

Abstract
Parkinson's disease (PD) is a common neurodegenerative disorder, for which there are no effective disease-modifying therapies. The transcription factor ATF4 (activating transcription factor 4) is induced by multiple PD-relevant stressors, such as endoplasmic reticulum stress and oxidative damage. ATF4 may exert either protective or deleterious effects on cell survival, depending on the paradigm. However, the role of ATF4 in the pathogenesis of PD has not been explored. We find that ATF4 levels are increased in neuromelanin-positive neurons in the substantia nigra of a subset of PD patients relative to controls. ATF4 levels are also upregulated in neuronal PC12 cells treated with the dopaminergic neuronal toxins 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenylpyridinium (MPP+). To explore the role of ATF4 in cell survival in PD-relevant contexts, we either silenced or overexpressed ATF4 in cellular models of PD. In neuronal PC12 cells, silencing of ATF4 enhanced cell death in response to either 6-OHDA or MPP+. Conversely, overexpression of ATF4 reduced cell death caused by dopaminergic neuronal toxins. ATF4 was also protective against 6-OHDA-induced death of cultured mouse ventral midbrain dopaminergic neurons. We further show that parkin, a gene associated with autosomal recessive PD, plays a critical role in ATF4-mediated protection. After treatment with 6-OHDA or MPP+, parkin protein levels fall, despite an increase in mRNA levels. ATF4 silencing exacerbates the toxin-induced reduction of parkin, whereas ATF4 overexpression partially preserves parkin levels. Finally, parkin silencing blocked the protective capacity of ATF4. These results indicate that ATF4 plays a protective role in PD through the regulation of parkin.
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder with dopaminergic neuron degeneration in the substantia nigra (SN) and accumulation of Lewy bodies. The mechanisms of neuronal loss in PD are incompletely clear, with several pathophysiologic mechanisms implicated. One of these is the endoplasmic reticulum stress (ERS) pathway, a conserved cellular response to various insults. Multiple studies indicate that the ERS response is active in PD models (Ryu et al., 2002; Holtz and O'Malley, 2003; Colla et al., 2012). One of the major effectors of the ERS response is ATF4 (activating transcription factor 4, or CREB2), a member of the ATF/CREB family of basic leucine zipper transcription factors. There are higher levels of phosphorylated eukaryotic initiation factor 2α (eIF2α), an upstream activator of ATF4, in the SN of PD patients compared with controls (Hoozemans et al., 2007). However, the involvement of ATF4 in PD pathogenesis has not been addressed.
The effect of ATF4 activation on neuronal survival is complex. ATF4 can promote either cell survival or death depending on the paradigm. For example, ATF4-null mice show less neuronal loss in stroke models (Lange et al., 2008), and ATF4-deficient neurons are more resistant to ERS (Galehdar et al., 2010), consistent with a pro-apoptotic effect. However, ATF4-null neurons are more sensitive to DNA-damaging agents (Galehdar et al., 2010), and activating mutations in ATF4 reduce glutamate toxicity (Lewerenz et al., 2012), implying a protective function. The role of ATF4 signaling in PD and whether it is beneficial or harmful are unknown.
ATF4 regulates expression of target genes involved in multiple cellular processes (Fels and Koumenis, 2006; Ameri and Harris, 2008; Ye and Koumenis, 2009), including the ubiquitin E3 ligase parkin, mutations in which cause early-onset, autosomal recessive PD (Dawson and Dawson, 2010). PD-linked mutations typically lead to a reduction in E3 ligase activity (Henn et al., 2005), consistent with a loss of parkin function in PD. Impairment of parkin function leads to neuronal loss in animal models (Cha et al., 2005; Shin et al., 2011). Furthermore, multiple posttranslational modifications of parkin, which are observed in PD models and autopsy material, lead to a reduction in parkin activity (Chung et al., 2004; Yao et al., 2004; Ko et al., 2010; Meng et al., 2011). ATF4 binds directly to the parkin promoter and increases its expression during ERS (Bouman et al., 2011).
In this study, we investigate the role of ATF4 in PD models. We find that ATF4 is upregulated in midbrain dopaminergic neurons in PD patients and in cellular PD models. ATF4 expression plays a protective role in cellular PD models, because knocking it down sensitized neuronal cells to dopaminergic neuronal toxins, whereas overexpression attenuates cell death. Finally, ATF4 exerts this protective effect via regulation of parkin expression. These studies show that ATF4–parkin signaling plays a role in limiting neuronal death in PD; enhancing this pathway represents a potential neuroprotective strategy in PD.
Materials and Methods
Antibodies and materials.
The following antibodies were used: anti-ATF4 for Western blot was commercially generated for our laboratory; anti-ATF4 for immunostaining, anti-tyrosine hydroxylase (TH), anti-ERK, anti-parkin (#4211), anti-GAPDH, and anti-CHOP (CCAAT/enhancer-binding protein-homologous protein) were from Santa Cruz Biotechnology; anti-parkin (#sc-32282) and anti-phospho-eIF2α were from Cell Signaling Technology; anti-GFP was from Invitrogen. Both anti-parkin antibodies (Cell Signaling Technologies and Santa Cruz Biotechnology) recognized overexpressed rat parkin in HEK293 cells by Western blotting, and the band was greatly reduced by cotransfecting a short-hairpin (sh) RNA against parkin (data not shown). MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal), chloroquine, ammonium chloride, and zVAD-fmk (benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) were purchased from Sigma.
Plasmids.
All shRNA constructs for transfection experiments were generated in pSIREN (Clontech) and coexpress ZsGreen. The following are the target sequences for ATF4 silencing constructs: shATF4-A, 5′-GCCTGACTCTGCTGCTTATAT-3′; shATF4-B, 5′-GCCCTCACTGGCGAGTGTA-3′. Rat shParkin was generated in pSIREN vector based on the following target sequence: 5′-ATCACCTGACAGTACAGAACT-3′. Two control shRNA constructs were used: (1) for transfection experiments, a mutant version of GATA2 silencing target sequence (Biswas et al., 2010), with no detectable homology to rodent genomic sequence, was used (5′-GCACCTGATGTCTTCTTCAACC-3′); (2) for lentiviral experiments, we used a mutated version of shATF4-A (mutated bases are underlined; 5′-GCCAGATTCAGCGGCCTACAT-3′). Rat and human ATF4 cDNAs were cloned into pCMS–eGFP, which coexpresses eGFP, for single transfection experiments, or into pcDNA3.1 for cotransfection experiments. These ATF4 constructs lack the 5′UTR that mediates enhanced translation by phospho-eIF2α. Myc-tagged rat parkin cDNA was obtained from Origene. For virus production, shRNA and cDNA inserts were subcloned into appropriate transfer plasmids (see below). All plasmids were sequenced to confirm the correct insert.
Lentivirus preparation.
All viral plasmids were obtained from Addgene. The transfer plasmid for overexpression was pWPI, which encodes transgene–IRES–GFP under the control of the EF1-α promoter. The transfer plasmid for shRNA was pLVTHM, which encodes the shRNA driven by the H1 promoter along with GFP driven by the EF1-α promoter. Viruses were prepared in HEK293T cells using a second-generation packaging system (pMD2G and psPAX2) using calcium phosphate transfection. Viral particles were concentrated by ultracentrifugation.
Cell culture and viability assays.
PC12 cells were cultured as described previously (Greene and Tischler, 1976). Briefly, cells were cultured on tissue culture plates coated with rat tail collagen (Roche). For neuronal differentiation, cells were grown in RPMI-1640 media supplemented with 1% horse serum, penicillin/streptomycin, and 50 ng/ml recombinant human nerve growth factor for 6–12 d. Media were changed every other day and immediately before toxin treatment. For transient transfections, cells were transfected on day 3–4 of differentiation, using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. For cotransfections, the ratio of shRNA–pSIREN/pcDNA3.1 plasmids was 1:3. For lentiviral infections, cultures were transduced at day 3–4 of differentiation at an approximate multiplicity of infection (MOI) of 5, and experiments were performed at least 3 d later. Using these conditions, the infection rate was reliably >90%.
For survival experiments, stock solutions of 6-hydroxydopamine (6-OHDA) (Tocris) or 1-methyl-4-phenylpyridinium (MPP+) (Sigma) were prepared fresh immediately before each experiment. Cells were treated for the indicated amount of time and fixed in 4% paraformaldehyde, and nuclei were stained with Hoescht 33328. Viable transfected (GFP+) cells were counted in a fixed area of each well. Triplicate cultures were used for each condition, and each experiment was performed at least three times. Two-tailed Student's t test was performed to analyze statistical differences.
Primary cortical neuron cultures from E18 rats were prepared as described previously (Lesuisse and Martin, 2002) and maintained in Neurobasal media supplemented with B-27 and glutamine. Cultures were maintained for 7 d, treated with dopaminergic neuronal toxins, and then collected for total RNA or whole-cell lysates as described below. Primary ventral midbrains neurons from P1–P2 mice were prepared as described previously (Staal et al., 2007). Cultures were infected with lentivirus at an approximate MOI of 10. After 3–4 d, cultures were treated with 50 μm 6-OHDA for 24 h. The cells were then fixed and immunostained for TH and GFP. Using these conditions, ≥95% of TH+ cells were infected, i.e., GFP+. Cell survival was measured as the total count of double-positive (TH+, GFP+) cells in each culture. For both ATF4 knockdown and overexpression, three independent experiments were performed, with each condition performed in triplicate in each experiment.
Western immunoblot and real-time PCR.
Whole-cell extracts were collected in cell lysis buffer (Cell Signaling Technology), followed by sonication. Lysates were prepared for SDS-PAGE with Laemmli's sample buffer and boiled for 10 min at 100°C before running. For preparation of detergent-soluble and -insoluble fractions, cells were lysed in cell lysis buffer containing 1% Triton X-100 and sedimented at 125,000 × g for 30 min at 4°C. The supernatant was saved as the detergent-soluble fraction (S). The pellet was washed once with cell lysis buffer before re-extraction with SDS buffer (PBS with 1% SDS). After sedimentation at 125,000 × g for 30 min at 4°C, the supernatants were saved as the detergent-insoluble fractions (P) (Wang et al., 2005a).
Proteins were transferred to PVDF membranes, which were blocked with 5% milk, incubated with the indicated primary antibody and the appropriate secondary antibody, and visualized with ECL reagent (Pierce).
Total cellular RNA was isolated using TRI Reagent (Ambion). cDNA was synthesized using first-strand cDNA synthesis kit (Origene) with 1 μg of total RNA. Quantitative real-time PCR was performed using FastStart SYBR Green Master Mix (Roche) and an Eppendorf Realplex Mastercyler. For the quantification of parkin or ATF4 mRNA, values were normalized to α-tubulin mRNA (Table 1).
Table 1.
Forward and reverse primers
| Gene | Forward primer | Reverse primer |
|---|---|---|
| ATF4 | CCTTCGACCAGTCGGGTTTG | CTGTCCCGGAAAAGGCATCC |
| Parkin | ACCACAGAGGAAAAGTCACG | GGCCTTTGCAGTAGACAAAA |
| α-Tubulin | TACACCATTGGCAAGGAG | GGCTGGGTAAATGGAGAA |
Statistical analysis of RT-PCR data is based on at least three independent experiments with triplicate samples.
Immunohistochemistry in human brain sections.
Postmortem brain samples from neuropathologically confirmed PD cases and age- and gender-matched controls were obtained from the New York Brain Bank at Columbia University (New York, NY). Midbrain sections (6 μm) were deparaffinized in xylene and rehydrated in an ethanol series. Sections were then heated in citrate buffer for 45 min in a rice cooker at 100°C. Sections were then blocked in goat serum for 20 min and incubated with anti-ATF4 (Santa Cruz Biotechnology) at 1:200 in blocking buffer overnight at 4°C. Sections were then washed and incubated with biotinylated anti-rabbit secondary antibody for 1 h at room temperature, washed, and incubated in ready-to use ABC complex solution at room temperature, and then SG substrate (Vector Laboratories) was added. Sections were counterstained with Nuclear Fast Red.
Results
ATF4 is induced by dopaminergic neuronal toxins
Previously, it has been shown that the ERS response, including elevation of ATF4, is strongly activated in cellular models by dopaminergic neuronal toxins (Ryu et al., 2002; Holtz and O'Malley, 2003). We sought to confirm and extend these observations regarding the regulation of ATF4 in cellular models of PD. We initially used differentiated PC12 cells, which resemble sympathetic neurons, a vulnerable population in PD (Amino et al., 2005). Cultures were treated with two different dopaminergic neuronal toxins: (1) 6-OHDA, a metabolite of dopamine that produces oxidative stress, or (2) MPP+, a complex I inhibitor that is the active metabolite of the dopaminergic neuronal toxin MPTP (Dauer and Przedborski, 2003). At the concentrations used, 20–60% of cells died at 24–48 h after treatment, with no visible signs of cell death before 12 h. We first measured ATF4 mRNA levels in neuronal PC12 cells treated with either 6-OHDA or MPP+ for varying times, using quantitative real-time PCR. We found that ATF4 mRNA levels were upregulated by ∼1.5- to 2-fold after 8–16 h of exposure to either toxin (Fig. 1A). Next, we tested the effect of dopaminergic neuronal toxins on ATF4 protein levels. ATF4 protein levels began to rise 8 h after 6-OHDA exposure and continued to rise up to 24 h (Fig. 1B). With MPP+ treatment, ATF4 protein began to increase as early as 2 h, peaking at 16–24 h (Fig. 1B). These results are consistent with previous studies (Ryu et al., 2002; Holtz and O'Malley, 2003). Maximal induction of ATF4 occurred with 100 μm 6-OHDA and 1 mm MPP+ (Fig. 1B). In both models, the magnitude of increase in ATF4 protein was greater than that of ATF4 mRNA, indicating a posttranscriptional contribution to the elevation of ATF4 levels. Both 6-OHDA and MPP+ lead to PERK (PRKR-like endoplasmic reticulum kinase) activation and eIF2α phosphorylation (Ryu et al., 2002; Holtz and O'Malley, 2003), which lead to increased translation of ATF4. Therefore, we assessed the level of eIF2α phosphorylation in our cultures. Indeed, both toxins led to a strong phosphorylation of eIF2α, which preceded the increase in ATF4. As a positive control, we treated cells with thapsigargin, a classic ERS inducer, and observed robust phosphorylation of eIF2α and elevation of ATF4 protein (Fig. 1B). Together, these results indicate that ATF4 is upregulated at both the transcriptional and translational levels in response to the dopaminergic neuronal toxins 6-OHDA and MPP+ at a time that precedes cell death.
Figure 1.

ATF4 mRNA and protein levels are upregulated by dopaminergic neuronal toxins. A, Neuronal PC12 cells were treated with either 150 μm 6-OHDA or 1 mm MPP+ for different times as indicated. Total RNA was isolated and used to prepare cDNA. ATF4 message was quantified by RT-PCR and normalized to the levels of α-tubulin. Results are from a representative experiment, with each time point performed in triplicate. This experiment was performed three times. *p < 0.05, **p < 0.01 compared with time = 0 h, by Student's t test. B, Neuronal PC12 cells were treated with varying doses of 6-OHDA, MPP+, or 10 μm thapsigargin (Tg) for the indicated amounts of time. Total cell lysates were prepared and subjected to SDS-PAGE and Western blotting with the indicated antibodies. These experiments were repeated three times.
ATF4 is increased in neuromelanin-positive neurons in PD
Next, we determined whether the elevation of ATF4 observed in cellular PD models also occurs in the brains of patients with PD. Although activation of PERK and eIF2α is higher in the SN of PD patients (Hoozemans et al., 2007), ATF4 levels had not been reported. We performed immunohistochemistry for ATF4 on sections of midbrain from both PD and control subjects. The presence of the brownish pigment neuromelanin served as a marker for dopaminergic SN neurons, and ATF4 immunostaining was visualized with the blue–gray SG substrate (as done previously by Malagelada et al., 2008) to best distinguish it from neuromelanin. In the midbrain of both control and PD brains, ATF4 staining is most prominent in the SN and the dorsal midbrain (Fig. 2A). Preabsorption of the ATF4 antibody with a blocking peptide abolished the staining, indicating the specificity of the observed signal (Fig. 2A,C). Most of the ATF4+ cells have the morphological features of neurons. In the SN, staining is primarily, although not exclusively, in neuromelanin-positive neurons. ATF4 is localized primarily to the cytoplasm (Fig. 2B). Some cells exhibit ATF4 staining at the periphery of the nucleus, with very rare cells displaying diffuse nuclear staining (Fig. 2D). In addition, there is prominent ATF4 immunostaining in the neuropil in the SN and the proximal processes of neuromelanin-positive neurons (Fig. 2E,F).
Figure 2.

ATF4 immunostaining in neuromelanin-positive SN neurons is elevated in a subset of PD patients. A–I, Hemi-midbrain sections from postmortem brain were immunostained for ATF4. A, Low-power view of adjacent sections from control midbrain. In the right section, the antibody was preabsorbed with blocking peptide. Dotted lines show boundaries of the SN. B, Higher-power view of SN from control, showing predominantly blue–gray cytoplasmic staining in neuromelanin-positive neurons (arrows). C, Serial section incubated with ATF4 antibody preabsorbed with blocking peptide. Arrows demonstrates cytoplasm of neuromelanin-positive cell. D, Control SN, showing one neuromelanin-positive neuron with intense nuclear ATF4 (arrow), surrounded by three neurons with cytoplasmic ATF4. E, PD SN, showing ATF4 staining of proximal neuronal process (arrowheads) and neuropil (arrows). F, PD SN, showing very intense ATF4 in the neuropil. G, Beaded neurites with ATF4 staining (arrows) in PD SN. H, Neuromelanin-positive neuron with four Lewy bodies (indicated by arrows), with varying degrees of ATF4 immunostaining. I, Representative fields from SN are shown from two controls (CTRL; left), one PD patient with low ATF4 levels (top right), and one PD patient with high ATF4 (bottom right). Scale bars: B–F, I, 50 μm; G, H, 25 μm. J, Semiquantitative assessment of ATF4 immunostaining in neuromelanin-positive neurons from controls (n = 9) and PD patients (n = 10). The intensity of ATF4 labeling in each neuron was graded, in a blinded manner, as absent, weak, or strong. For each case, 40–150 cells were analyzed. The results are graphed as the percentage of neuromelanin-positive neurons in each case that exhibited absent, weak, or strong ATF4 staining. PD patients were separated into groups showing low and high ATF4 expression. The mean percentage of neurons from each group with each staining intensity level is indicated by a solid bar.
To assess whether ATF4 levels are altered in PD, we performed a semiquantitative analysis of ATF4 immunostaining in individual neuromelanin-positive neurons from 10 PD and nine control brains (Fig. 2I,J). Immunostaining in individual neurons was scored as absent, weak, or strong in a blinded manner. In controls, most (average of 77%) neurons display weak ATF4 staining; the percentage of neurons in each case with strong staining averaged 16% (range of 0–35%). However, in 5 of 10 PD cases, nearly 80% of neurons show strong ATF4 staining, a level not seen in any of the controls (PD high ATF4 group; representative field in Fig. 2I and analysis in J). The remaining five PD cases show ATF4 staining similar to controls (PD low ATF4 group; representative field in Fig. 2I and analysis in J). The mean duration of disease in cases with stronger ATF4 immunostaining is significantly longer than in cases with lower ATF4 levels (mean of 21.4 vs 9.8 years, p = 0.047 by t test). Along with this, there is a trend toward a younger age of onset in cases with higher ATF4 levels (mean of 55.0 vs 69.2 years of age, p = 0.077 by t test). In contrast, there is no difference in age at death between the groups (mean of 76.4 vs 79.0 years of age, p = 0.294 by t test), nor are there differences in gender or postmortem interval (data not shown). We also found ATF4 in pathological structures associated with PD. There are beaded structures that appeared to be abnormal neuritic processes; these are seen predominantly in the SN of PD patients, although they are seen occasionally in control brains as well (Fig. 2G). In addition, some Lewy bodies are positive for ATF4, typically in the core (Fig. 2H). In summary, ATF4 is expressed in neuromelanin-positive neurons in the SN, and its expression in elevated in a subset of patients with PD relative to controls.
ATF4 protects cells from death induced by 6-OHDA and MPP+
ATF4 may have pro-apoptotic or anti-apoptotic effects depending on the paradigm studied. To explore the potential role of ATF4 in PD, we used a shRNA (shATF4-A) construct to knockdown endogenous ATF4 in our cellular models. Silencing ATF4 exacerbated PC12 cell death in response to either 6-OHDA or MPP+ (Fig. 3A). There was no effect on cell survival in the absence of toxin treatment. Similar results were obtained with an shRNA construct targeting a different sequence (shATF4-B; Fig. 4; data not shown for PC12 cells). To further demonstrate the specificity of our silencing construct, we made use of the fact that human ATF4 cannot be recognized and is not silenced by shATF4-A, targeted to rat ATF4 (Fig. 3B). Coexpression of human ATF4 with shATF4-A reversed the sensitization to toxin-mediated cell death observed with the shRNA construct alone (Fig. 3C).
Figure 3.

ATF4 protects PC12 cells against neuronal cell death induced by dopaminergic neuronal toxins. A, Neuronal PC12 cells were transfected with plasmids encoding shRNA against ATF4 (shATF4-A) or a scrambled sequence as a control (shCTRL) for 2 d and then treated with different concentrations of 6-OHDA for 20 h or MPP+ for 36 h. Survival was assessed by counting the number of viable GFP+ cells. B, shATF4-A recognizes the rat-ATF4 sequence but not human-ATF4. Plasmids encoding shATF4-A and either rat or human ATF4 cDNA (r-ATF4 and h-ATF4, respectively) were transfected into HEK293T cells, and cell lysates were blotted with ATF4 antibody. C, Neuronal PC12 cells were transfected with the indicated plasmids, treated with 6-OHDA and MPP+, followed by counting of GFP+ cells. D, Neuronal PC12 cells were transfected with plasmids encoding rat ATF4 cDNA and GFP (ATF4) or GFP alone (GFP). Cultures were treated with 6-OHDA for 20 h or MPP+ for 36 h, followed by counting of GFP+ cells. *p < 0.05, **p < 0.01, ***p < 0.001 by Student's t test for A and D and by ANOVA with Tukey's post hoc test for C. The results shown are from representative experiments, with each condition performed in triplicate. There were six, three, and five independent experiments performed for the studies shown in A, C, and D, respectively.
Figure 4.

ATF4 protects against 6-OHDA-induced cell death of primary midbrain dopaminergic neurons. Primary ventral midbrain cultures were infected with lentivirus carrying shRNA against either ATF4 (shATF4-B) or a mutant version of the targeted sequence (shCTRL) in A or with lentivirus carrying GFP alone (GFP) or GFP and rat ATF4 (ATF4) in B. Three days later, cells were treated with 50 μm 6-OHDA for 24 h and then fixed and immunostained for TH and GFP. Survival was measured by counting the number of double-positive (TH+, GFP+) cells. These are representative results from one experiment performed in triplicate; each experiment was repeated three times. *p < 0.01 compared with 6-OHDA-treated control condition by Student's t test.
If silencing ATF4 leads to an increase in toxin-induced PC12 cell death, then overexpression of ATF4 should improve survival. Indeed, ATF4 overexpression significantly attenuated cell death in response to both 6-OHDA and MPP+ (Fig. 3D). Similar to ATF4 silencing, overexpression of ATF4 had no effect on cell survival at baseline. Together, these data demonstrate that ATF4 plays a protective role in neuronal PC12 cells treated with dopaminergic neuronal toxins.
ATF4 protects primary ventral midbrain dopaminergic neurons against 6-OHDA
To extend our findings from PC12 cells, we examined the role of ATF4 in the survival of primary ventral midbrain dopaminergic neurons from early postnatal rats. These neurons correspond closely to the dopaminergic SN pars compacta neurons that are preferentially lost and account for the motor symptoms in PD. Using lentiviral delivery of shRNA against ATF4, we found that silencing ATF4 sensitized dopaminergic neurons to 6-OHDA-induced cell death (Fig. 4A), similar to the effect observed in neuronal PC12 cells. Conversely, lentiviral-mediated overexpression of ATF4 enhanced survival in response to 6-OHDA (Fig. 4B). In summary, ATF4 protects both primary midbrain dopaminergic neurons, as well as neuronal PC12 cells, against cell death induced by dopaminergic neuronal toxins.
Parkin is regulated by 6-OHDA and MPP+ in cellular PD models
Next, we sought to determine the mechanism by which ATF4 enhances neuronal survival. We chose to evaluate parkin as a potential downstream effector of ATF4 for several reasons. Loss-of-function mutations in parkin are a relatively common cause of autosomal recessive, early-onset PD. Parkin enhances neuronal survival in numerous model systems (Jiang et al., 2004). Finally, ATF4 binds to the parkin promoter and upregulates parkin expression in ERS paradigms (Bouman et al., 2011). Therefore, we hypothesized that ATF4 protects cells from death caused by 6-OHDA or MPP+ at least in part through parkin. First, we tested the effect of dopaminergic neuronal toxins on parkin mRNA and protein levels. Consistent with the elevation of ATF4 by dopaminergic neuronal toxins and the regulation of parkin by ATF4, parkin mRNA levels were increased approximately twofold in response to 6-OHDA and approximately fivefold by MPP+ (Fig. 5A). Despite the increase in its message, parkin protein levels were markedly reduced by both 6-OHDA and MPP+ (Fig. 5B). Some studies have found that parkin may aggregate and/or become insoluble after treatment with dopaminergic neuronal toxins (Wang et al., 2005b; Um et al., 2010). However, we did not observe parkin aggregation or a shift to detergent-insoluble fractions in our experimental conditions (Fig. 5C). Identical results were observed with a second parkin antibody (data not shown). To ensure that this toxin-induced reduction in parkin protein was not unique to PC12 cells, we repeated these experiments in cultures of primary cortical neurons, a population that develops Lewy body pathology in PD (Braak et al., 2003). In response to either 6-OHDA or MPP+, parkin mRNA was upregulated (Fig. 5D), whereas parkin protein levels were significantly reduced (Fig. 5E).
Figure 5.

Parkin message and protein are differentially affected by dopaminergic neuronal toxins. A, B, Differentiated PC12 cells were treated with 6-OHDA and MPP+ for the indicated times. Parkin mRNA levels were measured by qPCR (A) and normalized to α-tubulin levels. Parkin protein levels were analyzed by Western blotting (B). Results are representative of experiments that were performed three times; each condition was performed in triplicate in each experiment. *p < 0.01 compared with control (CTR) by Student's t test. C, Parkin protein levels were tested in detergent-soluble (S) and -insoluble (P) fractions of PC12 cells in response to 6-OHDA or MPP+. The blot is from a representative experiment, which was repeated three times. D, E, Primary cortical neuron cultures were treated with either 6-OHDA or MPP+ as indicated, and parkin mRNA and protein levels were tested by qPCR (D) and Western blotting (E). Each experiment was performed three independent times. The blot in E is from a representative experiment, and the densitometric quantification is an average of the three independent experiments. *p < 0.01 compared with control condition by Student's t test. F, Differentiated PC12 cells were treated with 100 μm 6-OHDA for 4 h, washed, and then incubated with the indicated inhibitors—ammonium chloride (NH4Cl), chloroquine (CQN), zVAD-fmk (zVAD), and MG132—for 4 h. Protein lysates were prepared and analyzed by Western blotting with the indicated antibodies. G, Differentiated PC12 cells were treated with 1 mm MPP+ for 16 h, washed, and then incubated with MG132 for 8 h. Samples analyzed as in F. H, Densitometric quantification of parkin protein levels after treatment with either 6-OHDA or MPP+, in either the presence or absence of MG132. *p < 0.05, **p < 0.001 compared with toxin-treated condition without MG132 by Student's t test. The experiments with MG132 were performed three times with each toxin, with representative blots shown; the other inhibitors were used in one experiment.
Parkin can be degraded by caspases (Kahns et al., 2002; Kahns et al., 2003) and the ubiquitin-proteasome system (Yu and Zhou, 2008). To assess the mechanism underlying the reduction in parkin protein by PD mimetics, we used pharmacologic inhibitors of caspases (zVAD-fmk), proteasomal function (MG132), or the lysosomal pathway (NH4Cl or chloroquine). The 6-OHDA-induced reduction in parkin protein was minimally affected by inhibition of either caspases or lysosomal function in PC12 cells (Fig. 5F). In contrast, MG132 significantly attenuated the loss of parkin protein in response to both 6-OHDA and MPP+ (Fig. 5F–H), without altering the amount of parkin in the detergent-insoluble fraction (data not shown). These data suggest that both toxins lead to a reduction in parkin protein, at least in part, via enhanced proteasomal degradation, although other mechanisms, such as reduced translation of parkin mRNA, may also play a role.
CHOP induction occurs independently of ATF4 in response to dopaminergic neuronal toxins
In many ERS paradigms, ATF4 induces expression of the transcription factor CHOP (Harding et al., 2000; Averous et al., 2004). Although CHOP typically exerts pro-apoptotic actions, it has also been reported to have neuroprotective effects (Halterman et al., 2010). We therefore assessed the role of ATF4 in CHOP induction in our models. As described previously (Ryu et al., 2002; Holtz and O'Malley, 2003), 6-OHDA and MPP+ both produce an increase in CHOP mRNA (data not shown) and CHOP protein (Fig. 6B,D; CHOP could not be consistently detected in the absence of toxin treatment). However, ATF4 knockdown does not diminish the toxin-mediated induction of CHOP mRNA (data not shown) or protein (Fig. 6B,D). In fact, ATF4 silencing leads to an even stronger induction of CHOP protein by 6-OHDA. Therefore, unlike other ERS paradigms, CHOP is not positively regulated by ATF4 in response to dopaminergic neuronal toxins.
Figure 6.
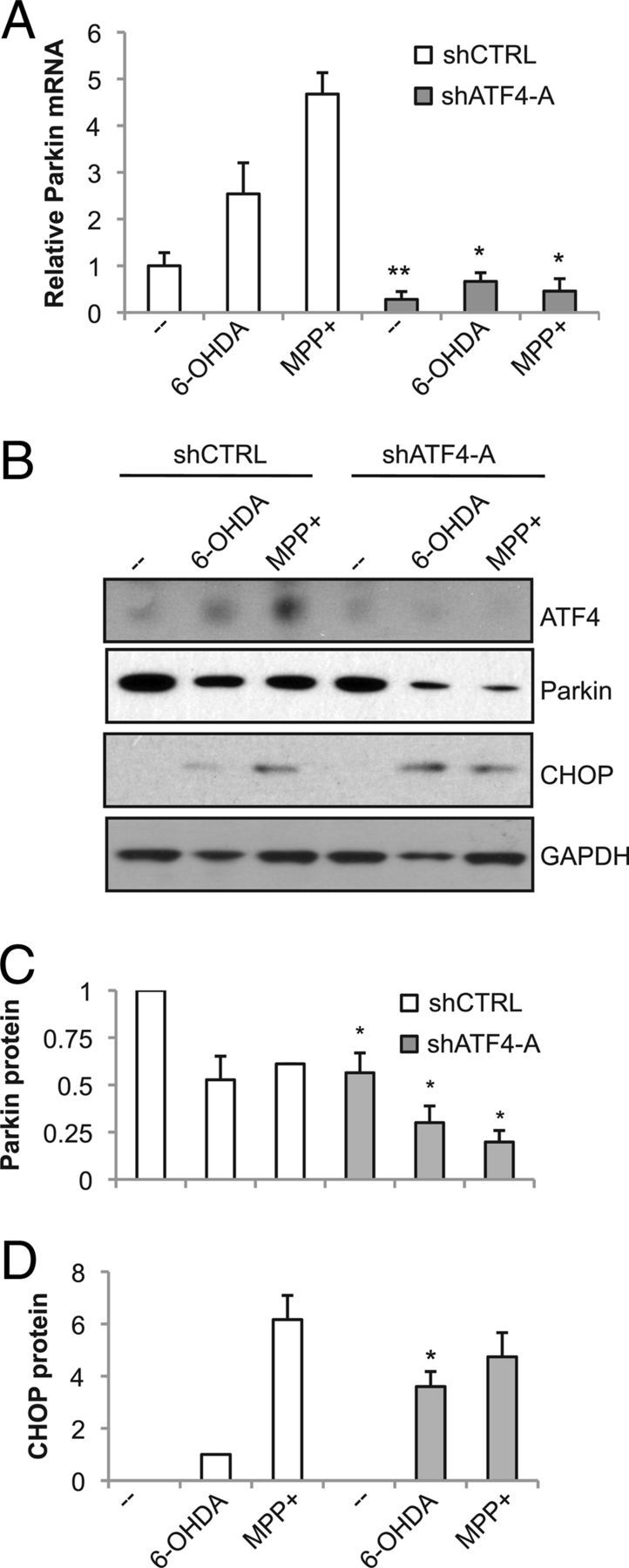
ATF4 knockdown exacerbates the loss of parkin levels but does not diminish CHOP induction after treatment with dopaminergic neuronal toxins. Neuronal PC12 cells were infected with lentivirus carrying shRNA against either ATF4 (shATF4-A) or a mutant version of the targeted sequence (shCTRL) for 3 d, treated with 100 μm 6-OHDA for 10 h or 1 mm MPP+ for 24 h, and then analyzed by qPCR (A) and Western blotting (B). Blots are representative of experiments performed in triplicate. Blots from the three independent experiments were quantified by densitometry for parkin (C) or CHOP (D), with values normalized to GAPDH. CHOP signal was not consistently detectable in untreated cells, so the intensity of CHOP in 6-OHDA-treated PC12 cells infected with control virus is used as a reference. *p < 0.05, **p < 0.01, compared with shCTRL with identical drug treatment, by Student's t test.
ATF4 limits the reduction in parkin levels caused by dopaminergic neuronal toxins
Next, we determined whether ATF4 regulates parkin in response to PD-relevant stressors. First, we tested the effect of silencing ATF4, using lentiviral delivery of ATF4 shRNA, in neuronal PC12 cells treated with either 6-OHDA or MPP+. Parkin mRNA levels were reduced below control levels when ATF4 was silenced, in either the presence or absence of toxins (Fig. 6A). Strikingly, the toxin-induced increase in parkin mRNA was completely abolished by silencing ATF4, suggesting that ATF4 is necessary for this response. ATF4 knockdown had a parallel effect on parkin protein (Fig. 6B,C). At baseline before toxin treatment, parkin protein levels were reduced after ATF4 knockdown. As anticipated, with the control shRNA, treatment with either 6-OHDA or MPP+ led to a reduction in parkin protein, despite the increase in parkin mRNA. Significantly, silencing ATF4 produced a much greater loss of parkin protein in response to dopaminergic neuronal toxins, likely as a result of the suppression of parkin mRNA.
Next, we tested parkin levels in PC12 cells overexpressing ATF4. Overexpression of ATF4 had little effect on parkin mRNA levels, in either controls or in response to dopaminergic neuronal toxins (Fig. 7A). However, overexpression of ATF4 attenuated the decline in parkin protein that occurs after 6-OHDA and MPP+ treatment (Fig. 7B,C). In summary, these results indicate that ATF4 plays a critical role in limiting the loss of parkin protein caused by PD-related stressors.
Figure 7.

ATF4 overexpression preserves parkin protein levels after treatment with dopaminergic neuronal toxins. Neuronal PC12 cells were infected with lentivirus carrying GFP alone (GFP) or GFP and rat ATF4 (ATF4) for 3 d, treated with 100 μm 6-OHDA for 10 h or 1 mm MPP+ for 24 h, and then analyzed by qPCR (A) and Western blotting (B). Blots are representative of experiments performed in triplicate. Blots from the three independent experiments were quantified by densitometry for parkin (C), with values normalized to GAPDH. *p < 0.05, compared with GFP with identical drug treatment, by Student's t test.
Parkin mediates the protective effect of ATF4
Because parkin has a protective role in PD cellular and animal models and is regulated by ATF4, we next assessed whether the ATF4-mediated increase in parkin levels plays a direct role in survival. First, we simultaneously silenced ATF4 and overexpressed parkin in neuronal PC12 cells and measured survival after treatment with 6-OHDA or MPP+. Overexpression of parkin reversed the deleterious effects of silencing ATF4 on toxin-induced PC12 cell death (Fig. 8B). Next, we performed the converse experiment, overexpressing ATF4 while silencing parkin. The parkin shRNA construct strongly silenced transfected rat parkin in HEK293 cells (Fig. 8A), as well as endogenous parkin in PC12 cells (data not shown). In this case, when parkin was silenced, overexpression of ATF4 had no effect on cell survival after 6-OHDA or MPP+ (Fig. 8C). Thus, in both overexpression and silencing experiments, reversing the effect of ATF4 on parkin abolished the effect of ATF4 on cell survival. This implies that parkin acts downstream of ATF4 and mediates it ability to enhance cell survival in PD cellular models.
Figure 8.

The effect of ATF4 on survival in PD cellular models is mediated through parkin. A, HEK293 cells were cotransfected with myc-tagged rat parkin and either shParkin or a control shRNA. Lysates were collected and blotted with myc antibody. B, C, Neuronal PC12 cells were cotransfected with either shATF4-A and rat parkin plasmids (B) or shParkin and rat ATF4 plasmids (C). Forty-eight hours later, cells were treated with 100 μm 6-OHDA for 20 h or 1 mm MPP+ for 36 h, and survival was assessed by counting GFP+ transfected cells. These are representative results from one experiment with each condition performed in triplicate; each experiment was repeated three times. *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA with Tukey's post hoc test.
Discussion
The goal of this study was to explore the role of the ERS-related transcription factor ATF4 in PD. We observed ATF4 upregulation in SN dopaminergic neurons in half of the PD patients studied. As reported previously for several PD models, we found ATF4 elevation in response to the dopaminergic neuronal toxins 6-OHDA and MPP+. Knockdown of ATF4 sensitized neuronal PC12 cells to 6-OHDA and MPP+, whereas ATF4 overexpression enhanced survival. ATF4 had a similar pro-survival effect on 6-OHDA-induced death of primary dopaminergic ventral midbrain neurons. Next, we found that ATF4 promoted survival via regulation of the PD-related gene parkin. Treatment with 6-OHDA or MPP+ leads to a loss of parkin protein, but ATF4 limits this decline. Furthermore, parkin is downstream of ATF4 with regard to enhancing cell survival. Together, these results identify ATF4 upregulation and subsequent maintenance of parkin as critical mediators of neuronal survival in PD models.
We observed elevation of ATF4 in SN dopaminergic neurons of PD cases, as well as in neuronal PC12 cells treated with 6-OHDA or MPP+. Our findings are consistent with higher levels of PERK and eIF2α phosphorylation observed in midbrain dopaminergic neurons of PD patients (Hoozemans et al., 2007) and that we and others have observed in cellular PD models (Ryu et al., 2002; Holtz and O'Malley, 2003). Interestingly, although ATF4 is a transcription factor, it localized primarily to the cytoplasm and processes of midbrain neurons. Previous studies suggest that ATF4 can be regulated in neuronal processes. ATF4 directly interacts with GABAB receptors, which may regulate ATF4 activity (Ritter et al., 2004). In cultured neurons, ATF4 is abundant in processes and synapses, in which it can be retrogradely transported to the nucleus (Lai et al., 2008). This observation is particularly interesting, given the defect in axonal transport that is posited to occur in PD (De Vos et al., 2008). We found that half of the PD cases tested showed stronger ATF4 immunostaining in SN neurons. This increase tended to be associated with longer disease duration. Although this observation needs to be confirmed in additional cases, one interpretation is that higher ATF4 levels contribute to the survival of these remaining SN neurons, given our finding that ATF4 has a protective function in cellular PD models. Alternatively, increased ATF4 levels may be an epiphenomenon, indicating higher levels of neuronal stress.
Using knockdown and overexpression experiments, we demonstrate that ATF4 protects neuronal PC12 cells and primary midbrain dopaminergic neurons against the dopaminergic neuronal toxins 6-OHDA and MPP+. This is the first study to directly assess the role of ATF4 in neuronal survival in PD-relevant models. Previous studies exploring ATF4 in other paradigms have yielded results supporting both pro-survival and pro-apoptotic functions. In support of a pro-apoptotic role, ATF4-null mice and primary neurons are more resistant to in vivo stroke, hypoxia, and ER stressors (Lange et al., 2008; Galehdar et al., 2010). This pro-apoptotic effect of ATF4 is linked with its transcriptional induction of CHOP, another transcription factor with predominantly (although not exclusively) pro-apoptotic effects (Galehdar et al., 2010; Halterman et al., 2010; Bromati et al., 2011). However, in our experiments, CHOP induction occurred independently of ATF4. Other studies suggest a pro-survival function for ATF4. DNA damage-induced death was enhanced in ATF4-null primary neurons (Galehdar et al., 2010). In neuronal cell lines, activating mutations in ATF4 lead to resistance against glutamate and β-amyloid toxicity (Lewerenz et al., 2012). Studies looking at upstream regulators of ATF4 are consistent with a protective function for ATF4 in PD models. Salubrinal inhibits eIF2α dephosphorylation, thus favoring ATF4 translation. Salubrinal protects against α-synuclein-induced cell death in vitro and improves survival in an α-synuclein transgenic mouse (Smith et al., 2005; Colla et al., 2012). Together, these studies suggest that, although ATF4 may have opposite effects on neuronal survival, it appears to be protective in the context of PD.
One factor likely to contribute to the differential effect of ATF4 on neuronal survival is the set of transcriptional targets regulated by ATF4. For example, as mentioned previously, ATF4 can regulate CHOP expression in some paradigms, although not in our model systems, or others (Zou et al., 2008). ATF4 functions as either a homodimer (Ameri and Harris, 2008) or a heterodimer with other basic leucine zipper transcription factors, such as c-Jun, c-Fos (Hai and Curran, 1991), or C/EBP (CCAAT/enhancer-binding protein) (Vallejo et al., 1993) proteins. Different combinations have different DNA recognition sequences and can either activate or repress transcription. Therefore, ATF4 can regulate different sets of downstream genes depending on the status of its binding partners, which in turn may be determined by the cell type and specific stressor. The suite of regulated genes may determine the ultimate effect of ATF4 on neuronal survival.
In our studies, ATF4 overexpression further protected cells against dopaminergic neuronal toxins, although the magnitude of protection was rather modest. One reason may have been reduced expression of the ATF4 transgene during ERS, because the transgene lacked the 5′UTR of the endogenous ATF4 that allows for enhanced translation by phospho-eIF2α. Another reason may be that ATF4 is already upregulated in response to dopaminergic neurotoxins, so that an additional increase in ATF4 does not provide substantial additional protective signaling. Another important consideration is that ATF4 overexpression before the initiation of toxin treatment may be critical for its protective effect, akin to the preconditioning effect seen with sublethal doses of various toxins. For example, pretreatment with low doses of ER stressors leads to an increase in ATF4 and a reduction in 6-OHDA-induced cell death, although the role of ATF4 in the observed neuroprotection was not evaluated (Hara et al., 2011). ATF4 might be central to the pro-survival effects of preconditioning regimens.
In our studies, dopaminergic neuronal toxins lead to a substantial fall in parkin protein levels, despite an elevation in parkin mRNA. We observe this effect in both differentiated PC12 cells and primary cortical neurons, demonstrating that this is not a cell-type-specific finding. Proteasome inhibition attenuates this toxin-induced decrease in parkin protein. Several studies have found reduced parkin activity secondary to dopaminergic neuronal toxins, although the postulated underlying mechanism varies. Some find an increase in the amount of detergent-insoluble and/or aggregated parkin (Wang et al., 2005b; Jensen et al., 2006; Um et al., 2010; Meng et al., 2011); we did not observe this in our experiments. Many of these studies examined overexpressed parkin rather than the endogenous protein. Others show that parkin can become posttranslationally modified, e.g., phosphorylation (Ko et al., 2010; Imam et al., 2011) or S-nitrosylation (Yao et al., 2004), with subsequent inhibition of its E3 ligase activity (with or without aggregation). Our results do not rule out effects on E3 ligase activity, which may occur in addition to the loss of parkin protein we observe. In summary, our data are consistent with previous studies showing a loss of parkin after treatment with dopaminergic neuronal toxins. Our results further suggest that parkin levels decrease, at least in part, via enhanced proteasomal degradation.
ATF4 knockdown led to a decrease in parkin mRNA, with a corresponding reduction in parkin protein. Specifically, the increase in parkin message seen with dopaminergic neuronal toxins was blocked by ATF4 silencing, leading to an even greater decline in parkin protein. These observations are consistent with previous studies showing that ATF4 transcriptionally regulates parkin (Bouman et al., 2011). Interestingly, ATF4 overexpression had little effect on parkin mRNA. Together, these data suggest that ATF4 is necessary but not sufficient for transactivation of parkin. As discussed above, ATF4 may function as a heterodimer with numerous transcription factors; thus, another rate-limiting transcription factor may function together with ATF4 to regulate parkin. Despite the lack of an effect on parkin mRNA, ATF4 overexpression lessened the toxin-induced loss of parkin protein. Given our finding that dopaminergic neuronal toxins increase proteasomal degradation of parkin, one possibility is that ATF4 overexpression impacts other proteins that in turn regulate parkin turnover.
By simultaneously overexpressing ATF4 and silencing parkin and vice versa, we found that parkin is downstream of ATF4 in terms of protecting cells against dopaminergic neuronal toxins. Parkin overexpression protects neurons, whereas parkin knockdown sensitizes them to different stressors (Dawson and Dawson, 2010). Parkin regulates mitochondrial fusion/fission, as well as autophagic clearance of damaged mitochondria (Poole et al., 2008; Vives-Bauza et al., 2010). ATF4 has been linked to the induction of autophagy via transcriptional regulation of LC3B, an essential autophagosome protein (Milani et al., 2009; Rzymski et al., 2010). ATF4 may also impact mitochondrial oxidative phosphorylation (Martínez-Reyes et al., 2012). Regulation of parkin by ATF4 may represent an additional mechanism by which ATF4 can modulate mitochondrial function and clearance, processes considered important in the pathogenesis of PD.
In conclusion, we find that ATF4 plays a previously undescribed protective role in PD-related neuronal death by maintaining levels of parkin. These findings have implications regarding potential disease-modifying strategies for PD. Manipulations that enhance ATF4 levels, such as salubrinal-induced eIF2α phosphorylation, enhance neuronal survival in PD model systems (Colla et al., 2012). Activation of the ATF4–parkin pathway may thus be a neuroprotective strategy in PD.
Footnotes
This work was supported in part by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant K08 NS070608 (O.A.L.), Udall Center Grant P50 NS38370 (L.A.G.), the William and Bernice Bumpus Foundation (X.S.), the Smart Foundation (O.A.L.), and the Parkinson's Disease Foundation (O.A.L., D.S. and L.A.G.). We thank the New York Brain Bank at Columbia University for providing postmortem tissue.
The authors declare no competing financial interests.
References
- Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Amino T, Orimo S, Itoh Y, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol. 2005;15:29–34. doi: 10.1111/j.1750-3639.2005.tb00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem. 2004;279:5288–5297. doi: 10.1074/jbc.M311862200. [DOI] [PubMed] [Google Scholar]
- Biswas SC, Zhang Y, Iyirhiaro G, Willett RT, Rodriguez Gonzalez Y, Cregan SP, Slack RS, Park DS, Greene LA. Sertad1 plays an essential role in developmental and pathological neuron death. J Neurosci. 2010;30:3973–3982. doi: 10.1523/JNEUROSCI.6421-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, Gasser T, Augustin R, Trümbach D, Irrcher I, Park DS, Wurst W, Kilberg MS, Tatzelt J, Winklhofer KF. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18:769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Bromati CR, Lellis-Santos C, Yamanaka TS, Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anh ê GF, Bordin S. UPR induces transient burst of apoptosis in islets of early lactating rats through reduced AKT phosphorylation via ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr Comp Physiol. 2011;300:R92–R100. doi: 10.1152/ajpregu.00169.2010. [DOI] [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic reticulum stress is important for the manifestations of synucleinopathy in vivo. J Neurosci. 2012;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson's disease. Mov Disord. 2010;25(Suppl 1):S32–S39. doi: 10.1002/mds.22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Fels DR, Koumenis C. The PERK/elF2 alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5:723–728. doi: 10.4161/cbt.5.7.2967. [DOI] [PubMed] [Google Scholar]
- Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem. 2010;285:21329–21340. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Kamiya T, Adachi T. Endoplasmic reticulum stress inducers provide protection against 6-hydroxydopamine-induced cytotoxicity. Neurochem Int. 2011;58:35–43. doi: 10.1016/j.neuint.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Henn IH, Gostner JM, Lackner P, Tatzelt J, Winklhofer KF. Pathogenic mutations inactivate parkin by distinct mechanisms. J Neurochem. 2005;92:114–122. doi: 10.1111/j.1471-4159.2004.02854.x. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M, Roberts JL, Kahle PJ, Clark RA, Li S. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci. 2011;31:157–163. doi: 10.1523/JNEUROSCI.1833-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LD, Vinther-Jensen T, Kahns S, Sundbye S, Jensen PH. Cellular parkin mutants are soluble under non-stress conditions. Neuroreport. 2006;17:1205–1208. doi: 10.1097/01.wnr.0000230511.63220.e3. [DOI] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Kahns S, Lykkebo S, Jakobsen LD, Nielsen MS, Jensen PH. Caspase-mediated parkin cleavage in apoptotic cell death. J Biol Chem. 2002;277:15303–15308. doi: 10.1074/jbc.M111534200. [DOI] [PubMed] [Google Scholar]
- Kahns S, Kalai M, Jakobsen LD, Clark BF, Vandenabeele P, Jensen PH. Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem. 2003;278:23376–23380. doi: 10.1074/jbc.M300495200. [DOI] [PubMed] [Google Scholar]
- Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A. 2010;107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KO, Zhao Y, Ch'ng TH, Martin KC. Importin-mediated retrograde transport of CREB2 from distal processes to the nucleus in neurons. Proc Natl Acad Sci U S A. 2008;105:17175–17180. doi: 10.1073/pnas.0803906105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol. 2002;51:9–23. doi: 10.1002/neu.10037. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Sato H, Albrecht P, Henke N, Noack R, Methner A, Maher P. Mutation of ATF4 mediates resistance of neuronal cell lines against oxidative stress by inducing xCT expression. Cell Death Differ. 2012;19:847–858. doi: 10.1038/cdd.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. 2008;28:14363–14371. doi: 10.1523/JNEUROSCI.3928-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Reyes I, Sánchez-Arag ó M, Cuezva JM. AMPK and GCN2-ATF4 signal the repression of mitochondria in colon cancer cells. Biochem J. 2012;444:249–259. doi: 10.1042/BJ20111829. [DOI] [PubMed] [Google Scholar]
- Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, Ma Y, Moosmann B, Masliah E, Lipton SA, Gu Z. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener. 2011;6:34. doi: 10.1186/1750-1326-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009;69:4415–4423. doi: 10.1158/0008-5472.CAN-08-2839. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter B, Zschuntzsch J, Kvachnina E, Zhang WQ, Ponimaskin EG. The GABA(B) receptor subunits R1 and R2 interact differentially with the activation transcription factor ATF4 in mouse brain during the postnatal development. Dev Brain Res. 2004;149:73–77. doi: 10.1016/j.devbrainres.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene. 2010;29:4424–4435. doi: 10.1038/onc.2010.191. [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- Staal RG, Rayport S, Sulzer D. Amperometric detection of dopamine exocytosis from synaptic terminals. In: Michael AC, Borland LM, editors. Electrochemical methods for neuroscience. Ed January 2011. Boca Raton, FL: CRC; 2007. [PubMed] [Google Scholar]
- Um JW, Park HJ, Song J, Jeon I, Lee G, Lee PH, Chung KC. Formation of parkin aggregates and enhanced PINK1 accumulation during the pathogenesis of Parkinson's disease. Biochem Biophys Res Commun. 2010;393:824–828. doi: 10.1016/j.bbrc.2010.02.090. [DOI] [PubMed] [Google Scholar]
- Vallejo M, Ron D, Miller CP, Habener JF. C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc Natl Acad Sci U S A. 1993;90:4679–4683. doi: 10.1073/pnas.90.10.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magran é J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005a;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005b;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Koumenis C. ATF4, an ER stress and hypoxia-inducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr Mol Med. 2009;9:411–416. doi: 10.2174/156652409788167096. [DOI] [PubMed] [Google Scholar]
- Yu F, Zhou J. Parkin is ubiquitinated by Nrdp1 and abrogates Nrdp1-induced oxidative stress. Neurosci Lett. 2008;440:4–8. doi: 10.1016/j.neulet.2008.05.052. [DOI] [PubMed] [Google Scholar]
- Zou W, Yue P, Khuri FR, Sun SY. Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res. 2008;68:7484–7492. doi: 10.1158/0008-5472.CAN-08-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]