

Abstract
Connective tissue growth factor (CTGF/CCN2) belongs to the CCN family of matricellular proteins, comprising Cyr61, CTGF, NovH and WISP1-3. The CCN proteins contain an N-terminal signal peptide followed by four conserved domains sharing sequence similarities with the insulin-like growth factor binding proteins, von Willebrand factor type C repeat, thrombospondin type 1 repeat, and a C-terminal growth factor cysteine knot domain. To investigate the role of CCN2 in breast cancer, we transfected MCF-7 cells with full-length CCN2, and with four mutant constructs in which one of the domains had been deleted. MCF-7 cells stably expressing full-length CCN2 demonstrated reduced cell proliferation, increased migration in Boyden chamber assays and promoted angiogenesis in chorioallantoic membrane assays compared to control cells. Deletion of the C-terminal cysteine knot domain, but not of any other domain-deleted mutants, abolished activities mediated by full-length CCN2. We have dissected the role of CCN2 in breast tumorigenesis on a structural basis.
Keywords: connective tissue growth factor, breast cancer, migration, angiogenesis
Introduction
Connective tissue growth factor (CTGF/CCN2) belongs to the CCN family of proteins (1–4) which has five additional members: cysteine rich 61 (Cyr61/CCN1), nephroblastoma overexpressed (NOV/CCN3) and the Wnt-inducible secreted proteins (WISP1-3/CCN4-6). With the exception of WISP2, CCN proteins contain an N-terminal signal peptide followed by four conserved modular domains, each encoded by a separate exon. Module I shares sequence similarities with the insulin-like growth factor binding proteins (IGFBPs). Module II has sequence homology with the von Willebrand factor type C repeat (VWC). Module III shares sequence similarity with the thrombospondin type 1 repeat (TSP). Module IV resembles the C-terminal growth factor cysteine knot domain (CT). Different isoforms of CCN proteins lacking one or more domains have been detected under both normal and disease conditions, arising from either alternative mRNA splicing or post-translational processing (5–7). Several lines of evidence demonstrate that these variants possess significant biological activity, suggesting that the production of truncated forms of CCN proteins influences the eventual physiological or patho-physiological outcome. Indeed, transformation of chick embryonic fibroblasts was induced by forced expression of amino-truncated, but not intact, CCN3 (8). In addition, plasmin cleavage of CCN1 released an N-terminal form into the culture media of MDA-MB-231 breast cancer cells, which stimulated endothelial cell migration at equivalent levels to that produced by the full-length protein (9). A plasmin digestion site is also present in the central hinge region of CCN2, and cleavage of the protein generates fragments that have distinct biological activities. The N-terminal portion containing modules I and II promoted myofibroblast differentiation and collagen production, whereas the C-terminal fragment containing modules III and IV stimulated fibroblast DNA synthesis (10). Additionally, a 10 kDa C-terminal fragment including the CT domain of CCN2 is present in uterine secretions, and was found to be mitogenic for fibroblasts and smooth muscle cells (6). In contrast to its mitogenic effects on fibroblasts, CCN2 has tumor suppressive activities in lung cancer (11,12).
The wide-range and often conflicting effects of CCN family members have been attributed in part to their multimodular structure, which enables different regions of the protein to fulfill distinct functions (3). Our laboratory has identified that module 4 of CCN1 is involved in cell spreading, migration and proliferation in breast cancer cells (13). In the present study, we generated a series of deletion mutants, containing CCN2 isoforms in which each of the modular domains had been deleted. These CCN2 deletion mutants were stably expressed in MCF-7 cells, and the cellular activities of these deletion mutants were compared to that of the full-length CCN2 in structure-function analysis. Additional experiments examined the influence of CCN2 on growth in vitro and in vivo of human breast cancer. Notably, CCN2 was important in breast cancer migration and angiogenesis.
Materials and methods
Cell culture
The breast cancer cell lines MCF-7, T47D, BT-474 were obtained from the American Type Tissue Culture Collection (Rockville, MD). Cells were cultured in Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal calf serum (Gemini Bio-Products, Calabasas, CA) and 10 units/ml penicillin-streptomycin (Life Technologies) and incubated at 37°C in 5% CO2. Deletion mutants of CCN2 were generated by PCR amplification using a full-length CCN2 expression vector as a template. Breast cancer cells were stably transfected with the constructs using Lipofectin (Invitrogen) according to manufacturer’s protocols. Stable cells were grown in complete culture medium with 500 μg/ml of G418.
Cell proliferation and apoptosis assays
For measurement of apoptosis, cells were labeled with FITC-conjugated Annexin V antibody and propidium iodide using Annexin V-FITC Apoptosis Detection kit I (BD Biosciences, San Jose, CA) according to the instructions from the manufacturer. Positive cells were detected by fluorescence-activated cell sorting. For cell cycle analysis, cells were washed and suspended in ice-cold phosphate-buffered saline (PBS), fixed in 75% chilled methanol at 4°C, and stained with propidium iodine. Cell cycle status was analyzed on a Beckton-Dickinson flow cytometer (BD Biosciences). To measure growth, cells were placed into 96-well plates at 1 × 103 cells per well. After incubation for the indicated time, cell number was assessed by the MTT assay (Sigma-Aldrich). Recombinant protein containing only the CT domain (CT-CCN2) was obtained from PeproTech (Rocky Hills, NJ). To measure DNA synthesis, [3H]-thymidine (0.25 μCi) was added to each well for the last 6 h of culture; cells were harvested onto filters, which were assayed for [3H]-thymidine content using the beta plate liquid scintillation counter (Amersham Pharmacia Biotech).
Western blot analysis
Protein from cells harvested for Western blotting was extracted with RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 7.5) containing a protease inhibitor cocktail (Roche Molecular Biochemicals). Protein lysates (40 μg) were boiled in Laemmli sample buffer (Bio-Rad), resolved by electrophoresis on 4–15% SDS-polyacrylamide gels, and transferred to PVDF membranes. Membranes were probed with antibodies (Santa Cruz) and developed using the SuperSignal West Pico Chemiluminescent Substrate (Pierce). For heparin-pull down experiments, cells were lysed in IP buffer (50 mM Tris pH 7.4, 150 mM NaCl, 0.5% NP-40), and 500 μg of protein was incubated with heparin-agarose beads (Sigma) at 4°C overnight with rotation. Beads were washed 3 times in PBS, boiled in Laemmli sample buffer and subjected to Western blotting as described above.
In vivo tumorigenesis assay
MCF-7 cells (1 × 106) were suspended in 100 μl Matrigel (BD Biosciences) for each tumor. Cells were injected into the mammary fat pad of immunodeficient BNX mice. Animals were supplemented with 17-estradiol pellets (Innovative Research of America, FL) that were implanted subcutaneously one week in advance. The size of the tumors was measured at regular intervals, and their volume was calculated by the following formula: A (length) × B (width) × C (height) × 0.5236. All mice were euthanized at the end of the study, and the final mass of the tumors was determined by weight after their dissection. Data were analyzed by Student’s t-test.
Cell adhesion, wound healing, and Boyden chamber assays
For cell adhesion assay, 96-well plates were coated with 50 μg/ml of fibronectin, vitronectin, or collagen overnight at 4°C. Cells were resuspended in serum-free medium and allowed to adhere to ECM-coated wells for 30 min. After the incubation period, wells were washed with PBS 3 times, and adherent cells were quantified by crystal violet staining and OD measurement at OD 540 nm. For wound healing assays, cells were grown to confluence and serum-starved overnight. Monolayer of cells was scratched with a sterile razor and images were captured at a fixed point along the cut surface. Width of the wound was calculated from five measurements of the distance between the migrating fronts in the captured images using the Datinf Measure software (Tubingen, Germany). The relative rates of total wound closure were calculated from 100% minus the percentage of (width of wound at 24 h/width of wound at 0 h). Boyden Chambers (Nunc, Rochester NY) with 8 mm pore size were coated on the underside of the membrane with 50 μg/ml fibronectin, vitronectin, or collagen for 1 h at room temp. Cells (2 × 105) in 300 μl serum-free media were added to the top of each chamber with the lower chamber containing 300 μl of serum-free medium plus 0.5% BSA. Chambers were placed in 24-well plates, and cells were allowed to migrate to the under-side of the top chamber for 12 h. The migratory cells that attached to the bottom surface of the membrane were stained with 0.25% crystal violet in 50% methanol for 15 min. The stained cells were extracted with 10% acetic acid. The number of migratory cells per membrane was determined by absorbance at 540 nm.
Chorioallantoic membrane angiogenesis assay
MCF-7 cells were seeded on the chorioallantoic membrane (CAM) of 7-day old quail embryos. After incubation for three additional days, CAMs were dissected and mounted on slides. The pattern of vessel branching was assessed by measurement of the fractal dimension (Df) in order to quantify the degree of angiogenesis.
Results
We first determined the impact of CCN2 on proliferation of human breast cancer cells in vitro and in vivo. Three breast cancer cell lines (MCF-7, T47D and BT-474) were transfected with either vector containing full-length CCN2 (pcDNA3.1-CCN2) or control empty vector (pcDNA3.1-Neo). Transfected cells were grown in selection medium, and cell number was assessed by measuring the absorbance of cells stained with crystal violet. CCN2 expression decreased the proliferation of all three cell lines (Fig. 1A). We then made stable CCN2-expressing breast cancer cells. MCF-7 cells in which CCN2 was stably expressed (MCF-7/CCN2) were used for further analysis in comparison to control cells (MCF-7/Neo). Two stable clones for each construct (Neo and CCN2) were selected, and their increased expression of CCN2 was demonstrated in these cells (Fig. 1B, left panel, #1 and #2). These cells also had decreased cell numbers similar to Fig. 1A (data not shown). We examined if this was caused by cell apoptosis. Annexin V staining showed no difference in the degree of apoptosis in control (Neo, 5%) vs. CCN2 expressing cells (CCN2, 6%) (Fig. 1B, right panel). On the other hand, cell cycle analysis showed a significant increase in the proportion of CCN2-transfected cells in the G0 − G1 phase (52%, CCN2 transfected vs. 36%, control) and decrease in the S phase (34%, CCN2 transfected vs. 51%, control) (Fig. 1C). These results demonstrated that CCN2-mediated growth inhibition of MCF-7 cells is primarily due to cell cycle arrest. The tumorigenic potency of the stably CCN2 transfected MCF-7 breast cancer cells was investigated in an orthotopic xenograft model in nude mice. In contrast to in vitro study, no significant difference was observed in the growth rate of CCN2-transfected MCF-7 cells orthotopi-cally implanted into the breast tissue of mice compared to controls (Fig. 1D).
Figure 1.
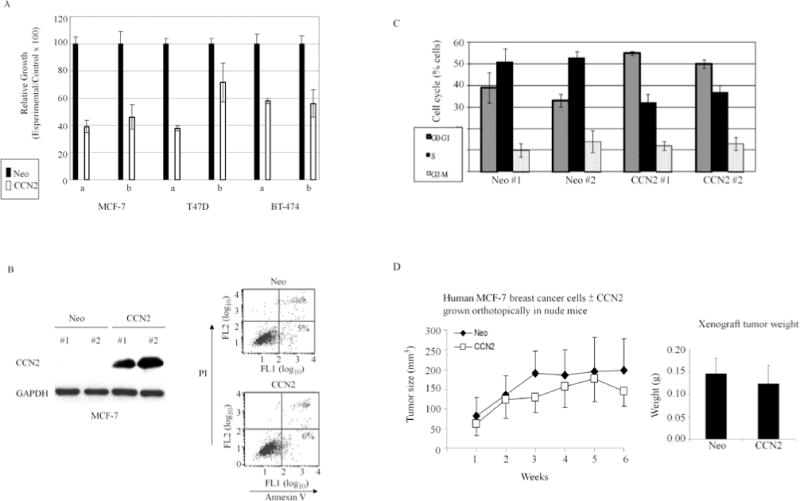
Effect of CCN2 on proliferation of human breast cancer cells in vitro and in vivo. (A) Breast cancer cell lines MCF-7, T47D and BT-474 transfected with either pcDNA3.1 empty vector (Neo) or pcDNA3.1-CCN2 (CCN2) were grown in the presence of 500 μg/ml G418 for 14 days. Crystal violet-stained cells were dissolved in methanol and absorbance was measured at 540 nm. Two independent transfection experiments (i and ii) were carried out for each cell line. Results represent the mean ± SD of triplicates. (B) Two clones of MCF-7 cells stably expressing CCN2 were selected (#1 and #2). Apoptotic cells were detected by Annexin-V assays. Representative results are shown. (C) Cell cycle analysis of MCF-7 cells stably-transfected with Neo and CCN2. (D) Left panel: MCF-7 cells ± stable expression of CCN2 were injected into the mammary fat pad of immunodeficient BNX mice. The size of the tumors was measured weekly. Results are presented as the mean ± SD of 6 tumors from each of MCF-Neo and MCF-CCN2 cells. Right panel: The final mean ± SD weight of the tumors was determined after their dissection.
To investigate the contribution of each of the structural domains of CCN2 to its biological properties, we generated four mutant constructs in which one of the modules (I, V, T, C) was deleted (Fig. 2A). A HA-tag was added to the C-terminus of the protein, enabling detection of each of the isoforms using an anti-HA antibody (Fig. 2B). Characteristic doublets of protein bands were detected, as previously observed by other investigators (14). CCN2 is a secreted heparin-binding protein. We used heparin-agarose beads to investigate the heparin-binding ability of each deletion mutants. Western blot analysis showed heparin-agarose beads could pull-down the CCN2 protein that was either full-length or missing module I, V, or T (Fig. 2B, FL, dI, dV, and dT), but not the MCF-7 CT-domain deletion mutant (Fig. 2B, dC). Overexpression of deletion mutants missing I, V, or T modules in MCF-7 cells showed comparable growth suppression to that of the full-length protein (Fig. 2C, dI, dV, dT vs. FL). In contrast, deletion of the CT domain prevented the CCN2-mediated growth inhibition of MCF-7 cells (Fig. 2C, dC vs. FL).
Figure 2.
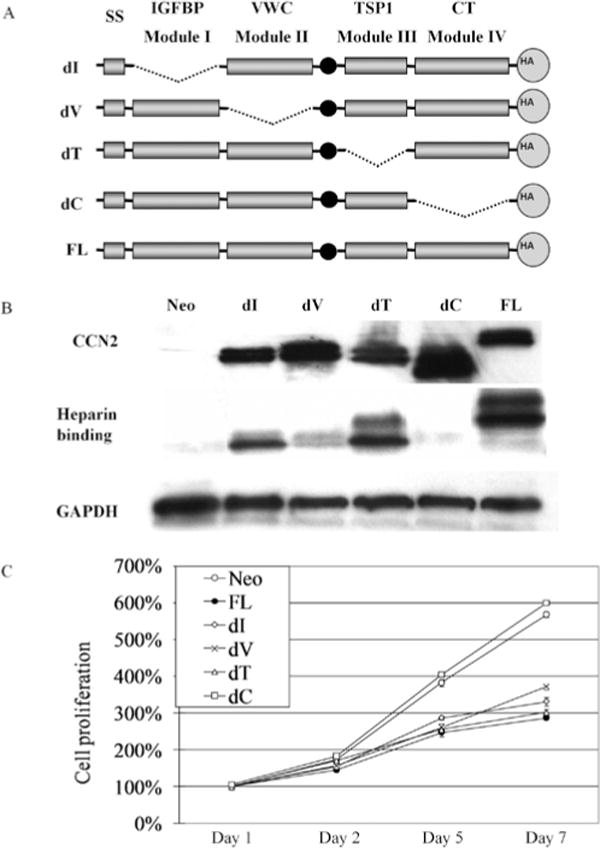
Stable expression of CCN2 deletion mutants in MCF-7 cells and their effect on human breast cancer cell proliferation. (A) Schematic diagram of CCN2 deletional mutants in which one of the modules [IGFBP (dI), von Willebrand factor type C repeat (dV), thrombospondin type 1 repeat (dT), and cysteine knot domain (dC)] has been deleted. (B) Forced expression of each of the mutant CCN2 genes in MCF-7 cells was detected by Western blot analysis using a HA antibody that recognizes the HA-tag on each protein. Cell lysates were incubated with heparin-agarose beads to pull down heparin-binding proteins and detected by HA antibody. (C) MTT assays showed CCN2 deletion mutants expressed in MCF-7 breast cancer cells. Results represent the mean ± SD of three experiments done in triplicates.
Cell adhesion of MCF-7 to extracellular matrix proteins was assessed on plates coated with either fibronectin, vitronectin, or collagen. Overexpression of CCN2 in these cells decreased their cell adhesion to both collagen and vitronectin, but not to fibronectin (Fig. 3A, Neo vs. FL). The decreased cell adhesion to collagen was abolished by deleting any of the different modules of CCN2 (Fig. 3A, dI, dV, dT, and dC vs. FL).
Figure 3.
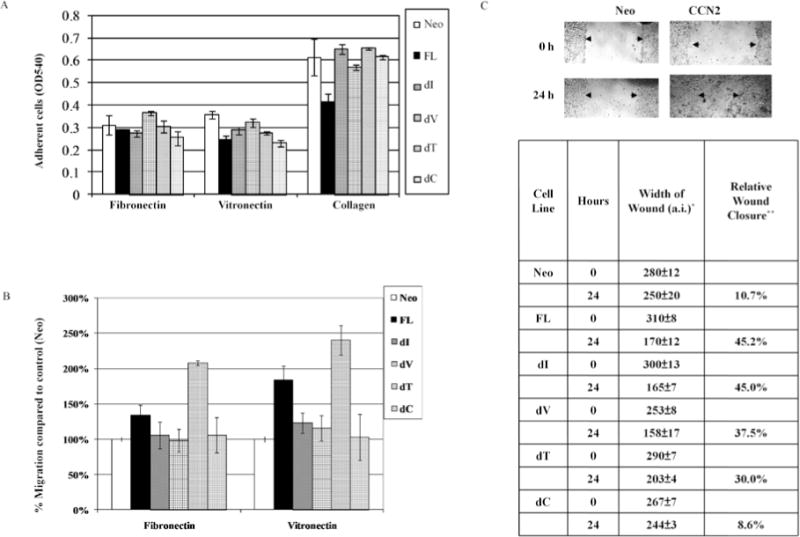
Effect of expression of CCN2 mutant protein on cell adhesion and migration of MCF-7 human breast cancer cells. (A) Breast cancer cells (MCF-7) in serum-free media were allowed to adhere to ECM-coated 96-well plates for 30 min. Adherent cells were quantified by staining with crystal violet and reading absorbance of the dissolved product at OD 540 nm. Results represent mean ± SD of three experiments done in triplicates. (B) MCF-7 breast cancer cells (2 × 105) were placed onto ECM-coated Boyden chambers and allowed to migrate for 12 h. The number of cells that migrated through the membrane was determined by absorbance of crystal violet-stained cells at 540 nm. Each bar represents the mean ± SD of triplicate experiments. (C) Confluent breast cancer (MCF-7) cells were scratched with a razor and grown in serum-free media. Cells were monitored for their ability to migrate into the wounded area. Images were taken after 24 h incubation. Quantification of the migration by each CCN deletion mutant is shown in the bottom panel. *a.i., artificial unit from Datinf measure software. Results represent the mean of three experiments; for each experiment, five measurements were made along the migrating fronts. **Relative wound closure = 100% minus percentage of (width of wound at 24 h/width of wound at 0 h).
To examine whether overexpression of CCN2 and its deletion mutants have an effect on the migratory ability of the MCF-7 breast cancer cells, ECM-coated Boyden chambers assays were performed. Overexpression of full-length CCN2 increased MCF-7 cell migration in both fibronectin- and vitronectin-coated Boyden chambers, with more significant increase on vitronectin-coated chambers (Fig. 3B, Neo vs. FL).
Deletion of the TSP domain of CCN2 enhanced even more the migration of the MCF-7 cells (Fig. 3B, FL and dT). The presence of the IGFBP, VWC, and CT domains were important for CCN2-mediated migration, as mutants in which these domains had been deleted migrated more slowly than those cells expressing full-length CCN2 (FL) (Fig. 3B, Neo, dI, dV, and dC). Next, wound healing assays were also used to measure motility of MCF-7 breast cancer cells. Overexpression of full-length CCN2 increased the migration of MCF-7 cells (Fig. 3C, upper panel). This increased migratory phenotype occurred with each of the deletion mutants dI, dV, and dT, except the mutant missing the CT domain (Fig. 3C, bottom panel).
To verify the significance of the CT domain in cell growth and migration, we treated MCF-7 cells with recombinant protein containing only the CT domain. This recombinant CT protein inhibited DNA synthesis of MCF-7 cells, as determined by [3H]-thymidine incorporation (Fig. 4A). Furthermore, recombinant CT protein promoted migration of MCF-7 cells in a collagen-coated Boyden chamber assay (Fig. 4B).
Figure 4.
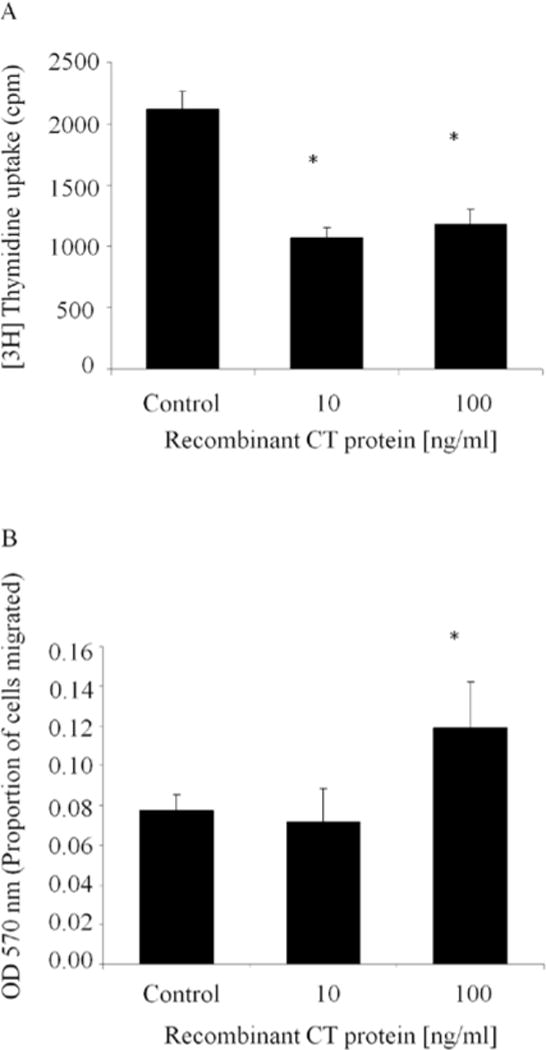
Recombinant CT domain of CCN2 inhibits proliferation and increases migration of MCF-7 cells. (A) Wild-type MCF-7 breast cancer cells were treated for 24 h with the CT domain of CCN2 as a recombinant protein. Proliferation was estimated by measuring [3H]-thymidine uptake for the last 6 h of incubation. Results represent the mean of triplicate wells done in two independent experiements. (B) Wild-type MCF-7 breast cancer cells (2 × 105) were placed onto collagen-coated Boyden chambers with either 0.10 or 100 ng/ml recombinant CT protein in the bottom layer of the filter. Cells were allowed to migrate for 12, and the number of cells that migrated through the membrane was determined by absorbance at 540 nm. Each bar represents the mean ± SD of triplicate experiments. *P < 0.05 vs. control.
To assess the effect of CCN2 on angiogenesis, the chorioallantoic membrane angiogenesis assay was used. Compared with blood vessel formation in fertilized quail eggs of control MCF-7 breast cancer cells (Fig. 5, panel Neo), blood vessels were prominently stimulated to grow when the MCF-7 cells overexpressed full-length CCN2 (Fig. 5, panel FL). MCF-7 breast cancer cells that overexpressed CCN2 with a deletion of the CT domain no longer had enhanced angiogenesis (Fig. 5, panel dC).
Figure 5.
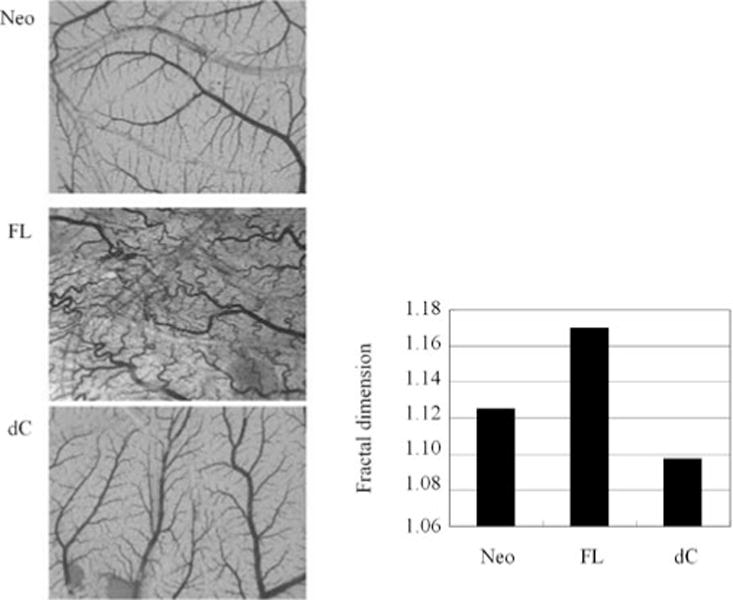
CCN2 stimulates angiogenesis. MCF-7 cells expressing full-length CCN2 (FL), CCN2 with deleted cysteine motif (dC) or neo-alone control (Neo) were seeded on chorioallantoic membrane (CAM) of 7-day old quail embryos. After incubation for three days CAMs were dissected and mounted on slides to assess the degree of angiogenesis (left panel). Bar graph shows the measurement of the fractal dimension as quantification of the pattern of vessel branching (right panel).
Discussion
Breast cancer is a multistep process in which the cells acquire a series of malignant characteristics, including increased proliferation and greater cell motility (15). In this study we demonstrated that CCN2 expression in breast cancer produced divergent effects within the same cell population. MCF-7 cells transfected with CCN2 proliferated less rapidly than control cells, yet their ability to migrate was enhanced. CCN2 is a TGFβ responsive gene (14), and as such, our results are consistent with the ambiguous role of TGFβ in breast cancer progression, as TGFβ possesses both tumor-suppressing and tumor-enhancing effects (16–18). The actions of TGFβ on breast cancer are both cell type- and context-dependent. TGFβ signaling inhibits the proliferation of epithelial cells, but it promotes invasion and metastasis as the cancer progresses. CCN2 expression is up-regulated by TGFβ in MCF-7 cells, where it contributes to the anti-proliferative actions of TGFβ (19). In addition, high levels of both CCN2 and TGFβ were associated with breast cancer metastasis to bone in an animal model (20,21). Similarly to CCN2, stable transfection of a CCN3 expression vector, both reduced proliferation and increased migration of an Ewing’s sarcoma cell line (22). However, divergent from our observations, a prior study found that overexpression of CCN2 in MCF-7 cells increased motility without affecting cell growth (23). Although we observed an anti-proliferative effect of CCN2 in vitro, growth of orthotopic human xenografts of MCF-7/CCN2 breast cancer cells was only slightly slower than for control tumors, but results were not statistically significant, suggesting that other factors present in vivo can overcome CCN2-mediated growth suppression. Since CCN2 expression is elevated in breast cancer patient samples and correlates with more advanced features of disease (24,25), the overall effect of CCN2 up-regulation is likely advantageous for the cancer. Moreover, a recent study showed that drug resistance was conferred in MCF7 cells over-expressing CTGF (26).
Biological activities of CCN proteins are domain-specific. Examples include: a novel variant of CCN4 lacking a VWC module was associated with progression of scirrhous gastric carcinoma (27). In myofibroblast, the N-terminal domain of CCN2 mediates migration, while the C-terminal region mediates proliferation (10). In contrast, the C-terminal fragment of CCN2 inhibits the proliferation of chondrocytes (28). CT module of CCN3 is involved in inhibition of cell growth in gliomas (29). The CT domain is important for the anti-metastatic properties of CCN2 in lung cancer, where a CT-domain deleted mutant had a reduced ability to inhibit lung cancer cell invasion compared to the full-length protein (11).
Our data highlight the importance of the C-terminal CT domain in mediating CCN2 activities in breast cancer cells. MCF-7 cells expressing CCN2 in which the CT domain was deleted, lost both the suppression of growth, and the enhancement of migration and angiogenesis. Furthermore, a recombinant CT domain peptide effectively reproduced the effects of the full-length protein as measure by DNA synthesis and migration. The C-terminal portion of CCN2 contains heparin-binding sites; and we show that deletion of this region, in particular the CT domain, prevents binding to heparin-agarose. Interaction of CCN2 with the cell surface is important for its regulation of growth factor signaling, as CCN2 chaperones other growth factors regulate their receptor-binding ability (12,30). Furthermore, heparin sulfate proteoglycans act as co-receptors for other integrin ligands, including CCN1 (31).
The CT domain of CCN2 contains a binding site for integrin ανβ3 (32). Integrin activation plays an important role in the development and progression of breast cancer, achieved through intracellular signaling via activation of downstream kinases such as ILK and MAPK (33–35). CCN1 has been shown to promote an aggressive breast cancer phenotype in an integrin ανβ3-dependent manner (36). A recent study in chondrosarcoma cells showed that CCN2 stimulates up-regulation of MMP-13 and migration via integrin ανβ3 (37). Anti-Integrin ανβ3 antibody has been shown to block CTGF-induced migration in MCF-7 cells (23).
Taken together, we have previously shown that high expression of CCN2 is associated with advanced breast cancer including metastasis to lymph nodes and a worse prognosis (25). Consistent with these findings, we now report that forced expression of CCN2 in breast cancer cells is associated with their increased migration and stimulation of angiogenesis. The migratory activity is dependent on the CT domain of CCN2. Furthermore, recombinant CT-CCN2 recapitulates the activity of the full-length protein. Therefore, the CT domain behaves as a critical regulator of CCN2 function and is a valid therapeutic target for breast cancer.
Acknowledgments
H.P. Koeffler holds the Mark Goodson Chair in Oncology Research and is a member of the Jonsson Cancer Center at UCLA. This research was supported in part by grants from National Institutes of Health RO1CA109290; U54 CA143930-01; NIH 5R011CA109295-05; A*STAR Investigator Award; and by grants from the Women’s Cancer Research Institute and Samuel Oschin Comprehensive Cancer Institute at Cedars-Sinai Medical Center.
References
- 1.Brigstock DR, Goldschmeding R, Katsube K, et al. Proposal for a unified CCN nomenclature. J Clin Pathol Mol Pa. 2003;56:127–128. doi: 10.1136/mp.56.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perbal B, Brigstock DR, Lau LF. Report on the second international workshop on the CCN family of genes. J Clin Pathol Mol Pa. 2003;56:80–85. doi: 10.1136/mp.56.2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perbal B. CCN proteins: multifunctional signaling regulators. Lancet. 2004;363:62–64. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- 4.De Winter P, Leoni P, Abraham D. Connective tissue growth factor: Structure-function relationships of a mosaic, multifunctional protein. Growth Factors. 2008;26:80–91. doi: 10.1080/08977190802025602. [DOI] [PubMed] [Google Scholar]
- 5.Hirschfeld M, zur Hausen A, Bettendorf H, Jager M, Stickeler E. Alternative splicing of Cyr61 is regulated by hypoxia and significantly changed in breast cancer. Cancer Res. 2009;69:2082–2090. doi: 10.1158/0008-5472.CAN-08-1997. [DOI] [PubMed] [Google Scholar]
- 6.Brigstock DR, Steffen CL, Kim GY, Vegunta RK, Diehl JR, Harding PA. Purification and characterization of novel heparin-binding growth factors in uterine secretory fluids. Identification as heparin-regulated Mr 10,000 forms of connective tissue growth factor. J Biol Chem. 1997;272:20275–20282. doi: 10.1074/jbc.272.32.20275. [DOI] [PubMed] [Google Scholar]
- 7.Perbal B, Martinerie C, Sainson R, Werner M, He B, Roizman B. The C-terminal domain of the regulatory protein NOVH is sufficient to promote interaction with fibulin 1C: A clue for a role of NOVH in cell-adhesion signaling. Proc Natl Acad Sci USA. 1999;96:869–874. doi: 10.1073/pnas.96.3.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joliot V, Martinerie C, Dambrine G, et al. Proviral rearrangements and overexpression of a new cellular gene (Nov) in myeloblastosis-associated virus type-1-induced nephroblastomas. Mol Cel Biol. 1992;12:10–21. doi: 10.1128/mcb.12.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pendurthi UR, Tran TT, Post M, Rao LVM. Proteolysis of CCN1 by plasmin: Functional implications. Cancer Res. 2005;65:9705–9711. doi: 10.1158/0008-5472.CAN-05-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grotendorst GR, Duncan MR. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 2005;19:729–738. doi: 10.1096/fj.04-3217com. [DOI] [PubMed] [Google Scholar]
- 11.Chang CC, Shih JY, Jeng YM, et al. Connective tissue growth factor and its role in lung adenocarcinoma invasion and metastasis. J Natl Cancer Inst. 2004;96:364–375. doi: 10.1093/jnci/djh059. [DOI] [PubMed] [Google Scholar]
- 12.Chien W, Yin D, Gui D, et al. Suppression of cell proliferation and signaling transduction by connective tissue growth factor in non-small cell lung cancer cells. Mol Cancer Res. 2006;4:591–598. doi: 10.1158/1541-7786.MCR-06-0029. [DOI] [PubMed] [Google Scholar]
- 13.O’Kelly J, Chung A, Lemp N, et al. Functional domains of CCN1 (Cyr61) regulate breast cancer progression. Int J Oncol. 2008;33:59–67. [PubMed] [Google Scholar]
- 14.Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ. 1996;7:469–480. [PubMed] [Google Scholar]
- 15.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Massague J. A very private TGF-beta receptor embrace. Mol Cell. 2008;29:149–150. doi: 10.1016/j.molcel.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Bierie B, Moses HL. TGF-[β] and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression (vol 29, pg 117, 2001) Nat Genet. 2001;29:351–351. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 19.Hishikawa K, Oemar BS, Tanner FC, Nakaki T, Luscher TF, Fujii T. Connective tissue growth factor induces apoptosis in human breast cancer cell line MCF-7. J Biol Chem. 1999;274:37461–37466. doi: 10.1074/jbc.274.52.37461. [DOI] [PubMed] [Google Scholar]
- 20.Kang YB, Siegel PM, Shu WP, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 21.Shimo T, Kubota S, Yoshioka N, et al. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J Bone Miner Res. 2006;21:1045–1059. doi: 10.1359/jbmr.060416. [DOI] [PubMed] [Google Scholar]
- 22.Benini S, Perbal B, Zambelli D, et al. In Ewing’s sarcoma CCN3(NOV) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene. 2005;24:4349–4361. doi: 10.1038/sj.onc.1208620. [DOI] [PubMed] [Google Scholar]
- 23.Chen PS, Wang MY, Wu SN, et al. CTGF enhances the motility of breast cancer cells via an integrin-ανβ3-ERK1/2-dependent S100A4-upregulated pathway. J Cell Sci. 2007;120:2053–2065. doi: 10.1242/jcs.03460. [DOI] [PubMed] [Google Scholar]
- 24.Frazier KS, Grotendorst GR. Expression of connective tissue growth factor mRNA in the fibrous stroma of mammary tumors. Int J Biochem Cell B. 1997;29:153–161. doi: 10.1016/s1357-2725(96)00127-6. [DOI] [PubMed] [Google Scholar]
- 25.Xie D, Nakachi K, Wang HM, Elashoff R, Koeffler HP. Elevated levels of connective tissue growth factor, WISP-1, and CYR61 in primary breast cancers associated with more advanced features. Cancer Res. 2001;61:8917–8923. [PubMed] [Google Scholar]
- 26.Wang MY, Chen PS, Prakash E, et al. Connective tissue growth factor confers drug resistance in breast cancer through concomitant up-regulation of Bcl-xL and cIAP1. Cancer Res. 2009;69:3482–3491. doi: 10.1158/0008-5472.CAN-08-2524. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka S, Sugimachi K, Saeki H, et al. A novel variant of WISP1 lacking a Von Willebrand type C module overexpressed in scirrhous gastric carcinoma. Oncogene. 2001;20:5525–5532. doi: 10.1038/sj.onc.1204723. [DOI] [PubMed] [Google Scholar]
- 28.Kubota S, Kawata K, Yanagita T, Doi H, Kitoh T, Takigawa M. Abundant retention and release of connective tissue growth factor (CTGF/CCN2) by platelets. J Biochem. 2004;136:279–282. doi: 10.1093/jb/mvh126. [DOI] [PubMed] [Google Scholar]
- 29.Bleau AM, Planque N, Lazar N, et al. Antiproliferative activity of CCN3: Involvement of the C-terminal module and post-translational regulation. J Cell Biochem. 2007;101:1475–1491. doi: 10.1002/jcb.21262. [DOI] [PubMed] [Google Scholar]
- 30.Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grzeszkiewicz TM, Lindner V, Chen NY, Lam SCT, Lau LF. The angiogenic factor cysteine-rich 61 (CYR61, CCN1) supports vascular smooth muscle cell adhesion and stimulates chemotaxis through integrin alpha(6)beta(1) and cell surface heparan sulfate proteoglycans. Endocrinology. 2002;143:1441–1450. doi: 10.1210/endo.143.4.8731. [DOI] [PubMed] [Google Scholar]
- 32.Gao RP, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin α(ν)β(3) and heparan sulfate proteoglycan. J Biol Chem. 2004;279:8848–8855. doi: 10.1074/jbc.M313204200. [DOI] [PubMed] [Google Scholar]
- 33.Taddei I, Faraldo MM, Teuliere J, Deugnier MA, Thiery JP, Glukhova MA. Integrins in mammary gland development and differentiation of mammary epithelium. J Mammary Gland Biol. 2003;8:383–394. doi: 10.1023/B:JOMG.0000017426.74915.b9. [DOI] [PubMed] [Google Scholar]
- 34.Felding-Habermann B, O’Toole TE, Smith JW, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu CY, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;155:505–510. doi: 10.1083/jcb.200108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menendez JA, Mehmi I, Griggs DW, Lupu R. The angiogenic factor CYR61 in breast cancer: molecular pathology and therapeutic perspectives. Endocr Relat Cancer. 2003;10:141–152. doi: 10.1677/erc.0.0100141. [DOI] [PubMed] [Google Scholar]
- 37.Tan TW, Lai CH, Huang CY, et al. CTGF enhances migration and MMP-13 up-regulation via ανβ3 integrin, FAK, ERK, and NF-kappa B-dependent pathway in human chondrosarcoma cells. J Cell Biochem. 2009;107:345–356. doi: 10.1002/jcb.22132. [DOI] [PubMed] [Google Scholar]