light source was either a blue laser or LED (∼473 nm) driven by pulses of 0.2–5 ms duration (or longer to test whether the cell responds directly to the light). The blue-light intensity was controlled by adjusting the output range of the light source or the duration of the pulse, and could be easily monitored by recording the evoked short-latency FP response. In addition, the intensity (irradiance) of the light beam exiting the optic fiber was measured by flashing a photodiode power sensor placed in the location of the slice. The approximate ranges for low, medium, and high intensities were 0.2–1.5, 1.5–3, and 3–8 mW/mm2, respectively. The FP electrode was first used to identify the cortical region with the strongest and shortest latency response evoked by thalamic electrical stimulation. Intracellular recordings were then obtained adjacent to the FP electrode. Thalamocortical EPSPs met two criteria: they depressed at 20 Hz and had short latencies (<2.5 ms). The intracortical electrical stimulating electrode was placed lateral to the recording electrodes (∼400 μm) in layers III-II. Drugs were dissolved in the ACSF at the indicated concentrations.
Electrical or blue-light stimulation were applied at a minimum 5 s apart between each other and recurred at an interval of 5 s or longer. During each stimulus, the membrane potential (Vm) could be set at different potentials by applying negative and positive current pulses (>200 ms before the synaptic stimulus onset), up to the level that produced spontaneous firing. This enabled us to derive the reversal potential for each point of the synaptic response, and to estimate the excitatory and inhibitory synaptic conductance (Gsynexc and Gsyninh, 0 and −81 mV reversal potential, respectively) before and during a drug application (for details, see Favero et al., 2012). Reversal potential for each point was estimated by calculating the y-intercept of the best fit line between the baseline-corrected (values subtracted 5 ms before the stimulus) Vm values (x-axis) and the Vm values (y-axis). Total conductance for each point was estimated by calculating the inverse slope of the best fit line between the injected current (x-axis) and the Vm values (y-axis). The synaptic conductance (Gsyn) is the total conductance minus the baseline conductance. Gsyninh for each point was estimated as the product of the reversal potential and the synaptic conductance divided by the Nernst equilibrium potential for inhibitory (Cl− is −81 mV) currents. Gsynexc for each point was Gsyn minus Gsyninh. We assumed that membrane capacitance was constant for each cell during our experiments and that, during the period we measured (<15 ms poststimulus), the synaptic conductance was composed primarily of glutamatergic excitation and Cl−-mediated inhibition.
Up-states were typically detected offline by using a threshold detector in the FP recording (Rigas and Castro-Alamancos, 2007, 2009). Relatively rare transient FP events that were not up-states were easily rejected by setting the detection algorithm to reject short-duration events (<50 ms). In addition, all detected events were sent to a sorting algorithm (similar to those used to sort spikes) and this enabled us to classify all detected up-states based on several projections (e.g., principal components, etc.). Finally, all detected events (selected or unselected as up-states) were inspected by eye to ensure that the procedure was adequate.
For statistical analyses, data were first tested for normality using the Shapiro–Wilk test. If the data were considered normally distributed, parametric statistics were applied (ANOVA repeated measures or t test paired). Otherwise, we applied nonparametric statistics (Wilcoxon signed ranks for paired comparisons, Mann–Whitney for nonpaired comparisons, Kruskal–Wallis for multiple groups).
Results
Regulation of spontaneous up-states by glutamate receptors
The thalamus and neocortex generate a variety of electrical activities during different behavioral states that have profound consequences on signals flowing through them (Castro-Alamancos and Connors, 1997; Castro-Alamancos, 2004b, 2009; Harris and Thiele, 2011). During slow-wave sleep and anesthesia, neocortical networks typically produce slow oscillations characterized by rhythmic cycles of synaptically mediated depolarization and increased firing (up-states) followed by decrease of synaptic inputs leading to membrane hyperpolarization and cessation of firing (down-states) (Metherate et al., 1992; Steriade et al., 1993a, 1993b). Up-states correspond to recurrent synaptic network activity generated within neocortex and maintained by balanced excitatory and inhibitory conductances (Steriade et al., 1993b; Shu et al., 2003).
As in previous studies (Rigas and Castro-Alamancos, 2007, 2009; Favero et al., 2012), we conducted simultaneous intracellular (whole-cell) and FP recordings in somatosensory cortex slices of adult mice that display spontaneous persistent network activity resembling slow oscillations (up-states and down-states). The up-state was characterized by barrages of postsynaptic potentials that depolarize the recorded neuron by 5–15 mV for the duration of the network event, which was always clearly reflected in the FP recording. During the down-state, cortical cells were relatively hyperpolarized and there was little synaptic or network activity. Figure 1A,B shows examples of spontaneous slow oscillations consisting of up-states and down-states in two different cells and simultaneous FP recordings in layer III (the cell in Fig. 1B is cell5 reconstructed in Fig. 3A). Although block of AMPA or NMDA receptors has been reported to suppress spontaneous up-states (McCormick et al., 2003), the relative importance of these receptors for spontaneous and evoked up-states is not well understood.
Figure 1.
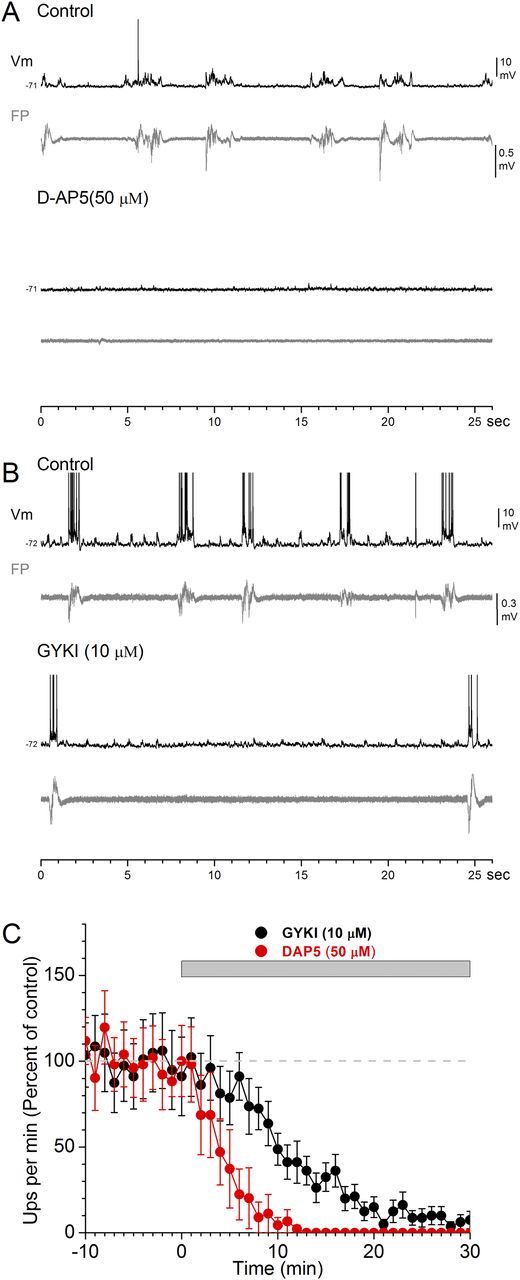
Effect of AMPA-receptor and NMDA-receptor antagonists on spontaneous slow oscillations in thalamocortical slices. A, B, Whole-cell intracellular (Vm) and FP recordings from layer V cells of somatosensory cortex before and during application of the NMDA-receptor antagonist d-AP5 (A) or the AMPA-receptor antagonist GYKI (B). Note the complete abolishment of spontaneous up-states during d-AP5 but not during GYKI, which is reflected in both Vm and FP recordings. C, Population data showing the incidence of spontaneous up-states per minute during control and during application of d-AP5 or GYKI.
Figure 3.

Histological verification of recorded cells and ChR2 expression. A, Identification of some of the cells studied. In some cases, during histological processing, the apical dendrite was truncated and lost by the resectioning (80 μm) of the 400 μm slices. B, Typical thalamocortical slice obtained from a Thy1-ChR2-eYFP-line18 mouse (1 resectioned 80 μm section is shown). EYFP, green; DAPI, blue. Note the robust eYFP fluorescence (ChR2 expression) of layer V cells and their apical dendrites, and the typical sparseness of expression in layer IV, indicating lack of ChR2 expression in thalamocortical fibers. C, Three successive slices from a CD-1 mouse cut in the thalamocortical plane (1 resectioned 80 μm section is shown) that had been injected with AAV-hSyn-ChR2-eYFP in somatosensory thalamus. Note the robust ChR2 expression of thalamocortical fibers in layer IV, and somewhat in layer VI, of somatosensory cortex. The inset strip in the middle slice shows a neurobiotin-filled cell (red) recorded in that slice overlapped with the ChR2 expression (green); this is cell9 reconstructed in A. EYFP, green; DAPI, blue; DyLight594, red. wm, white matter.
We found that application of the NMDA-receptor antagonist d-AP5 (50 μm) rapidly and completely abolished the incidence of spontaneous up-states (Fig. 1A). In contrast, application of the AMPA-receptor antagonist GYKI35348 (GYKI; 10 μm) strongly suppressed the incidence of spontaneous up-states but did not completely abolish them. The same result was obtained when another AMPA-receptor antagonist (CNQX; 10 μm) was applied. Results from these two AMPA-receptor antagonists are combined and referred to as GYKI from here on. Figure 1C shows population data of the effects of d-AP5 and GYKI on the incidence of spontaneous up-states. Block of NMDA receptors rapidly abolished the incidence of spontaneous up-states, so that 15 min after application, the incidence of up-states was nil (n = 8). In contrast, blockage of AMPA receptors suppressed more slowly the incidence of spontaneous up-states. However, 15 min after GYKI, the incidence of spontaneous up-states was still significant (n = 12). The spontaneous up-states that persisted in the presence of GYKI were always rapidly and completely abolished by application of d-AP5 (data not shown but see below), indicating that they were mediated by NMDA receptors. As expected from blocking fast excitation, the NMDA-mediated up-states were significantly shorter in duration than those that occurred during control (the difference is quantified below for evoked up-states). The results show that spontaneous cortical up-states depend critically on the slow excitation provided by NMDA receptors, without which they do not occur. Moreover, in the absence of fast excitation provided by AMPA receptors, NMDA receptors can mediate spontaneous up-states, albeit at a much slower rate and of shorter duration than when fast excitation is present. But as shown below, purely NMDA-mediated up-states can be readily driven by afferent activity. We next focused on evoked up-states.
Block of AMPA receptors unmasks intracortical-evoked up-states
In thalamocortical slices, electrical stimulation of the thalamus produces a stereotypical response in neocortex that consists of a short-latency response (<10 ms) followed by a longer-latency up-state (>10 ms) that persists with the highest stimulus intensities. In contrast, intracortical electrical stimulation >20 μA (typically in layers III-II) produces a strong short-latency response followed by a period of nil network activity for hundreds of milliseconds [i.e., up-states are not evoked by intracortical stimuli unless low intensities (<20 μA) are used] (Rigas and Castro-Alamancos, 2007). Examples of these responses can be found in our previous work (Rigas and Castro-Alamancos, 2007, 2009; Favero et al., 2012) and in Fig. 2A (top, control). We tested the effect of NMDA-receptor and AMPA-receptor antagonists on short-latency and long-latency (up-state) responses evoked by thalamocortical and intracortical electrical stimulation (>20 μA). Suppression of AMPA receptors (GYKI) abolished thalamocortical-evoked up-states (Fig. 2A, left column), but unmasked up-states triggered by intracortical stimulation (Fig. 2A, right column). As the AMPA antagonist washed in (Fig. 2A, early), the thalamocortical-evoked up-state and the short-latency responses were suppressed but, at the same time, a robust up-state triggered by intracortical stimulation was unmasked. During complete block of AMPA receptors (Fig. 2A, later), thalamocortical-evoked up-states and the short-latency intracortical responses were completely abolished, but the unmasked intracortical-evoked up-states were still present. The evoked up-states unmasked by GYKI were always completely abolished by d-AP5, and were significantly shorter in duration than those evoked during control at low intensities (n = 10 cells; control, 410 ± 46 ms; GYKI, 250 ± 27 ms; p < 0.01). Many of the recorded cells were filled with neurobiotin and identified (Fig. 3A). Despite the differences in laminar location and morphological type, there was no obvious difference in the results described above among different cells. This is expected because up-states correspond to network activity that spreads through all, or most, cells in the cortical network.
Figure 2.
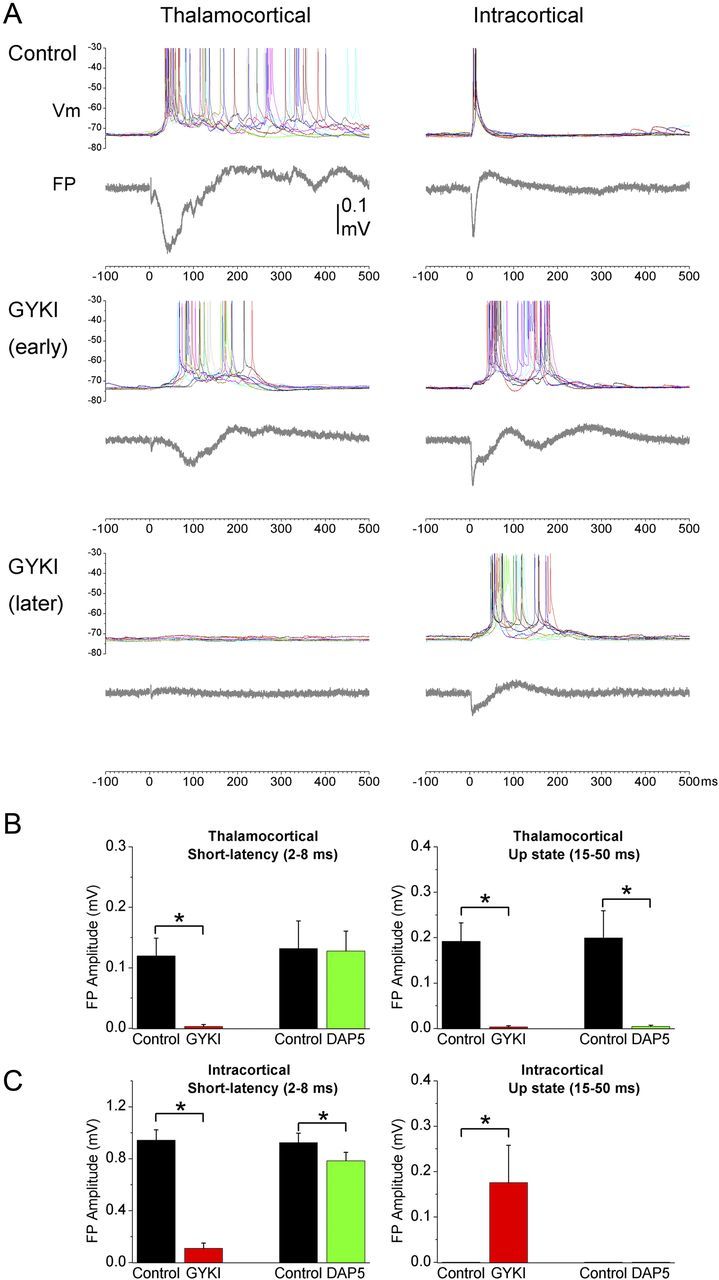
Effect of an AMPA-receptor antagonist (GYKI) on up-states evoked by electrical stimulation of thalamocortical and intracortical pathways. A, The intensity was set so that, during control, electrical stimulation of the thalamus evokes up-states in somatosensory cortex (left), while stimulation within the cortex does not (right). Application of GYKI abolishes up-states evoked by thalamocortical stimulation but at the same time unmasks up-states evoked by intracortical stimulation. This effect is reflected in both intracellular and FP recordings. B, Population data of the effects of GYKI or d-AP5 on thalamocortical-evoked short-latency (2–8 ms) and long-latency (15–50 ms) FP responses. The long-latency response reflects the up-state. C, Population data of the effects GYKI or d-AP5 on intracortical-evoked short-latency (2–8 ms) and long-latency (15–50 ms) responses. *p < 0.01.
Since evoked up-states are network (population) events, they are easily quantified using FP recordings by measuring the peak amplitude of the long-latency (15–50 ms) response, while the short-latency synaptic response is quantified by measuring the peak amplitude between 2 and 8 ms excluding the fiber volley (Rigas and Castro-Alamancos, 2007, 2009; Favero et al., 2012). Figure 2B,C shows population data of the effects of GYKI (n = 12) and d-AP5 (n = 6) on the short-latency and long-latency evoked FP responses. GYKI abolished up-states and short-latency responses evoked by thalamocortical stimuli (Fig. 2B, p < 0.01). GYKI also suppressed short-latency responses evoked by intracortical stimulation, but at the same time increased the long-latency response corresponding to the unmasking of intracortical-evoked up-states (Fig. 2C, p < 0.01). The unmasked up-state was rapidly and completely abolished by subsequent application of d-AP5 (n = 6; data not shown, but see below). d-AP5 also rapidly abolished thalamocortical-evoked up-states (p < 0.01) with little effect on short-latency responses triggered by thalamocortical stimulation (Fig. 2B). d-AP5 had a small but significant effect on short-latency responses triggered by intracortical stimulation (Fig. 2C), but did not unmask intracortical-evoked up-states in any experiment; there was no discernible long-latency peak to measure corresponding to an up-state during control or d-AP5.
Stimulation of intracortical excitatory synapses using light
The previous results indicate that relatively strong intracortical electrical stimulation does not trigger up-states under control conditions. However, when fast excitation is suppressed by blocking AMPA receptors, an NMDA-mediated intracortical-evoked up-state is readily unmasked. Obviously, electrical stimulation within neocortex is fairly unselective; it directly excites the membranes of all excitatory and inhibitory cells surrounding the stimulating electrode. To address this confound, we used slices from line18 mice (Fig. 3B), which primarily express ChR2 in excitatory cortical cells (Wang et al., 2007). In somatosensory cortex, layer V pyramidal cells robustly express ChR2 (Fig. 3B), which is targeted to the plasma membrane so that the intensity of the YFP signal is highest where the membrane has a high surface/volume ratios, such as in dendrites and axons (Wang et al., 2007). A spot of blue light will activate all ChR2-expressing membranes located in the spot. In these slices, application of a short spot of blue light through an optic fiber centered in layer IV produces synaptic responses that resemble those evoked by electrical stimulation (Fig. 4). Moreover, similar to the effect of intracortical electrical stimuli, the intensity of the blue light determines whether up states are evoked. Low-intensity blue-light stimulation triggers up-states reliably in both intracellular (Fig. 4A,B) and FP recordings (Fig. 4C). Increasing the light intensity (medium and high) leads to a significant increase in the short-latency synaptic response and a concomitant abolishment of evoked up-states (p < 0.01; Fig. 4D; n = 6).
Figure 4.

Activation of intracortical synapses that express ChR2 in slices from Thy1-ChR2-line18 mice, using different intensities of blue light, produces effects similar to intracortical electrical stimulation; up-states are evoked at low intensities but suppressed at high intensities. A, B, Examples from two different cells showing the effects of three different blue-light intensities on short-latency responses and up-states. Note that low-intensity blue light evokes up-states reliably, while increasing the intensity of the blue light leads to larger short-latency responses and abolishment of evoked up-states. The cell in A is cell13 reconstructed in Figure 3A. C, Effect of blue-light intensity on FP responses in somatosensory cortex; up-states are only evoked at low intensities. D, Population data showing the effect of intracortical blue-light intensity on short-latency (2–8 ms) and long-latency (15–50 ms) FP responses. The long-latency response reflects the up-state. *p < 0.01 versus low intensity.
We next determined whether GYKI unmasks up-states for intracortical responses evoked by pulses of blue light at intensities that do not evoke up-states during control. Indeed, as shown in Figure 5, GYKI unmasked up-states for intracortical stimuli evoked by blue-light pulses, and the unmasked up-states were abolished by subsequent d-AP5. In addition, the cell shown in Figure 5 appears to express ChR2 as shown by the responses to long pulses of blue light in the presence of GYKI, d-AP5, and TTX (Fig. 5B). Similar results were obtained in other cells (n = 5; data not shown). Thus, selective intracortical stimulation with pulses of blue light in slices that express ChR2 in excitatory cells leads to results similar to those obtained with electrical stimulation. The results indicate that, for intracortical pathways, strong synaptic cooperativity mediated by fast excitation suppresses evoked up-states. Conversely, during strong synaptic cooperativity, block of fast synaptic excitation mediated by AMPA receptors unmasks up-states purely mediated by NMDA receptors.
Figure 5.
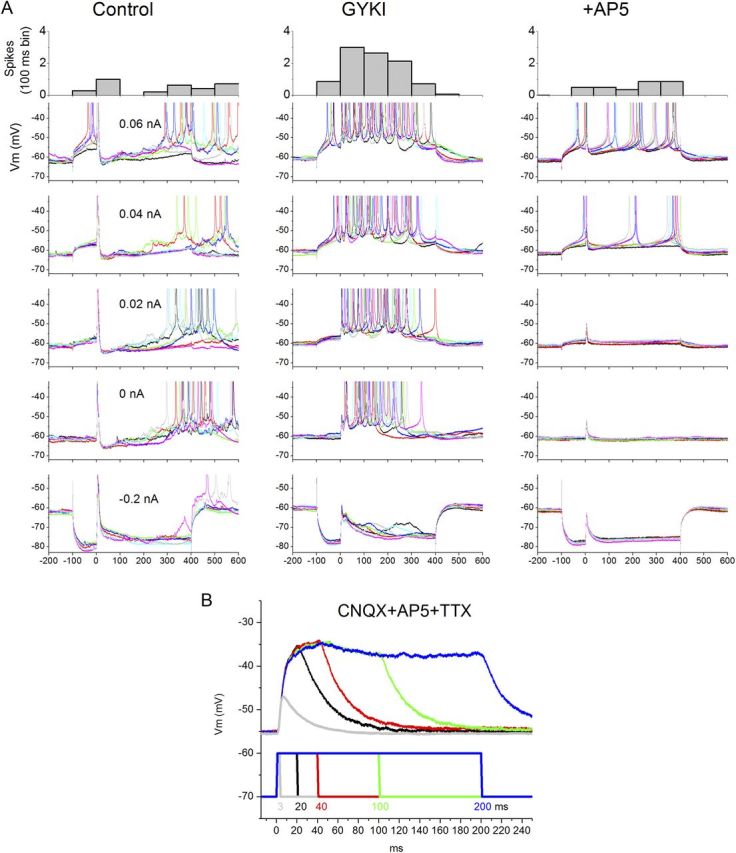
Effect of GYKI on intracortical responses evoked by high-intensity blue-light stimuli in Thy1-ChR2-line18 slices. A, Typical example from a cell recorded at different Vm levels set by intracellular current pulses (500 ms; blue light occurs 100 ms after current pulse onset). The high-intensity blue-light stimulus evokes a sharp short-latency response curtailed by a robust IPSP that can lead to a rebound excitation. GYKI strongly suppresses the short-latency response (but does not abolish it; see below), but at the same time it unmasks an up-state during the period where previously the IPSP was evoked. B, Subsequent application of d-AP5 completely abolishes the unmasked up-state, leaving intact a short-latency response caused by direct activation of ChR2 channels in this cortical cell; it survived block of glutamate receptors and Na+ channels. The blue-light duration is clearly reflected in the depolarization it causes, indicating that the cell expresses ChR2. In A (top), the number of spikes evoked by the depolarizing current pulses (> 0 nA) are calculated (100 ms bin). Note the unmasking of spikes during GYKI. The cell is cell10 reconstructed in Figure 3A.
Why does thalamocortical stimulation not suppress up-states?
Why does thalamocortical stimulation not suppress up-states? One possibility is that the difference between thalamocortical and intracortical stimulation is the lower synaptic cooperativity that can be recruited in slices by thalamic stimulation. Several observations indicate that thalamocortical fiber recruitment is lower in slices compared with in vivo. First, retrograde neurobiotin labeling in thalamocortical slices shows that few thalamocortical fibers remain intact between thalamus and cortex (Rigas and Castro-Alamancos, 2007, 2009). Thus, when the stimulating electrode is placed within the thalamus in slices, only a small subset of the total thalamocortical fibers that innervate a particular cortical area can be stimulated and mediate cortical responses. In contrast, intracortical electrical stimulation obviously recruits many more fibers. Second, the difference in synaptic cooperativity is apparent by measuring the amplitudes of short-latency FP (population) responses evoked by thalamocortical and intracortical stimulation in slices (Rigas and Castro-Alamancos, 2007, 2009); short-latency thalamocortical FP responses are much smaller even when much stronger intensities are used. Finally, thalamocortical FP responses evoked in slices are not only smaller but are also less complex than thalamocortical responses observed in vivo (Castro-Alamancos and Connors, 1996a, 1997; Castro-Alamancos and Oldford, 2002; Oldford and Castro-Alamancos, 2003; Hirata and Castro-Alamancos, 2006, 2011). For instance, thalamocortical FP responses in vivo produce a primary short-latency response that contains multiple inflections at different latencies corresponding to the propagating current flow through different layers (Castro-Alamancos and Connors, 1996a; Castro-Alamancos and Oldford, 2002; Castro-Alamancos, 2004a). In contrast, thalamocortical FP responses in slices are much simpler. They consist of a single component, which reflects weak thalamocortical EPSPs that rarely reach firing threshold during down-states (Rigas and Castro-Alamancos, 2007, 2009).
According to our previous arguments, up-states in slices are suppressed by robust intracortical stimulation, but not by robust thalamocortical stimulation, because of the limited synaptic cooperativity that can be recruited by thalamic stimulation in thalamocortical slices. To test this hypothesis, we set out to increase the number of thalamocortical synapses available for recruitment in slices. This was accomplished by infusing AAV into the somatosensory thalamus of CD-1 mice (Fig. 3C) to express ChR2 in thalamocortical fibers (Zhang et al., 2006, 2007). This way, thalamocortical synapses can be stimulated directly in the somatosensory cortex regardless of whether their axons have been truncated during slice preparation (Cruikshank et al., 2010). Figure 6A shows a typical thalamocortical slice expressing ChR2 in thalamocortical synapses. Application of pulses of blue light in the thalamus (Fig. 6B, top) produces thalamocortical FP responses in cortex that behave much like electrical stimulation within the thalamus; weak short-latency responses that evoke up-states at all high intensities tested. In contrast, moving the light stimulus to the cortex (Fig. 6B, bottom), where it can directly recruit many more thalamocortical synapses regardless of whether their axons reach the thalamus intact in the slice, leads to much stronger short-latency synaptic responses and up-states are only evoked at low intensities. Importantly, the thalamocortical FP responses evoked in slices by blue-light stimuli applied in cortex resemble the thalamocortical FP responses evoked in vivo by electrical stimulation of the thalamus or fast whisker deflections (Castro-Alamancos and Connors, 1996a, 1997; Castro-Alamancos and Oldford, 2002; Hirata and Castro-Alamancos, 2006).
Figure 6.
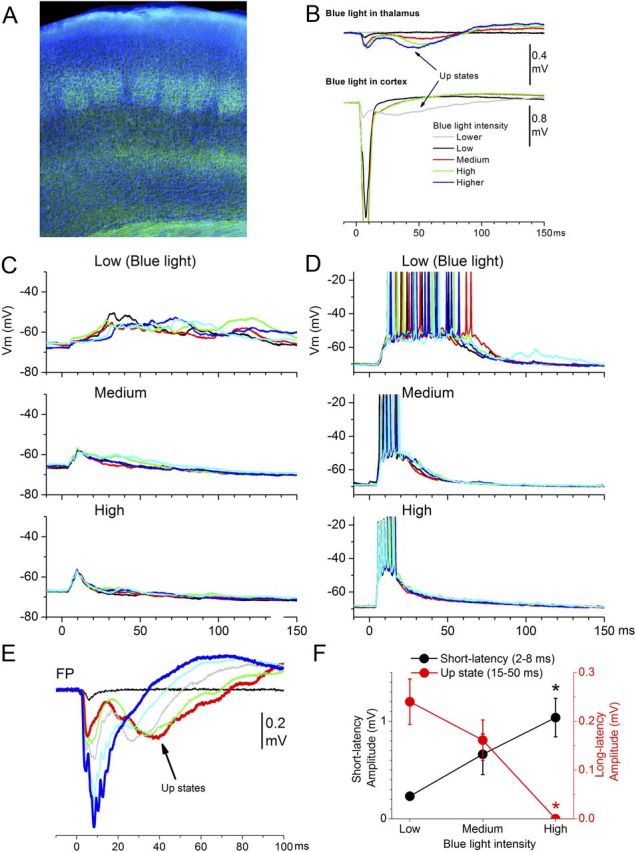
Activation of thalamocortical synapses that express ChR2 in slices from mice injected with AAV-hSyn-ChR2-eYFP in somatosensory thalamus, using different intensities of blue light, produces effects similar to intracortical electrical or blue-light stimulation but different from electrical stimulation of the thalamus; up-states are evoked at low intensities and suppressed at high intensities. A, Example of thalamocortical ChR2 expression (eYFP) in the somatosensory (note the barrels in layer IV) cortex of a typical thalamocortical slice from a mouse injected with AAV-hSyn-ChR2-eYFP in the somatosensory thalamus. EYFP, green; DAPI, blue. B, Different effects of application of blue light into the thalamus or into the somatosensory cortex on FP responses recorded in cortex. Blue light in thalamus results in small short-latency responses that evoke up-states (like thalamocortical electrical stimulation). Application of blue light in cortex of the same slice, which recruits only thalamocortical synapses regardless of whether their axons reach the thalamus in the slice, evoked up-states only at low intensities. However, increasing the intensity recruits many more thalamocortical synapses, which lead to sharper short-latency responses that abolish evoked up-states. C, D, Examples from two different cells showing the effects of three different blue-light intensities on short-latency responses and up-states. Note that low-intensity blue light evokes up-states reliably, while increasing the intensity of the blue light leads to larger short-latency responses and abolishment of evoked up-states. The cell in D is cell9 reconstructed in Figure 3A. E, Effect of blue-light intensity on FP responses in somatosensory cortex; up-states are only evoked at low intensities. F, Population data showing the effect of intracortical blue-light intensity on short-latency (2–8 ms) and long-latency (15–50 ms) FP responses. The long-latency response reflects the up-state. *p < 0.01 versus low intensity.
Figure 6C,D shows typical intracellular responses obtained in two different cells by blue-light pulses of different intensities delivered in cortex. In all cases (n = 7 cells), a low-intensity blue-light stimulus, which barely produced short-latency responses, evoked up-states very reliably. Increasing the intensity of the light stimulus increased the short-latency response but abolished the evoked up-state. This behavior is also clearly visible in FP recordings (Fig. 6E). Figure 6F shows population data (n = 7) demonstrating that optogenetic activation of thalamocortical synapses in cortex (which, compared to electrical stimulation of the thalamus, allows more synapses in slices to be recruited) triggers up-states at low intensities but not at high intensities. Thus, when the cooperativity of thalamocortical synapses is increased, thalamocortical stimulation behaves pretty much like intracortical stimulation.
Furthermore, we tested whether application of GYKI during high-intensity thalamocortical stimulation would also unmask up-states and found that this was also the case. Figure 7 shows a cell recorded at different Vm in which we alternated between electrical stimulation of the thalamus (Fig. 7A) and blue-light stimulation of the cortex (Fig. 7B) at high intensities. Application of GYKI resulted in the complete abolishment of up-states evoked by electrical stimulation of the thalamus. At the same time, GYKI abolished the short-latency response evoked by high-intensity blue-light stimulation of cortex and unmasked a longer latency up-state that was completely abolished by subsequent application of d-AP5 (Fig. 7B). This behavior is also clearly visible in FP recordings (Fig. 7C). Figure 7D shows population data (n = 7) revealing that GYKI abolished the short-latency response evoked by blue-light stimuli in the cortex and unmasked a long-latency (up-state) FP response that is abolished by d-AP5.
Figure 7.

If synaptic cooperativity is increased, GYKI also unmasks up-states in thalamocortical pathways. A, B, Effect of GYKI on thalamocortical responses evoked by electrical stimulation of the thalamus (A) and by high-intensity blue-light stimuli in slices from mice injected with AAV-hSyn-ChR2-eYFP in somatosensory thalamus (B). Responses in A and B are from the same cell, in which electrical and blue-light stimulation alternated. Evoked responses are recorded at different Vm levels set by intracellular current pulses. High-intensity electrical stimulation of the thalamus evokes an up-state during control, while high-intensity blue-light stimulus evokes a sharp short-latency response but no up-state. GYKI abolished the up-state evoked by electrical stimulation of the thalamus and the short-latency response evoked by blue-light pulses. At the same time, GYKI unmasks an up-state evoked by blue light. Subsequent application of d-AP5 completely abolishes the unmasked up-state (gray trace). C, The same effects are observed in FP recordings evoked by high-intensity blue-light stimuli. D, E, Population data of the effects of GYKI and subsequent application of d-AP5 on thalamocortical-evoked short-latency (2–8 ms) and long-latency (15–50 ms) FP responses. The long-latency response reflects the up-state. *p < 0.01 GYKI versus control.
Possibly, AAV injection in the thalamus leads to transsynaptic expression of ChR2 in cortical cells, which means that the intracortical light stimulus is not only directly recruiting thalamocortical synapses but also directly stimulating intracortical cells and their synapses. However, previous studies have not found this to occur. Moreover, we found no cell bodies labeled with eYFP in the somatosensory cortex, where only fibers were labeled; cell body staining is clearly recognized in the injection sites. We checked all the sections of the slices used in this study (resectioned at 80 μm) and none had eYFP-labeled cells in somatosensory cortex. Also, in these animals, we never encountered a cortical cell that directly responded to the light stimulus. Both intracellular and FP responses evoked by the light were abolished by CNQX plus AP5; note that in the line18 mice, which express ChR2 in cortical cells, FP responses evoked by light were not completely abolished by CNQX plus AP5. In some AAV slices, a fiber volley could be observed only in the FP recordings after CNQX plus AP5. This component was almost completely (∼90%) abolished by TTX (1 μm), and the remaining component is likely due to direct depolarization of thalamocortical fibers by ChR2 channels.
Together these results demonstrate that the ability to drive up-states by neocortical pathways depends on the level of cooperativity that can be recruited in the pathway and not on the origin of the afferents.
Block of AMPA receptors suppresses feedforward inhibition
We have previously argued that strong intracortical electrical stimulation does not evoke up-states because it drives robust feedforward inhibition (Rigas and Castro-Alamancos, 2007). But why would blocking AMPA receptors unmask up-states? Feedforward inhibition is driven by excitation, which depolarizes inhibitory neurons (Gibson et al., 1999; Gabernet et al., 2005; Sun et al., 2006; Cruikshank et al., 2007). Consequently, block of AMPA receptors may result in a suppression of feedforward inhibition. To determine whether suppressing AMPA receptors relieves feedforward inhibition evoked by intracortical electrical stimulation, we studied the effect of GYKI on intracellular responses evoked at different Vm set by variable current pulses. This provides two measures. First, the synaptic potentials were used to estimate the excitatory and inhibitory synaptic conductance evoked by intracortical stimuli. Second, the number of spikes evoked by positive current pulses during synaptic stimulation should depend on feedforward inhibition. If GYKI suppresses feedforward inhibition, we would expect an increase in the number of evoked spikes during the time course of evoked feedforward inhibition due to less shunting.
In these experiments, we realized that it was necessary to use slices that did not produce spontaneous or evoked up-states because these network events would interfere with our measurements. This issue is evident in Figure 8A, which shows negative (hyperpolarizing) and positive (depolarizing) intracellular current pulses (500 ms duration) with intracortical electrical stimulation starting 100 ms after each current pulse. During control, the intracortical stimulus evokes a short-latency EPSP truncated by a longer-lasting IPSP, which is particularly noticeable on the depolarizing current pulses. Application of GYKI unmasked intracortical-evoked up-states that were evoked at all Vm, which interferes with estimating the synaptic conductances. Also, most of the spikes during GYKI are driven by the unmasked up-state per se and not the current pulse.
Figure 8.

GYKI unmasks an NMDA-mediated slow excitation and relieves shunting inhibition evoked by intracortical electrical stimulation. A, In slices that evoke up-states, it is difficult to study what GYKI unmasks to trigger up-states because the response is overwhelmed by the unmasked up-state. B, To overcome this problem, we used slices that do not produce spontaneous or evoked up-states but that otherwise appear to have normal short-latency responses. Under these conditions, GYKI unmasked a slow NMDA-mediated excitation not apparent during control (see arrows); it was abolished by d-AP5 (Fig. 9). C, Population data of the effect of GYKI on spikes evoked by positive current pulses (> 0 nA) before (−100–0 ms bin) and after the intracortical stimulus (0–400 ms bins) in slices that do not produce up-states. During control, the IPSP evoked by the intracortical stimulus shunts the spikes evoked by the current pulse for the period after the intracortical stimulus, but not before the stimulus. GYKI relieved this effect; the number of spikes increased during the period after the stimulus but not before. *p < 0.01 GYKI versus control per 100 ms bin.
To address these confounds, we tested the impact of GYKI on synaptic responses in the absence of up-states by using slices that randomly did not express them. During control, these slices had short-latency responses indistinguishable from slices that expressed up-states. As shown in Figure 8B, application of GYKI abolished the short-latency response and revealed two effects. First, a slow depolarization that was not obvious during control emerged (arrows in Fig. 8B). Second, consistent with a reduction in inhibitory shunting, the number of spikes evoked during the depolarizing current pulses increased for the period after the synaptic stimulus (0–400 ms), but not for the period before the synaptic stimulus −100–0 ms). Figure 8C shows a peristimulus time histogram (100 ms bins; n = 5 cells) of the number of spikes evoked by the same depolarizing current pulses during control and GYKI. Note that GYKI significantly increased (p < 0.01) the number of evoked spikes for the period corresponding to feedforward inhibition (after the intracortical stimulus) but not before.
To determine the nature of the slow synaptic potentials evoked by intracortical stimulation during GYKI, we estimated the excitatory (Gsynexc) and inhibitory (Gsynexc) synaptic conductances from synaptic potentials evoked at different Vm set by variable current pulses (Fig. 9A). Figure 9B,C shows Gsynexc and Gsyninh evoked in two different cells by intracortical stimuli before and during GYKI, and during subsequent d-AP5. Population data (n = 10 cells) of the short-latency (2–8 ms) and long-latency (15–50 ms) peak amplitudes of the evoked responses are shown in Figure 9D. As expected, GYKI produced a strong suppression of Gsynexc (p < 0.01; Fig. 9D), but a slow Gsynexc response was still present during GYKI (Fig. 9B,C, left red traces). This slow response was completely abolished by d-AP5 (p < 0.01; Fig. 9D), indicating that it was entirely mediated by NMDA receptors (Fig. 9B,C, left green traces). At the same time, GYKI robustly suppressed both the fast (short-latency; 2–8 ms) and the slower (long-latency; 15–50 ms) Gsyninh responses (p < 0.01; Fig. 9D). The same results were obtained using blue-light stimulation in line18 slices (n = 4 cells); GYKI robustly suppressed both fast and slow Gsyninh responses (p < 0.01). Subsequent application of d-AP5 did not result in any additional significant suppression of Gsyninh. The effect on the longer-latency Gsyninh was somewhat variable between cells (Fig. 9B,C), possibly because of nonlinearities in the longer-latency synaptic responses. During experiments with GYKI plus d-AP5, cortical cells showed a small pure Gsyninh response driven by electrical intracortical stimulation, which is likely caused by direct stimulation of GABAergic synapses.
Figure 9.
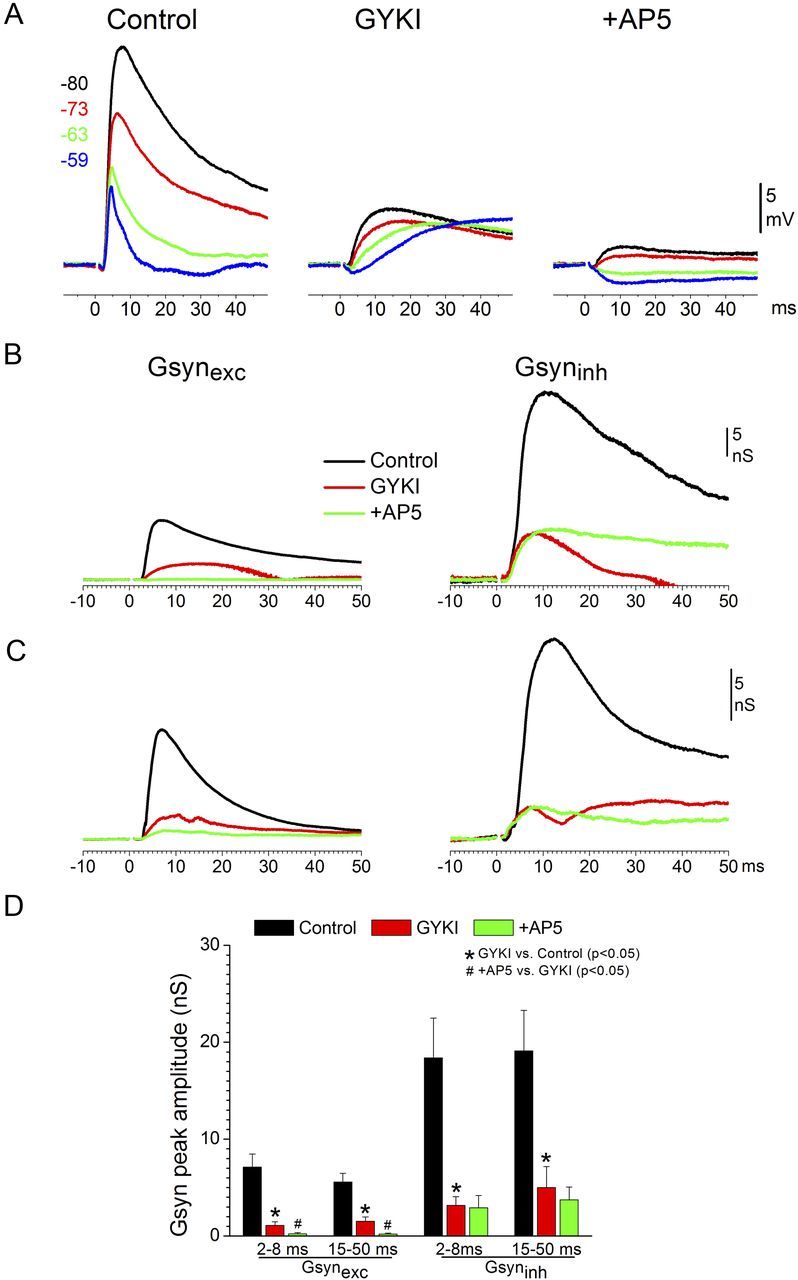
GYKI suppresses the inhibitory synaptic conductance (feedforward inhibition) evoked by intracortical electrical stimulation. A, Example showing the effect of GYKI and subsequent d-AP5 on intracortical-evoked postsynaptic potentials recorded at different Vm set by intracellular current injection in slices that do not evoke up-states. B, C, Excitatory and inhibitory synaptic conductance derived from postsynaptic potentials (as in A) in two different cells during control, GYKI, and subsequent d-AP5. D, Population data showing the effects of GYKI and subsequent d-AP5 on the short-latency (2–8 ms) and long-latency (15–50 ms) excitatory and inhibitory synaptic conductances evoked by intracortical electrical stimulation.
The results indicate (Fig. 9D) that feedforward inhibition is mostly driven by fast excitation (AMPA receptors). Thus, blocking AMPA receptors strongly suppresses feedforward inhibition and reveals a slow NMDA-mediated excitation, which drives the expression of up-states.
Discussion
The results demonstrate that the ability to trigger up-states in neocortical pathways depends critically on the level of synaptic cooperativity driven by fast excitation (i.e., AMPA-receptor mediated) and not on the origin of the pathway. Somewhat counterintuitively, weak synaptic cooperativity reliably triggers up-states. Conversely, strong synaptic cooperativity suppresses up-states because it recruits stronger feedforward inhibition driven by fast excitation.
The results also point toward a prominent role of NMDA receptors in the generation and/or expression of up-states in slices. The up-states produced in the absence of fast AMPA-mediated excitation are driven purely by slow NMDA-mediated excitation. These NMDA up-states are similar to normal up-states (i.e., a network event) but are predictably shorter in duration and occur spontaneously at very low rates due to the lack of fast excitation. Importantly, even when fast excitation is intact, both spontaneous and evoked up-states depend critically on NMDA receptors; up-states are abolished by blocking NMDA receptors, but not by blocking AMPA receptors. Several models have emphasized a critical role for NMDA-receptor dendritic spikes (Schiller et al., 2000) in the generation of up-states (Milojkovic et al., 2005; Kepecs and Raghavachari, 2007; Antic et al., 2010). Also, NMDA receptors can mediate persistent sensory-evoked responses in neocortex (Miller et al., 1989). Moreover, cortical synapses can produce NMDA-mediated responses at resting Vm in the absence of AMPA-mediated excitation, particularly when buffers (containing ∼1 mm [Mg2+]o) that resemble the CSF are used (Kanter et al., 1996; Espinosa and Kavalali, 2009). Normally, afferent activity produces fast AMPA-mediated excitation that drives feedforward inhibition, which suppresses up-states. However, in the absence of fast AMPA-mediated excitation, feedforward inhibition is strongly weakened, and afferent activity produces only slow NMDA-mediated excitatory responses capable of driving network events resembling up-states. Thus, block of fast excitation weakens feedforward inhibition and relieves the expression of up-states mediated by the remaining slow excitation.
The previous conclusion raises questions about the source of feedforward inhibition that controls evoked up-states. There are two obvious sources of feedforward inhibition driven by fast excitation in principal cells of neocortex: intrinsic and synaptic inhibition. The first source, intrinsic inhibition, is the aftereffect of action potentials driven by fast synaptic excitation; the action potentials produce after-hyperpolarizing potentials, due to activation of K+ conductances that vary in amplitude and duration among different cells (Storm, 1990; Azouz et al., 1996). The second source, synaptic inhibition, is caused when fast excitation drives the firing of GABAergic cells, which then release GABA and activate both GABAA (fast inhibition) and GABAB (slow inhibition) receptors (Connors et al., 1988). Blocking fast synaptic excitation (e.g., GYKI) will suppress the evoked short-latency action potentials in principal and GABAergic cells, and consequently will suppress intrinsic and synaptic inhibition, respectively. Future work will have to tease apart the relative influence of these sources of inhibition in controlling evoked up-states. It is already known that pharmacological suppression of either synaptic or intrinsic inhibition affects spontaneous slow oscillations in neocortex (Castro-Alamancos, 2000; Castro-Alamancos and Rigas, 2002; Shu et al., 2003; Castro-Alamancos et al., 2007; Sanchez-Vives et al., 2010). However, teasing apart their relative influence on evoked up-states may be difficult because their suppression can readily lead to epileptiform activity.
Cooperativity and up-states
The ability of thalamocortical electrical stimulation to trigger up-states in thalamocortical slices throughout the stimulus intensity range (Rigas and Castro-Alamancos, 2007) has always been hard to reconcile with the fact that relatively robust thalamocortical electrical stimulation in vivo produces sharp short-latency responses truncated by strong long-lasting IPSPs and are devoid of up-states during this period; rebound network excitation can occur at longer (>150 ms) latencies (Castro-Alamancos and Connors, 1996a,b; 1997; Castro-Alamancos, 1997). Another incongruence is that FP (population) responses in slices, even those driven by the strongest thalamic stimulation, are usually weaker and less complex than those observed in vivo (Rigas and Castro-Alamancos, 2009); activity spreads very effectively from thalamocortical-recipient layers to adjacent layers in vivo but not in slices. The present results reconcile these differences by showing that the amount of thalamocortical cooperativity that can be achieved by stimulating within the thalamus in slices is limited compared with in vivo, but this limitation can be overcome using optogenetics. By expressing ChR2 in thalamocortical fibers, one need not worry about whether thalamocortical axons are intact between thalamus and neocortex because thalamocortical synapses can be specifically recruited by applying light in the neocortex. The in vivo-like thalamocortical responses driven by optogenetics in slices reveal that thalamocortical activity behaves much like intracortical activity to drive up-states; the stronger the synaptic cooperativity mediated by fast excitation, the more effective suppression of up-states. Also, stronger cooperativity mediated by fast excitation inhibits up-states because it drives strong feedforward inhibition. Blocking fast excitation relieves feedforward inhibition, unmasking robust up-states purely mediated by slow, NMDA receptor, excitation.
Functional relevance
At a functional level, the present results indicate that up-states are most effectively driven by sparse synaptic inputs resulting from limited cooperativity. In contrast, strong inputs resulting from high cooperativity will tend to suppress up-states. The activity from a single cortical cell, which obviously produces low intracortical cooperativity, is apparently capable of driving changes in cortical network state in vivo (Li et al., 2009). We are not aware of in vivo studies that have investigated the effect of thalamocortical (or sensory) cooperativity on the ability to trigger cortical up-states; studies have focused instead on how up-states affect afferent inputs (for reviews, see Castro-Alamancos, 2009; Harris and Thiele, 2011). However, in the somatosensory (barrel) cortex, up-states should probably be most effectively driven by low-velocity (slow) whisker deflections that recruit fewer synchronous thalamocortical cells than high-velocity (fast) deflections (Temereanca and Simons, 2003). Indeed, fast whisker deflections (high thalamocortical cooperativity) are well known to evoke sharp short-latency cortical responses truncated by robust IPSPs, lasting for hundreds of milliseconds and devoid of up-states (Hirata and Castro-Alamancos, 2008, 2011), just like robust electrical stimulation of the thalamus (see above).
The fact that up-states are most effectively triggered by weak inputs raises questions about their functional roles (Castro-Alamancos, 2009; Harris and Thiele, 2011). For example, tonic but weakly cooperative activity in thalamocortical pathways may serve to drive the neocortex into an activated state (Hirata and Castro-Alamancos, 2010). Conversely, strong cooperativity may serve to stop persistent activity (Shu et al., 2003). Control of network excitability by the cooperativity of synaptic afferents seems like a useful mechanism to rapidly engage and disengage cortical networks between information processing modes as dictated by behavioral state (Castro-Alamancos, 2004a, b; Stoelzel et al., 2009; Harris and Thiele, 2011).
Footnotes
This work was supported by the National Institutes of Health. We thank the Deisseroth Lab for providing us with access to the AAV material.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Correlation between intrinsic firing patterns and thalamocortical synaptic responses of neurons in mouse barrel cortex. J Neurosci. 1992;12:319–329. doi: 10.1523/JNEUROSCI.12-01-00319.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso JM, Swadlow HA. Thalamocortical specificity and the synthesis of sensory cortical receptive fields. J Neurophysiol. 2005;94:26–32. doi: 10.1152/jn.01281.2004. [DOI] [PubMed] [Google Scholar]
- Antic SD, Zhou WL, Moore AR, Short SM, Ikonomu KD. The decade of the dendritic NMDA spike. J Neurosci Res. 2010;88:2991–3001. doi: 10.1002/jnr.22444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno RM, Sakmann B. Cortex is driven by weak but synchronously active thalamocortical synapses. Science. 2006;312:1622–1627. doi: 10.1126/science.1124593. [DOI] [PubMed] [Google Scholar]
- Bruno RM, Simons DJ. Feedforward mechanisms of excitatory and inhibitory cortical receptive fields. J Neurosci. 2002;22:10966–10975. doi: 10.1523/JNEUROSCI.22-24-10966.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Short-term plasticity in thalamocortical pathways: cellular mechanisms and functional roles. Rev Neurosci. 1997;8:95–116. doi: 10.1515/revneuro.1997.8.2.95. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Origin of synchronized oscillations induced by neocortical disinhibition in vivo. J Neurosci. 2000;20:9195–9206. doi: 10.1523/JNEUROSCI.20-24-09195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Absence of rapid sensory adaptation in neocortex during information processing states. Neuron. 2004a;41:455–464. doi: 10.1016/s0896-6273(03)00853-5. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Dynamics of sensory thalamocortical synaptic networks during information processing states. Prog Neurobiol. 2004b;74:213–247. doi: 10.1016/j.pneurobio.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Cortical up and activated states: implications for sensory information processing. Neuroscientist. 2009;15:625–634. doi: 10.1177/1073858409333074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Spatiotemporal properties of short-term plasticity sensorimotor thalamocortical pathways of the rat. J Neurosci. 1996a;16:2767–2779. doi: 10.1523/JNEUROSCI.16-08-02767.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Cellular mechanisms of the augmenting response: short-term plasticity in a thalamocortical pathway. J Neurosci. 1996b;16:7742–7756. doi: 10.1523/JNEUROSCI.16-23-07742.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Thalamocortical synapses. Prog Neurobiol. 1997;51:581–606. doi: 10.1016/s0301-0082(97)00002-6. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Oldford E. Cortical sensory suppression during arousal is due to the activity-dependent depression of thalamocortical synapses. J Physiol. 2002;541:319–331. doi: 10.1113/jphysiol.2002.016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Rigas P. Synchronized oscillations caused by disinhibition in rodent neocortex are generated by recurrent synaptic activity mediated by AMPA receptors. J Physiol. 2002;542:567–581. doi: 10.1113/jphysiol.2002.019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Rigas P, Tawara-Hirata Y. Resonance (approximately 10 Hz) of excitatory networks in motor cortex: effects of voltage-dependent ion channel blockers. J Physiol. 2007;578:173–191. doi: 10.1113/jphysiol.2006.119016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW, Malenka RC, Silva LR. Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol. 1988;406:443–468. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Kavalali ET. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol. 2009;101:2290–2296. doi: 10.1152/jn.90754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero M, Varghese G, Castro-Alamancos MA. The state of somatosensory cortex during neuromodulation. J Neurophysiol. 2012;108:1010–1024. doi: 10.1152/jn.00256.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Harris KD, Thiele A. Cortical state and attention. Nat Rev Neurosci. 2011;12:509–523. doi: 10.1038/nrn3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata A, Castro-Alamancos MA. Relief of synaptic depression produces long-term enhancement in thalamocortical networks. J Neurophysiol. 2006;95:2479–2491. doi: 10.1152/jn.01145.2005. [DOI] [PubMed] [Google Scholar]
- Hirata A, Castro-Alamancos MA. Cortical transformation of wide-field (multiwhisker) sensory responses. J Neurophysiol. 2008;100:358–370. doi: 10.1152/jn.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata A, Castro-Alamancos MA. Neocortex network activation and deactivation states controlled by the thalamus. J Neurophysiol. 2010;103:1147–1157. doi: 10.1152/jn.00955.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata A, Castro-Alamancos MA. Effects of cortical activation on sensory responses in barrel cortex. J Neurophysiol. 2011;105:1495–1505. doi: 10.1152/jn.01085.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter ED, Kapur A, Haberly LB. A dendritic GABAA-mediated IPSP regulates facilitation of NMDA-mediated responses to burst stimulation of afferent fibers in piriform cortex. J Neurosci. 1996;16:307–312. doi: 10.1523/JNEUROSCI.16-01-00307.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Raghavachari S. Gating information by two-state membrane potential fluctuations. J Neurophysiol. 2007;97:3015–3023. doi: 10.1152/jn.01242.2006. [DOI] [PubMed] [Google Scholar]
- Li CY, Poo MM, Dan Y. Burst spiking of a single cortical neuron modifies global brain state. Science. 2009;324:643–646. doi: 10.1126/science.1169957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Shu Y, Hasenstaub A, Sanchez-Vives M, Badoual M, Bal T. Persistent cortical activity: mechanisms of generation and effects on neuronal excitability. Cereb Cortex. 2003;13:1219–1231. doi: 10.1093/cercor/bhg104. [DOI] [PubMed] [Google Scholar]
- Metherate R, Cox CL, Ashe JH. Cellular bases of neocortical activation: modulation of neural oscillations by the nucleus basalis and endogenous acetylcholine. J Neurosci. 1992;12:4701–4711. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KD, Chapman B, Stryker MP. Visual responses in adult cat visual cortex depend on N-methyl-D-aspartate receptors. Proc Natl Acad Sci. 1989;86:5183–5187. doi: 10.1073/pnas.86.13.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic BA, Radojicic MS, Antic SD. A strict correlation between dendritic and somatic plateau depolarizations in the rat prefrontal cortex pyramidal neurons. J Neurosci. 2005;25:3940–3951. doi: 10.1523/JNEUROSCI.5314-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldford E, Castro-Alamancos MA. Input-specific effects of acetylcholine on sensory and intracortical evoked responses in the “barrel cortex” in vivo. Neuroscience. 2003;117:769–778. doi: 10.1016/s0306-4522(02)00663-2. [DOI] [PubMed] [Google Scholar]
- Rigas P, Castro-Alamancos MA. Thalamocortical up states: differential effects of intrinsic and extrinsic cortical inputs on persistent activity. J Neurosci. 2007;27:4261–4272. doi: 10.1523/JNEUROSCI.0003-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigas P, Castro-Alamancos MA. Impact of persistent cortical activity (up states) on intracortical and thalamocortical synaptic inputs. J Neurophysiol. 2009;102:119–131. doi: 10.1152/jn.00126.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Mattia M, Compte A, Perez-Zabalza M, Winograd M, Descalzo VF, Reig R. Inhibitory modulation of cortical up states. J Neurophysiol. 2010;104:1314–1324. doi: 10.1152/jn.00178.2010. [DOI] [PubMed] [Google Scholar]
- Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993a;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993b;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Stoelzel CR, Bereshpolova Y, Swadlow HA. Stability of thalamocortical synaptic transmission across awake brain states. J Neurosci. 2009;29:6851–6859. doi: 10.1523/JNEUROSCI.5983-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA. Barrel cortex microcircuits: thalamocortical feedforward inhibition in spiny stellate cells is mediated by a small number of fast-spiking interneurons. J Neurosci. 2006;26:1219–1230. doi: 10.1523/JNEUROSCI.4727-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temereanca S, Simons DJ. Local field potentials and the encoding of whisker deflections by population firing synchrony in thalamic barreloids. J Neurophysiol. 2003;89:2137–2145. doi: 10.1152/jn.00582.2002. [DOI] [PubMed] [Google Scholar]
- Wang H, Peca J, Matsuzaki M, Matsuzaki K, Noguchi J, Qiu L, Wang D, Zhang F, Boyden E, Deisseroth K, Kasai H, Hall WC, Feng G, Augustine GJ. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci. 2007;104:8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3:785–792. doi: 10.1038/nmeth936. [DOI] [PubMed] [Google Scholar]
- Zhang F, Aravanis AM, Adamantidis A, de Lecea L, Deisseroth K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8:577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]