

Abstract
Tricyclic antidepressants exert their pharmacological effect - inhibiting the reuptake of serotonin, norepinephrine and dopamine - by directly blocking neurotransmitter transporters (SERT, NET and DAT, respectively) in the presynaptic membrane. The drug-binding site and the mechanism of this inhibition are poorly understood. We determined the crystal structure at 2.9 Å of the bacterial leucine transporter (LeuT), a homolog of SERT, NET and DAT, in complex with leucine and the antidepressant desipramine. Desipramine binds at the inner end of the extracellular cavity of the transporter and is held in place by a hairpin loop and by a salt bridge. This binding site is separated from the leucine-binding site by the extracellular gate of the transporter. By directly locking the gate, desipramine prevents conformational changes and blocks substrate transport. Mutagenesis experiments on human SERT and DAT indicate that both the desipramine-binding site and its inhibition mechanism are probably conserved in the human neurotransmitter transporters.
Na+/Cl−-dependent neurotransmitter transporters for serotonin (SERT), norepinephrine (NET) and dopamine (DAT) in the presynaptic plasma membrane terminate neuronal signal transmission in the central nervous system through a reuptake mechanism (1–6). These systems have been shown to modulate mood, emotion, sleep and appetite (7). Depression, arguably the most prevalent psychiatric disorder, is directly associated with perturbation of serotonergic neurotransmission (8, 9), and drugs blocking serotonin reuptake have been used successfully for its treatment. One class of these drugs, tricyclic antidepressants (TCAs) such as desipramine and imipramine, binds to serotonin and norepinephrine transporters with affinities of nanomolar to tens of nanomolar concentrations and blocks transport activity (10). The response rate of patients to TCAs is typically 60–70% (11). More recently, highly selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine (Prozac) have also been developed and are increasingly prescribed to treat depression (12). The molecular pharmacology of TCAs and SSRIs has been well defined, and their in vivo pharmacological effects appear to be mediated almost exclusively by serotonin and norepinephrine reuptake inhibition. Despite extensive investigations, however, whether the substrate-binding and drug-binding sites are overlapping and whether the drug inhibition mechanism is of a competitive nature remain controversial (13).
The human SERT, DAT and NET proteins all belong to a family of transporters for amino acids and their derivatives, the Neurotransmitter:Sodium Symporter (NSS) family (2–5, 14). Whilst the dopamine transporters from human, bovine or rat are inhibited by TCAs at a Ki of micromolar concentrations, the DAT proteins from C. elegans (15) and D. melanogaster (16) are inhibited by TCAs at a Ki of nanomolar and sub-micromolar concentrations, respectively (17). As bacterial NSS proteins share up to 30 % sequence identity with human SERT and NET as well as worm and fly DATs, we hypothesized that bacterial NSS proteins also possess high binding affinity to TCAs and could provide opportunities for studying protein-drug interactions. We therefore chose a bacterial NSS protein, the leucine transporter (LeuT) from Aquifex aeolicus, to study the molecular mechanism of neurotransmitter transporter binding to TCAs (18). LeuT shares 20–25% sequence identity and 40–45% similarity with human neurotransmitter transporters. The crystal structure of LeuT, which was previously determined (19), revealed a shot- glass-shaped bundle of twelve transmembrane α-helices (TM1–12), with a substrate leucine and two sodium ions bound at the center of the protein. The substrate-binding site is closed off from the extracellular space by a gate. There is an extracellular cavity in the protein, into which protrudes a helix hairpin formed by loop EL4.
We screened for binding of various tricyclic and other types of antidepressants to LeuT using a scintillation proximity assay (20). Several tricyclic compounds (imipramine, nortriptyline, protriptyline, amitriptyline and doxepin) showed binding affinity (Fig. S1), but desipramine bound LeuT most tightly, with an IC50 of 80 μM (Fig. 1A). In addition, fluoxetine also showed measurable binding (Figs. S1&1A). Desipramine was found to inhibit leucine binding to LeuT by decreasing its maximal binding capacity without changing its binding affinity (Fig. 1B), indicating a mechanism that does not involve competitive inhibition. To test if desipramine binding also inhibits substrate transport, we measured the leucine transport activity of LeuT in reconstituted proteoliposomes and found that at a concentration of 200 μM desipramine indeed completely abolished LeuT’s transport activity (Fig. S2).
Fig. 1.
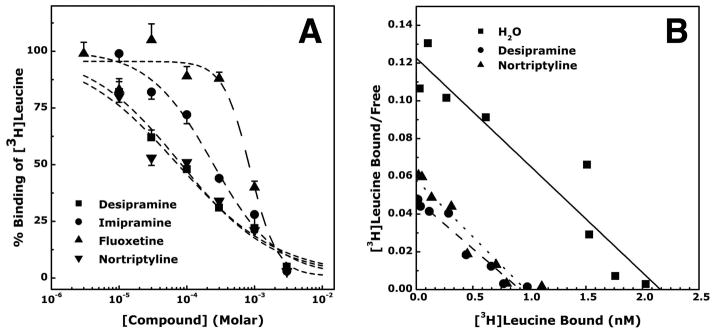
Binding of various antidepressants and other compounds to LeuT. (A). Affinity of desipramine, imipramine, fluoxetine and nortriptyline. Data are shown as mean ± SEM (vertical bars, N = 3). The IC50 values for inhibition [3H]leucine binding to LeuT were 80±5, 244±12, 858±64 and 75±14μM, respectively. (B). Mechanism of inhibition of [3H]leucine binding to LeuT by desipramine and nortriptyline. The plot shows that desipramine and nortriptyline are not competitive inhibitors of leucine binding to LeuT. A representative experiment is shown (N = 3).
To investigate the molecular basis of TCA binding to LeuT, we co-crystallized the transporter with desipramine and, by directly refining the diffraction data against the TCA-free LeuT structure (19) (Table S1), determined the crystal structure at 2.9 Å resolution (Figs. 2A&S3). The overall structure of the LeuT-desipramine complex (Fig. 2) is similar to that of the protein in the absence of desipramine (19), with an r.m.s.d. of 0.2 Å for all the non-hydrogen atoms. Neither the leucine substrate nor the two Na+ ions had moved. However, a 5-σ Fo-Fc electron density peak was observed at the inner end of the extracellular cavity of the protein (Fig. 2A, 2B&S3), which fits well with a desipramine molecule, an interpretation consistent with the inhibitory effect of the TCA molecule on LeuT’s transport activity (Fig. S2) and the evidence that desipramine is not a competitive inhibitor (Fig. 1B).
Fig. 2.

Structure of the LeuT-desipramine complex and molecular mechanism of LeuT inhibition by desipramine. (A). Structure shown as ribbon diagram viewed from within the membrane plane. An Fo-Fc map contoured at 3 σ is superimposed on the structural model. The EL4 hairpin is colored green, and the rest of the protein pink. The helices TM6 and TM11 are removed for clarity. (B). 2Fo-Fc map contoured at 1 σ showing the desipramine binding site in LeuT, viewed from within the membrane plane. Residues R30, Y108 and F253 form the extracellular gate that separates the leucine substrate from the bound desipramine. (C). Local structural changes of LeuT induced by desipramine binding. The structure with desipramine bound is shown in pink and green, without desipramine bound in cyan and blue. Upon desipramine binding, the side chain of R30 rotates toward D404 and forms a salt bridge with the latter, and the EL4 hairpin, along with A319 and F320, is pushed toward to the extracellular space. (D). Molecular contacts between LeuT and the bound desipramine molecule. The chemical structure of desipramine is shown together with LeuT residues that are in direct contact with the drug. Residues from the EL4 hairpin are shown in the green box, while residues from the rest of the protein are shown in pink boxes.
The bound desipramine molecule sits at the inner end of the extracellular cavity in LeuT with its three rings tilted at a 40° angle to the membrane plane (Fig. 2B). The “tail” of the molecule, an extended methylaminopropyl chain, projects toward the extracellular space. The desipramine molecule is separated from the substrate leucine by the extracellular gate of the transporter, which consists of residues R30, Y108 and F253. Importantly, the desipramine-binding site and the leucine-binding site are non-overlapping, but they do share F253 as a common residue. While F253 and Q34 make contact with the first and third rings of desipramine (Figs. 2B&2D, Table S2), respectively, R30 from TM1 forms cation-π interactions (21) with both the third desipramine ring and the phenylalanine ring of F253. On the extracellular side, desipramine is held in place by A319 and F320 of the turn of the EL4 helix hairpin, with the side chain of F320 engaging in hydrophobic interactions with the desipramine azepine ring and making contact with its tail. The desipramine tail also makes contact with residues L400 and D401 from the extracellular end of TM10. With a pKa of 10.2 (22) and with its nitrogen N2 atom being only 2.76 Å away from D401 (Fig. 2D, Table S2), desipramine probably forms a salt bridge with the aspartate residue. In total, 393 out of 469 Å2 the surface area of the desipramine molecule is buried by the protein. There is however still a substantial space in the protein on the extracellular side of the desipramine tail (Fig 2. 2B&3A), which could accommodate the tail of other types of TCA molecule.
Fig. 3.

Homology models and electrostatic surface potential of desipramine-binding sites in human SERT, NET and DAT. (A). Desipramine-binding site in the LeuT-desipamine crystal structure. Homology model and electrostatic surface potential of desipramine-binding site in (B). hSERT, (C). hNET and (D). hDAT, viewed from within the membrane plane. The equivalent residues of those in LeuT that are in direct contact with desipramine are indicated.
While the overall LeuT structure does not change upon desipramine binding several residues at this binding site as well as the backbone of the EL4 hairpin do move (Figs. 2A&C), via a typical “induced-fit” mechanism. Specifically, the guanidinium group of R30 is rotated by 170° about its Cδ–Nε bond towards D404, and two water molecules that were located between these two residues in the absence of bound desipramine (19) are no longer observed. As a result, R30 and D404, a pair of conserved charged residues, form a salt bridge, effectively sealing the substrate leucine off from the extracellular space. Also in TM10, the side chain of D401 is rotated towards desipramine. Finally, the bound desipramine pushes the EL4 hairpin toward the extracellular direction by ~1 Å. Coupled with a ~16 ° rotation of its phenylalanine ring, F320 effectively pins desipramine in place.
The crystal structure of LeuT-desipramine complex immediately suggests a mechanism for inhibition of substrate transport by the TCA molecule (Figs. 2A&S4). The desipramine molecule is held in place by a salt bridge it forms with residue D401 and by interactions with residues of the EL4 hairpin loop. Desipramine also directly binds to the extracellular gate of the transporter and locks the gate by inducing formation of a salt bridge between R30 of TM1 and D404 of TM10. The formation of this salt bridge prevents tilting of TM1, which is believed to be required for substrate release to the cytosol (19). Thus, no substrate transport can occur. This inhibition mechanism is in contrast from the competitive inhibition of the aspartate transporter Glt by threo-β-benzyloxyaspartate, in which the inhibitor binds partially to the substrate-binding site and concomitantly keeps the extracellular gate in an open position (23).
Human NSS proteins, as well as C. elegans and Drosophila DATs, share significant sequence homology with LeuT (Fig. S5). Given that they all bind desipramine, we built three-dimensional homology models for the human proteins in complex with the drug by directly threading their sequences onto our LeuT-desipramine structure (Fig. 3). Both the residues of the extracellular gate and the topology of the EL4 helix hairpin are conserved (Fig. S5). The TCA binding pocket in between is therefore likely to be conserved, and the homology models, which all show an acidic pocket at this position as in LeuT (Fig. 3B–D), support this. The difference in binding affinity to TCAs among NSS proteins is likely to depend upon sequence variations at the tip of the EL4 hairpin and at the extracellular end of TM10 (Fig. S5). Indeed, previous experiments of both loss-of-function mutagenesis for hNET (17) and gain-of-function mutagenesis (24) for hDAT in terms of TCA binding support a binding site at just this position for the human NSS proteins. Thus, we hypothesized that both the desipramine-binding site and the inhibition mechanism of substrate uptake by LeuT were conserved also in the human neurotransmitter transporters.
We tested the above two hypotheses by mutating key residues at the presumed TCA-binding sites in the human neurotransmitter transporters SERT and DAT, followed by measuring their transport inhibition by desipramine in human embryonic kidney cells. The IC50s of desipramine for inhibiting uptake by DAT, SERT and NET are 82,000 nM, 64 nM and 4.2 nM, respectively (10). We performed gain-of-function mutagenesis in terms of desipramine binding for both hDAT and hSERT proteins by mutating their key residues to those found in the sequence of hNET, the protein with the highest affinity to desipramine. We focused on residues at the tip of the EL4 hairpin and the extracellular end of the TM10 (Figs. 2B&D, S5). As a control experiment, we first mutated P387 in hDAT (the equivalent of F320 in LeuT) to an alanine. Since either a proline (as in C. elegans DAT) or an alanine (as in hNET) at this position can confer nanomolar affinity for TCAs, a proline -> alanine mutation should not further increase hDAT’s affinity for TCAs. Indeed, hDAT-P387A mutant showed little increase in inhibition compared with the wild-type at either 10 μM desipramine or 30 μM desipramine (Fig. 4), with no change in IC50 (Table S3). However, when I390 in the EL4 hairpin (equivalent of G323 in LeuT) and F472 in TM10 (equivalent of L400 in LeuT) were mutated into their corresponding residues in hNET (valine and leucine, respectively), the IC50 for inhibition of [3H]dopamine uptake by desipramine decreased by 2.8 and 4.8 fold, respectively (Fig. 4, Table S3). Similarly, in hSERT when K490 (corresponding to D401 in LeuT) was mutated into a threonine as found in hNET, a 2-fold decrease in IC50 of inhibition of [3H]serotonin uptake by desipramine was observed (Fig. 4, Table S3). Such gain-of-function mutagenesis data clearly demonstrate that desipramine binds to the same site in both hDAT and hSERT (Figs. 3B&D) as it does in LeuT (Fig. 3A) and inhibits transport activity in the same manner (Fig. 2A).
Fig. 4.

Measurements of inhibition of [3H]dopamine and [3H]serotonin uptake by desipramine for human DAT and SERT mutants in HEK-293 cells. Wild-type (WT) hDAT, hSERT and various mutant constructs are denoted on the left. Results are expressed as percent of uptake measured with vehicle (% control). Data are shown as mean ± SEM (horizontal bars, N = 3–5). Asterisk indicates P < 0.05 (compared with corresponding WT at the same concentration of desipramine, one-way analysis of variance followed by Dunnett multiple comparison test for hDAT, and Student’s t-test for hSERT).
Taken together, our results show that the TCA-binding site is probably conserved from bacterial to mammalian NSS proteins. Therefore, in the human Na+/Cl−-dependent neurotransmitter transporters SERT, NET and DAT it is likely that tricyclic antidepressants as well as selective serotonin reuptake inhibitors also bind between the extracellular gate and EL4 hairpin, thereby inhibiting neurotransmitter reuptake at the synapse. In combination with homology modeling (25) and molecular docking, the identification of this drug-binding site will allow studies on the interactions of SERT-, NET- and DAT-specific inhibitors (26) with these transporters and may aid in the structure-based design of more effective neurotransmitter reuptake inhibitors as antidepressants. As membrane proteins constitute 60–70% of the targets for small molecule drugs (27), the strategy employed in this work can probably be used in studying protein-drug interaction for other systems.
Supplementary Material
Acknowledgments
We are grateful to the staff at the X29 beamline at NSLS in Brookhaven National Laboratory for assistance in x-ray diffraction data collection. We thank B. Czyzewski, C. Soudant, A. Waight, J. Wu and R. Yang for helpful discussions and assistance. N.K.K. is a recipient of an American Heart Association Postdoctoral Fellowship. This work was financially supported by the NIH Roadmap and PSI-II Initiatives (GM075936 and GM075026 to D.N.W.) and NIH (DA019676 and DA013261 to M.E.A.R.). Atomic coordinates and structure factors have been deposited with the Protein Data Bank under access code 2QJU. The authors dedicate this work to the memory of Professor Kehsin Kuo.
Footnotes
Materials and Methods
References
References and Notes
- 1.Hertting G, Axelrod J. Nature. 1961;192:172. doi: 10.1038/192172a0. [DOI] [PubMed] [Google Scholar]
- 2.Amara SG, Kuhar MJ. Annu Rev Neurosci. 1993;16:73. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 3.Nelson N. J Neurochem. 1998;71:1785. doi: 10.1046/j.1471-4159.1998.71051785.x. [DOI] [PubMed] [Google Scholar]
- 4.Torres GE, Gainetdinov RR, Caron MG. Nat Rev Neurosci. 2003;4:13. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- 5.Blakely RD, Defelice LJ, Galli A. Physiology (Bethesda) 2005;20:225. doi: 10.1152/physiol.00013.2005. [DOI] [PubMed] [Google Scholar]
- 6.Iversen L. Br J Pharmacol. 2006;147:S82. doi: 10.1038/sj.bjp.0706428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schloss P, Williams DC. J Psychopharmacol. 1998;12:115. doi: 10.1177/026988119801200201. [DOI] [PubMed] [Google Scholar]
- 8.Schafer WR. Cell. 1999;98:551. doi: 10.1016/s0092-8674(00)80042-2. [DOI] [PubMed] [Google Scholar]
- 9.Klimek V, et al. J Neurosci. 1997;17:8451. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eshleman AJ, et al. J Pharmacol Exp Ther. 1999;289:877. [PubMed] [Google Scholar]
- 11.Gumnick JF, Nemeroff CB. J Clin Psychiatry. 2000;61(Suppl 10):5. [PubMed] [Google Scholar]
- 12.Brunton LL, Lazoskip JS, Parker KL. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. McGraw-Hill; Columbus, OH: 2005. p. 451. [Google Scholar]
- 13.Chen NH, Reith MEA. In: Neurotransmitter Transporters: Structure, Function, and Regulation. Reith MEA, editor. Humana Press; Totowa, NJ: 2002. pp. 53–109. [Google Scholar]
- 14.Saier MH., Jr Microbiology. 2000;146:1775. doi: 10.1099/00221287-146-8-1775. [DOI] [PubMed] [Google Scholar]
- 15.Jayanthi LD, et al. Mol Pharmacol. 1998;54:601. [PubMed] [Google Scholar]
- 16.Pörzgen P, Park SK, Hirsh J, Sonders MS, Amara SG. Mol Pharmacol. 2001;59:83. doi: 10.1124/mol.59.1.83. [DOI] [PubMed] [Google Scholar]
- 17.Roubert C, et al. J Biol Chem. 2001;276:8254. doi: 10.1074/jbc.M009798200. [DOI] [PubMed] [Google Scholar]
- 18.Materials and methods are available supporting materials on Science Online.
- 19.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Nature. 2005;437:215. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 20.Quick M, Javitch J. Proc Natl Acad Sci U S A. 2007;104:3603. doi: 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallivan JP, Dougherty DA. Proc Natl Acad Sci U S A. 1999;96:9459. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantu MD, Hillebranda S, Carrilho E. J Chromatogr A. 2005;1068:99. doi: 10.1016/j.chroma.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Nature. 2007;445:387. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 24.Mortensen OV, Amara SG. J Neurochem. 2006;98:1531. doi: 10.1111/j.1471-4159.2006.04060.x. [DOI] [PubMed] [Google Scholar]
- 25.Forrest LR, Tavoulari A, Zhang YW, Rudnick G, Honig B. Proc Natl Acad Sci U S A. 2007;104:12761. doi: 10.1073/pnas.0705600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olivier B, Soudijn W, van Wijngaarden I. Prog Drug Res. 2000;54:59. doi: 10.1007/978-3-0348-8391-7_3. [DOI] [PubMed] [Google Scholar]
- 27.Drews J. Science. 2000;287:1960. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.