

Abstract
In the mouse model of Fragile X syndrome, the Fmr1 knock-out, local excitation of layer 4 fast-spiking (FS) inhibitory neurons is robustly decreased by 50%, but the mechanisms mediating this change are unknown. Here, we performed recordings in acutely prepared slices obtained from Fmr1 “mosaic” mice, where Fmr1 is deleted in about half of all neurons, and we found that loss of presynaptic, but not postsynaptic, Fmr1 fully recapitulates the deficit. The change in connection strength is primarily due to a decrease in release probability indicating that FMRP normally positively regulates these processes. This change in presynaptic neurotransmitter release is observed both in the mosaic mice and in the constitutive Fmr1 knock-out mice. Manipulations in release probability enabled both the mimic and rescue of the impaired function in this synaptic pathway. Loss of presynaptic Fmr1 has no effect on excitatory synapses onto excitatory neurons, indicating a target cell-specific function for presynaptic FMRP. Finally, we demonstrate that the excitation decrement onto FS neurons also exists in layer 5 of the Fmr1 knock-out, suggesting a widespread role for presynaptic Fmr1 in the excitation of inhibitory neurons. In summary, we identify a novel function for presynaptic FMRP in promoting presynaptic neurotransmitter release, and we show that loss of this function accounts for impaired excitation of neocortical FS inhibitory neurons. These changes may contribute to the cognitive dysfunction and circuit hyperexcitability associated with Fragile X syndrome, including patients with complete deletion of FMRP and those with mosaic expression of FMRP.
Introduction
Fragile X syndrome (FXS) is the leading cause of intellectual disability and the most common monogenic form of Autism (Bassell and Warren, 2008). It is caused by the transcriptional silencing of FMR1, which is located on the X-chromosome. FMR1 codes for FMRP, an RNA-binding protein that is involved in the translational regulation of its mRNA targets. Patients with FXS present with cognitive and social deficits, seizures, sensory hypersensitivity, and hyperactivity (Berry-Kravis, 2002; Hagerman et al., 2009). Many of these impairments are reproduced in the FXS mouse model, the Fmr1 knock-out (KO) mouse (Musumeci et al., 2000; Nielsen et al., 2002; Spencer et al., 2005). FXS not only includes patients who lack FMRP in all cells, but also those who are “mosaic” in FMRP expression (Hagerman et al., 2009).
Alterations in synaptic plasticity and synaptic structure have been observed in FXS patients and in Fmr1 KO mice (Pfeiffer and Huber, 2009). Also, “baseline” synaptic function and connectivity measured electrophysiologically are altered in acute cortical slices obtained from Fmr1 KO mice (Bureau et al., 2008; Gibson et al., 2008; Harlow et al., 2010; Olmos-Serrano et al., 2010; Testa-Silva et al., 2012). But the mechanisms underlying these baseline functional alterations remain completely unknown.
FMRP has both presynaptic and postsynaptic function in invertebrates (Zhang et al., 2001; Till et al., 2010). To date, most work in rodents has focused on postsynaptic mechanisms (Pfeiffer and Huber, 2009). But FMRP is also expressed in presynaptic structures (Antar et al., 2006; Christie et al., 2009; Till et al., 2012), and its mRNA targets include approximately one-third of the presynaptic proteome (Darnell et al., 2011). Little is known about its presynaptic FMRP function. Studies in the Fmr1 KO mouse indicate it may regulate neurotransmitter release (Deng et al., 2011; Klemmer et al., 2011). A study in slice culture demonstrated that hippocampal CA3 neurons without presynaptic FMRP had a reduced connection probability onto neighboring neurons (Hanson and Madison, 2007). But for all of these reported changes, their mechanisms and their relevance to circuit connectivity alterations in the Fmr1 KO remain unknown.
We previously demonstrated a 50% decrease in local excitation onto neocortical fast-spiking (FS) inhibitory neurons (Gibson et al., 2008). This deficit persists until at least 1 month of age. FS neurons represent ∼50% of all cortical inhibitory neurons, and through the feedback inhibition they provide, they decrease the rate and increase the synchrony of spiking in postsynaptic target neurons (Bartos et al., 2001; Galarreta and Hestrin, 2001; Atallah et al., 2012). Consistent with an impairment in the excitation and activation of FS neurons, we have observed both longer and less synchronous persistent activity states in the neocortex of the Fmr1 KO (Gibson et al., 2008; Hays et al., 2011). Similarly, decreased synchrony and inhibitory tone could underlie cognitive disabilities, sensory hypersensitivity, and epilepsy associated with FXS (Berry-Kravis, 2002; Uhlhaas and Singer, 2006).
Here, we examine the mechanisms underlying the decrease in excitation of FS inhibitory neurons and find that it is entirely due to presynaptic deletion of Fmr1. The deficit is primarily due to a decrease in presynaptic glutamate release. We make these observations with simultaneous recordings of presynaptic and postsynaptic neurons, which provide precise measurements of cell-to-cell synaptic function and connectivity in an isolated synaptic pathway.
Materials and Methods
Mice.
Congenic Fmr1 KO mice on the C57BL/6 background were originally obtained from Dr. S. Warren (Emory University, Atlanta, GA) (Dutch-Belgian Fragile X Consortium, 1994). X-linked GFP mice were originally composed of a mixed genetic background (Jackson Laboratory) (Hadjantonakis et al., 2001), but we have backcrossed them onto the C57BL/6 background for at least four generations. GFP/Fmr1 mosaic females did not express FMRP in approximately half of all neurons due to X inactivation and were made by breeding X-linked GFP males with Fmr1 KO females. G42 mice expressed GFP in a subset of parvalbumin-expressing neurons and are comprised of a mixed C57BL/6 and CB6F1/J background (J. Huang, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) (Chattopadhyaya et al., 2004). To control for genetic and litter variability, only within-litter comparisons were used for comparing Fmr1 KO versus wild-type (WT) data. The Fmr1 KO phenotype examined in this study—the decrease in excitation of FS inhibitory neurons—is robust to various genetic mouse strains and breeding strategies, because between this study and a previous study (Gibson et al., 2008), the phenotype has been observed in three different mouse breeding paradigms involving three different background strains. For all Fmr1 KO experiments, we used males, since this sex was used in our previous study (Gibson et al., 2008), and this makes littermate comparisons of KOs versus WTs more feasible.
Electrophysiology.
Mice [postnatal day 13 (P13)–P16] were anesthetized with Euthasol and the brains removed following protocols approved by the University of Texas Southwestern. Thalamocortical slices, 350 μm thick, were cut at ∼4°C in dissection buffer, placed in normal artificial CSF (ACSF) at 35°C for 30 min, and slowly cooled to 21°C over the next 30 min. This slice preparation preserves thalamic input to the barrel cortex, interlayer projections within neocortex, as well as the local connectivity in all layers, and therefore it is a good preparation to examine circuit changes in mutant mice (Agmon and Connors, 1991). In the barrel field of somatosensory cortex, whole-cell recordings were performed in either layer 4 (inside a barrel) or layer 5 using infrared-differential interference contrast visualization and an Olympus FV300 confocal microscope. The use of a confocal microscope enabled more reliable identification of GFP-negative neurons since background GFP fluorescence was high (∼50% of cells expressed GFP). Recordings were performed at 21°C (see Figs. 1–6) and at 32°C (see Figs. 7–9). Data were collected with a 10 kHz sampling rate and a 3 kHz Bessel filter.
Figure 1.
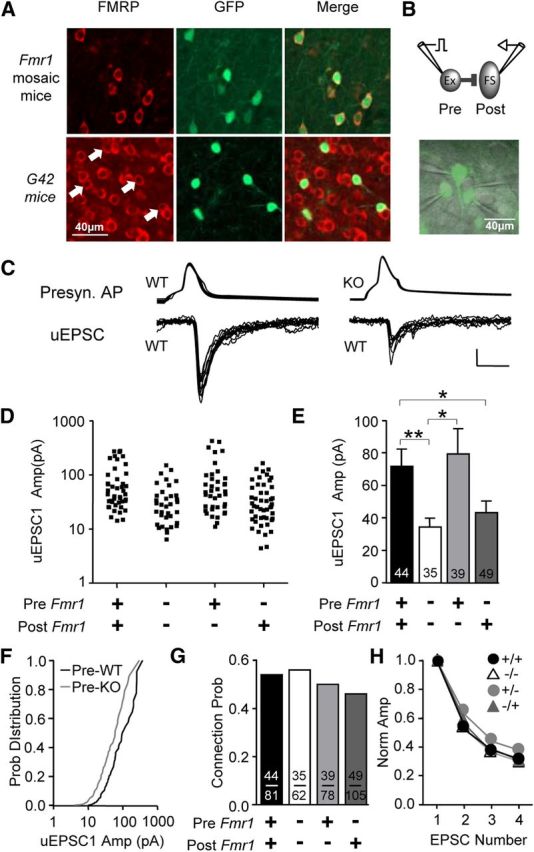
Presynaptic deletion of Fmr1 in mosaic mice results in decreased unitary excitation of FS inhibitory neurons. A, Immunohistochemistry for GFP and FMRP in somatosensory neocortex at P14. Top, In GFP/Fmr1 mosaic mice, GFP marks neurons that express FMRP, while lack of GFP indicates no FMRP. Bottom, In WT mice expressing GFP in parvalbumin-positive FS neurons, FMRP colocalizes with GFP indicating that FS neurons express FMRP (arrows). B, Top, Diagram of glutamatergic connection examined. Bottom, In live mosaic slices, GFP-positive and GFP-negative neurons were clearly identified. C, Example traces obtained from simultaneous recordings show that uEPSCs observed in FS neurons and evoked from presynaptic KO excitatory neurons were smaller. Calibration: 50 mV, 20 pA; 5 ms. D, E, Scatter plot and bar graph showing that average uEPSC1 amplitude was smaller whenever Fmr1 was deleted from the presynaptic excitatory neuron. F, Cumulative distribution of uEPSC1 amplitude for merged presynaptic KO and merged presynaptic WT groups. G, No detectable change in connection probability was observed. H, Average normalized uEPSC amplitude during the train of four presynaptic action potentials was unchanged. *p < 0.05; **p < 0.01.
Figure 2.

Unitary excitation of postsynaptic excitatory neurons was not affected by Fmr1 deletion in mosaic mice. A, Top, Diagram of the connection examined. Bottom, Example traces of simultaneous recordings of excitatory neurons. Calibration: 50 mV, 10 pA; 5 ms. B, C, Scatter plot and bar graph showing that average uEPSC1 amplitude was not affected by Fmr1 deletion. D, Connection probability was unchanged. E, Average normalized uEPSC amplitude during the train of 4 presynaptic action potentials was unchanged.
Figure 3.

Increases in the CV and unitary failures with presynaptic Fmr1 deletion indicate quantal content is decreased. A, Left, middle, Example uEPSC1s from WT/WT and KO/WT pairs. Right, Same KO/WT traces scaled to match average WT/WT uEPSC1 and to highlight variability. Black traces are averages. B, Plot of CV versus genotypic combination of pre- and postsynaptic neurons. CV is higher with presynaptic deletion of Fmr1. C, Examples of unitary “failures” from a KO/WT pair. D, The proportion of unitary failures was higher in KO/WT pairs compared to WT/WT pairs. E, Scatter plot of CV versus uEPSC1 amplitude. F, Data from E merged into presynaptic KO versus presynaptic WT groups and binned. *p < 0.05; **p < 0.01. Individual uEPSC traces in A and C are a running average of three samples. Calibrations: 20 pA, 2 ms.
Figure 4.

Quantal amplitude does not depend on presynaptic Fmr1 expression. A–D, Examples of uEPSCs before (A, B) and after (C, D) wash-in of CdCl2 to lower the probability of release to the point where it is assumed that isolated single quanta are observed. Note the decreased uEPSC amplitude and increased failures after wash-in. Black and gray lines indicate successes and failures, respectively. Calibrations: (in A) A, B, 50 mV, 20 pA; 2 ms; (in C) C, D, 10 pA, 2 ms. E, uEPSCs were averaged during CdCl2 application (excluding failures) to obtain an estimate of the average quantal amplitude. This was unchanged. F, uEPSC amplitude before wash-in was smaller in the KO/WT pairs in these experiments. *p < 0.05. uEPSC traces are a running average of three samples.
Figure 5.
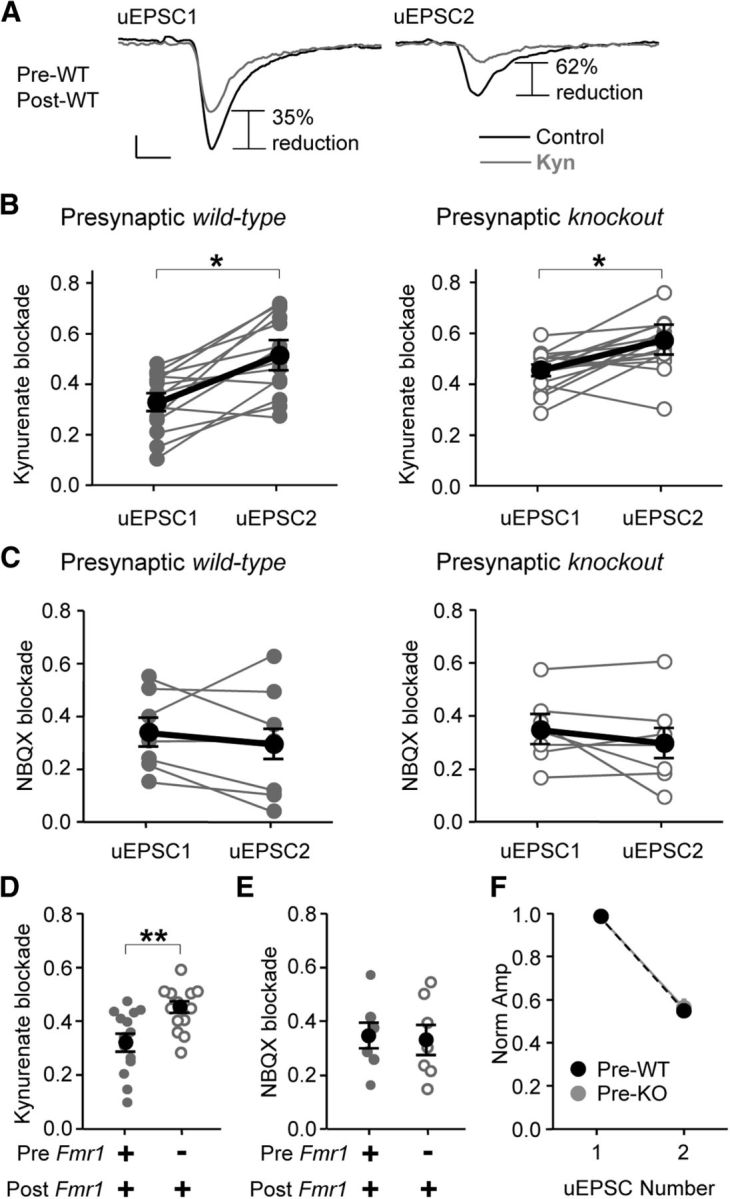
Presynaptic Fmr1 deletion causes reduced multivesicular release. A, Example average traces obtained from a WT/WT pair demonstrating the effect of the AMPAR competitive antagonist, kynurenate (Kyn, 250 μm). Note the greater blockade of uEPSC2 compared to blockade of uEPSC1, indicating multivesicular release. Calibration: 20 pA, 2 ms. B, Paired line plots showing Kyn blockade for uEPSC1 and uEPSC2 with presynaptic WT (left) and presynaptic KO expression (right). Dark lines indicate average data. C, Paired line plots showing blockade by the noncompetitive AMPAR antagonist NBQX (75 nm). NBQX had uniform affects across uEPSC number. D, Blockade by Kyn of uEPSC1 was greater with presynaptic KO, indicating less multivesicular release. E, Blockade by NBQX of uEPSC1 was not different with presynaptic KO. F, No change in short-term plasticity as observed by the average normalized uEPSC amplitude during the paired pulse train. *p < 0.05; **p < 0.01.
Figure 6.

Presynaptic Fmr1 deletion causes reduced release probability. A, Individual examples of use-dependent NMDAR blockade by MK-801. Curves are best fits of the data to single exponential decay functions. Inset, Average NMDAR-mediated uEPSCs acquired during baseline measurement. Calibration: 10 pA, 40 ms. B, Binned and averaged responses from each individual experiment were then averaged across all experiments (see Materials and Methods). Four intervals are higher in the KO data (two-way ANOVA followed by multiple comparisons; WT, n = 8; KO, n = 6). C, The average time constant derived from each individual experiment is longer with presynaptic Fmr1 deletion. *p < 0.05; **p < 0.01.
Figure 7.

The presynaptic Fmr1 deletion phenotype can be mimicked with a decrease in release probability in presynaptic WT pairs. A, Left, Example average traces before and after 4 μm CdCl2. Right, We obtained the relationship between CdCl2 (Cd+2) concentration and blockade of uEPSC1. Data were fitted to a sigmoid curve. Dashed lines mark the [Cd+2] (2.6 μm) that corresponds to the average fractional decrease of uEPSC1 amplitude found for presynaptic Fmr1 deletion (0.40). B, Example average traces obtained from a WT/WT pair in the presence of 2.6 μm CdCl2 before and after kynurenate (Kyn; 250 μm) application. C, Average blockade by kynurenate with 2.6 μm CdCl2 closely matched the blockade for presynaptic KO pairs. D, No change in short-term plasticity with Cd2+ application. WT/WT and KO/WT data in C and D are replotted from Figure 5. *p < 0.05. Calibrations: 20 pA, 2 ms.
Figure 8.

The presynaptic Fmr1 deletion phenotype can be rescued with an increase in release probability in presynaptic KO pairs. A, Left, Example average traces before and after 6 mm CaCl2. Right, We obtained the relationship between CaCl2 (Ca+2) concentration and increase of uEPSC1 amplitude. Data were fitted to a sigmoid curve. Dashed lines mark the [Ca+2] (6 mm) that corresponds to the average fractional increase of uEPSC1 amplitude found for WT/WT pairs over KO/WT pairs (0.62). B, Example average traces obtained from a KO/WT pair in the presence of 6 mm CaCl2 before and after kynurenate (250 μm) application. C, Average blockade by kynurenate with 6 mm CaCl2 closely matched the blockade for presynaptic WT pairs. D, More short-term depression in high Ca2+. KO/WT and WT/WT data in C and D are replotted from Figure 5. *p < 0.05. Calibrations: 20 pA, 2 ms.
Figure 9.
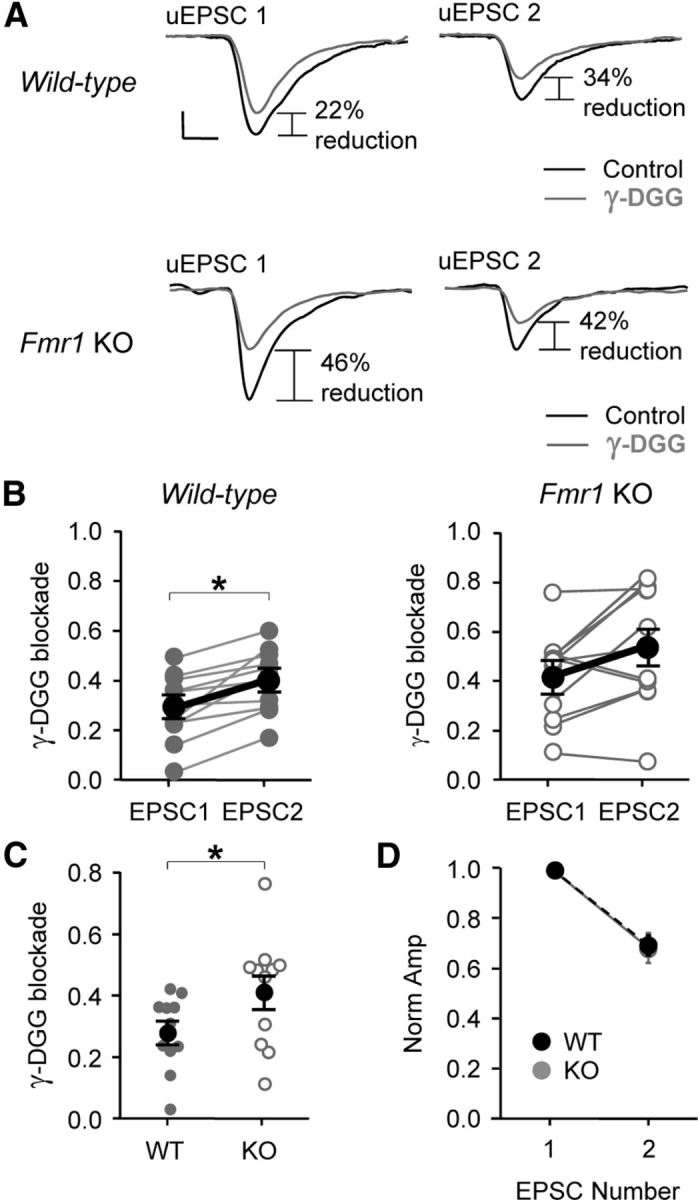
Decreased multivesicular release exists in the constitutive Fmr1 KO. A, Example average traces from Fmr1 WT and KO slices demonstrating the blockade caused by the AMPAR competitive antagonist, γ-DGG (500 μm). Note the greater blockade at uEPSC2 when compared to blockade at uEPSC1, indicating multivesicular release. B, Paired line plots showing blockade for uEPSC1 and uEPSC2 in Fmr1 WT (left) and KO slices (right). Dark lines indicate averaged data. Calibrations: 20 pA, 2 ms. C, Average blockade for uEPSC1 was greater in KO slices indicating reduced multivesicular release. D, No difference observed in short-term plasticity. *p < 0.05.
Layer 4 FS cells were identified by their large size and nonadapting, high firing rates (Connors and Gutnick, 1990; Gibson et al., 1999). The rare occurrence of FS neurons necessitated a high number of both Fmr1 WT and KO neurons found in GFP/Fmr1 mosaic females since we needed to find FS/excitatory pairs in the four possible Fmr1 expression configurations that were close in distance where the probability of connection is highest. Layer 5 FS neurons in the Fmr1 KO line were identified by GFP fluorescence by cross-breeding with the G42 mouse line.
Electrophysiology solutions.
The ACSF contained the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 25 dextrose, and 2 CaCl2. All slices were prepared in a sucrose dissection buffer containing the following (in mm): 75 sucrose, 87 NaCl, 3 KCl, 1.25 NaH2PO4, 7 MgSO4, 26 NaHCO3, 20 dextrose, and 0.5 CaCl2. All solutions were pH 7.4. ACSF was saturated with 95% O2/5% CO2. The pipette solution consisted the following (in mm): 130 K-gluconate, 6 KCl, 3 NaCl, 10 HEPES, 0.2 EGTA, 4 ATP-Mg, 0.3 GTP-Tris, 14 phosphocreatine-Tris, and 10 sucrose. This was adjusted to pH 7.25 and 290 mOsm. The junction potential was ∼10 mV and was not corrected.
Unitary PSCs.
Unitary EPSCs and IPSCs (uEPSCs and uIPSCs) were measured at a holding potentials of −60 and −55 mV, respectively. For data in Figures 1–3, a train of four pulses was applied once every 10 s. For data in Figures 7–9, a train of two pulses was applied once every 15 s. Average uPSC amplitude was always determined from the first uPSC in the train (uPSC1). A connection was considered to exist when the average amplitude was >2 pA. Individual presynaptic action potentials were elicited with an 8 ms depolarizing current step (generally 600 pA). Short-term plasticity analysis was performed when the average uPSC1 was (1) >10 pA in layer 4 FS neurons and (2) >5 pA in layer 5 FS neurons and layer 4 excitatory neurons. An experiment was analyzed if the postsynaptic recording of the inhibitory neuron met the following criteria: series resistance was <20 MΩ, and both series and input resistance did not deviate by >25% during the recording. Criteria for excitatory neuron recordings were the same, except series resistance was less that 40 MΩ.
The coefficient of variation (CV) was calculated as the square root of (VAREPSC1 − VARbaseline) divided by the meanEPSC1, where VAREPSC1 and VARbaseline are the variance of uEPSC1 and baseline noise respectively, and meanEPSC1 is the mean of uEPSC1 (Faber and Korn, 1991; Markram et al., 1997). Only responses >10 pA were used to avoid significant contamination by noise. uEPSC amplitude measurements <1.6 times root mean square noise were considered “failures.” CV and failures were measured on traces further filtered by a three-point running average, which had no detectable impact on response amplitude.
For Figures 7A and 8A, the fitted sigmoid function was a three-parameter Hill function (SigmaPlot; Systat Software).
Cd2+ experiments measuring quantal amplitude.
Experiments were performed in 5 μm CdCl2. As in a previous study (Gibson et al., 2009), long action potential trains were applied, and the number (10–30) of action potentials was modified at a frequency of 20 Hz to reduce the percent of action potentials evoking a uEPSC to <20%. These uEPSCs are referred to as “successes” and were only analyzed if they occurred within ±0.3 ms of the average latency. The successes are considered to be “putative” quantal events. Systematic error in average quantal amplitude measurements was calculated in a previous study, where the overestimate in quantal amplitude (q) was dependent on the number of release sites, ranging from 0% for 1 release site to 18% for 10 release sites, and the error in the relative difference in q between WT/WT and KO/WT pairs would be <5% (Gibson et al., 2009). This analysis was performed on traces further filtered by a three-point running average.
MK-801 experiments.
This protocol is based on previous studies (Rosenmund et al., 1993; Castro-Alamancos and Connors, 1997). All train intervals were 10 s, and the following compounds were part of a modified ACSF to isolate and promote NMDAR-mediated currents: 20 μm DNQX (Sigma), 3 mm Ca2+, 0 mm Mg2+, and 5 μm glycine (Sigma). The postsynaptic FS neuron was held at −50 mV. An initial train of eight uEPSCs was acquired to obtain an average baseline uEPSC amplitude. Then MK-801 (40 μm) was applied. After a 10 min wait to allow the compound to completely permeate the slice, a train of 75 uEPSCs was acquired. uEPSC amplitudes were mediated by NMDARs and averaged around 15 pA. All uEPSC amplitudes during MK-801 were normalized to the average baseline amplitude. We then performed two sets of analyses. First, we fit a single exponential decay to the data obtained from each experiment (Fig. 5A), and then compared the time constant for these decays between WTs and KOs. Fitting was performed in SigmaPlot 12 using the Levenberg–Marquardt method. Second, we binned and averaged the responses in each experiment based on the following intervals (in ms): 1–5, 6–16, and every interval of 10 up to 75 (Fig. 5B). Then we averaged this data across experiments and performed a two-way ANOVA (presynaptic genotype by time interval), with the time interval dimension being repeated measures. We examined the differences between WTs and KOs at each interval to confirm the changes in time constant derived for each experiment individually.
Immunocytochemistry.
Mice (P14–P20) were anesthetized with Euthasol and transcardially perfused following protocols approved by the University of Texas Southwestern. The perfusion rate was 1.7 ml/min and began with ice cold saline for 1 min followed by 15 min of 4% paraformaldehyde. The brain was then removed and postfixed in 4% paraformaldehyde for 30 min. We subsequently followed procedures described previously, which included antigen retrieval for FMRP staining (Hays et al., 2011). Antibodies were for GFP (1:1500, chick; Aves Labs, #GFP-1020; secondary, Alexa488; Invitrogen) and FMRP (1:200, mouse, 2F5; gift from J. Darnell, Rockefeller University, New York, NY; secondary, Alexa555; Invitrogen). Specificity for the FMRP antibody has been described previously (Hays et al., 2011). Images were collected on an inverted Zeiss LSM confocal microscope.
Drugs.
For quantal and wild-type mimic experiments, we reduced release probability using cadmium chloride (CdCl2; Sigma). Quantal experiments also contained the GABAA receptor antagonist picrotoxin (50 μm; Sigma) and the NMDA receptor antagonist AP5 [d-2-amino-5-phosphonovaleric acid; 50 μm; Sigma]. For multivesicular release experiments, we used kynurenic acid (Sigma), γ-d-glutamylglycine (γ-DGG; Tocris Bioscience) and NBQX (Sigma). For miniature EPSC experiments, action potentials were blocked with tetrodotoxin (1 μm; Sigma), and GABAA receptors were blocked with picrotoxin.
Statistics.
All statistics were performed using SigmaPlot (Systat Software), and statistical significance was at p < 0.05. Unless stated otherwise, we used the nonparametric unpaired t test (Mann–Whitney) when comparing two groups. For more than two groups, Kruskal–Wallis one-way ANOVA on ranks was performed followed by a post hoc comparisons test (Dunn's method). Sample number (n) is the cell or synaptically connected pair number. Data are represented by the mean ± SEM. Connectivity frequency changes were detected using the χ2 test, and a Fishers exact p value was used to determine significance.
Results
Excitation of fast-spiking neurons decreases with presynaptic Fmr1 deletion
We used GFP/Fmr1 mosaic females where cells either coexpress GFP and FMRP or express neither, which we refer to as Fmr1 WT and Fmr1 KO neurons, respectively. FMRP is expressed in layer 4 barrel cortex at this age (Harlow et al., 2010). However, we wanted to validate that GFP expression in GFP/Fmr1 mosaic mice faithfully reports FMRP expression. Immunohistochemistry indicated that GFP reliably reported FMRP expression in the neocortex at the ages examined in this study (Fig. 1A): 159 of 159 GFP-positive (GFP+) neurons were also FMRP-positive (FMRP+), and conversely, 159 of 168 FMRP+ neurons were GFP+. We next determined whether FMRP is expressed in parvalbumin-positive inhibitory neurons, since these are the biochemical identity of the FS neurons we examines in this study (Cauli et al., 1997; Gibson et al., 1999). Using the G42 mouse line, which selectively expresses GFP in parvalbumin-positive neurons, we found that 54 of 55 GFP+ neurons in the neocortex also express FMRP (in layer 4, 17 of 17; Fig. 1A).
We performed simultaneous whole-cell recordings of presynaptic excitatory and postsynaptic FS inhibitory neurons where we knew the genotype of all neurons (GFP indicating Fmr1 WT; Fig. 1B). Up to four neurons could be recorded simultaneously. We examined uEPSCs in FS neurons in response to action potentials evoked in a single presynaptic excitatory neuron (Fig. 1C). We compared the amplitude of the first uEPSC in a train (uEPSC1; 4 at 20 Hz) between all four groups of presynaptic and postsynaptic genotypic combinations and found that Fmr1 deletion in the presynaptic excitatory neuron greatly decreased uEPSC1 amplitude (Fig. 1D,E). The two presynaptic KO groups were both significantly different from the WT/WT (pre/post) group (p < 0.05 and p < 0.05; WT/WT, KO/KO, and KO/WT, 71.8 ± 10.1, 34.4 ± 5.1, and 43.1 ± 7.2 pA; n = 44, 35, and 49, respectively), and the average amplitude of both presynaptic KO groups merged was only 54% of the WT/WT group. On the other hand, no difference was detected with postsynaptic Fmr1 deletion (71.8 ± 10.1, 79.2 ± 15.5; WT/WT vs WT/KO; n = 46 and 39). The difference between presynaptic Fmr1 KO and WT groups represented a uniform shift in the distribution as indicated by the cumulative probability distribution (Fig. 1F). If we restrict our analysis to experiments in which we recorded presynaptic KO and WT neurons simultaneously and both had connections to the same WT postsynaptic FS neuron, we observe the same decrement with presynaptic Fmr1 deletion (p < 0.05; 117.5 ± 26.2, 50.8 ± 9.2 pA; WT/WT and KO/WT, n = 10 and 10, paired t test), indicating that our results are not due to heterogeneity of FS neurons or our biases in targeting FS neurons.
Both the probability of the occurrence of functional connections and short-term depression during the stimulus train were not detectably different among the four groups (Fig. 1G,H). No changes in the following uEPSC1 properties were observed: rise time (WT/WT, KO/KO, WT/KO, and KO/WT, 0.66 ± 0.1, 0.60 ± 0.1, 0.68 ± 0.1, and 0.66 ± 0.2 ms; n = 24, 17, 16, and 25, respectively), decay time-constant (6.8 ± 0.6, 6.0 ± 0.6, 5.9 ± 0.4, and 6.5 ± 0.4 ms; n = 24, 17, 16, and 25), or onset latency (0.97 ± 0.04, 1.08 ± 0.05, 0.96 ± 0.06, and 0.93 ± 0.03 ms; n = 29, 30, 25, and 23). If we quantify connection probability in all WT/WT and KO/WT pairs in this study, the higher sample number allows us to detect an 18% decrease with presynaptic Fmr1 deletion (p < 0.05, 66 vs 54%; WT/WT, 85 of 129; KO/WT, 86 of 160; does not include MK-801 experiments). We did not perform control experiments testing the effect of GFP alone in GFP mosaic mice since a previous study using the same GFP/Fmr1 mosaic strategy to study synaptic transmission performed this control and observed no effect on synaptic transmission between hippocampal pyramidal neurons (Hanson and Madison, 2007).
To determine whether presynaptic deletion of Fmr1 selectively affected excitation of FS neurons, we also examined the local excitation onto postsynaptic excitatory neurons in the GFP/Fmr1 mosaic mice (Fig. 2A). In this instance, we did not detect any effect on uEPSC1 amplitude or connection probability under any combination of Fmr1 deletion (Figs. 2B–E). Therefore, while the decrease in excitation is due to Fmr1 deletion in presynaptic excitatory neurons, this decrease is postsynaptic specific and occurs only at FS inhibitory neurons. These results implicate a target cell-specific function for presynaptic Fmr1.
We next examined the role of selective presynaptic and postsynaptic deletion of Fmr1 on local inhibitory synaptic function of FS neurons. Similar to what we observed in the complete Fmr1 KO (Gibson et al., 2008), we did not detect any changes in uIPSCs originating from FS neurons as a function of presynaptic or postsynaptic deletion of Fmr1 (WT/WT, KO/KO, WT/KO, and KO/WT for excitatory/FS neurons, amplitude, 14.2 ± 2.4, 16.1 ± 3.1, 16.4 ± 3.5, and 13.0 ± 1.4 pA; n = 16, 17, 25, and 15; connection probability, 34, 33, 31, and 26%; n = 47, 51, 80, and 57, respectively). These results suggest that the cell autonomous role of presynaptic Fmr1 is specific for excitatory synaptic transmission.
The results concerning both uEPSCs and uIPSCs very closely follow those found in the constitutive Fmr1 KO mouse where the identical synaptic connections have been examined (Gibson et al., 2008). On the other hand, the increased membrane excitability found in L4 excitatory neurons in the Fmr1 KO (Gibson et al., 2008) was not observed in KO neurons in the mosaic. No changes in input resistance (WT, 594 ± 42 MΩ; KO, 592 ± 31 MΩ), resting potential (WT, −63 ± 1 mV; KO, −64 ± 1 mV), the number of action potentials evoked with 15 and 30 pA steps (data not shown), or threshold current to evoke an action potential (WT, 30 ± 4 pA; KO 32 ± 3 pA) were observed in excitatory neurons (WT, n = 46; KO, n = 47). Similarly, no changes were observed in FS neurons (n = 63 and 68; data not shown). Therefore, while we cannot rule out a role for sex since the Fmr1 KO data were collected from males, changes in intrinsic membrane properties in the constitutive Fmr1 KO appear to not be cell autonomous.
Quantal content is decreased with presynaptic Fmr1 deletion
To determine the mechanism underlying the decrease in uEPSC size, we used a simple classical model of neurotransmission that defines a unitary response: uEPSC amplitude = m * q, where m is the quantal content (the total number of quanta released over all synapses), and q is the average quantal size over all synapses (Johnston and Wu, 1995). Therefore, a decrease in uEPSC amplitude may be due to a decrease in either m or q. We first examined quantal content (m). According to the model above, if quantal content is decreased with presynaptic deletion of Fmr1, the CV (see Materials and Methods) of the uEPSC1 amplitude must increase. Indeed, the CV was significantly increased with presynaptic Fmr1 deletion (Fig. 3A,B; WT/WT vs KO/KO, p < 0.05; WT/WT vs KO/WT, p < 0.05; WT/WT, KO/KO, and KO/WT, 0.24 ± 0.01, 0.40 ± 0.03, and 0.42 ± 0.02; n = 28, 22, and 29, respectively), but no difference was detected with postsynaptic deletion (WT/WT, 0.24 ± 0.01, n = 28; WT/KO, 0.25 ± 0.02, n = 23).
We also examined the failure rate of uEPSC1s—the proportion of trials in which a presynaptic action potential evoked no uEPSC1 (Fig. 3C). As expected with decreased quantal content, failure rate was increased with presynaptic deletion (Fig. 3D; p < 0.01; WT/WT vs KO/WT, 0.02 ± 0.01 vs 0.11 ± 0.02; n = 44 and 49, respectively). Finally, the relationship of CV to uEPSC1 amplitude was unchanged with presynaptic deletion (Fig. 3E,F), indicating that the CV change can be best explained by the shift in uEPSC1 amplitude.
While the decrease in uEPSC amplitude with presynaptic deletion of Fmr1 involves a decrease in quantal content, average quantal amplitude (q) may also play a role. To investigate this possibility, in a subset of recordings involving only WT/WT and KO/WT genotypic combinations, we washed in the general Ca2+-channel blocker Cd2+ (5 μm CdCl2) to reduce release probability to a point where a train of presynaptic action potentials (10–30 at 20 Hz; see Materials and Methods) successfully evoked a uEPSC only 20% of the time or less (Fig. 4C,D). Under these conditions, we consider “successes” to be putative quantal events (i.e., the release of a single vesicle) (Gil et al., 1999; Gibson et al., 2009). We found no genotypic differences in success amplitude with application of Cd2+ (Fig. 4E; WT/WT vs KO/WT, 9.9 ± 1.2 vs 10.5 ± 1.5 pA; n = 18 and 14, respectively), even though average uEPSC amplitude before application was decreased with presynaptic Fmr1 deletion (Fig. 4A,B,F). In conclusion, the decrease in uEPSC amplitude is most likely due to a decrease in quantal content and not quantal amplitude (q).
Multivesicular release is decreased with presynaptic Fmr1 deletion
Multivesicular release is the ability for a single action potential to induce the fusion of multiple vesicles at a single locus with the resulting release of neurotransmitter binding to a common pool of postsynaptic receptors (Wadiche and Jahr, 2001; Foster et al., 2002; Oertner et al., 2002). From now on, we refer to this single release locus and postsynaptic receptor pool as a single synapse. Multivesicular release occurs at excitatory synapses targeting FS neurons in somatosensory cortex (Watanabe et al., 2005; Bagnall et al., 2011), and therefore, the average quantal content released at a single synapse (msyn) can be >1. In our simple model of neurotransmission, quantal content (m) can be described as a function of the number of synapses (Nsyn) and msyn: m = Nsyn * msyn [modified from the study by Johnston and Wu (1995)]. Measuring a change in Nsyn in the context of paired recordings would be difficult, but measuring a change in msyn might be relatively straightforward. In the above model, msyn is directly proportional to release probability at the synapse (Psyn). The lack of change in short-term plasticity might suggest no change in Psyn, but these two processes are not always simply linked (Brody and Yue, 2000; Kraushaar and Jonas, 2000; Regehr and Stevens, 2001). We used measurements of multivesicular release (msyn >1) at this synaptic connection to probe for changes in Psyn as a possible mechanism underlying the decrease in uEPSC amplitude. Consistent with our logic, it has been shown that Psyn controls the extent of multivesicular release at this synaptic connection (Watanabe et al., 2005).
Multivesicular release is usually inferred by varying release probability and observing changes in both the transient glutamate concentration and the degree of postsynaptic receptor saturation (Wadiche and Jahr, 2001; Foster et al., 2002). Changes in transient glutamate concentration and receptor saturation at individual synapses are assayed by the amount of blockade of the EPSC by a low-affinity competitive AMPA receptor (AMPAR) antagonist, such as kynurenate (Wadiche and Jahr, 2001). To determine whether the decrement in excitation of FS neurons was due to changes in multivesicular release, we applied kynurenate (250 μm) to the slices and varied release probability by applying two presynaptic action potentials per trial (50 ms interval) (Rudolph et al., 2011). For each uEPSC, we measured the amount of blockade by kynurenate [(uEPSCbefore − uEPSCantag)/uEPSCbefore]. Because of short-term depression and vesicle depletion, multivesicular release, when it exists, is more prominent during uEPSC1 than during uEPSC2, and therefore blockade of uEPSC2 would be less than blockade of uEPSC1 due to less glutamate being in the cleft during uEPSC2. Indeed, recordings with WT/WT pairs showed greater blockade of uEPSC2 compared to uEPSC1 (Fig. 5A,B; p < 0.02; uEPSC1 vs uEPSC2, 0.32 ± 0.03 vs 0.51 ± 0.04; n = 14), which indicated a higher transient glutamate concentration in the synaptic cleft, and hence multivesicular release, during uEPSC1. The same was true for KO/WT pairs since the blockade of uEPSC2 was greater (Fig. 5B; p < 0.01; uEPSC1 vs uEPSC2, 0.47 ± 0.02 vs 0.58 ± 0.02; n = 13). This effect was specific for the competitive nature of kynurenate since a noncompetitive antagonist, NBQX (75 nm), decreased uEPSC1 and uESPC2 equally (Fig. 5C). When comparing the block of uEPSC1 between WT/WT and KO/WT pairs, the blockade was greater in KO/WT pairs (Fig. 5D; p < 0.01), indicating that less glutamate was released and less multivesicular release occurred with presynaptic Fmr1 deletion. No genotypic difference was observed with NBQX blockade of uEPSC1 (Fig. 5E).
In addition to multivesicular release, changes in spillover from neighboring synapses and the efficacy of single quanta (q) could account for the differences in cleft glutamate concentration. We observed no change in q (Fig. 4E). Inadequate pore-fusion during normal synaptic transmission could also change q, but this is probably not occurring since both rise time and decay time constants were not detectably altered (rise times, WT/WT, 0.40 ± 0.03 ms; KO/WT, 0.43 ± 0.02 ms; decay times, WT/WT, 4.6 ± 0.6 ms; KO/WT, 4.8 ± 0.5 ms; WT/WT, n = 11; KO/WT, n = 9) (Christie and Jahr, 2006). Spillover has not yet been observed in cortical structures at the low-frequency stimulation that we applied, nor under any circumstance during AMPAR-mediated glutamate transmission (Arnth-Jensen et al., 2002; Christie and Jahr, 2006; Sun and Beierlein, 2011). A role for spillover is also unlikely since kynurenate did not affect the decay times of the uEPSCs in WT/WT pairs (before vs during kynurenate, 4.6 ± 0.6 vs 4.5 ± 0.7 ms; n = 14) (DiGregorio et al., 2002). Therefore, we attribute the effects of presynaptic Fmr1 deletion on kynurenate sensitivity to a decrease in multivesicular release (or in other words, a decrease in msyn).
As explained above, a decrease in multivesicular release reflects a decrease in Psyn since fewer vesicles are released per action potential. As in Figure 1H, we did not observe a change in short-term plasticity (Fig. 5F). Receptor saturation was not masking a change since short-term depression was not different between presynaptic WT and KO conditions during kynurenate application (normalized EPSC2, pre-WT, 0.40 ± 0.02; pre-KO, 0.42 ± 0.02). AMPAR desensitization likely did not confound our results for three reasons: (1) we observed no desensitization since 50 μm cyclothiazide did not alter paired pulse ratio (EPSC2/EPSC1; 0.43 ± 0.09 vs 0.48 ± 0.05; before vs after cyclothiazide; n = 7), which is consistent with previous studies using train stimulation frequencies (Galarreta and Hestrin, 1998; Rozov et al., 2001) (but see Watanabe et al., 2005), (2) the NBQX results reveal the specificity of the kynurenate results for competitive blockade, and (3) our conclusions based on uEPSC1 would not be affected by desensitization.
Presynaptic GABABR activation is known to decrease release probability at glutamatergic synapses (Isaacson et al., 1993; Chalifoux and Carter, 2010), and GABAergic synaptic transmission is known to be altered in some brain structures of the Fmr1 KO (D'Hulst and Kooy, 2007; Olmos-Serrano et al., 2010). Therefore, it is possible that increased GABABR activation at presynaptic terminals mediated the effect, but we found this to not be the case since a comparison of uEPSC amplitude before and after wash-in of a GABABR antagonist ([(2S)-3-[[(1S)-1-(3,4-Dichlorophenyl)ethyl]amino]-2-hydroxy-propyl](phenylmethyl)phosphinic acid hydrochloride (CGP55845); 2 μm) had no effect at either presynaptic WT or KO connections (WT, 67.5 ± 13 vs 66.9 ± 12; n = 10; KO, 51.7 ± 13 vs 49.9 ± 11, n = 10; before vs after). The lack of an effect was not due to ineffective CGP55845, since we demonstrated that it could bring uEPSC amplitude back to normal after suppression with the GABABR agonist baclofen (500 nm; n = 2; data not shown).
Release probability is decreased with presynaptic Fmr1 deletion
To confirm our interpretation that a decrease in Psyn is occurring with presynaptic Fmr1 deletion, we made a second, independent measure of Psyn by isolating NMDAR-mediated uEPSCs and determining how quickly the irreversible, open-channel NMDAR antagonist MK-801 (40 μm) could block responses during a 75 pulse stimulus train (Rosenmund et al., 1993; Castro-Alamancos and Connors, 1997) (see Materials and Methods; Fig. 6A). If Psyn decreases with presynaptic Fmr1 deletion, then the rate to full blockade by MK-801 should be slower. Single exponential decay functions were fit to the data for each experiment (Fig. 6A). Two forms of analysis of this data indicated that the rate was indeed slower with presynaptic Fmr1 deletion. First, the binned and averaged data from each experiment were averaged across all experiments (see Materials and Methods; Fig. 6B), and four of the points were higher with presynaptic Fmr1 deletion, indicating that the rate to full blockade was slower. Second, the average time constant, derived from each individual experiment, was higher with presynaptic deletion (Fig. 6C). It is unlikely that the difference in MK-801 blockade was due to different NMDAR subtypes being expressed since the decay time constant of the uEPSC was unchanged (29.4 ± 5.5 vs 26.8 ± 4.5 ms; WT, n = 8; KO, n = 6).
The presynaptic Fmr1 KO phenotype is mimicked with a decrease in release probability
If decreased release probability (Psyn) is the main process underlying the two key phenotypes of decreased uEPSC amplitude and decreased multivesicular release, we should be able to simultaneously mimic these two phenotypes by simply decreasing Psyn in a WT/WT pair. We did this by applying the Ca2+-channel blocker Cd2+ (CdCl2). First, we experimentally determined the relationship between Cd2+ concentration and the fractional block of uEPSC1 (Fig. 7A; two pulses, 50 ms interval). We fit a sigmoid curve (see Materials and Methods) to the raw data points and found that ∼2.6 μm Cd2+ reduced the WT/WT uEPSC1 by a fraction equal to that between the average uEPSC1 derived from WT/WT and KO/WT pairs (0.40; Fig. 7A, dashed lines; based on data from Figs. 1, 5). Importantly, the average quantal content at this concentration (mCd) is assumed to equal that of the KO/WT pairs in normal ACSF (mKO) since we have demonstrated that q is unchanged (Fig. 4).
Next, we performed experiments in 2.6 μm Cd2+ and examined multivesicular release during uEPSC1 by again washing in kynurenate (250 μm; Fig. 7B). There are three possible results when comparing blockade of uEPSC1 (BCd) with that of KO/WT data (BKO), and we interpret these results based on the equation, m = N * msyn (as defined above with N = Nsyn for brevity), and on the assumption that Cd2+ does not alter N (NCd = NWT): (1) BCd > BKO indicates msyn_Cd < msyn_KO and NWT > NKO; (2) BCd = BKO indicates msyn_Cd = msyn_KO and NWT = NKO; (3) BCd < BKO indicates msyn_Cd > msyn_KO and NWT < NKO. Therefore, this experiment potentially also reveals the relative roles of release probability and functional synapse number to the change in uEPSC amplitude.
Our results most closely matched scenario (2), where blockade closely mimicked that of the KO/WT uEPSC1s (Fig. 7C; WT/WT vs WT/WT + Cd2+, p < 0.01; WT/WT, KO/WT, and WT/WT + Cd2+, 0.32 ± 0.03, 0.44 ± 0.02, and 0.45 ± 0.02; n = 14, 15, and 11, respectively). These data support three conclusions: (1) the difference in multivesicular release is a direct result of changes in Psyn, (2) a simple decrease in Psyn primarily accounts for the decrease in uEPSC1 amplitude, and (3) a decrease in the number of functional synapses (Nsyn) plays a less significant, undetectable role. Interestingly, we observed no change in short-term plasticity in 2.6 μm Cd2+ (Fig. 7D), further indicating that release probability and short-term plasticity are not strongly linked at this synapse, at least under these specific conditions. Rise time and decay time constants were not different from WT or KO reported above (rise time, 0.47 ± 0.03 ms; decay time, 4.8 ± 0.05 ms; n = 11).
The presynaptic Fmr1 KO phenotype is rescued with an increase in release probability
We next attempted the complimentary test by determining whether the KO/WT uEPSC amplitude and multivesicular release can be simultaneously rescued to WT characteristics. In this instance, we increased Psyn in KO/WT pairs by applying higher Ca2+ concentrations. First, we experimentally determined the relationship between Ca2+ concentration and the fractional increase in uEPSC1 (Fig. 8A; two pulses, 50 ms interval), and we found that 6 mm Ca2+ increased uEPSC1 to a similar degree as the average fractional increase from KO/WT to WT/WT groups (0.62 increase; based on data from Figs. 1, 5). We assume this increases the quantal content to the WT level (mCa = mWT). This manipulation has a possible confound since AMPARs at glutamatergic synapses targeting FS neurons are Ca2+-permeable, but we found this possibility to have minimal impact since mEPSC amplitude in FS cells was not detectably altered with 6 mm Ca2+ wash-in (17.8 ± 0.26 vs 17.3 ± 0.24 pA, before vs after wash-in; n = 5).
We proceeded to perform experiments in 6 mm Ca2+ and examined multivesicular release by again washing in kynurenate (250 μm; Fig. 8B). As stated in the mimic experiment above, there are three possible results when comparing blockade of uEPSC1 with that of WT/WT data, each with an interpretation based on m = N * msyn (again, N = Nsyn) and assuming NCa = NKO. The kynurenate block very closely matched that found in WT/WT pairs, indicating that a rescue to normal multivesicular release underlies the rescue to normal uEPSC1 amplitude (Fig. 8C; p < 0.02 for KO/WT vs KO/WT + Ca2+; WT/WT, KO/WT, and KO/WT + Ca2+, 0.32 ± 0.03, 0.44 ± 0.02, and 0.33 ± 0.04; n = 14, 15, and 7, respectively). Rise time and decay time constants were not different from WT or KO reported above (rise time, 0.36 ± 0.06 ms; decay time, 4.9 ± 1 ms; n = 7). With this manipulation, we did observe a change in short-term plasticity where depression was slightly more pronounced (Fig. 8D), and therefore, the rescue could not mimic this aspect of synaptic transmission.
In summary, results from both the mimic and rescue experiments support three conclusions: (1) the difference in multivesicular release is a direct result of changes in Psyn, (2) a simple decrease in Psyn primarily accounts for the decrease in uEPSC1 amplitude with Fmr1 deletion in the presynaptic neuron, and (3) a decreased Nsyn plays a less significant, undetectable role. These data all support a model whereby FMRP plays a prominent role in regulating glutamate release (Psyn) at excitatory synapses contacting FS inhibitory neurons.
Multivesicular release is decreased in the constitutive Fmr1 KO
The similar decrement in uEPSC amplitude in the mosaic and constitutive KO suggests that both mouse models may share a common mechanism of impairment. If this is true, then we would predict that the excitatory connection onto FS neurons in the Fmr1 male KO should also have decreased Psyn. Because of experimental ease, we again used the multivesicular release assay as a measure of Psyn. We performed the same experiment as in the mosaic (two pulses, 50 ms interval) except that we used a more specific compound for competitively blocking AMPA receptors, γ-DGG (500 μm; Fig. 9A). We observed that the glutamate cleft concentration during uEPSC1 was greater in WT connections since the amount of block was less for uEPSC1 compared to uEPSC2 (Figs. 9A,B; p < 0.05; WT, 0.28 ± 0.04 vs 0.41 ± 0.04; n = 11). As in the mosaic, γ-DGG blocked uEPSC1 in the Fmr1 KO to a greater extent (Fig. 9C; p < 0.05; WT vs KO, 0.28 ± 0.04 vs 0.41 ± 0.05; n = 11 and 11, respectively), indicating that cleft glutamate concentration, multivesicular release, and Psyn were indeed decreased in the Fmr1 KO. We observed no change in short-term plasticity in the Fmr1 KO slices (Fig. 9D), which is consistent with the data above and our previous study (Gibson et al., 2008).
Impaired excitation of FS neurons in layer 5 in the Fmr1 KO mouse
To begin to determine whether the impairment in the excitation of FS neurons exists outside of layer 4, we examined the same connection in layer 5. If it occurs there as well, this would increase the relevance of our findings above by opening the possibility that it occurs throughout the neocortex. We evoked action potentials in layer 5 pyramidal neurons (four with a 50 ms interval) and measured uEPSCs in neighboring FS neurons (identified by GFP; see Materials and Methods) and compared uEPSC amplitude and connectivity between constitutive KO (Fmr1 KO) and WT male mice. As we found previously for the same connection in layer 4 (Gibson et al., 2008), uEPSC1 amplitude was decreased in the Fmr1 KOs (p < 0.05; WT, 27.5 ± 3.2, n = 23; KO, 19.2 ± 2.9 pA, n = 20). We observed no alteration in connection probability (WT, 41%, n = 23 of 56; KO, 41%, n = 20 of 48) or short-term plasticity [(average of uEPSC3 and uEPSC4)/uEPSC1; WT, 0.26 ± 0.02, n = 20; KO, 0.25 ± 0.03, n = 18].
For the uIPSC originating from the FS neuron and measured in the excitatory neuron, no change in uIPSC amplitude was observed (11.0 ± 3 vs 9.4 ± 1; WT, n = 6; KO, n = 15), but there was increased connectivity in the Fmr1 KO (p = 0.02; 11 vs 31%; WT, 6 of 56; KO, 15 of 48). Together with the lack of any uIPSC change at this connection in layer 4, these data indicate that GABAergic synaptic input originating from nearby neocortical FS neurons is either normal or slightly enhanced in the Fmr1 KO.
Discussion
We find that presynaptic Fmr1 deletion causes a ∼50% decrease in the local excitation of layer 4 FS inhibitory neurons. Underlying this change is a decrease in the release probability (Psyn) of glutamate into the synaptic cleft and a decrease in the multivesicular release that normally occurs at this synapse. These changes occur in both the Fmr1 mosaic mouse and in the Fmr1 KO mouse, indicating that similar impairments in this synaptic pathway occur in both disease models. The decrease in excitation is specific for glutamatergic synapses targeting FS neurons since glutamatergic synapses targeting other excitatory neurons are not affected. We make these observations in acute slices that preserve many facets of native brain circuitry, structure, and function, and therefore our observations are very relevant to conditions in vivo. In summary, we find a novel mechanism for FMRP regulation of synaptic function where presynaptic FMRP positively regulates baseline glutamatergic transmission at synapses targeting FS neurons, and when FMRP is no longer expressed, effective strength in this synaptic pathway is decreased due to a decrease in release probability.
Regulation of synaptic function by presynaptic Fmr1
In hippocampal slice cultures made from the same GFP/Fmr1 mosaic mice that we use here, unitary excitatory synaptic transmission among CA3 pyramidal neurons was reduced with presynaptic Fmr1 deletion (Hanson and Madison, 2007). This was due to reduced connection frequency, while the uEPSC amplitude among existing connections was unchanged. We find a similar theme of pathway weakening with presynaptic Fmr1 deletion, but the main underlying mechanism is a reduction in release probability. The difference may reflect a differential role for FMRP in different synaptic pathways, or reflect experimental differences between acute slices and culture preparations. Our findings support a simple model whereby Psyn is decreased with presynaptic Fmr1 deletion, resulting in both decreased multivesicular release and decreased uEPSC amplitude. This model is supported by our ability to mimic and rescue both KO phenotypes (amplitude and multivesicular release decreases) by manipulating release probability uniformly across all synapses.
While a decrease in release probability with presynaptic Fmr1 deletion might be expected to alter short-term plasticity, we did not observe this (Regehr and Stevens, 2001). This is not a significant inconsistency since release probability and short-term plasticity are not always linked (Dobrunz et al., 1997; Brody and Yue, 2000; Kraushaar and Jonas, 2000; Luthi et al., 2001). Changes in other synaptic properties such as vesicle number, recycling, residual Ca2+, and core release mechanisms could offset a decrease in Psyn, resulting in unchanged short-term plasticity. Indeed, a number of alterations in presynaptic structure and function have been reported in the Fmr1 KO mouse (Deng et al., 2011; Klemmer et al., 2011). In our recordings, we also observe a clear component of release-independent depression, which would further weaken the link between Psyn and short-term plasticity. Specifically, we found that the prior occurrence of an action potential with no resulting EPSC decreases the probability of an EPSC occurring 50 ms later (73 ± 0.04 vs 44 ± 0.07%; probability of EPSC, first vs second action potential, p < 0.05; n = 10). We also observed that a 40% decrease in release probability induced by Cd2+ application did not change short-term plasticity (Fig. 7).
Our observation of a small decrease in connectivity frequency with presynaptic Fmr1 deletion suggests there could be a synapse number decrease (calculated from all pairs in this study), but our mimic and rescue data (Figs. 7, 8) indicate that such a change, if it exists, would play a less significant role. Obtaining reliable anatomical data for synapse number for the specific connection in our study would be a laborious task, but existing evidence is consistent with these assertions. First, total synapse number in layer 4 barrel cortex at this age in the Fmr1 KO mouse is unchanged (Till et al., 2012). Second, there is no change in the frequency of spontaneous, action potential-independent EPSCs (miniature EPSCs) in layer 4 FS neurons (Gibson et al., 2008), which is often interpreted as a lack of change in synapse number. Examining the axonal tree of layer 4 excitatory neurons, while not definitive, may provide a clue to the role of synapse number in the excitation decrement. Layer 4 neurons do have altered axonal trees in the Fmr1 KO by becoming more dispersed in their projection to layer 2/3 (Bureau et al., 2008), but a link to our phenotype is mitigated by five points concerning these individual axonal trees: (1) no decrease in total axon length was observed; (2) the altered axonal tree did not account for the functionally assayed decrease in strength of the projection; (3) the projection phenotype, both anatomically and functionally, disappeared by 3 weeks of age (a time similar to that in which the full 50% decrement in excitation still exists at FS neurons); (4) no alterations in total axon length were detected within layer 4, the region where one would expect most of the synaptic contacts onto layer 4 FS neurons to occur (4.8 ± 0.3 vs 4.2 ± 0.5 mm; WT, n = 14; KO, n = 11); and (5) no alterations in the axonal length density as a function of horizontal or vertical distance in layer 4 were observed [points 4 and 5 are unpublished data from the same data set used in the study by Bureau et al. (2008)]. Therefore, any axonal structure change is unlikely to play a large role in the decreased excitation of layer 4 FS neurons.
Mechanisms of regulation
FMRP is associated with, and perhaps regulates, a large set of mRNAs that code for presynaptic function (Darnell et al., 2011), and a subset of presynaptic proteins have altered levels in the Fmr1 KO (Klemmer et al., 2011). The proteins through which FMRP regulates transmitter release in this study remain unknown, and it is not known whether FMRP functions in the vicinity of the presynaptic terminal, where recent studies have reported its expression (Antar et al., 2006; Christie et al., 2009; Till et al., 2012), or in the soma of the presynaptic neuron. Since the presynaptic phenotype depends on the postsynaptic neuron type, the mechanism probably involves postsynaptic proteins as well.
Very little is known about how FMRP regulates presynaptic neurotransmitter release. Structural and vesicle configuration changes have been reported in hippocampal glutamatergic synapses of the Fmr1 KO, which may affect release probability (Deng et al., 2011; Klemmer et al., 2011). While no changes in presynaptic structure or vesicle content have been observed in synapses in the barrel cortex at the age examined here (Till et al., 2012), no ultrastructural study specifically examining excitatory synapses targeting FS neurons has been performed. In addition, increases in augmentation and vesicle recycling rate have been reported to underlie increases in neurotransmitter release from hippocampal CA3 to CA1 synapses in the Fmr1 KO during prolonged, high-frequency action potential trains (Deng et al., 2011). We did not examine this aspect of neurotransmission, but future studies should be performed to determine whether similar changes exist and if similar mechanisms might be involved in these diverse synaptic phenotypes.
Mosaic vs constitutive Fmr1 mouse models
The connectivity changes are similar in both the global deletion and mosaic Fmr1 mouse models (a selective decrease in the excitation of FS neurons and no change in short-term plasticity), but there are some differences. The uEPSC amplitude decrease in the mosaic mouse was more pronounced and more easily detectable compared to the Fmr1 KO—40% versus 25% (Gibson et al., 2008)—which we attribute either to different mouse strains used or to different processes occurring in mosaic circuits. In our previous study, the CV of uEPSCs was reported as unaltered, implying no quantal content change (Gibson et al., 2008), and this would be inconsistent with our results here. Because uEPSC amplitude in the Fmr1 mosaic mouse was roughly double compared to what we observed in the Fmr1 KO in the previous study, this likely made it more feasible to observe CV changes in mosaic mice. Furthermore, the amplitude criterion for inclusion in the CV analysis in the previous study, in hindsight, was too conservative. If we reanalyze these data examining CV and failure rates using the same criteria used in the current study, a more clear trend is observed (for CV, 0.37 ± 0.03 vs 0.44 ± 0.04, p = 0.13; for failures, 0.04 ± 0.02 vs 0.08 ± 0.02; p = 0.16; WT, n = 26; KO, n = 31). While inconclusive, these data are consistent with increased quantal content in the Fmr1 KO mouse. Ultimately, our observation of a decrease in multivesicular release using kynurenate blockade in both mosaic and Fmr1 KO slice is the most compelling evidence for the occurrence of a common mechanism in both mouse models.
Mosaic mice probably have subtly altered circuit function considering that half of all neurons are Fmr1 KOs, and this could confound our results. This possibility is diminished by that fact that deficits in mosaic FXS patients are not as severe (Hagerman et al., 2009), suggesting that cortical circuit function may not be profoundly changed. At the very least, the fact that we observe the same alterations in both mosaic and KO mice suggests that our observations in the mosaic are not due to a competitive process. It should be noted that our mosaic mice were female and our KO mice were male, but three factors suggest that this did not significantly confound our results: (1) most phenotypes appear similar in males and females (Qin et al., 2005; Baker et al., 2010), (2) the synaptic phenotypes in our study existed in both mouse models, and (3) the mice were too young to be in the estrus cycle, which could affect neuronal properties.
In conclusion, our data suggest that a common impairment in the excitation of neocortical FS inhibitory neurons exists in both forms of FXS: full mutation and mosaic forms. Therefore, the normal functioning of FS neuron circuitry would be expected to be deficient in similar ways across these variants of FXS, have a common consequence for cognitive function, and require a common strategy for treatment.
Footnotes
This work was supported by NIH Grant HD056370 and a linked American Recovery and Reinvestment Act administrative supplement (J.R.G.), and NIH Grants HD052731 (KMH), GM008203 (training grant; S.A.H.), and 1T32NS069562 (training grant; A.B.P.). Support was also provided by Autism Speaks (K.M.H.). We thank Lorea Ormazabel and Nicole Cabalo for technical assistance with the mice. We thank Ege Kavalali for the suggestion of examining multivesicular release.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall MW, Hull C, Bushong EA, Ellisman MH, Scanziani M. Multiple clusters of release sites formed by individual thalamic afferents onto cortical interneurons ensure reliable transmission. Neuron. 2011;71:180–194. doi: 10.1016/j.neuron.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KB, Wray SP, Ritter R, Mason S, Lanthorn TH, Savelieva KV. Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes Brain Behav. 2010;9:562–574. doi: 10.1111/j.1601-183X.2010.00585.x. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Frotscher M, Geiger JR, Jonas P. Rapid signaling at inhibitory synapses in a dentate gyrus interneuron network. J Neurosci. 2001;21:2687–2698. doi: 10.1523/JNEUROSCI.21-08-02687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44:724–728. doi: 10.1017/s0012162201002833. [DOI] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Distinct forms of short-term plasticity at excitatory synapses of hippocampus and neocortex. Proc Natl Acad Sci U S A. 1997;94:4161–4166. doi: 10.1073/pnas.94.8.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S, Rossier J. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci. 1997;17:3894–3906. doi: 10.1523/JNEUROSCI.17-10-03894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SB, Akins MR, Schwob JE, Fallon JR. The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29:1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Sojka D, Klyachko VA. Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci. 2011;31:10971–10982. doi: 10.1523/JNEUROSCI.2021-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30:425–431. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 2002;35:521–533. doi: 10.1016/s0896-6273(02)00787-0. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci U S A. 1997;94:14843–14847. doi: 10.1073/pnas.94.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, Kreitzer AC, Regehr WG. Interaction of postsynaptic receptor saturation with presynaptic mechanisms produces a reliable synapse. Neuron. 2002;36:1115–1126. doi: 10.1016/s0896-6273(02)01106-6. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat Neurosci. 1998;1:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Spike transmission and synchrony detection in networks of GABAergic interneurons. Science. 2001;292:2295–2299. doi: 10.1126/science.1061395. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Huber KM, Südhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci. 2009;29:13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/s0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis AK, Cox LL, Tam PP, Nagy A. An X-linked GFP transgene reveals unexpected paternal X-chromosome activity in trophoblastic giant cells of the mouse placenta. Genesis. 2001;29:133–140. doi: 10.1002/gene.1016. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, Kronk R, Delahunty C, Hessl D, Visootsak J, Picker J, Gane L, Tranfaglia M. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123:378–390. doi: 10.1542/peds.2008-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Madison DV. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J Neurosci. 2007;27:4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Huber KM, Gibson JR. Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci. 2011;31:14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Johnston D, Wu S. Foundations of cellular neurophysiology. Cambridge, MA: MIT; 1995. [Google Scholar]
- Klemmer P, Meredith RM, Holmgren CD, Klychnikov OI, Stahl-Zeng J, Loos M, van der Schors RC, Wortel J, de Wit H, Spijker S, Rotaru DC, Mansvelder HD, Smit AB, Li KW. Proteomics, ultrastructure, and physiology of hippocampal synapses in a fragile X syndrome mouse model reveal presynaptic phenotype. J Biol Chem. 2011;286:25495–25504. doi: 10.1074/jbc.M110.210260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi A, Di Paolo G, Cremona O, Daniell L, De Camilli P, McCormick DA. Synaptojanin 1 contributes to maintaining the stability of GABAergic transmission in primary cultures of cortical neurons. J Neurosci. 2001;21:9101–9111. doi: 10.1523/JNEUROSCI.21-23-09101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41:19–23. doi: 10.1111/j.1528-1157.2000.tb01499.x. [DOI] [PubMed] [Google Scholar]
- Nielsen DM, Derber WJ, McClellan DA, Crnic LS. Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 2002;927:8–17. doi: 10.1016/s0006-8993(01)03309-1. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci. 2002;5:657–664. doi: 10.1038/nn867. [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30:9929–9938. doi: 10.1523/JNEUROSCI.1714-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15:549–567. doi: 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Kang J, Smith CB. A null mutation for Fmr1 in female mice: effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience. 2005;135:999–1009. doi: 10.1016/j.neuroscience.2005.06.081. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF. Physiology of synaptic transmission and short-term plasticity. In: Cowan WM, Sudhof TC, Stevens CF, editors. Synapses. Baltimore: Johns Hopkins UP; 2001. pp. 135–175. [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N. AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci. 2001;21:8062–8071. doi: 10.1523/JNEUROSCI.21-20-08062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph S, Overstreet-Wadiche L, Wadiche JI. Desynchronization of multivesicular release enhances Purkinje cell output. Neuron. 2011;70:991–1004. doi: 10.1016/j.neuron.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 2005;4:420–430. doi: 10.1111/j.1601-183X.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Sun YG, Beierlein M. Receptor saturation controls short-term synaptic plasticity at corticothalamic synapses. J Neurophysiol. 2011;105:2319–2329. doi: 10.1152/jn.00942.2010. [DOI] [PubMed] [Google Scholar]
- Testa-Silva G, Loebel A, Giugliano M, de Kock CP, Mansvelder HD, Meredith RM. Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb Cortex. 2012;22:1333–1342. doi: 10.1093/cercor/bhr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till SM, Li HL, Miniaci MC, Kandel ER, Choi YB. A presynaptic role for FMRP during protein synthesis-dependent long-term plasticity in Aplysia. Learn Mem. 2010;18:39–48. doi: 10.1101/lm.1958811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till SM, Wijetunge LS, Seidel VG, Harlow E, Wright AK, Bagni C, Contractor A, Gillingwater TH, Kind PC. Altered maturation of the primary somatosensory cortex in a mouse model of fragile X syndrome. Hum Mol Genet. 2012;21:2143–2156. doi: 10.1093/hmg/dds030. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Watanabe J, Rozov A, Wollmuth LP. Target-specific regulation of synaptic amplitudes in the neocortex. J Neurosci. 2005;25:1024–1033. doi: 10.1523/JNEUROSCI.3951-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]