

Abstract
To date, while various diagnostic approaches for pathogen detection have been proposed, most are too expensive, lengthy or limited in specificity for clinical use. Nanoparticle systems with unique material properties, however, circumvent these problems and offer improved accuracy over current methods. Herein, we present novel magneto-DNA probes capable of rapid and specific profiling of pathogens directly in clinical samples. A nanoparticle hybridisation assay, involving ubiquitous and specific probes that target bacterial 16S rRNAs, was designed to detect amplified target DNAs using a miniaturised nuclear magnetic resonance device. Ultimately, the magneto-DNA platform allowed both universal and specific detection of various clinically relevant bacterial species, with sensitivity down to single bacteria. Furthermore, the assay was robust and rapid, simultaneously diagnosing a panel of 13 bacterial species in clinical specimens within 2 hours. The generic platform described could be used to rapidly identify and phenotype pathogens for a variety of applications.
Keywords: bacteria, 16S rRNA, magnetic nanoparticles, μNMR
Rapid and sensitive detection of pathogenic bacteria is crucial for improving patient care with proper antibiotic treatment, preventing the spread of disease, and identifying the source of infection in hospitals, home or field settings1–3. To date, a variety of diagnostic approaches have been proposed, each varying in sensitivity, specificity, cost, and efficacy4–7. In particular, strategies based on the polymerase chain reaction (PCR) and sequencing have shown promise as highly sensitive tools for microbiological identification8–11. However, real-time PCR-based systems are often still too expensive for resource limited environments12, and current sequencing techniques still lack practical applicability to patient care5. Bacterial culture and biochemical staining remain the clinical gold standard despite long procedural times (up to several days) and limitations in identifying certain species. There is thus a need for generic, accurate and point-of-care platforms that allow both pathogen detection and phenotyping. Such systems could have far-reaching benefits to other sectors including food industries, shipping and export businesses, defense, and agriculture.
We herein report a new diagnostic platform for rapid detection and phenotyping of common clinical pathogens. The assay utilises magnetic nanoparticles and oligonucleotide probes to specifically detect target nucleic acids from the pathogen. In particular, we hypothesised that ribosomal RNA (rRNA) sequence information from microorganisms could be used in a robust magneto-DNA assay. Because this magnetic detection strategy allows near background-free sensing, the assay steps are greatly simplified and detection is much faster. For bacterial detection, we selected 16S rRNA, a component of the 30S small subunit of bacterial ribosomes13, as the target marker, since a single bacterium contains many 16S rRNA strands (103 – 105 strands)14. Furthermore, the strands have a high degree of sequence consensus across species (important for general bacterial detection) as well as species-specific variable regions (important for species typing)15,16. For bacterial phenotyping (e.g., identifying drug resistance), targeting of specific mRNA sequences was done in parallel with species detection. In this study, rather than sequencing the whole RNA strand, we established a series of primers and probes for amplification and detection of specific regions of interest within common bacterial types. For signal readout, we used a miniaturised micro-nuclear magnetic resonance (μNMR) system that requires only small volumes of sample for processing (~2 μL) and is also capable of supporting rapid, high-throughput operations in point-of-care settings17–19.
Design and validation of assay
The magneto-DNA assay is based on a sandwich hybridisation technique wherein two oligonucleotide probes bind to each end of the target nucleic acid (Figure 1a). Total RNA is extracted from a specimen, and target regions within the 16S rRNA are amplified by asymmetric RT-PCR to produce large numbers of single-strand DNA of only the sense (or antisense) sequences. The resultant DNAs are then captured by polymeric microspheres conjugated with probe oligonucleotides (bead-capture probe). Subsequently, the overhanging edges of the target DNA is hybridised with magnetic nanoparticle (MNP)-detection probe conjugates (MNP-detection probe). In turn, these magnetically-labeled beads shorten the transverse relaxation rate (R2) of samples, which is then detected by a miniaturised μNMR device. The developed detection method is both robust and highly sensitive not only because of the multiple 16S rRNA strands per bacterium (as opposed to a single strand of genomic DNA) but also because of the 3-steps of signal amplification, including: i) PCR amplification of the target nucleic acids; ii) bead capture and enrichment of target nucleic acids; and iii) magnetic amplification,(since a single MNP can affect billions of surrounding water molecules17).
Figure 1. Magneto-DNA assay for the detection of bacterial 16S rRNA.
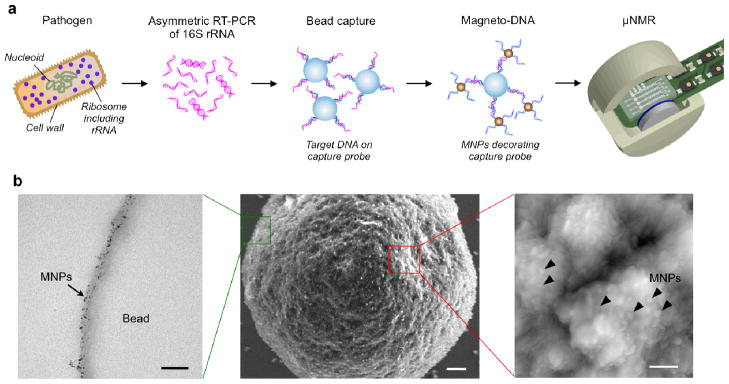
a, Schematic of the assay procedure. Total RNA is extracted from the specimen, and the 16S rRNA is amplified by asymmetric real time-PCR. Single-strand DNA of the amplified product is then captured by beads conjugated to capture probes, before hybridising with MNPs to form a magnetic sandwich complex. Samples are subsequently analyzed using a μNMR system. b, Hybridised probe complexes, as observed by transmission electron microscopy (left, size bar 100 nm), scanning electron microscopy (center, size bar 300 nm), and atomic force microscopy (right, size bar 100 nm).
Probes specific to each bacterial target were designed through comparative analyses of 16S rRNA gene sequences from different types of bacterial species (see Supplementary Table S1 for details). By aligning multiple sequences from several different genera, we identified both conserved as well as variable regions; both regions were subsequently selected as targets. Primers were designed to flank the target sequences for hybridisation. For each type of bacteria, an optimal primer set was screened in order to maximise the amount of single-stranded DNA produced by asymmetric PCR. Two oligonucleotide sequences that were complementary to (or near) the 5′ end and 3′ end of the amplicon sequence were selected as probes. One probe type was used as the capture probe and conjugated onto polystyrene beads (diameter, 3 μm). The other detection probe was conjugated to MNPs (diameter, 20 nm) for magnetic detection by the NMR system. The numbers of probes per bead and MNP were 300,000 ~ 800,000 and 16 ~ 29, respectively (Supplementary Table S2). After target binding, the beads were densely covered with MNPs, as confirmed by both electron and atomic force microscopy (Figure 1b, S1–3). The estimated MNP number per targeted bead was ~3 × 105. Control samples, on the other hand, showed negligible MNPs on the bead surface, which validated the high specificity of the sequence hybridisation.
We first determined the detection sensitivity of the magneto-DNA assay, using Staphylococcus aureus (S. aureus) as the model organism. Both synthetic oligonucleotides and amplified target amplicons from bacteria-derived RNA were prepared. Titration experiments revealed that the limit of detection (LOD) was ≈ 0.5 pM [DNA] (Figure 2a; see Methods for details). Since the sample volume for the current assay was 50 μl, the minimum amount of target DNA needed for detection was 25 amol or 1.5 × 106 molecules. When samples with varying numbers of S. aureus were used, the bacterial detection sensitivity was close to a single bacterium (Figure 2b). Furthermore, depending on the pathogen density, the PCR cycles could be adjusted to achieve optimal dynamic ranges for detection. Note that errors at lower bacterial concentration were higher, reflecting sampling errors from serial dilution. Similar results were shown for detection sensitivity in blood using Escherichia coli (E. coli), one of the major pathogens that cause sepsis, for which early diagnosis by detecting only a few bacteria in blood is crucial. When serial dilutions of E. coli were spiked into whole blood and processed for detection, we were able to detect as low as 1–2 bacteria per 10 ml of blood (Figure 2c). Importantly, we were also able to estimate bacterial load over several log orders. The high sensitivity and robustness of the assay is attributed to the abundance of 16S rRNA (103 – 105) per bacterium, which can be easily amplified and detected despite inevitable loss during sample processing. Indeed, because of this high detection sensitivity, an extremely small amount of sample was needed (~0.1% of total volume per sample) not only for detection but also for further characterization by other assays (e.g., standard culture, real-time PCR).
Figure 2. Detection sensitivity of the magneto-DNA system.
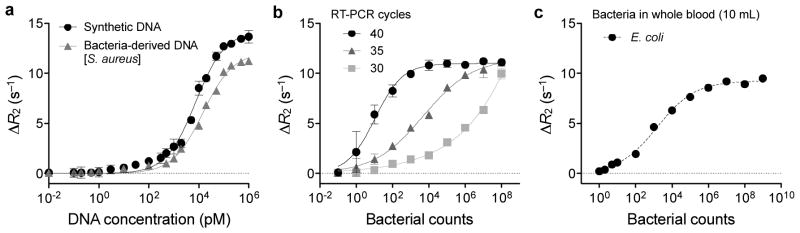
a, Serial dilutions of synthetic DNA or bacteria-derived DNA were used as detection targets. Bacteria-derived DNA molecules were obtained via asymmetric RT-PCR of S. aureus 16S rRNA (35 cycles); synthetic DNA had the same sequence as bacteria-derived DNA. The detection limit was ~ 0.5 pM. b, Bacterial detection by the magneto-DNA assay. Samples with varying numbers of S. aureus were used. Total RNA was extracted and target sequences were amplified by 30, 35, and 40 cycles of RT-PCR. Amplified target DNA were detected using the probe set in a. The observed detection limit was a single bacterium, and the dynamic range of detection could be controlled by changing the PCR cycle number. c, Bacterial detection in blood. Serial dilutions of E. coli were spiked into human blood and processed by first lysing the red blood cells and then extracting the RNA using the same procedure as described above. All experiments were done in triplicate. All ΔR2 values were obtained by subtracting the relaxation rate values of the hybridised probe complexes in the presence of target DNA (R2,target), with the relaxation rate values of the beads alone (R2,control). Data are expressed as mean ± SD.
Universal and species-specific detection of pathogens
We next adapted the assay as a generic platform that can comprehensively detect the presence of different pathogens. A set of universal probes were developed by identifying a highly conserved region of 16S rRNA genes from a large pool of different bacterial species (Figure 3a). Two sequences with a single base difference were selected to serve as the capture probe, and another (single) sequence was selected for detection. When the probe was tested on individual species (Figure 3b), the observed R2 values were highly consistent, indicating that different species could be detected using a common probe. Mixtures of different bacterial types could also be detected as a whole (Figure 3c). The R2 values normalised by bacterial numbers were within a close range for each mixture; this confirms the universal nature of the probes in quantifying the total bacterial load of a given sample.
Figure 3. Universal detection of bacteria using the magneto-DNA system.

a, Sequences of universal probes targeting a conserved region of bacterial 16S rRNA. Two probe sequences with a single-base difference were blended and used for capture. (R, Y) = (A, T) for Staphylococcus, Escherichia, Pseudomonas, Klebsiella, Enterobacter, Haemophilus, Stenotrophomonas; (R, Y) = (G, C) for Streptococcus, Enterococcus, Acinetobacter, Proteus, Lactobacillus. b, Thirteen different bacterial species could each be detected using the universal probes. c, Mixtures containing different bacterial types were detected by the universal probes. The observed ΔR2 values were consistent with average ΔR2 value (dotted line) from single species. Data are expressed as mean ± SD. All samples for the assay were prepared in triplicate.
We further extended the magneto-DNA assay to the identification of different target pathogens. A panel of probes were thus designed to target a hypervariable region within the 16S rRNA gene sequences of different bacterial species (Supplementary Table S1, Figure S4). To minimise non-specific hybridisation, we ensured that the homology of the sequences between each genus type was less than 50%. Figure 4a shows an example of bacterial detection. Using specific probes for Staphylococcus, the amplified DNA from S. aureus 16S rRNA could be detected with negligible background signals from other species. Likewise, all specially-designed probes for each bacterial type showed high selectivity with minimal off-target binding (Figure 4b, c). Notably, the specificity of the magneto-DNA assay was superior to that of quantitative real-time PCR (qPCR; Figure 4d, S5), a result that can be attributed to the beneficial aspects of the sandwich assay. For magneto-DNA sensing, both capture and detection probes must bind to their target to generate a signal. The incubation conditions (e.g., stringency) can also be controlled to minimise non-specific binding. Finally, the washing steps following each hybridisation cycle in our assay allow removal of any unbound/non-specifically bound targets or probes. The high specificity of the magneto-DNA assay enabled reliable and rapid bacterial typing. Even in mixtures of different bacterial species, we could identify and quantify specific bacterial types (Figure S6). Furthermore, by expanding the assay to the detection of mRNA, we could differentiate phenotypes (e.g., drug resistance) within closely related species. For example, we were able to detect the mRNA of mecA and Panton-Valentine leukocidin (PVL) genes20,21, which in turn enabled us to identify Methicillin-resistant Staphylococcus aureus (MRSA; Figure S7).
Figure 4. Differential detection using the magneto-DNA system.

Probes targeting hypervariable regions of bacterial 16S rRNA sequences were used to specifically detect various bacterial types. RNA was extracted from bacterial cultures, amplified by asymmetric RT-PCR (35 cycles) using specific primers for each species, and detected using the corresponding probe conjugates. a, Probes specific for Staphylococcus were used for detecting S. aureus (DNA amount equivalent to 50,000 CFU). Target DNA from other bacterial species were added as controls to test off-target binding of the probes. b, Relaxation rates for differential detection of various bacterial types. Note the high specific signals and low background noise against other bacteria. Data are expressed as mean ± SD. All samples for the assay were prepared in triplicate. c,d, Heat maps comparing the specificity of the magneto-DNA assay with that of qPCR. Specificities in c were based on ΔR2 values from the magneto-DNA assay shown in b. Specificities in d are relative target amounts obtained from qPCR in Figure S5. Significant signals were marked as positive: positive signals for specific target bacteria were regarded as ‘true-positive’, while positive signals from non-targeted samples were classed as ‘false-positive’.
Clinical testing
We evaluated the clinical utility of the magneto-DNA assay using patient specimens. Aspirated samples from patients with suspected infections were collected and analyzed by conventional culture (procedural time: 3–5 days) as well as the magneto-DNA assay (procedural time: 2 hours). Figure 5 shows the results from the magneto-DNA assay. To place these findings in perspective, Table 1 compares our magneto-DNA assay results to those obtained using current gold standard methods. As made clear by this table, the magneto-DNA assay showed excellent accuracy, detecting all bacterial species identified by standard culture. Interestingly, however, the magneto-DNA assay was also able to identify other species (Citrobacter in sample 4 and Acinetobacter in sample 6) that were left undetected by standard culture. Considering that these organisms could be detected by qPCR (Table 1), it is possible that growth inhibition of species may have occurred during processing. Both Citrobacter and Acinetobacter belong to a group of gram-negative bacteria that are poorly targeted by commonly used third generation cephalosporins due to their capacity to produce inducible beta-lactamase. Thus, the clinical implications of being able to detect such organisms (undetected by standard culture) with the magneto-DNA assay are significant. Furthermore, by having the ability to detect pathogens directly in the specimen, the magneto-DNA assay not only overcomes problems of growth competition, but also minimises sample contamination. Diagnostic accuracy is thus maximised. Whilst these results are encouraging, further in-depth studies on these culture-negative organisms are clearly needed. This will require developing the magneto-DNA assay for even higher-throughput, and performing larger prospective clinical trials.
Figure 5. Diagnosis of clinical samples.

Detection of pathogens by the magneto-DNA nanoparticle system using universal (a), and specific (c) probes for each bacteria type. b, Heatmap of obtained ΔR2 values for universal and specific detection. Clinical specimens (300 μl for each sample) were processed to extract total RNA. This was followed by asymmetric RT-PCR (35 cycles) with universal and specific primers for all bacterial types. The PCR products (equivalent to 0.3 μl volume of sample) were magnetically labeled and detected by μNMR. a, Two of the 9 clinical samples tested negative, which correlated well with standard culture results. The other 7 samples were positive. ND represents samples with no pathogens detected. c, Identification of pathogen types within each sample. Note that some samples were infected with more than one pathogen, and the identified bacterial types correlated well with standard culture (see also Supplementary Table S3). Data are expressed as mean ± SD. All samples for the assay were prepared in triplicate.
Table 1.
Comparison of bacterial detection in clinical samples: the magneto-DNA system versus gold standard procedures.
| Patient no. | Type of specimen | Magneto-DNA* | Gold standard | ||
|---|---|---|---|---|---|
| Universal | Differential | Bacterial culture** | qPCR | ||
| 1 | Urine | Positive (5.9) | Staphylococcus (3.8) Enterococcus (3.6) Proteus (2.0) |
Staphylococcus (+) Enterococcus (++) Proteus mirabilis (+++) |
Culture results confirmed |
| 2 | Pleural fluid | Positive (6.8) | Enterococcus (2.3) Enterobacter (7.2) |
Enterococcus (+++) Enterobacter (+++) |
Culture results confirmed |
| 3 | Kidney urine | Negative (< 0.1) | None | None | Negative |
| 4 | Kidney urine | Positive (6.4) | Escherichia (5.0) Citrobacter (3.9) |
Escherichia (+++) |
Culture results confirmed and also Citrobacter |
| 5 | Biliary fluid | Positive (2.4) | Enterococcus (2.4) | Enterococcus (++) | Culture results confirmed |
| 6 | Pelvic abscess | Positive (7.2) | Stenotrophomonas (3.0) Staphylococcus (2.5) Lactobacillus (1.5) Acinetobacter (2.1) |
Stenotrophomonas (+++) Staphylococcus (+++) Lactobacillus (+) |
Culture results confirmed and also Actinobacter |
| 7 | Kidney urine | Positive (6.5) | Escherichia (5.7) | Escherichia (+) | Escherichia |
| 8 | Kidney urine | Positive (4.3) | Enterococcus (2.0) Escherichia (5.5) Lactobacillus (1.8) |
Enterococcus (++) Escherichia (+) Lactobacillus (++) |
Culture results confirmed |
| 9 | Ascitic fluid | Negative (< 0.1) | None | None | Negative |
Values for magneto-DNA assay represent ΔR2 values. Higher ΔR2 values indicate higher bacterial abundance.
Detection levels: abundant (> 100,000 CFU/ml) for +++, moderate (10,000 to < 100,000 CFU/ml) for ++, and few (100 to < 10,000 CFU/ml) for +.
Conclusions
In the current study, we designed a dual probe-nanoparticle system capable of detecting and phenotyping common human pathogens. The method is robust, fast (< 2 hours), sensitive, accurate and potentially adaptable to a wide variety of other pathogens. It thus has the potential for guiding decisions across various clinical scenarios. We used NMR as the primary read-out for detecting targets labeled with MNPs17,22. While nanoparticle based magnetic detection methods of whole bacteria have been previously reported, they have largely relied on the use of either small molecule affinity ligands or antibodies19,23,24. These approaches tend to have very limited sensitivity and are not comprehensive enough to be adapted to broad clinical use (i.e., they are unable to detect gram-negative bacteria, resistant strains or specific species). The magneto-genetic assay presented not only overcomes these issues, but also offers a platform technology that can be easily applied to the clinic as well as other point-of-care settings.
Nucleic acid-based techniques allow the specific typing of species with high enough sensitivity to circumvent lengthy culturing processes. 16S rRNA sequence information has been used for bacterial classification and taxonomy, and thus typing methods based on these databases have emerged as the preferred technique for microbial identification over traditional culture and biochemical assays. Since the advent of genomic sequencing technologies, extensive databases have been established25, and these have been widely used to develop real-time PCR assays for identification of specific targets. Metagenomic studies also represent a novel approach for identifying microbial communities in heterogenous samples, and could thus be useful for the diagnosis of clinical specimens26,27. Despite such advances, however, the development of a robust diagnostic platform for systematically detecting bacteria in point-of-care settings has remained a challenge28,29. Various technical and practical problems, such as the propensity for false positives (especially for standard PCR) along with prohibitive cost issues (e.g., Taqman assays, LightCycler probes, DNA/RNA sequencing, etc.) have been major obstacles. Here, using validated, readily available bacterial sequence information, we designed a nucleic acid probe-based magnetic detection assay to identify some relevant human pathogens.
In view of the high sensitivity exhibited by the magneto-DNA nanoparticle system (down to single bacteria), it could potentially be used for early diagnosis or detection of rare pathogens in dilute samples. In addition, the method has proven to be rapid and robust, with high specificity as well as low background. Moreover, the magneto-DNA approach has considerable advantages over standard culture and real-time PCR systems, particularly in terms of assay time and cost; it can also be easily applied to point-of-care scenarios. Going forward, we envision that this generic approach could have far-reaching applications, possibly in conjunction with newly emerging single cell magnetic detection methods30.
Methods
Primer and probe design
To select a target region for amplification and hybridisation, we used the 16S rRNA gene sequences of 30 different bacterial genera (from the NCBI nucleotide database). All sequences were aligned using the MegAlign software (DNASTAR, Inc, Madison, WI), and a high consensus region was selected for the universal target, and a low consensus (variable) region for the differential target. For detecting antibiotic resistance, specificity regions within the mRNA sequences of mecA and Panton-Valentine leukocidin (obtained from NCBI database) were selected as the target sequences. For targeting the 16S rRNA of 13 different genus types (Staphylococcus, Streptococcus, Enterococcus, Escherichia, Pseudomonas, Klebsiella, Enterobacter, Citrobacter, Acinetobacter, Proteus, Haemophilus, Stenotrophomonas, Lactobacillus) as well as the mRNAs of the two drug resistance-related genes for MRSA, pairs of specific oligonucleotide probes (length 18 – 22 nucleotides) were designed to be complementary to sequences within the target regions, with one hybridising to a 5′ end of the target and the other hybridising to the 3′ end (see Supplementary Table S1). Multiple adenine residues (AAAAA or AAAAAA, oligoA) were then added to the external end of the probes, with a thiol group at the terminal end of the oligoA linker. Primers were designed so that all amplicons (50 ~ 70 nucleotides in length) would include the target regions. All oligonucleotides used for the primers and probes were custom-synthesised and provided by Integrated DNA Technologies (Coralville, IA). Probe specificity was tested by polyacrylamide gel electrophoresis (See Supplementary methods).
Probe conjugations
For bead-capture probe conjugation, 3 mg of amine-functionalised polystyrene beads (diameter, 3 μm; Polysciences) were first reacted with 292 μg of sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo SMCC, Thermo Scientific, Rockford, IL) in phosphate buffered saline (PBS) with 10 mM sodium bicarbonate (4 h, room temperature). The activated beads were then extensively washed with PBS. 50 nmoles of thiol-modified oligonucleotide probes (capture probes) were treated with 125 mM dithiothreitol in PBS with 10 mN sodium hydroxide for 2 h, and the mixture was purified by illustra Microspin G-25 columns (GE Healthcare, Pittsburgh, PA). Sulfo SMCC-activated beads and the deprotected capture probes were then mixed and reacted for 12 h at 4 °C. Unreacted probes were removed by extensive washing with PBS. To prepare magnetic nanoparticle (MNP)-detection probe conjugates, we used dextran-coated iron oxide nanoparticles with a size of 21 nm (r2= 51 s−1mM−1Fe)31. 0.5 mg of MNPs, which had 22 free amine groups per particle, were activated with 472 μg of sulfo-SMCC for 4 h, and extensively washed using Amicon Ultra centrifugal filters (Millipore, Billerica, MA). Thiol-modified detection probes were pre-treated for deprotection as described above. The sulfo SMCC-activated MNPs and the detection probes were mixed and reacted for 12 h at 4 °C. The mixture (MNP-detection probe) was purified using Sephadex G-100 columns (Millipore). The amount of oligonucleotides conjugated onto the beads and MNPs were quantified using the Qubit DNA quantification kit (see Supplementary methods).
Bacteria culture and RNA extraction
All bacteria were purchased from American Type Culture Collection (ATCC, Manassas, VA). Bacteria were each seeded and cultured in suspension using the following medium: Staphylococcus aureus (S. aureus; #25923) in Staphylococcus broth (BD Biosciences, Sparks, MD); Streptococcus pneumoniae (S. pneumoniae; #6318) and Enterococcus faecalis (E. faecalis; #29212) in trypticase soy broth containing 5% defibrinated sheep lood (Hemostat Laboratories, Dixon, CA); Escherichia coli (E. coli; #25922) in Luria-Bertani (LB) media (BD Biosciences); Pseudomonas aeruginosa (P. aeruginosa; #142), Klebsiella pneumoniae (K. pneumoniae; #43816), and Methicillin-resistant Staphylococcus aureus (MRSA-mecA+; #BAA-1720 and MRSA-VPL+; BAA-1707) in trypticase soy broth; Enterobacter aerogenes (E. aerogenes; #13048), Citrobacter freundii (C. freundii; #6879), Acinetobacter baumannii (A. baumannii, #15149), and Proteus mirabilis (P. mirabilis, #7002) in nutrient broth (BD Biosciences); Haemophilus influenzae (H. influenzae, 49247) in trypticase soy broth containing 5% lysed blood; Stenotrophomonas maltophilia (S. maltophilia, #17671) in trypticase soy broth; and Lactobacillus gasseri (L. gasseri, #4963) in Lactobacilli MRS broth (BD Biosciences). For RNA extraction, bacteria were first centrifuged (8,000 rpm, 10 min) and pellets were treated with Max Bacterial Enhancement Reagent (Life Technologies, Grand Island, NY), and then lysed using TRIzol (Life Technologies). After solvent extraction of the RNA followed by precipitation and washing, the final RNA yield was measured using Nanodrop 1000 (Thermo Scientific). To determine detection sensitivity in blood, human blood specimens were collected from healthy donors and serial dilutions of cultured E. coli were added. For RNA extraction, red blood cells were first removed using the ACK (Ammonium-Chloride-Potassium) Lysing Buffer (MPBio) and then treated with the Max Enhancement reagent. The procedure for RNA extraction then continued as for pure bacterial cultures.
Clinical samples
This proof-of-principle study was approved by the Partners Institutional Review Board. Excess and discarded samples were collected from nine subjects with clinical suspicion for infected bodily fluid or abscess and referred for drainage of such specimens. Specimens were collected using routine image guided approaches by MGH Interventional Radiology physicians and analyzed blindly with the magneto-DNA assay, before being compared to conventional culture results. For RNA extraction, 300 μl volume of the specimens were centrifuged to form pellets, and treated as described above. In the case of specimens containing a high content of blood, samples were repeatedly centrifuged and pellets were treated with the ACK Lysing Buffer to remove the red blood cells, before treating with the Max Enhancement reagent.
RT-PCR
The cDNA from each bacterial RNA or RNA from the clinical samples were synthesised using the Reverse Transcription System (Promega, Madison, WI) with thermal cycling conditions of 42 °C for 60 min, followed by 70 °C for 5 min (MasterCycler, Eppendorf). For asymmetric PCR, cDNAs were amplified using Taq DNA Polymerase (Qiagen, Valencia, CA) and specific primers for each bacteria type (Supplementary Table S1). Either the forward or reverse primers were added in excess (relative to the other primer). The following thermal cycling conditions were used: initiation (94 °C, 5 min); 30 – 40 cycles of denaturation (94 °C, 30 sec), annealing (55 °C, 30 sec), extension (94 °C, 30 sec); and termination (72 °C, 7 min). The final PCR products were validated by polyacrylamide gel electrophoresis.
Magnetic sandwich assay
For sandwich hybridisation, 1 – 3 μl of the PCR reaction mixture solution were first mixed with the bead-capture probe conjugates and incubated in hybridization buffer (DIG Hyb, Roche Diagnostics) at 40 °C for 15 min. Unbound nucleic acids were removed by washing the beads with hybridization buffer. MNP-detection probe conjugates were then added (50 μg/ml) and the mixture was incubated in the same hybridisation buffer at 40 °C for 15 min. The bead-MNP complexes were washed again with hybridisation buffer and finally with PBS. The μNMR measurements were performed using a previously reported miniaturised μNMR device32. Transverse relaxation times were measured using Carr–Purcell–Meiboom–Gill pulse sequences with the following parameters: echo time, 3 ms; repetition time, 4 s; number of 180° pulses per scan, 900; number of scans, 7. All measurements were done in triplicate, and data are expressed as mean ± standard deviation (SD). Limit of detection (LOD) values were determined by calculating 3×(SD of background signal), and limit of quantification (LOQ) values were determined by calculating 10×(SD of background signal)33.
Supplementary Material
Acknowledgments
We would like to thank Y. Fisher-Jeffes for review of the manuscript, N. Sergeyev for synthesis of MNPs, S. Chen and M. Mckee for help with the electron microscopy, C. Min for help with the μNMR device, and J. Chung, K. S. Yang, H. Shao, and A. V. Ullal for many helpful discussions. The work was funded in parts by National Institute of Health grants R01EB004626, R01EB010011, HHSN268201000044C, R01HL113156.
Footnotes
Author contributions
H.J.C. designed and performed the research, and co-wrote the manuscript. R.W. and H.L. designed the research and wrote the manuscript. R.W. provided overall guidance, and H.L. reviewed the magnetic resonance measurement data. C.M.C. provided guidance and assistance on the clinical studies. H.I. performed the scanning electron microscopy and atomic force microscopy. All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interests.
Supplementary information accompanies this paper at www.nature.com/naturenanotechnology.
Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Allegranzi B, et al. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis. The Lancet. 2011;377:228–241. doi: 10.1016/S0140-6736(10)61458-4. [DOI] [PubMed] [Google Scholar]
- 2.Polin RA, et al. Epidemiology and Diagnosis of Health Care–Associated Infections in the NICU. Pediatrics. 2012;129:e1104–e1109. doi: 10.1542/peds.2012-0147. [DOI] [PubMed] [Google Scholar]
- 3.Klompas M, Yokoe DS, Weinstein RA. Automated surveillance of health care–associated infections. Clin Infect Dis. 2009;48:1268–1275. doi: 10.1086/597591. [DOI] [PubMed] [Google Scholar]
- 4.Giljohann DA, Mirkin CA. Drivers of biodiagnostic development. Nature. 2009;462:461–464. doi: 10.1038/nature08605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loman NJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 6.Sauer S, Kliem M. Mass spectrometry tools for the classification and identification of bacteria. Nat Rev Microbiol. 2010;8:74–82. doi: 10.1038/nrmicro2243. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Cu YT, Luo D. Multiplexed detection of pathogen DNA with DNA-based fluorescence nanobarcodes. Nat Biotechnol. 2005;23:885–889. doi: 10.1038/nbt1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dark PM, Dean P, Warhurst G. Bench-to-bedside review: the promise of rapid infection diagnosis during sepsis using polymerase chain reaction-based pathogen detection. Crit Care. 2009;13:217. doi: 10.1186/cc7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pechorsky A, Nitzan Y, Lazarovitch T. Identification of pathogenic bacteria in blood cultures: comparison between conventional and PCR methods. J Microbiol Methods. 2009;78:325–330. doi: 10.1016/j.mimet.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Ottesen EA, Hong JW, Quake SR, Leadbetter JR. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science. 2006;314:1464–1467. doi: 10.1126/science.1131370. [DOI] [PubMed] [Google Scholar]
- 11.Loman NJ, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 12.Niemz A, Ferguson TM, Boyle DS. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011;29:240–250. doi: 10.1016/j.tibtech.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward DM, Weller R, Bateson MM. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature. 1990;345:63–65. doi: 10.1038/345063a0. [DOI] [PubMed] [Google Scholar]
- 14.Yang S, et al. Quantitative multiprobe PCR assay for simultaneous detection and identification to species level of bacterial pathogens. J Clin Microbiol. 2002;40:3449–3454. doi: 10.1128/JCM.40.9.3449-3454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajendhran J, Gunasekaran P. Microbial phylogeny and diversity: small subunit ribosomal RNA sequence analysis and beyond. Microbiol Res. 2011;166:99–110. doi: 10.1016/j.micres.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Woo PC, Lau SK, Teng JL, Tse H, Yuen KY. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 2008;14:908–934. doi: 10.1111/j.1469-0691.2008.02070.x. [DOI] [PubMed] [Google Scholar]
- 17.Lee H, Sun E, Ham D, Weissleder R. Chip-NMR biosensor for detection and molecular analysis of cells. Nat Med. 2008;14:869–874. doi: 10.1038/nm.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Bioorthogonal chemistry amplifies nanoparticle binding and enhances the sensitivity of cell detection. Nat Nanotechnol. 2010;5:660–665. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Yoon TJ, Figueiredo JL, Swirski FK, Weissleder R. Rapid detection and profiling of cancer cells in fine-needle aspirates. Proc Natl Acad Sci USA. 2009;106:12459–12464. doi: 10.1073/pnas.0902365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wada M, et al. Quantitative reverse transcription-PCR assay for the rapid detection of methicillin-resistant Staphylococcus aureus. J Appl Microbiol. 2010;108:779–788. doi: 10.1111/j.1365-2672.2009.04476.x. [DOI] [PubMed] [Google Scholar]
- 21.Tristan A, et al. Global distribution of Panton-Valentine leukocidin--positive methicillin-resistant Staphylococcus aureus, 2006. Emerg Infect Dis. 2007;13:594–600. doi: 10.3201/eid1304.061316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez JM, Josephson L, O’Loughlin T, Hogemann D, Weissleder R. Magnetic relaxation switches capable of sensing molecular interactions. Nat Biotechnol. 2002;20:816–820. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]
- 23.Chung HJ, et al. Ubiquitous Detection of Gram-Positive Bacteria with Bioorthogonal Magnetofluorescent Nanoparticles. ACS Nano. 2011;5:8834–8841. doi: 10.1021/nn2029692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budin G, Chung HJ, Lee H, Weissleder R. A magnetic Gram stain for bacterial detection. Angew Chem Int Ed Engl. 2012;51:7752–7755. doi: 10.1002/anie.201202982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser CM, Eisen JA, Salzberg SL. Microbial genome sequencing. Nature. 2000;406:799–803. doi: 10.1038/35021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tringe SG, et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 27.Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55:856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ince J, McNally A. Development of rapid, automated diagnostics for infectious disease: advances and challenges. Expert Rev Med Devices. 2009;6:641–651. doi: 10.1586/erd.09.46. [DOI] [PubMed] [Google Scholar]
- 29.Maurer JJ. Rapid detection and limitations of molecular techniques. Annu Rev Food Sci Technol. 2011;2:259–279. doi: 10.1146/annurev.food.080708.100730. [DOI] [PubMed] [Google Scholar]
- 30.Issadore D, et al. Ultrasensitive clinical enumeration of rare cells ex vivo using a micro-hall detector. Sci Transl Med. 2012;4:141ra92. doi: 10.1126/scitranslmed.3003747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Josephson L, Tung CH, Moore A, Weissleder R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug Chem. 1999;10:186–191. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- 32.Issadore D, et al. Miniature magnetic resonance system for point-of-care diagnostics. Lab Chip. 2011;11:2282–2287. doi: 10.1039/c1lc20177h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacDougall D, Crummett WB. Guidelines for data acquisition and data quality evaluation in environmental chemistry. Anal Chem. 1980;52:2242–2249. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.