

Abstract
The expression of the gut tumor suppressor gene adenomatous polyposis coli (Apc) and its role in the oligodendroglial lineage are poorly understood. We found that immunoreactive APC is transiently induced in the oligodendroglial lineage during both normal myelination and remyelination following toxin-induced, genetic, or autoimmune demyelination murine models. Using the Cre/loxP system to conditionally ablate APC from the oligodendroglial lineage, we determined that APC enhances proliferation of oligodendroglial progenitor cells (OPCs) and is essential for oligodendrocyte differentiation in a cell-autonomous manner. Biallelic Apc disruption caused translocation of β-catenin into the nucleus and upregulated β-catenin-mediated Wnt signaling in early postnatal but not adult oligodendroglial lineage cells. The results of conditional ablation of Apc or Ctnnb1 (the gene encoding β-catenin) and of simultaneous conditional ablation of Apc and Ctnnb1 revealed that β-catenin is dispensable for postnatal oligodendroglial differentiation, that Apc one-allele deficiency is not sufficient to dysregulate β-catenin-mediated Wnt signaling in oligodendroglial lineage cells, and that APC regulates oligodendrocyte differentiation through β-catenin-independent, as well as β-catenin-dependent, mechanisms. Gene ontology analysis of microarray data suggested that the β-catenin-independent mechanism involves APC regulation of the cytoskeleton, a result compatible with established APC functions in neural precursors and with our observation that Apc-deleted OPCs develop fewer, shorter processes in vivo. Together, our data support the hypothesis that APC regulates oligodendrocyte differentiation through both β-catenin-dependent and additional β-catenin-independent mechanisms.
Introduction
Germ-line truncated mutations of the tumor suppressor gene, adenomatous polyposis coli (Apc, 15 exons, encoding a 312 kDa protein), cause familial adenomatous polyposis, and somatic Apc mutations are frequent in sporadic gut neoplasms. In colorectal cancers, APC regulates Wnt/β-catenin signaling via its β-catenin domain, and cytoskeletal configuration via its β-catenin, microtubule, EB1, and DLG1-binding domains (Näthke, 2006).
In the CNS, in situ hybridization demonstrated a largely neuronal localization of Apc mRNA (Bhat et al., 1994), but immunohistochemistry (IHC) using a monoclonal antibody developed against an APC immunogen [clone CC1 (CC1)] suggested predominant expression of APC in oligodendrocytes (Bhat et al., 1996). This paradox was later resolved by proof that CC1, though a useful “marker” for oligodendrocytes, recognizes an antigen other than APC (Brakeman et al., 1999). This left unresolved, however, whether APC is expressed in the oligodendroglial lineage and, if so, at what developmental stages and with what functional significance.
Prior reports indicated that APC in the CNS regulates process formation and extension by radial glia, astroglia, neurons, proliferation of neuroblasts, and trophic interactions between Bergmann glia and Purkinje cells (Yokota et al., 2009; Imura et al., 2010; Wang et al., 2011). A recent study (Fancy et al., 2009) demonstrated that oligodendroglial differentiation and remyelination are delayed in adult Apcmin/+ mice (Apc multiple intestinal neoplasia, 1-allele truncated mutation of Apc, lacking β-catenin-binding domain) (Moser et al., 1993) following lysolecithin-induced demyelination, and suggested, but did not prove, that this delay was attributable to dysregulation of Wnt/β-catenin signaling in these Apc-haploinsufficient mice. That study did not address several important questions. Does single-allele APC truncation lead to upregulation of Wnt/β-catenin signaling in the CNS? Since axonal activity regulates oligodendroglial progenitor cell (OPC) differentiation (Barres and Raff, 1999), were the delays in oligodendrogenesis and remyelination secondary to axonal dysfunction elicited by the Apc mutation (Zhou et al., 2004; Chen et al., 2011; Wang et al., 2011), or does APC exert cell-autonomous effects on oligodendroglial differentiation?
Using an antibody specific to APC, we observed that immunoreactive APC is expressed transiently in the oligodendroglial lineage during normal oligodendroglial development and oligodendroglial regeneration, in sharp contrast to the pattern of oligodendrocyte marker CC1. We found that biallelic APC disruption decreases OPC proliferation and inhibits OPC process formation, and that APC is essential for oligodendrocyte differentiation through both β-catenin-dependent and β-catenin-independent mechanisms, the latter likely affecting the structure of the oligodendroglial cytoskeleton.
Materials and Methods
Animals.
NG2 (Cspg4)-Cre (JAX stock #008533) (Zhu et al., 2008), Apcmin/+ (JAX stock #002020) (Moser et al., 1993), BAT-lacZ (Wnt/β-catenin signaling reporter, JAX stock #005317) (Maretto et al., 2003), Rosa-EYFP (JAX stock #006148), Ctnnb1exon2-6flox/flox (Ctnnb1fl/fl, JAX stock #004152) (Brault et al., 2001) were purchased from The Jackson Laboratory. Cnp-Cre (Lappe-Siefke et al., 2003), Olig2-Cre-ERT2 (Takebayashi et al., 2002), and ASPANur7 mutant mice (Traka et al., 2008) were kindly provided by Klaus-Armin Nave (Max Planck Institute of Experimental Medicine) via Judith Grinspan (Children's Hospital of Philadelphia), Hirohide Takebayashi (Kumamoto University) via Chengji Zhou (University of California, Davis) and Brian Popko (University of Chicago), respectively. Apc-flox mice (National Cancer Institute strain #01XAA, LoxP sites flanking exon 14) (Kuraguchi et al., 2006) were purchased from the National Cancer Institute. Upon Cre-mediated deletion of Apc exon 14, the transcription of Apc in these mice results in a shift in the normal reading frame, thus encoding a possible truncated 580 aa polypeptide that lacks central β-catenin-binding domain and C-terminal microtubule-binding, EB1-binding, and DLG1-binding domains. These transgenic mice were bred to produce transgenic study mice and corresponding control mice. Both male and female mice were used in our study. All mice were maintained on the C57BL/6 background.
Tamoxifen and ethynyl deoxyuridine injection.
For conditional gene deletion (either Apc or Ctnnb1 or both) by inducible Olig2-Cre-ERT2 (OCE), neonatal pups received one intraperitoneal tamoxifen injection (dose, 100 μg of tamoxifen/g body weight) per day on postnatal day (P) 6 and P7. The spinal cord and forebrain were analyzed at different time points as indicated in the text and figure legends. Mice received a single intraperitoneal injection of ethynyl deoxyuridine (EdU, 100 μg/g body weight) 2 h before kill.
Primary OPC culture and differentiation.
Primary mixed glial (MG) cultures were prepared from mouse and rat forebrains by methods modified from our previous reports (Itoh et al., 2002; Horiuchi et al., 2010). Forebrains were harvested between ages P0 and P2. The tissues were dissociated using the Papain Dissociation System (PDS) Kit (Worthington) supplemented with DNase I (250 U/ml) and d-(+)-glucose (0.36%; Sigma-Aldrich) for 90 min in a humidified incubator at 33°C/10% CO2. Next, the tissue was immersed in PDS Kit-Inhibitor solution [10 mg/ml ovomucoid protease inhibitor, 10 mg/ml bovine serum albumin (BSA) and 125 U/ml DNase I in Dulbecco's PBS; Worthington]. Tissue chunks were triturated, and the cell suspension supernatant was collected. After centrifugation [220 relative centrifugal force, 15 min], cells were plated on a poly-d-lysine (PDL)-coated (Sigma-Aldrich) 10 cm dish in medium containing 10% heat-inactivated fetal bovine serum, glutamax, and penicillin/streptomycin (P/S) in high-glucose DMEM (all Invitrogen). After 24 h in vitro, serum-free “growth medium” (GM) was added to stimulate OPC proliferation. GM consisted of seven parts N1 medium (5 μg/ml insulin, 50 μg/ml transferrin, 100 μm putrescine, 30 nm sodium selenite, 20 nm progesterone, 10 ng/ml biotin; Sigma-Aldrich) and three parts B104 neuroblastoma-conditioned N1 medium together with P/S. The cultures were provided fresh GM at 72 h in vitro and, after 96 h total in vitro, the MG cultures were immunopanned to produce OPC-enriched cultures. Rat cultures were immunopanned according to our published methods (Itoh et al., 2002; Horiuchi et al., 2010). Before panning, mouse cultures were resuspended in “panning solution” (0.1% BSA in N1 medium). Mouse cultures were negatively panned once with rat anti-Thy1.2 antibody (American Type Culture Collection, clone 30H12) and positively panned once with rabbit anti-NG2 antibody (Millipore, AB5320). OPC-enriched cultures were grown on PDL-coated plates. Rat OPC-enriched cultures were expanded in GM with recombinant murine PDGF-A chain homodimer (PDGF-AA, 1 ng/ml) and FGF2 (5 ng/ml) (Peprotech). Mouse OPC-enriched cultures were expanded in GM with PDGF-AA (2 ng/ml) and FGF2 (10 ng/ml), forskolin (50 μm; Enzo Life Sciences) (Horiuchi et al., 2010), and glutamax. To induce differentiation, the medium was switched to “differentiation medium,” which consisted of 12.5 μg/ml insulin, 50 μg/ml transferrin, 100 μm putrescine, 24 nm sodium selenite, 10 nm progesterone, 10 ng/ml biotin, 30 ng/ml 3,3′,5-triiode-l-thyronine, 40 ng/ml l-thyrozine (all Sigma-Aldrich), glutamax, and 0.3% d-(+)-glucose in F12/high-glucose DMEM, 1:1 medium (Invitrogen).
Remyelination models: myelin oligodendrocyte glycoprotein peptide 35–55 induced experimental autoimmune encephalomyelitis, ASPANur7 murine Canavan disease, and cuprizone-induced corpus callosum demyelination.
Myelin oligodendrocyte glycoprotein peptide (MOG-peptide) 35–55 experimental autoimmune encephalomyelitis (EAE) was induced in 3-month-old C57BL/6 mice of both sexes (Guo et al., 2011, 2012). Four-month postnatal ASPANur7 homozygous mutant male mice were analyzed; these mice showed demyelination and partial remyelination, together with microcyst formation, in forebrain and cerebellum (Traka et al., 2008). For cuprizone-induced demyelination, 2-month-old C57BL/6 mice of both sexes were maintained on 0.25% (w/w) diet for 6 weeks, causing selective depletion of oligodendroglia in corpus callosum; following return to a normal diet, surviving OPCs proliferated, and differentiated into myelinating oligodendrocytes (Mason et al., 2004), with remyelination completed by 6 weeks thereafter (Kipp et al., 2009). The cuprizone-fed mice were killed at the end of 6 week cuprizone diet, or 4 or 6 weeks later.
Tissue preparation.
Tissue processing and sectioning were conducted as described in our previous studies (Guo et al., 2009, 2010, 2011, 2012). Fourteen-micron-thick frozen sections were prepared for immunohistochemistry and mRNA in situ hybridization.
Primary antibodies.
The primary antibodies used in this study were described in our previous studies (Guo et al., 2009, 2010, 2011, 2012) except for the following: APC [rabbit, Santa Cruz Biotechnology, #sc-896, 1:100 on IHC and immunocytochemistry (ICC), 1:1000 on Western blot (WB)], APC immunogen peptide (Santa Cruz Biotechnology, #sc-896P, specific to sc-896 APC antibody), CC1 (mouse, Calbiochem, #OP-80, 1:200 on IHC), DLG1 (rabbit, Thermo Scientific, #PA1-741, 1:200 on paraffin section, 1:1000 on WB; mouse, BD PharMingen, #610874, immunoprecipitation), β-catenin (mouse, BD PharMingen, #610153, 1:1000 on WB, 1:200 on IHC), acetylated α-tubulin (mouse, Sigma-Aldrich, #T7451, 1:10,000 on WB, 1:200 on ICC), α-tubulin (mouse, Invitrogen, #A11126, 1:200 on ICC; rabbit, Cell Signaling Technology, #2144, 1:1000 on WB), phosphorylated histone H3, PH3 (rabbit, Millipore, #06-570, 1:200 on IHC), Ki67 (mouse, Vector, #VP-K452, 1:100 on IHC), active caspase 3 (rabbit, Promega, #G748A, 1:200 on IHC), β-galactosidase, β-gal (rabbit, Cappel, #55978, 1:200 on IHC).
IHC and confocal microscopy.
Immunostaining and confocal imaging were conducted according to our published protocol (Guo et al., 2009, 2010, 2011).
mRNA in situ hybridization.
Probe preparation and hybridization were performed as described in our previous study (Guo et al., 2012).
Coimmunoprecipitation.
Rat OPC-enriched cultures were seeded at 1.8 × 106 cells per 75 cm2 PDL-coated culture flask, and differentiation was induced the next morning. At day two, cultures were rinsed with ice-cold PBS and extracted with coimmunoprecipitation (CoIP) lysis buffer containing 10 mm Tris buffer, pH 7.4, 140 mm NaCl, 0.5% Nonidet P-40 (Igepal CA-630), 1 mm EDTA, 1 mm EGTA (all Sigma-Aldrich), complete protease inhibitor mixture (Roche), and PhosSTOP phosphatase inhibitor mixture (Roche). The cell lysate was gently homogenized by P1000 micropipetting 30 times and was incubated on ice for 15 min. Then, the lysate was centrifuged at 10,000 rpm for 10 min at 4°C. The supernatant was collected and the protein concentration was adjusted to ∼1 mg/ml, and some adjusted lysate was set aside for Western blot analysis (“Input” sample). For immunoprecipitation (IP), protein G magnetic beads (New England Biolabs) were used, and the samples were precleared and immunoprecipitated following the manufacturer's protocol except using CoIP lysis buffer. Antibodies used for IP included control IgG (eBioscience), anti-APC (Millipore), and anti-Dlg1 (BD PharMingen) antibodies. The bead-IP complexes were eluted with NuPAGE lithium dodecylsulfate (LDS) sample buffer with reducing agent (Invitrogen) and the IP samples were analyzed for CoIP by Western blot.
Protein extraction and Western blots.
Protein extractions were performed using radioimmunoprecipitation analysis lysis buffer system with protease inhibitors (Santa Cruz Biotechnology, #sc-24948). Protein concentrations were measured using BCA kits (Thermo Scientific, #23235). NuPAGE LDS sample buffer with reducing agent (Invitrogen) was used to denature 10 μg of protein extract. For analysis of APC expression and for CoIP experiments, samples were separated by electrophoresis in NuPAGE Novex 3–8% gels. For analysis of tubulin acetylation, 4–12% Bis-Tris gels were used (Invitrogen). Electrophoresed proteins were transferred to nitrocellulose membranes (Invitrogen). Membranes were blotted and imaged using the LI-COR Infrared scanner system (LI-COR Biosciences) according to the manufacturer's protocol with LI-COR Blocking Buffer and with the secondary antibodies goat anti-rabbit IRDye 800CW and goat anti-mouse IRDye 680RD, each diluted 1:10,000 (LI-COR Biosciences). LI-COR Image Studio 2.0 software was used to analyze band-signal intensity with subtraction of the median background outside the band selection perimeter.
RNA isolation and quantitative real-time PCR.
Total RNA extraction, cDNA preparation and quantitative real-time PCR (qRT-PCR) were performed as described in a previous study (Guo et al., 2012). The copy number of each gene of interest was normalized to that of internal control glyceraldehyde-3-phosphate dehydogenase (GADPH), and, for analysis, the expression level of each gene in wild-type mice was set at a value of 1.
The primers for qRT-PCR were obtained mainly from the PrimerBank at Harvard University (http://pga.mgh.harvard.edu/primerbank/). The primer sets were as follows (sequences are aligned from 5′ to 3′, forward/reverse primers): Mbp (ACACGAGAACTACCCATTATGGC/CCAGCTAAATCTGCTGAGGGA), Cnp (TTTACCCGCAAAAGCCACACA/CACCGTGTCCTCATCTTGAAG), Plp (CCAGAATGTATGGTGTTCTCCC/GGCCCATGAGTTTAAGGACG), GADPH (AGGTCGGTGTGAACGGATTTG/TGTAGACCATGTAGTTGAGGTCA), Tcf7l2 (TCACGCCTCTCATCACGTACA/GTGCGGAGGTGGATTTCCC), Axin2 (AACCTATGCCCGTTTCCTCTA/GAGTGTAAAGACTTGGTCCACC), Naked1 (CAGCTTGCTGCATACCATCTAT/GTTGAAAAGGACGCTCCTCTTA), Notum (GGACAGCTTTATGGCGCAAG/TCACCGACGTGTTCAGCAG), Dlg1 (CAACACAGACAGCTTAGAGACAC/ACCCGAATTTCCCCTTTCAAG), Casr (AGCAGGTGACCTTCGATGAGT/ACTTCCTTGAACACAATGGAGC), Kif19a (TCTATGTTCGGACCCTCAATGA/ATCACCCCTTTGGAGTCTTCC), Sox10 (ACACCTTGGGACACGGTTTTC/TAGGTCTTGTTCCTCGGCCAT), Rnf122 (CACCCATTCCAGTGGTGTAAC/GCACAGGTCCCATAGAGCTG), MAG (CTGCCGCTGTTTTGGATAATGA/CATCGGGGAAGTCGAAACGG), Fyn (ACCTCCATCCCGAACTACAAC/CGCCACAAACAGTGTCACTC), CXCR4 (GAAGTGGGGTCTGGAGACTAT/TTGCCGACTATGCCAGTCAAG), Cx3cl1 (ACGAAATGCGAAATCATGTGC/CTGTGTCGTCTCCAGGACAA), Ctnnb1 (ATGGAGCCGGACAGAAAAGC/CTTGCCACTCAGGGAAGGA), cyclin D1 (GCGTACCCTGACACCAATCTC/CTCCTCTTCGCACTTCTGCTC).
Microarray and gene ontology analysis.
Total RNA was isolated from the cervical spinal cord by RNeasy minikits (Qiagen, see RNA isolation and quantitative real-time PCR). RNA concentrations were measured by NanoDrop spectrometry, and RNA integrity was evaluated with the Agilent 2100 Bioanalyzer. Microarray experiments and analysis were performed by the University of California Davis Cancer Center Genomics and Expression Resource. Genome-wide expression profiling using Mouse Gene 1.0 ST GeneChip arrays (Mouse Gene 1.0 ST, Affymetrix) was performed according to the manufacturer's protocols. Data analysis was performed with the GeneSpring GX (version 11) software suite (Agilent Technologies). Briefly, exon RMA 16 was used for probe summarization and normalization of background-adjusted, normalized, and log-transformed perfect-match probe intensity values from the .cel raw data files. Comparison analysis was then performed to identify genes that were differentially expressed in the different treatment groups. Criteria for the selection of genes exhibiting significant expression changes included an average fold change of ≥1.2 between groups and p values of ≤0.05 while adjusting for multiple testing corrections using the Benjamini–Hochberg false discovery rate (FDR). Potential functions and pathways affected by the Apc deletion were queried by organizing the genes based upon gene ontology (GO) annotations for biological process using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang da et al., 2009a,b).
Sholl analysis.
Sholl analysis (Rajasekharan et al., 2009) of OPC processes was conducted by the Sholl ImageJ plugin, using 10 μm optical thickness confocal images with a 100× objective. Parameter settings were as follows: starting radius, 10 μm; ending radius, 50 μm; radius step size, 10 μm. The number of intersections at each radius circle was used to compare wild-type and Apc KO OPCs.
Data collection.
Mean intensities of myelin basic protein (MBP) immunostaining were measured by National Institutes of Health ImageJ software. Cell number per section was calculated by counting all marker-positive cells in 14-μm-thick frozen section under a 10× objective. Cell density (cell number per mm2) was calculated by dividing total cell number by the total area counted. At least four 40× confocal projection images (with 10 μm optical depth using confocal z-stack) from each 14-μm-thick frozen section were counted, and at least three frozen sections from each animal and at least three animals from each group were counted. Axons in the corticospinal tract were counted using ImageJ software as previously described (Soulika et al., 2009; Guo et al., 2012).
Statistics.
All quantification data in this study are expressed as means ± SD. Statistics were conducted using GraphPad Prism 5. Two-tail unpaired Student's t tests were used to compare two-group data, and one-way ANOVA with Bonferroni post-test was used to compare three-group data (*p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant).
Results
APC is transiently expressed in oligodendroglial lineage cells during normal development
We used a well characterized APC antibody (sc-896, Santa Cruz Biotechnology) (Sasaki et al., 2000; Jimbo et al., 2002; Wen et al., 2004; Mili et al., 2008) to study the expression of APC in oligodendroglial lineage cells. First, we used peptide-antibody preabsorptions to verify the specificity of the sc-896 antibody for APC. Consistent with a previous publication (Imura et al., 2010), APC was abundantly expressed in the molecular layer and dentate gyrus in the hippocampus (Fig. 1A), and this immunostaining was abolished when APC antibody was preincubated with the APC peptide (sc-896P, Santa Cruz Biotechnology) (Fig. 1B). Second, gene knock-out (KO) experiment also proved the specificity of the APC antibody we used (Fig. 2, compare A1, B2).
Figure 1.

APC is transiently expressed in oligodendroglial lineage cells during normal development and after CNS demyelination. A–B, Peptide-antibody preabsorption experiment showing specificity of the APC antibody (#sc-896) by IHC in the hippocampus. Mo, molecular layer of hippocampus; GL, granular layer of hippocampus; DG, dentate gyrus of hippocampus. C, Double IHC of APC and CC1 in the adult P60 neocortex. Note that APC stains cells with characteristic cortical neuron morphology (arrowhead), whereas CC1 stains small oligodendrocytes (arrows). D–H, Transient expression of APC in spinal cord and its colocalization with oligodendrocyte marker CC1. E, arrowhead, APC+ cell. F, G, arrowheads, APC+CC1+ cells. F–H, arrows, APC−CC1+ cells. I, Densities of APC+ and CC1+ cells in the WM of spinal cord at different ages (n = 3 in each group). J, Double IHC of APC and panoligodendroglial marker Sox10. K, APC+ cells were positive for Tcf7l2, an essential activator of Wnt/β-catenin signaling pathway in oligodendroglial lineage cells (Fancy et al., 2009). L, Experimental design for primary OPC culture and differentiation (top) and Western blot showing APC protein was low in purified OPCs (G0) and was upregulated in differentiating oligodendrocytes (D1, D2, and D4) (bottom). Note that mature oligodendrocyte marker ASPA (Traka et al., 2008) was essentially absent in OPCs on G0. M, Immunocytochemistry depicting the APC expression in OPCs on G0 and in differentiating oligodendrocytes on D2 and D4 in vitro. N1–N4, APC+ cells in the corpus callosum of mice on a normal diet (N1), 6 weeks on a cuprizone diet (N2), 4 weeks postcuprizone (N3), and 6 weeks postcuprizone (N4). Arrows in N1 indicate APC+ neurons in the cortex and arrowheads in N2–N4 point to APC+ cells in corpus callosum. Insert in N2 showing APC+ cells colabeled with oligodendroglial lineage marker Sox10. N5, Quantification of APC+ cells in corpus callosum (n = 4 in normal diet group, n = 3 in other groups). O–P, APC+ cell (arrowheads) in the cerebellar WM of ASPAnur7 homozygous mutant and ASPA WT mice. Boxed area in O was showing in orthogonal view (right) colabeling with CC1. Scale bars: all except J, K, 50 μm; J, K, 10 μm.
Figure 2.

Apc disruption results in hypomyelination and inhibition of oligodendrocyte differentiation. Tamoxifen was injected in OCE/Apcfl/fl pups and littermates at P6 and P7 (see Materials and Methods) and CNS tissues were analyzed as indicated. A1, A2, APC staining in P14 OCE/Apcfl/fl, EYFP (KO), and Apcfl/fl, EYFP (WT) mice showing most APC+ cells in the spinal WM were absent in KO mice. Dotted areas delineate APC+ motor neurons in the ventral horn. B1, B2, EYFP expression as a surrogate for APC disruption. C, EYFP expression in NG2+ OPCs in the spinal cord of OCE/Apcfl/fl, EYFP mice on P8. D1–E2, MBP (myelin) and SMI312 (axons) double IHC showing hypomyelination in spinal cord (D1–D4) and cerebellar WM (E1, E2) at the level of confocal microscopy on P14. Boxed areas in D1 and D3 (corticospinal tract) are shown at higher power in D2 and D4, respectively. Note many SMI312+/MBP− unmyelinated axons in KO (compare D4 with D2). F, Quantifications of MBP mean density (n = 4 spinal cord, n = 3 cerebellum) in spinal cord and cerebellum. G, Densities of Sox10+ oligodendroglial lineage cells in WM and GM of spinal cord (n = 10 in WT, n = 7 in KO). H, MRNA levels of Sox10 in P14 Apc WT (Apcfl/fl, n = 10), one-allele KO (OCE/Apcfl/+, n = 4), and two-allele KO (OCE, Apcfl/fl n = 7) spinal cord. I1–I3, Representative confocal images and quantification of CC1 immunostaining on P14 spinal cord (n = 10, 4, and 7, respectively). J, GST-π+ oligodendrocytes per section in each genotype of P14 spinal cord (n = 10, 4, and 7, respectively). K, Densities of Sox10+/CC1+ oligodendrocytes in the WM and GM of P14 spinal cord in Apcfl/fl (n = 10) and OCE/Apcfl/fl (n = 7) mice. L, Representative images showing Plp in situ hybridization and the number of Plp mRNA+ cells per section in P14 Apcfl/fl (n = 10), OCE/Apcfl/+ (n = 4), and OCE/Apcfl/fl (n = 7) mice. M, MRNA levels of Mbp, Plp, and Cnp in P14 Apcfl/fl (n = 10) and OCE, Apcfl/fl (n = 7) spinal cord. N, MRNA levels of Mbp, Plp, and Cnp in P14 Apcfl/fl (n = 5) and OCE/Apcfl/fl (n = 4) spinal cord without tamoxifen treatment (no Apc deletion). Scale bars: A1, A2, D1, D3, E1, E2, I1, I2, 50 μm; B1–C, D4, 10 μm; L, 500 μm.
By double IHC with APC and CC1 antibodies, we showed that in the adult neocortex, the immunostaining pattern of APC was completely different from that of CC1 (Fig. 1C). APC was expressed in neurons (Fig. 1C, arrowheads) (Zhou et al., 2004), whereas CC1 was expressed in oligodendrocytes (Fig. 1C, arrows). These data confirmed that immunohistochemistry by CC1, though useful for identifying oligodendroglia (Bhat et al., 1996), does not reflect endogenous APC expression.
Is APC expressed in oligodendroglial lineage cells? We found that immunoreactive APC was not detectable in murine spinal cord white matter (WM) until embryonic day 18.5 (Fig. 1D,E), transiently peaked during active myelination, and was markedly downregulated in the adult (Fig. 1F–H). This pattern of APC expression was in sharp contrast to the temporal patterns of CC1 immunostaining (Fig. 1I). Double IHC identified APC+ cells in WM as Sox10+ (a panoligodendroglial lineage cell marker) oligodendroglial lineage (Fig. 1J) rather than GFAP+ (an astrocyte marker) astrocytic lineage (data not shown). APC immunoreactivity was barely detectable in NG2+ OPCs (data not shown), and substantially upregulated in CC1+ oligodendrocytes (Fig. 1F–H). Interestingly, virtually all APC+ oligodendroglial cells were immunopositive for the Wnt/β-catenin signaling effector Tcf7l2, and virtually all Tcf7l2 cells expressed immunoreactive APC, independent of mouse age (Fig. 1K). Consistent with these in vivo results, APC protein was detected at a very low level in highly purified rodent primary OPC cultures (Horiuchi et al., 2010) (Fig. 1L, first lane; Fig. 1M, left), and was substantially upregulated as they differentiated to oligodendrocytes (Fig. 1L,M). Thus, both in vivo and in vitro data indicated that (1) APC expression is low at the early OPC stage and upregulated upon differentiation, and (2) APC expression in the oligodendroglial lineage is cell-autonomously regulated. Together, our study demonstrates that immunoreactive APC is transiently expressed in oligodendroglial lineage cells during normal oligodendrocyte development and myelination.
APC is transiently reinduced in oligodendroglial lineage cells after CNS injury
The transient expression of APC during normal oligodendroglial myelination prompted us to investigate whether APC is re-expressed during remyelination after CNS injury. In wild-type adult mice maintained on a normal diet, APC+ cells were barely detectable in corpus callosum (Fig. 1N1), but APC+ cells had substantially increased by 6 weeks on a cuprizone diet (Fig. 1N2,N5), when oligodendrocyte differentiation was active (Kipp et al., 2009), and then markedly decreased by 6 weeks postwithdrawal of cuprizone diet (Fig. 1N4,N5), when remyelination was completed (Kipp et al., 2009). In 4-month-postnatal ASPANur7 mutant mice, an animal model of Canavan disease, in which the lack of oligodendroglial aspartoacylase (ASPA) causes continuous demyelinative and remyelinative activity in cerebellum (Traka et al., 2008), APC+ cells were significantly increased in the cerebellar WM of ASPANur7 (Fig. 1O), in sharp contrast to ASPA wild-type littermate controls, in which few APC+ cells were observed in cerebellar WM (Fig. 1P) (34 ± 11 in WT, 155 ± 43 in ASPANur7, n = 3, p < 0.01). Thirty-five days after induction of MOG-peptide EAE (Soulika et al., 2009; Guo et al., 2012), APC+ cell numbers were significantly higher in spinal cord WM than in control mice given complete Freund's adjuvant (CFA) without MOG peptide (32 ± 12 in CFA, 103 ± 48 in MOG, n = 4, p < 0.05). These increases in numbers of CNS APC+ cells in toxin-induced, genetic, and autoimmune demyelination/remyelination models suggest that APC reinduction is a common feature of remyelination after CNS injury.
Loss of oligodendroglial APC results in hypomyelination
To investigate the role of APC in oligodendroglial differentiation, we bred Cnp-Cre mice (Lappe-Siefke et al., 2003) with Apc-floxed (Kuraguchi et al., 2006) mice to conditionally ablate APC expression from the Cnp+ OPCs and oligodendrocytes. Most Cnp-Cre/Apcfl/fl mice died within 24 h after birth, and the remaining mice died before weaning. Using the Cre reporter transgene Rosa-EYFP, we found that enhanced yellow fluorescent protein (EYFP) was expressed abundantly in lung, heart, and thymus, as well as in cortical neurons and spinal cord motor neurons (data not shown) at P1; we suspect that the early death of the Cnp-Cre/Apcfl/fl mice was due to the deletion of Apc's critical functions in these visceral organs and/or neurons (Gounari et al., 2005; Kuraguchi et al., 2006; Qian et al., 2008; Li et al., 2010). We demonstrated that the number of APC+ cells in the WM of spinal cord at P7 was ∼30% less in Cnp-Cre/Apcfl/fl mice than that in Apcfl/fl control mice, and that the number of Plp+ oligodendrocytes per section (14 μm thick, here and hereafter) by mRNA in situ hybridization was significantly decreased in the Cnp-Cre/Apcfl/fl mice (1097 ± 80 Apcfl/fl, 834 ± 67 Cnp-Cre/Apcfl/fl per section on P14, p < 0.01, n = 4), indicating that oligodendrocyte differentiation is inhibited upon Apc disruption.
To further examine the cell-autonomous role of APC in postnatal oligodendroglial lineage cells, we crossed tamoxifen-inducible OCE (Takebayashi et al., 2002) with Apc-floxed (Kuraguchi et al., 2006) mice and administered tamoxifen on P6 and P7 (see Materials and Methods). As shown in Figure 2A1,A2, this resulted in the loss of most APC+ cells in both spinal cord WM and GM, whereas APC expression in motor neurons within ventral horn (Fig. 2A1, A2, dotted areas) was intact, thus demonstrating the specificity of Apc deletion in Olig2+ OPCs and their progeny. Using EYFP expression to report Cre-mediated Apcfl/fl recombination (Fig. 2B1,B2, arrowheads) (Guo et al., 2012), we determined that 47 ± 5% of Sox10+ oligodendroglial lineage cells were EYFP+, 52 ± 8% of NG2+ OPCs were EYFP+ (Fig. 2C, arrowheads), and 75 ± 9% of EYFP+ cells were NG2+ OPCs on P8, thus indicating that approximately half of oligodendroglial lineage cells were Apc-deficient, and that most Apc-deficient cells were initially OPCs. Compared with Apcfl/fl control mice, the mean intensity of MBP+ myelin fibers was significantly lower in OCE/Apcfl/fl Apc KO mice in spinal cord (Fig. 2D1,D3,F), cerebellum (Fig. 2E1,E2,F), and forebrain (data not shown). While numbers of SMI312+ (panaxonal marker) axonal fibers were similar (Fig. 2D2,D4), numbers of SMI312+/MBP− unmyelinated axons in corticospinal tract were significantly higher in OCE/Apcfl/fl mice compared with that in Apcfl/fl control mice (2099 ± 286 WT, 2522 ± 250 KO, p < 0.05, n = 4). These data indicate that two-allele Apc disruption in neonatal oligodendroglial lineage cells results in CNS hypomyelination.
APC controls oligodendrocyte differentiation
We used immunohistochemistry to test the hypothesis that the hypomyelination elicited by biallelic conditional deletion of oligodendroglial lineage Apc resulted from impaired oligodendroglial differentiation. Sox10 is a transcription factor ubiquitously expressed throughout oligodendrocyte development (Stolt et al., 2002; Rivers et al., 2008), and we found that the densities of Sox10+ oligodendroglial lineage cells were significantly decreased in both spinal cord WM and GM in OCE/Apcfl/fl Apc KO mice (Fig. 2G). We confirmed this result by showing that Sox10 mRNA was also decreased by approximately twofold, compared with controls, in OCE/Apcfl/fl mice (Fig. 2H). Using CC1 as an oligodendrocyte marker (Bhat et al., 1996), we demonstrated that the number of CC1+ oligodendrocytes per spinal cord section (Fig. 2I1–I3) and the density of Sox10+/CC1+ oligodendrocytes (Fig. 2K) in OCE/Apcfl/fl mice were significantly lower than those in Apcfl/fl mice. Quantification of cells expressing another oligodendrocyte marker, GST-π, also showed significantly decreased oligodendrocyte numbers in the spinal cord (Fig. 2J). Because Olig2-Cre-ERT2 is a knock-in transgene, replacing one of two endogenous Olig2 alleles (Takebayashi et al., 2002), we evaluated the effect of the transgene knock-in itself on oligodendrocyte differentiation, and found that the number of oligodendrocytes was not affected in OCE/Apcfl/fl mice that had not received tamoxifen (i.e., no Apc deletion) compared with Apcfl/fl controls (1261 ± 103 in Apcfl/fl, 1242 ± 93 in OCE/Apcfl/fl, n = 6, p = 0.74).
We used in situ hybridization to visualize and quantify the frequency of proteolipid protein (Plp) mRNA+ oligodendrocytes and found that, upon Apc disruption, Plp mRNA+ cells were significantly decreased in OCE/Apcfl/fl KO mice (Fig. 2L). QRT-PCR also demonstrated that levels of mRNAs encoded by the myelin genes Plp, Cnp, and Mbp were significantly lower in OCE/Apcfl/fl KO mice than in Apcfl/fl controls (Fig. 2M). In contrast, mRNA levels of Mbp, Plp, and Cnp were similar in OCE/Apcfl/fl and Apcfl/fl mice that had not been given tamoxifen (i.e., no Apc deletion) (Fig. 2N), again indicating that the OCE knock-in transgene does not affect oligodendrocyte development. In summary, our data indicate that oligodendrocyte differentiation is inhibited in Apc two-allele-deficient mice.
Normal oligodendrocyte differentiation in Apc one-allele KO mice
We analyzed Apc one-allele KO mice (OCE/Apcfl/+ treated with tamoxifen at P6 and P7) to determine whether Apc haploinsufficiency impedes oligodendrocyte development. In sharp contrast to Apc two-allele KO mice (OCE/Apcfl/fl treated with tamoxifen), the number of CC1+ oligodendrocytes and Sox10 mRNA levels in one-allele Apc KO mice were not significantly different from those in Apcfl/fl controls (Fig. 2H,I3) at P14. Also, the number of Plp mRNA+ cells in one-allele Apc KO did not differ from Apcfl/fl controls (Fig. 2L), but was significantly higher than that in two-allele Apc KO mice (Fig. 2L). Thus, oligodendrocyte differentiation is not perturbed in Apc one-allele KO mice.
APC disruption decreases OPC numbers by diminishing OPC proliferation
The frequency of Sox10+/NG2+ OPCs at P14 was significantly lower in OCE/Apcfl/fl Apc KO mice in both spinal cord WM and GM (Fig. 3C). At this point in postnatal development, >90% of phosphorylated histone 3 (PH3+) or Ki67+ mitotic cells in CNS are Sox10+/NG2+ OPCs (Fig. 3A1,A2; ≥30 PH3+ OPCs from 3 mice). In OCE/Apcfl/fl Apc KO mice, numbers of Ki67+/PH3+ cycling cells per section were significantly below those in Apcfl/fl controls (Fig. 3A5). Furthermore, the number of EdU+ cells (2 h labeling) (Fig. 3B1,B2) was also significantly decreased in OCE/Apcfl/fl Apc KO mice at P14 (74 ± 23 WT, 33 ± 10 KO, n = 3, p < 0.05). Cyclin D1 is required for the progression of cell cycle (Baldin et al., 1993; Stacey, 2003), and both cyclin D1 mRNA (Fig. 3E, right) and protein (Fig. 3D,3E, left) expression levels were approximately twofold lower in OCE/Apcfl/fl Apc KO mice than in WT controls. These results indicate that biallelic disruption of Apc inhibits OPC division. By observing no significant difference in activated caspase-3+ cells, we determined that heightened OPC apoptosis did not contribute to the diminution in numbers of OPCs caused by biallelic APC disruption (WT, 1.8 ± 0.9 per section; KO, 1.4 ± 0.8 per section; p = 0.56, n = 4).
Figure 3.

Apc deletion reduces OPC proliferation. Tamoxifen was injected in pups at P6 and P7 and CNS tissues were analyzed as indicated. A1–A5, Confocal images of PH3/Ki67 and PH3/SOX10/NG2 immunostaining and quantifications of cells positive for these markers in Apcfl/fl WT and OCE/Apcfl/fl KO (n = 3 WT, 3 KO at P8; n = 10 WT, 7 KO at P14). B1, B2, Representative confocal images of EdU (2 h labeling). C, The number of Sox10+/NG2+ OPCs in the spinal WM and GM of P14 Apcfl/fl WT (n = 10) and OCE/Apcfl/fl KO mice (n = 7). D, Western blot of cyclin D1 in P14 spinal cord. E, Quantification of cyclin D1 protein (left) and mRNA (right) levels in P14 Apcfl/fl WT (n = 4) and OCE/Apcfl/fl KO (n = 4) mice. Scale bars: A1–A4, 10 μm; B1, B2, 50 μm.
Inhibition of oligodendrocyte differentiation persists in adult Apc KO mice
We conditionally ablated Apc at P6/P7 and analyzed oligodendrocyte differentiation at P40, yielding mildly tremorous mice. Sox10 and CC1 double immunohistochemistry (Fig. 4A) demonstrated that the densities of Sox10+ panoligodendroglial lineage cells and CC1+ oligodendrocytes (Fig. 4A) were significantly reduced in adult OCE/Apcfl/fl Apc KO mice, compared with Apcfl/fl controls (Fig. 4B). Furthermore, mRNA levels of the myelin genes Plp and Cnp were significantly decreased in adult OCE/Apcfl/fl Apc KO mice (Fig. 4C).
Figure 4.

Inhibition of oligodendrocyte differentiation and myelination persists in the adult CNS. Tamoxifen was injected in pups at P6 and P7 and CNS tissues were analyzed at P40. A, Representative confocal images showing Sox10 and CC1 double immunostaining in P40 spinal cord. B, Quantifications of Sox10+ oligodendroglial lineage cells (left) and CC1+ oligodendrocytes (right) in P40 Apcfl/fl WT (n = 6) and OCE/Apcfl/fl KO (n = 4) spinal cord. C, Plp and Cnp mRNA expression levels in P40 Apcfl/fl WT (n = 6) and OCE/Apcfl/fl KO (n = 4) spinal cord by qRT-PCR. D, Proportions of Sox10+/NG2+ OPCs and Sox10+/CC1+ OLs among total Sox10+ oligodendroglial lineage cells in P40 Apcfl/fl WT spinal cord (n = 6) (left bar) and proportions of EYFP+/Sox10+/NG2+ OPCs and EYFP+/Sox10+/CC1+ OLs among total EYFP+/Sox10+ in OCE/Apcfl/fl/Rosa-EYFP KO spinal cord (n = 4) (right bar) at P40. E, The single surviving P50 adult Cnp-Cre, Apcfl/fl mouse displayed hind-limb paralysis (left), whereas a Cnp-Cre/Apcfl/+ littermate did not (right). F, G, MBP and SMI312 double IHC revealed severe hypomyelination (SMI312+/MBP− naked axon fibers, green when overlaid) in the spinal cord dorsal horn (arrowheads) of the Cnp-Cre/Apcfl/fl mouse (G), compared with normal myelination in a Cnp-Cre/Apcfl/+ littermate (F, SMI312+/MBP+ axons, yellow when overlaid). Scale bars: 50 μm, applied to all.
Next, we quantified the ratio of OPCs to oligodendrocytes in Apc KO and control mice. In adult Apcfl/fl WT mice, the ratio of NG2+ OPC number to CC1+ oligodendrocyte number among Sox10+ oligodendroglial lineage (OL) cells (Apc-intact) was ∼1:11.8 (7.9% OPCs, 92.9% OLs) (Fig. 4D, left bar). However the ratio of EYFP+NG2+ OPCs to EYFP+CC1+ oligodendrocytes among EYFP+Sox10+ oligodendroglial lineage cells (Apc-deficient) increased to 1:1.7 (37.2% EYFP+ OPCs, 62.6% EYFP+ OLs) (Fig. 4D, right bar) in adult OCE/Apcfl/fl Rosa-EYFP mice. This marked shift in ratio of OPCs to OLs among EYFP+, Apc-deficient oligodendroglial lineage cells (see Fig. 7D) again suggests that differentiation of OPCs to oligodendroglia is inhibited in adult biallelic Apc KO mice and that the decreased oligodendrocyte density and number (Fig. 2) are not only due to the reduced OPC number (Fig. 3) but also due to the compromised differentiation.
Figure 7.
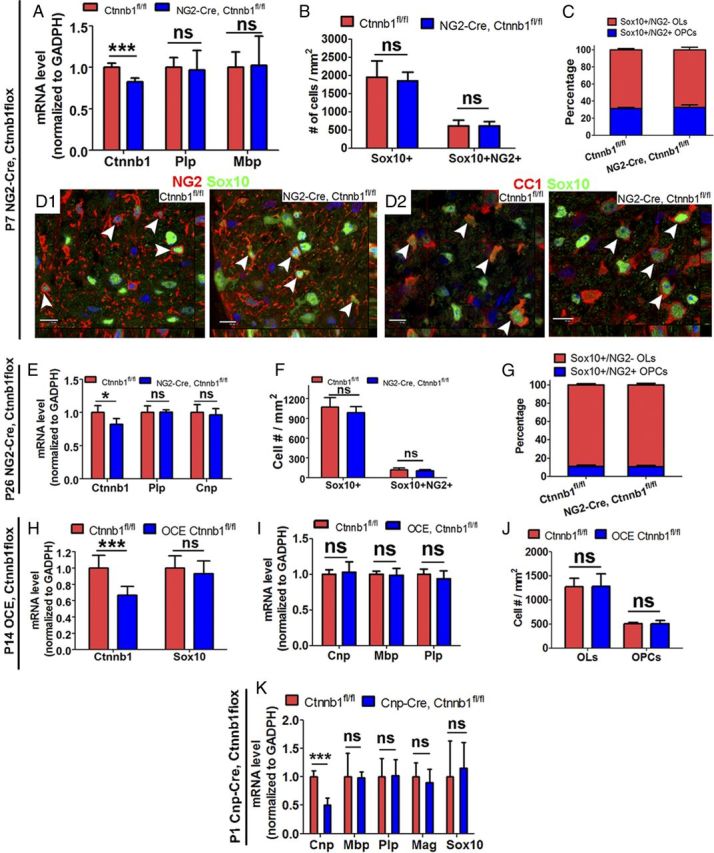
β-Catenin is dispensable for postnatal oligodendrocyte differentiation. A, QRT-PCR quantification of mRNAs encoded by the Ctnnb1 (gene for β-catenin) and the myelin genes, Plp and Mbp, in P7 spinal cord (n = 8 Ctnnb1fl/fl, n = 5 NG2-Cre/Ctnnb1fl/fl). B, Densities of Sox10+ and Sox10+/NG2+ cells (n = 4 Ctnnb1fl/fl, n = 3 NG2-Cre/Ctnnb1fl/fl) in P7 spinal cord. C, Proportions of OLs and OPCs in P7 spinal cord of Ctnnb1fl/fl (n = 4) and NG2-Cre/Ctnnb1fl/fl (n = 3) mice. D1, D2, Representative confocal images showing Sox10+/NG2+ OPCs (D1, arrowheads) and Sox10+/CC1+ OLs (D2, arrowheads) in the spinal WM of P7 Ctnnb1fl/fl and NG2-Cre/Ctnnb1fl/fl mice. E, MRNA levels of Ctnnb1 and myelin genes Plp and Cnp (n = 5 for each group) in P26 Ctnnb1fl/fl and NG2-Cre/Ctnnb1fl/fl spinal cord. F, G, Quantifications of Sox10+ oligodendroglial lineage cells and Sox10+/NG2+ OPCs (F), and the proportions of OPCs and OLs in the spinal cord at P26 (G) (n = 5 for each group) (K). H, I, Quantifications of mRNA level of Ctnnb1, Sox10 (H), and myelin genes Cnp, Mbp, and Plp (I) in the spinal cord of P14 Ctnnb1fl/fl (n = 6) and OCE/Ctnnb1fl/fl (n = 8) mice that received tamoxifen at P6 and P7 (see Material and Methods). J, Quantification of Sox10+/NG2+ OPCs and Sox10+/NG2− OLs in the spinal cord of P14 Ctnnb1fl/fl (n = 4) and OCE/Ctnnb1fl/fl (n = 3) mice that received tamoxifen at P6 and P7. K, QRT-PCR quantification of Cnp, Mbp, Plp, Mag, and Sox10 in the spinal cord of P1 Ctnnb1fl/fl (n = 4) and Cnp-Cre/Ctnnb1fl/fl (n = 4) pups. Scale bars, 10 μm.
As mentioned above, virtually all Cnp-Cre, Apcfl/fl mice died before weaning. Only one Cnp-Cre, Apcfl/fl mouse survived into adulthood during extensive attempts at breeding these bitransgenic mice. As shown in Figure 4E, this mouse was much smaller than Cnp-Cre, Apcfl/+ littermates, and displayed ataxia and hind-limb paralysis. Double immunohistochemistry with MBP (for myelin) and SMI312 (for axons) showed that most SMI312+ axons in the spinal dorsal horn of this adult Cnp-Cre/Apcfl/fl mouse were still unmyelinated (green when merged) (Fig. 4F, arrowhead), whereas almost all the SMI312+ axons were myelinated (yellow when merged) in a Cnp-Cre/Apcfl/+ littermate (Fig. 4G, arrowhead). Together, these data indicate that oligodendroglial differentiation and myelination are inhibited in adult Apc KO mice, and that one-allele Apc ablation does not affect oligodendrocyte development.
Wnt/β-catenin signaling is transiently upregulated in Apc two-allele KO mice
Upon activation of canonical Wnt signaling, β-catenin is translocated into the nucleus, where it binds to the Tcf/Lef transcription factor, leading to transcription of the Wnt target genes Axin2, Naked1, and Notum (Fancy et al., 2009, 2011). Therefore, immunohistological demonstration of nuclear accumulation of β-catenin in conjunction with assays for mRNA levels of Wnt target genes provide reliable hallmarks for activation of Wnt signaling. We found that β-catenin accumulated in the nuclei of EYFP+ Apc-deficient oligodendroglial lineage cells (Fig. 5A–C) in P14 OCE/Apcfl/fl Rosa-EYFP mice. Approximately 85% of nuclear β-catenin+ cells were EYFP+/PDGFRa+ OPCs (Fig. 5H), while the remainder were EYFP+/CC1+ differentiated oligodendrocytes (data not shown). Consistently, mRNA levels of Wnt target gene Axin2 and Naked1 were also significantly upregulated in Apc two-allele KO mice (Fig. 5G). In adult mice, in contrast, although we observed β-catenin nuclear accumulation in the EYFP+ cells (Fig. 5I), the mRNA levels of Axin2 and Naked1 were similar between P40 OCE/Apcfl/fl/Rosa-EYFP mice and Apcfl/fl/Rosa-EYFP controls (Fig. 5J, red bars), thus arguing that Apc ablation does not affect Wnt/β-catenin downstream signaling in these adult mice. Together, our data reveal that early postnatal Apc deletion leads to only a transient increase of Wnt/β-catenin signaling in oligodendroglial lineage cells.
Figure 5.

β-Catenin-dependent Wnt signaling is upregulated in Apc two-allele deleted oligodendroglial lineage cells. Tamoxifen was injected in pups at P6 and P7 and CNS tissues were analyzed as indicated. A–F, β-Catenin accumulated in the nuclei of EYFP+/SOX10+ cells in Apc two-allele (A–C) but not in Apc one-allele KO (D, E) or WT spinal cord at P14 (F). G, MRNA levels of Wnt target genes Axin2 and Naked1 in Apc two-allele (n = 7), one-allele (n = 4), and WT (n = 10) spinal cord at P14. H, Triple IHC showing nuclear accumulation of β-catenin in EYFP+/PDGFRα+ OPCs from two-allele (top) but not one-allele (bottom) Apc-KO mice. I, Double IHC of EYFP and β-catenin in P40 OCE/Apcfl/fl/Rosa-EYFP, two-allele Apc KO spinal cord. J, Axin2 and Naked1 expression levels in P40 Apcfl/fl (red bars, n = 6) and OCE, Apcfl/fl (blue bars, n = 4) spinal cord. Scale bars: A–F, 50 μm; H, 5 μm; I, 10 μm.
Unexpectedly, we found that the transient upregulation of Wnt/β-catenin signaling occurred in two-allele but not one-allele Apc KO oligodendroglial lineage cells. As shown in Figure 5D,E, there were no EYFP+ cells with nuclear β-catenin in one-allele KO spinal cord, similar to that in Apcfl/fl/Rosa-EYFP control mice (Fig. 5F). In accord with the absence of nuclear β-catenin, the mRNA levels of Axin2 and Naked1 in one-allele KO mice (OCE/Apcfl/+) were not significantly different from those in WT controls (Fig. 5G, green and red bars), suggesting Apc one-allele ablation is not sufficient to dysregulate Wnt/β-catenin signaling in oligodendroglial lineage cells. To support and confirm this conclusion, we evaluated Wnt/β-signaling in heterozygous Apcmin/+ mice (Moser et al., 1993), in which one-allele nonsense mutation in codon 850 of the Apc gene leads to a truncated APC polypeptide that lacks the β-catenin-binding domain. Consistently, mRNA levels of the Wnt/β-catenin signaling target genes Axin2, Naked1, and Notum in the spinal cord of Apcmin/+ mice were similar to those in Apc+/+ WT littermates at P7 (Fig. 6A) and in the adult (data not shown). Using Wnt-signaling reporter BAT-lacZ mice (Fig. 6B) (Maretto et al., 2003), we found that the number of β-gal+ (lacZ gene product) cells in the ventral spinal cord of Apcmin/+/BAT-lacZ double-transgenic mice (25 ± 5 per section, n = 4) was not significantly different from that in Apc+/+/BAT-lacZ littermates (23 ± 6 per section, n = 3) at P7 (Fig. 6C) and in the adult (data not shown). Collectively, these data indicate that (1) Apc one-allele KO is not sufficient to dysregulate Wnt/β-catenin signaling, and (2) Apc two-allele ablation leads to transient upregulation of this signaling in the early postnatal CNS, but not in the adult.
Figure 6.

Wnt signaling and oligodendrocyte differentiation are not perturbed in Apcmin/+ mice. A, MRNA levels of Wnt target genes Axin2, Naked1, and Notum in Apcmin/+ (n = 4) and Apc+/+ WT (n = 4) spinal cord at P7. B, Schematic drawing showing the mechanism of β-gal expression in BAT-lacZ Wnt reporter mice. C, β-Gal expression in APC+ cells from BAT-lacZ/Apc+/+ (top) and BAT-lacZ/Apcmin/+ mice (bottom) at P7. D–E, Representative confocal images showing Sox10+ oligodendroglial lineage cells and Sox10+/PDGFRα+ OPCs in the WM of Apc+/+ and Apcmin/+ spinal cord at P7. F, Schematic image depicting the sampling locations for G–I. V-WM, ventral WM; L-WM, lateral WM; CST, corticospinal tract. G–I, Quantifications of the frequency of Sox10+ panoligodendroglial lineage cells (G), Sox10+/PDGFRα+ OPCs (H), and Sox10+/CC1+ oligodendrocytes (I) in the spinal cord at P7 (n = 4 in each group). J, MRNA expression level of Cnp, Mbp, Plp, and cyclin D1 by qRT-PCR in P7 Apc+/+ and Apcmin/+ spinal cord (n = 4 in each group). Scale bars: C, 5 μm; D, E, 10 μm.
Oligodendrocyte differentiation is not perturbed in the Apcmin/+ mice
Our data demonstrate that neither Apc one-allele nonsense truncated mutation (Fig. 6A–C) nor one-allele conditional KO (Fig. 5A–G) dysregulates Wnt/β-catenin signaling in oligodendroglial lineage cells. These findings led us to assess whether oligodendrocyte development is perturbed in Apcmin/+ mice. To this end, we evaluated oligodendroglial differentiation in Apcmin/+ and Apc+/+ littermate control mice at P7, a time point when oligodendroglial Wnt/β-catenin signaling is most active (Fancy et al., 2009, 2011).
Double immunohistochemistry with the panoligodendroglial lineage marker Sox10 and the OPC marker PDGFRα revealed comparable immunostaining patterns in the P7 spinal cord of Apcmin/+ (Fig. 6E) and Apc+/+ mice (Fig. 6D). Quantification in different areas of spinal cord—ventral WM, lateral WM, corticospinal tract, and GM (Fig. 6F)—showed that the densities of Sox10+ oligodendroglial lineage cells (Fig. 6G), Sox10+PDGFRα+ OPCs (Fig. 6H), and Sox10+PDGFRα− oligodendrocytes (Fig. 6I) were comparable between Apcmin/+ and Apc+/+ mice (Fig. 6F), suggesting that the oligodendroglial population and differentiation are not affected in Apcmin/+ mice. This was further supported by qRT-PCR showing similar expression of the myelin genes Cnp, Mbp, and Plp in Apcmin/+ and Apc+/+ mice (Fig. 6J). Moreover, in contrast to decreased cyclin D1 levels (Fig. 3D,E) in Apc two-allele conditional KO, comparable cyclin D1 mRNA levels between Apcmin/+ and Apc+/+ controls (Fig. 6J) indicated that oligodendroglial proliferation was not affected in Apcmin/+ mice. We also did not detect differences in oligodendroglial differentiation between P30 Apcmin/+ and Apc+/+ control littermates (data not shown). These data suggest that oligodendroglial development and differentiation are normal in Apcmin/+ mice.
β-Catenin is dispensable for postnatal oligodendrocyte differentiation
Dominant active β-catenin (Cre-LoxP-mediated deletion of exon 3 of Ctnnb1, which results in a mutated β-catenin escaping degradation) inhibits oligodendrocyte differentiation (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009). Our data suggested that β-catenin-mediated Wnt-signaling overactivation contributed, at least in part, to the biallelic Apc-disruption-elicited inhibition of oligodendroglial differentiation (Fig. 5) (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009). To determine whether Apc disruption inhibits oligodendroglial differentiation exclusively through a Wnt/β-catenin-dependent mechanism, we simultaneously deleted Apc and Ctnnb1 (gene for β-catenin), and unexpectedly found that β-catenin is dispensable for postnatal oligodendrocyte differentiation.
When intercrossed with a Cre driver, β-catenin is successfully inactivated in Ctnnb1exon2-6flox/flox mice (Ctnnb1fl/fl) (Brault et al., 2001). Using NG2-Cre (Zhu et al., 2008; Guo et al., 2012), we confirmed that Ctnnb1 mRNA in the spinal cord of NG2-Cre/Ctnnb1fl/fl mice was reduced by ∼18% compared with Ctnnb1fl/fl controls (Fig. 7A). Based on the previous literature, we postulated that early postnatal ablation of Ctnnb1, when Wnt/β-catenin signaling is most active in the oligodendroglial lineage, would elicit precocious OPC differentiation. Some of the NG2-Cre/Ctnnb1fl/fl mice displayed loss of hair and smaller body-size phenotypes (data not shown). However, expression of the myelin genes Plp and Mbp (Fig. 7A), densities of Sox10+ panoligodendroglial lineage cells and Sox10+NG2+ OPCs (Fig. 7B), and the proportions of Sox10+NG2+ OPCs (Fig. 7D1) and Sox10+NG2− OLs (Fig. 7D2) were similar in NG2-Cre/Ctnnb1fl/fl mice compared with Ctnnb1fl/fl controls (Fig. 7C). Thus, disruption of Ctnnb1 does not perturb early postnatal OPC differentiation. We obtained similar results when we evaluated OPC differentiation at P26 in NG2-Cre/Ctnnb1fl/fl and Ctnnb1fl/fl control mice (Fig. 7E–G). Thus, β-catenin is dispensable for postnatal OPC differentiation.
Since the NG2 promoter and NG2-Cre transgene are active not only in OPCs but also in vascular pericytes (Zhu et al., 2008), we next used inducible OCE to specifically ablate Ctnnb1 in oligodendroglial lineage cells. Using the same tamoxifen injection paradigm as in OCE/Apcfl/fl neonates (Fig. 2) (see Materials and Methods), we found that Ctnnb1 mRNA levels were 33% lower in spinal cord of OCE/Ctnnb1fl/fl mice than in Ctnnb1fl/fl controls at P14 (Fig. 7H, left), but OPC differentiation was not altered (Fig. 7H–J).
A previous study used Cnp-Cre to successfully ablate Ctnnb1 in mouse embryos (Ye et al., 2009). We postulated that if Ctnnb1 ablation promotes OPC differentiation, as it promotes precocious OPC generation from neural stem cells in embryos (Ye et al., 2009), we would expect to see increased numbers of Plp mRNA+ oligodendrocytes and elevated levels of myelin gene expression at P1, the time point at which OPC differentiation is initiated. However, we did not detect a difference in Plp mRNA+ cells by in situ hybridization in the spinal cord of P1 Cnp-Cre/Ctnnb1fl/fl pups, compared with Ctnnb1fl/fl controls (data not shown). QRT-PCR quantification showed that levels of myelin Mbp, Plp, and Mag mRNAs and levels of oligodendroglial marker Sox10 mRNA in P1 Cnp-Cre/Ctnnb1fl/fl pups to Ctnnb1fl/fl were similar to those in controls. However, Cnp mRNA was decreased by ∼50% (Fig. 7K). This lower Cnp mRNA level was presumably due to the haploinsufficiency of Cnp gene transcription, since Cnp-Cre is a knock-in transgene replacing one of the two endogenous Cnp alleles (Lappe-Siefke et al., 2003).
Collectively, our data demonstrate that oligodendroglial differentiation is not perturbed upon Ctnnb1 disruption, and suggest that β-catenin is dispensable for postnatal oligodendroglial differentiation under physiological conditions, although its overactivation inhibits oligodendroglial differentiation (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009; Chew et al., 2011).
APC regulates oligodendroglial differentiation through additional β-catenin-independent mechanisms
Because β-catenin ablation does not affect postnatal oligodendroglial development (Fig. 7), we used Apc and Ctnnb1 conditionally double KO (dKO) mice to dissect the β-catenin-independent role of Apc in oligodendroglial differentiation. We demonstrated that β-catenin-mediated Wnt signaling was no longer elevated in dKO mice (Fig. 8E). Quantification data showed that the density of nuclear β-catenin+ cells in dKO mice was significantly reduced compared with that in Apc KO mice (Fig. 8A–C) and was indistinguishable from that in WT mice (Fig. 8C). In both WT (Fig. 8D) and dKO spinal cord (Fig. 8D), many nuclear β-catenin+ cells viewed at low magnification proved at higher magnification to be closely associated with blood vessels, as evidenced by the association of nuclear β-catenin and the pericyte marker PDGFRβ (Fig. 8D, arrowheads). Few nuclear β-catenin+ cells observed in normal postnatal development belonged to the Sox10+ oligodendroglial lineage (data not shown). In contrast, >95% of nuclear β-catenin+ cells in Apc KO mice were EYFP+ Apc-deficient oligodendroglial lineage cells (Fig. 8A). Consistent with the loss of β-catenin, the elevated Axin2 mRNA level observed in Apc KO mice was abolished in dKO mice (Fig. 8F). Together, our results demonstrate that dKO of Apc and Ctnnb1 provides an in vivo model with which to dissect the β-catenin-independent role of APC in oligodendroglial lineage cells.
Figure 8.

Inhibition of oligodendrocyte differentiation in Apc and Ctnnb1 double-conditional KO mice. Tamoxifen was injected in pups at P6 and P7 and tissues were analyzed at P14. A, B, Double IHC showed β-catenin accumulation in the nuclei of EYFP+ cells in OCE/Apcfl/fl/Rosa-EYFP mice (Apc deletion) (A, arrowheads), whereas β-catenin nuclear accumulation was abolished in Apc and Ctnnt1 dKO mice (B, arrowheads). C, Quantification of nuclear β-catenin+ cells among each genotype at P14 (n = 3 for each group). D, Nuclear β-catenin+ cells in the WT spinal cord were associated with PDGFRβ-expressing pericytes (arrowheads). E, Quantification of Ctnnb1 mRNA in each genotype at P14 (n = 3 in WT and dKO, n = 4 in Ctnnb1 KO). F, The expression level of Wnt target gene Axin2 was comparable between dKO and WT mice (n = 3 in WT and dKO; n = 7 in Apc KO). G, QRT-PCR quantification showed that expression of myelin genes Plp, Mbp, and Cnp was significantly reduced in spinal cords of P14 dKO mice (n = 7 in WT; n = 5 in dKO). H, Densities of Sox10+ oligodendroglial lineage cells and Sox10+CC1+ oligodendrocytes in the spinal cord of P14 WT and dKO mice (n = 3 in each group). Scale bars: A, B, 50 μm; D, 10 μm.
If the inhibition of oligodendroglial differentiation mediated by Apc disruption (Fig. 2) is caused solely by dysregulated β-catenin-mediated Wnt signaling, one would expect that oligodendroglial differentiation in dKO mice would resemble that in WT mice. On the contrary, we found that expression levels of three major myelin genes, Plp, Mbp, and Cnp (Fig. 8G), were significantly lower in dKO spinal cord compared with spinal cord of WT controls, in sharp contrast to their normal expression in Ctnnb1 KO mice (Fig. 7I). In addition, Sox10 and CC1 immunostaining showed that the densities of both Sox10+ panoligodendroglial lineage cells and Sox10+CC1+ oligodendrocytes (Fig. 8H) were significantly decreased in dKO mice, compared with WT mice, suggesting that oligodendroglial differentiation is disturbed in dKO mice. This is consistent with the finding that Apc disruption causes hypomyelination that persists into adulthood (Fig. 4), when β-catenin stabilization is not sufficient to activate Wnt signaling (Fancy 2009). Collectively, our experiments demonstrate a β-catenin-independent role of APC in regulating oligodendroglial differentiation.
Microarray analysis reveals cytoskeleton dysregulation upon Apc disruption
We used microarray gene expression profiling to search for genes and gene clusters that were dysregulated by Apc deletion. Analyses of raw microarray data obtained from four WT and four Apc KO spinal cords (see Materials and Methods) revealed 336 genes with altered expression of ≥1.2-fold change and p ≤ 0.05 with FDR correction (gene list available on request). Next, we used qRT-PCR to verify the microarray data by quantifying the expression levels of genes that were upregulated (Fig. 9B, red) and downregulated (Fig. 9B, black) in Apc KO mice, and the genes with both most-marked (Fig. 9B, Casr) and least-marked changes (Fig. 9B, Cx3cl1). Our results showed that all the qRT-PCR data were consistent with those from the microarray analysis (Fig. 9B), thus supporting the validity of the microarray profiling data.
Figure 9.
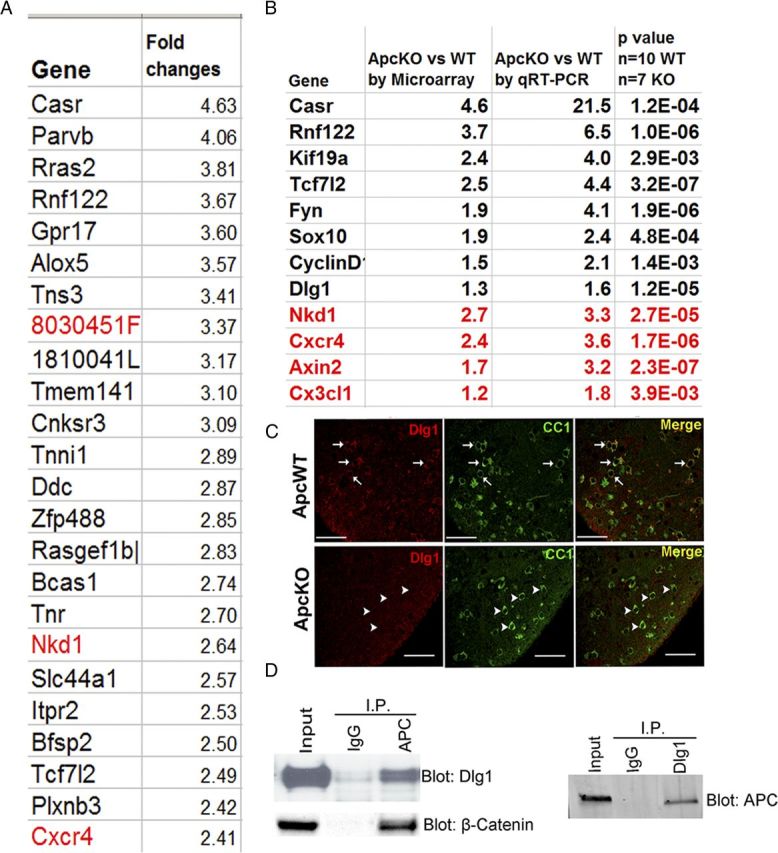
Genes with altered expression derived from microarray data analysis and their validations. A, Representative top 24 genes with altered expression upon Apc ablation (red, upregulated in Apc KO) mediated by Olig2-Cre-ERT2 with tamoxifen treatment at P6 and P7 and analysis at P14. B, Comparison and validation of microarray profile dysregulated genes by qRT-PCR. C, To provide validation at the protein level, double IHC showing that DLG1 protein expression was barely detectable in CC1+ oligodendrocytes in P14 Apc-KO (OCE/Apcfl/fl) (arrowheads), whereas there was robust expression in WT mice (Apcfl/fl) (arrows). Tamoxifen was injected at P6 and P7. D, Coimmunoprecipitation indicating APC and DLG1 physically interact with each other, with β-catenin as a positive control, using protein extracted from day 2 OPC cultures. Scale bars, 50 μm.
We next used the DAVID program (Huang da et al., 2009b,c) to explore the potential functions and pathways affected by oligodendroglial Apc ablation. The analysis showed that oligodendrocyte differentiation, regulation of Wnt receptor signaling and cell proliferation were significantly enriched when these 336 altered genes were queried against the mouse GO database (Table 1), which is consistent with our data showing that Apc disruption affects oligodendrocyte differentiation, proliferation, and Wnt signaling (Figs. 2–5). Interestingly, we also found that regulation of the cytoskeleton was significantly enriched upon Apc disruption (Table 1), suggesting that APC regulates oligodendrocyte differentiation by additionally controlling cytoskeleton structure (Näthke, 2006). Of note, one of the altered genes derived from microarray data analysis and verified by qRT-PCR and IHC was Dlg1 (Fig. 9B) (Chung, 2011). Previous studies reported that physical interaction between scaffolding protein DLG1 [Discs large homolog 1, also termed synapse-associated protein 97 (SAP97)] and APC is required for polarization of the microtubule cytoskeleton in astrocytes (Etienne-Manneville et al., 2005; Näthke, 2006). Our data showed that DLG1 is also expressed in oligodendrocytes, and that DLG1-immunoreactive signals were markedly decreased in the CC1+ oligodendrocytes of Apc KO spinal cord (Fig. 9C, arrowheads) compared with their extensive colocalization in WT mice (Fig. 9C, arrows). Coimmunoprecipitation demonstrated that APC physically interacted with DLG1 in oligodendrocytes, in addition to binding β-catenin (Fig. 9D). Our results suggest that the APC binding partner DLG1 regulates oligodendrocyte differentiation, possibly via modulation of the oligodendroglial cytoskeleton, but further studies are needed to define its role in oligodendroglial lineage cells.
Table 1.
Representative biological processes of gene ontology that are enriched among the altered gene expression by Apc gene disruption analyzed by DAVID program
| Terms of biological processes | Gene count | p value | Fold enriched |
|---|---|---|---|
| Oligodendrocyte differentiation | 4 | 0.0036 | 12.7 |
| Negative regulation of microtubule polymerization or depolymerization | 3 | 0.0254 | 11.9 |
| Myelination | 5 | 0.0026 | 8.6 |
| Axon ensheathment | 5 | 0.0031 | 7.1 |
| Negative regulation of cytoskeleton organization | 5 | 0.0067 | 6.6 |
| Regulation of Wnt receptor signaling pathway | 4 | 0.0229 | 6.5 |
| Regulation of actin polymerization or depolymerization | 5 | 0.0077 | 6.3 |
| Regulation of actin filament length | 5 | 0.0083 | 6.2 |
| Actin filament organization | 5 | 0.0114 | 5.7 |
| Regulation of actin filament polymerization | 4 | 0.0333 | 5.6 |
| Glial cell differentiation | 4 | 0.0392 | 5.3 |
| Regulation of actin filament-based process | 5 | 0.0153 | 5.2 |
| Regulation of cytoskeleton organization | 8 | 0.0009 | 5.1 |
| Regulation of cell migration | 7 | 0.0032 | 4.8 |
| Actin filament-based process | 11 | 0.0005 | 4.0 |
| Actin cytoskeleton organization | 10 | 0.0012 | 3.8 |
| Negative regulation of cell differentiation | 10 | 0.0023 | 3.5 |
| Positive regulation of cell proliferation | 12 | 0.0052 | 2.7 |
| Cell proliferation | 10 | 0.0160 | 2.6 |
| Cytoskeleton organization | 13 | 0.0054 | 2.5 |
| Regulation of cell proliferation | 18 | 0.0048 | 2.1 |
| Cell adhesion | 17 | 0.0154 | 1.9 |
Microarray analysis was used to identify 336 genes with FDR-corrected p value <0.05 and with 1.2-fold change cutoff. Note that oligodendrocyte differentiation and myelination, Wnt signaling pathway, and cytoskeleton regulation were significantly enriched as a result of oligodendroglial Apc disruption.
OPCs develop fewer and shorter processes in Apc deletion mice
APC stabilizes the microtubule cytoskeleton (Näthke, 2006), which is critical for process extension (Lunn et al., 1997; Bauer et al., 2009). There are two pools of microtubules: dynamic microtubules and stabilized microtubules (Saxton et al., 1984; Schulze and Kirschner, 1987). Acetylated α-tubulin is a marker of stabilized microtubules (Bloom, 2004; Yokota et al., 2009). We showed that α-tubulin and acetylated α-tubulin were colabeled with APC+ cell bodies and processes (Fig. 10A,B) in our culture system, a similar microtubule pattern obtained from β-tubulin (Lunn et al., 1997). Using primary OPC cultures, we found that the ratio of acetylated α-tubulin to total α-tubulin increased upon OPC differentiation into oligodendrocytes (Fig. 10C,D), indicating a role of microtubule stabilization during OPC differentiation. Unfortunately, unlike cultured OPCs with dominant active β-catenin (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009), cultured Apc-ablated OPCs did not survive well (data not shown); this precluded in vitro analysis of process extension by Apc-deficient OPCs.
Figure 10.

Apc disruption affects the morphological complexity of OPCs and localizes to stable microtubules in oligodendrocytes. A, B, Immunostaining of APC, α-tubulin, and acetylated α-tubulin on cultured oligodendrocytes with 2 d differentiation. C, D, Western blot of total α-tubulin and acetylated α-tubulin in OPCs (G0) and oligodendrocytes at 4 d differentiation (D4) and quantification. E–H, Sholl analysis (G) on the process complexity and branching of EYFP−/NG2+ Apc-intact OPC (E) and EYFP+/NG2+ Apc-deficient OPCs (F) in the spinal cord of P40 OCE, Apcfl/fl, EYFP mice (tamoxifen injection at P6 and P7) and quantification of intersection numbers at each radius (1–5) (H, ≥30 OPCs each group from 4 adult OCE, Apcfl/fl mice with tamoxifen treatment at P6, P7). Scale bars, 10 μm.
In vivo, OPCs undergo a morphological transition from a few simple processes to a complex process meshwork. We hypothesized that disruption of Apc would affect adult OPC process extension, an event that requires microtubule stability. In the spinal cord of adult OCE/Apcfl/fl, EYFP mice (tamoxifen treatment at P6 and P7), there was a mosaic pattern of OPCs: EYFP-negative Apc-WT OPCs and EYFP-positive Apc-deficient OPCs (Fig. 10E,F) due to incomplete Cre-mediated recombination. We used Sholl analysis (Rajasekharan et al., 2009) (see Materials and Methods) to quantify and compare the number and length of process branches of individual OPCs (Fig. 10G). Our data showed that Apc-KO (EYFP+) OPCs extended shorter and less complex processes compared with Apc-WT (EYFP−) OPCs (Fig. 10H), indicating that APC might regulate the formation of the complex process network of OPCs. Alternatively, since our data showed that Apc ablation inhibited OPC differentiation, the lesser complexity of OPC processes could also possibly result from the blockage of OPC differentiation at some step before complex process formation.
Discussion
Transient APC expression in oligodendroglial lineage cells
Double IHC using APC antibody and CC1 displayed completely nonoverlapping staining patterns in the adult neocortex (Fig. 1C), arguing strongly against the equivalence of APC with oligodendrocyte marker CC1. One of the interesting findings in our study is that APC is transiently expressed in oligodendroglial lineage cells during normal myelination and again during remyelination after CNS injury (Fig. 1). Immunoreactive APC was very low in OPCs but transiently upregulated in myelinating oligodendrocytes. We also demonstrated that APC and the Wnt-signaling effector Tcf7l2 were transiently and dynamically expressed in the same population of oligodendrocytes (Fig. 1K). A previous study showed that activity of the Wnt/β-catenin pathway increased after lysolecithin-induced demyelination, as evidenced by elevated expression of Tcf7l2 (Fancy et al., 2009). Since canonical Wnt/β-catenin signaling proceeds via Tcf7l2 to initiate transcriptional activity in oligodendroglial lineage cells (Fancy et al., 2009), the overlapping patterns of APC and Tcf7l2 (Fig. 1K) suggest that activation of Wnt/β-catenin signaling after lysolecithin-induced demyelination (Fancy et al., 2009) occurs in APC+ cells, and argues for the existence of a tightly regulated, negative feedback of APC and Wnt/β-catenin signaling during oligodendrocyte differentiation. Indeed, we observed completely overlapping patterns of APC and Tcf7l2 reinduction in both cuprizone-induced and MOG-peptide-induced demyelinative models (our unpublished data).
In the present study, genetic Apc ablation was initiated either by constitutive cre transgenes, or by activation of an inducible cre transgene at the end of the first postnatal week; thus, initial Apc recombination took place largely in OPCs, rather than in oligodendroglia. In future studies, we plan also to ablate Apc at later time points to determine the effects of APC deficiency in differentiated oligodendroglia on myelination and remyelination.
Wnt/β-catenin signaling and oligodendrocyte differentiation in Apcmin/+ mice
Though a prior publication speculated that Wnt/β-catenin signaling is overactivated in Apcmin/+ mice (Fancy et al., 2009), we found that both conditional KO of one-allele Apc (OCE/Apcfl/+ mice) (Fig. 5D–G) and nonsense one-allele truncated mutation of Apc (Apcmin/+ mice) (Fig. 6A–C) have similar levels of Wnt/β-catenin signaling to those in WT mice. These results argue against the assumption that canonical Wnt/β-catenin signaling is dysregulated in Apcmin/+ mice. We also found that postnatal oligodendrocyte development is normal in Apcmin/+ mice (Fig. 6D–G). These data suggest that the delayed oligodendroglial differentiation observed in Apcmin/+ mice with lysolecithin-induced demyelinative injury (Fancy et al., 2009) probably results from a Wnt/β-catenin-independent effect of monoallelic Apc truncation or deletion in an injury-specific manner.
β-Catenin disruption does not affect oligodendrocyte differentiation in early postnatal or adult mice
Prior data obtained from dominant activation of oligodendroglial lineage β-catenin suggested that canonical Wnt/β-catenin signaling inhibits early postnatal oligodendrocyte differentiation (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009). However, those reports did not conclusively address whether β-catenin is important in oligodendrocyte differentiation under physiological conditions, since dominant active β-catenin may affect signaling pathways or target genes not normally controlled by β-catenin-mediated Wnt signaling. In support of this caveat, β-catenin deletion does not affect hematopoiesis and lymphopoiesis (Cobas et al., 2004; Prlic and Bevan, 2011), although dominant activation of β-catenin suggests the opposite (Gounari et al., 2001).
Thus far, the best documented evidence for modulation of development of the oligodendroglial lineage by physiological β-catenin-mediated Wnt signaling was obtained by β-catenin loss of function (Ye et al., 2009) and manipulation of Wnt ligands (Langseth et al., 2010) in the embryo. These studies conclusively showed that Wnt/β-catenin signaling is important in the generation of OPCs from embryonic neural stem cells (Ye et al., 2009; Langseth et al., 2010). Using the same breeding paradigm as the one in a previous study (Ye et al., 2009), we showed that oligodendrocyte differentiation was similar in Cnp-Cre/Ctnnb1fl/fl neonates to that in Ctnnb1fl/fl controls (Fig. 7K). This observation was further supported by the results we obtained by β-catenin loss of function mediated by constitutive NG2-Cre (Fig. 7A–G) or inducible Olig2-Cre-ERT2 (Fig. 7H–J). Our data suggest that, though important in embryonic OPC generation, β-catenin is dispensable for postnatal oligodendrocyte differentiation under physiological conditions. Nevertheless, this does not preclude the possibility that manipulation of oligodendroglial Wnt signaling might promote oligodendrocyte remyelination under injury conditions, as suggested by a recent study (Fancy et al., 2011). It has been reported that γ-catenin (plakoglobin) can compensate for the loss of β-catenin in cardiomyocytes (Zhou et al., 2007) and hepatocytes (Wickline et al., 2011). Whether this compensatory mechanism applies to CNS oligodendroglial lineage cells needs to be determined.
Role of APC in oligodendrocyte development: β-catenin-dependent and β-catenin-independent mechanisms
The transient expression of APC suggests its role in oligodendroglial differentiation and myelination. To elucidate this role under physiological conditions, we used Cre-LoxP-mediated Apc loss of function specifically in oligodendroglial lineage cells. We found that Apc deletion inhibits oligodendrocyte differentiation and myelination (Fig. 2), and that this inhibition persists into adult oligodendroglial lineage cells (Fig. 4), suggesting that APC is critical in oligodendroglial development.
Why does Apc disruption inhibit oligodendrocyte differentiation? One of the mechanisms responsible for this effect is the elevation of canonical Wnt/β-catenin signaling. In the colorectal cancer research field, it is well established that APC negatively regulates canonical β-catenin-mediated Wnt signaling by targeting β-catenin for subsequent proteasome-mediated degradation (Näthke, 2006). According to this well established concept, we postulate and present compelling data to show that Apc loss of function results in dysregulation of oligodendroglial Wnt/β-catenin signaling (Fig. 5), which has previously been shown to affect early postnatal oligodendrocyte differentiation (Fancy et al., 2009; Feigenson et al., 2009; Ye et al., 2009).
Another interesting finding of our study is that Apc ablation reduced OPC proliferation (Fig. 3). Overactivation of Wnt/β-catenin signaling inhibits oligodendrocyte differentiation but does not perturb proliferation (Fancy et al., 2009). If the effects of Apc deletion on the oligodendroglial lineage were due solely to overactivation of Wnt/β-catenin signaling, we would not expect to observe a decrease in OPC proliferation in Apc KO mice. Our observation of a reduced rate of OPC proliferation in homozygous Apc KO mice is reminiscent of the slowed proliferation of Apc-deficient radial glia demonstrated in a prior study and ascribed to abnormal organization of microtubules in mitotic spindles (Yokota et al., 2009). Furthermore, the inhibition of oligodendrocyte differentiation and myelination persists in the adult CNS (Fig. 4), a phenotype that is different from that in dominant active β-catenin transgenic mice, in which oligodendrocyte differentiation is normal in the adult (Fancy et al., 2009). These phenotypic discrepancies between Apc KO and β-catenin stabilization suggest an additional, β-catenin-independent role of APC in regulating oligodendrocyte differentiation. To prove the existence of this β-catenin-independent role of APC in oligodendrocytes, we conditionally ablated Apc and Ctnnb1 genes simultaneously. Since our data showed that loss of function of β-catenin does not affect oligodendrocyte differentiation (Fig. 7), the inhibition of oligodendrocyte differentiation in Apc and Ctnnb1 dKO mice (Fig. 8) indicates that APC regulates oligodendrocyte differentiation via an additional novel β-catenin-independent mechanism.
What pathway accounts for this β-catenin-independent mechanism of APC? We explored this issue by GO analysis of a list, derived from microarray data, of 336 genes with altered expression upon Apc deletion. As a positive control to validate the efficacy of GO analysis, oligodendrocyte differentiation, and myelination, Wnt signaling and cell proliferation were all significantly enriched upon Apc deletion (Table 1), which is in line with APC's Wnt/β-catenin signaling modulation (Aoki and Taketo, 2007) and also with the data from our current study (Figs. 2–5). We also found that the biological process of GO, termed regulation cytoskeleton stability, was significantly enriched (Table 1), which suggests that APC's regulation of cytoskeleton stability might be one of the β-catenin-independent mechanisms, although we cannot exclude the possibility that this dysregulation of cytoskeletal stability could also result from β-catenin-mediated Wnt-signaling overactivation. Using an in vitro culture system, we showed that the stabilized microtubule component, acetylated α-tubulin (Fig. 10B) (Shintani et al., 2009), was significantly increased upon OPC differentiation (Fig. 10C,D), a result consistent with our in vivo observation that Apc-deficient OPCs developed less and shorter process branching (Fig. 10H), an event in which microtubule stability plays an important role (Bauer et al., 2009). Nevertheless, more studies are needed to investigate how APC modulates microtubule cytoskeleton stability and regulates oligodendrocyte differentiation by controlling process extension.
In conclusion, our study shows that APC is expressed transiently in the oligodendroglial lineage cells during normal oligodendrocyte development and during oligodendrocyte regeneration. Loss-of-function experiments suggest that APC regulates oligodendrocyte differentiation via both β-catenin-dependent and novel β-catenin-independent mechanisms. In addition, our findings have revealed that β-catenin is dispensable for postnatal oligodendrocyte differentiation under physiological conditions, and that Apc one-allele deficiency is not sufficient to dysregulate β-catenin-mediated Wnt signaling in oligodendroglial lineage cells.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS025044, National Multiple Sclerosis Society Grant RG4397-A-5, the Shriners Hospitals for Children, and the California Institute for Regenerative Medicine.
References
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–3335. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Axonal control of oligodendrocyte development. J Cell Biol. 1999;147:1123–1128. doi: 10.1083/jcb.147.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer NG, Richter-Landsberg C, Ffrench-Constant C. Role of the oligodendroglial cytoskeleton in differentiation and myelination. Glia. 2009;57:1691–1705. doi: 10.1002/glia.20885. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Baraban JM, Johnson RC, Eipper BA, Mains RE. High levels of expression of the tumor suppressor gene APC during development of the rat central nervous system. J Neurosci. 1994;14:3059–3071. doi: 10.1523/JNEUROSCI.14-05-03059.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bloom K. Microtubule composition: cryptography of dynamic polymers. Proc Natl Acad Sci U S A. 2004;101:6839–6840. doi: 10.1073/pnas.0401266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakeman JS, Gu SH, Wang XB, Dolin G, Baraban JM. Neuronal localization of the adenomatous polyposis coli tumor suppressor protein. Neuroscience. 1999;91:661–672. doi: 10.1016/s0306-4522(98)00605-8. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Chen Y, Tian X, Kim WY, Snider WD. Adenomatous polyposis coli regulates axon arborization and cytoskeleton organization via its N-terminus. PLoS One. 2011;6:e24335. doi: 10.1371/journal.pone.0024335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Shen W, Ming X, Senatorov VV, Jr, Chen HL, Cheng Y, Hong E, Knoblach S, Gallo V. SRY-box containing gene 17 regulates the Wnt/β-catenin signaling pathway in oligodendrocyte progenitor cells. J Neurosci. 2011;31:13921–13935. doi: 10.1523/JNEUROSCI.3343-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AY, Kim S, Kim H, Bae YK, Park HC. Microarray screening for genes involved in oligodendrocyte differentiation in the zebrafish CNS. Exp Neurobiol. 2011;20:85–91. doi: 10.5607/en.2011.20.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, Radtke F. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–229. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Manneville JB, Nicholls S, Ferenczi MA, Hall A. Cdc42 and Par6-PKCzeta regulate the spatially localized association of Dlg1 and APC to control cell polarization. J Cell Biol. 2005;170:895–901. doi: 10.1083/jcb.200412172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJ, Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson K, Reid M, See J, Crenshaw EB, 3rd, Grinspan JB. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci. 2009;42:255–265. doi: 10.1016/j.mcn.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Gounari F, Aifantis I, Khazaie K, Hoeflinger S, Harada N, Taketo MM, von Boehmer H. Somatic activation of beta-catenin bypasses pre-TCR signaling and TCR selection in thymocyte development. Nat Immunol. 2001;2:863–869. doi: 10.1038/ni0901-863. [DOI] [PubMed] [Google Scholar]
- Gounari F, Chang R, Cowan J, Guo Z, Dose M, Gounaris E, Khazaie K. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat Immunol. 2005;6:800–809. doi: 10.1038/ni1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Ma J, McCauley E, Bannerman P, Pleasure D. Early postnatal proteolipid promoter-expressing progenitors produce multilineage cells in vivo. J Neurosci. 2009;29:7256–7270. doi: 10.1523/JNEUROSCI.5653-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Xu J, Horiuchi M, Miers L, Vaccarino F, Pleasure D. Pyramidal neurons are generated from oligodendroglial progenitor cells in adult piriform cortex. J Neurosci. 2010;30:12036–12049. doi: 10.1523/JNEUROSCI.1360-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Delgado M, Sohn J, Miers L, Ko EM, Bannerman P, Xu J, Wang Y, Zhou C, Takebayashi H, Pleasure D. Macroglial plasticity and the origins of reactive astroglia in experimental autoimmune encephalomyelitis. J Neurosci. 2011;31:11914–11928. doi: 10.1523/JNEUROSCI.1759-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ko EM, Delgado M, Horiuchi M, Soulika A, Miers L, Burns T, Itoh T, Shen H, Lee E, Sohn J, Pleasure D. Disruption of NMDA receptors in oligodendroglial lineage cells does not alter their susceptibility to experimental autoimmune encephalomyelitis or their normal development. J Neurosci. 2012;32:639–645. doi: 10.1523/JNEUROSCI.4073-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Lindsten T, Pleasure D, Itoh T. Differing in vitro survival dependency of mouse and rat NG2+ oligodendroglial progenitor cells. J Neurosci Res. 2010;88:957–970. doi: 10.1002/jnr.22262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Zheng X, Yang J, Imamichi T, Stephens R, Lempicki RA. Extracting biological meaning from large gene lists with DAVID. Curr Protoc Bioinformatics Chapter. 2009c;13 doi: 10.1002/0471250953.bi1311s27. Unit 13.11. [DOI] [PubMed] [Google Scholar]
- Imura T, Wang X, Noda T, Sofroniew MV, Fushiki S. Adenomatous polyposis coli is essential for both neuronal differentiation and maintenance of adult neural stem cells in subventricular zone and hippocampus. Stem Cells. 2010;28:2053–2064. doi: 10.1002/stem.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Beesley J, Itoh A, Cohen AS, Kavanaugh B, Coulter DA, Grinspan JB, Pleasure D. AMPA glutamate receptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J Neurochem. 2002;81:390–402. doi: 10.1046/j.1471-4159.2002.00866.x. [DOI] [PubMed] [Google Scholar]
- Jimbo T, Kawasaki Y, Koyama R, Sato R, Takada S, Haraguchi K, Akiyama T. Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nat Cell Biol. 2002;4:323–327. doi: 10.1038/ncb779. [DOI] [PubMed] [Google Scholar]
- Kipp M, Clarner T, Dang J, Copray S, Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118:723–736. doi: 10.1007/s00401-009-0591-3. [DOI] [PubMed] [Google Scholar]
- Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, Kucherlapati M, Maas RL, Kucherlapati R. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006;2:e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langseth AJ, Munji RN, Choe Y, Huynh T, Pozniak CD, Pleasure SJ. Wnts influence the timing and efficiency of oligodendrocyte precursor cell generation in the telencephalon. J Neurosci. 2010;30:13367–13372. doi: 10.1523/JNEUROSCI.1934-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Li A, Xing Y, Chan B, Heisterkamp N, Groffen J, Borok Z, Minoo P, Li C. Cell type-specific expression of adenomatous polyposis coli in lung development, injury, and repair. Dev Dyn. 2010;239:2288–2297. doi: 10.1002/dvdy.22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn KF, Baas PW, Duncan ID. Microtubule organization and stability in the oligodendrocyte. J Neurosci. 1997;17:4921–4932. doi: 10.1523/JNEUROSCI.17-13-04921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Toews A, Hostettler JD, Morell P, Suzuki K, Goldman JE, Matsushima GK. Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am J Pathol. 2004;164:1673–1682. doi: 10.1016/S0002-9440(10)63726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S, Moissoglu K, Macara IG. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008;453:115–119. doi: 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AR, Mattes EM, Dove WF, Lindstrom MJ, Haag JD, Gould MN. ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc Natl Acad Sci U S A. 1993;90:8977–8981. doi: 10.1073/pnas.90.19.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näthke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer. 2006;6:967–974. doi: 10.1038/nrc2010. [DOI] [PubMed] [Google Scholar]
- Prlic M, Bevan MJ. Cutting edge: beta-catenin is dispensable for T cell effector differentiation, memory formation, and recall responses. J Immunol. 2011;187:1542–1546. doi: 10.4049/jimmunol.1100907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med. 2008;205:2163–2175. doi: 10.1084/jem.20080578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekharan S, Baker KA, Horn KE, Jarjour AA, Antel JP, Kennedy TE. Netrin 1 and Dcc regulate oligodendrocyte process branching and membrane extension via Fyn and RhoA. Development. 2009;136:415–426. doi: 10.1242/dev.018234. [DOI] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Horie Y, Bachmaier K, Fata JE, Li M, Suzuki A, Bouchard D, Ho A, Redston M, Gallinger S, Khokha R, Mak TW, Hawkins PT, Stephens L, Scherer SW, Tsao M, Penninger JM. Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature. 2000;406:897–902. doi: 10.1038/35022585. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Stemple DL, Leslie RJ, Salmon ED, Zavortink M, McIntosh JR. Tubulin dynamics in cultured mammalian cells. J Cell Biol. 1984;99:2175–2186. doi: 10.1083/jcb.99.6.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze E, Kirschner M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 1987;104:277–288. doi: 10.1083/jcb.104.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Ihara M, Tani S, Sakuraba J, Sakuta H, Noda M. APC2 plays an essential role in axonal projections through the regulation of microtubule stability. J Neurosci. 2009;29:11628–11640. doi: 10.1523/JNEUROSCI.2394-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:14965–14979. doi: 10.1523/JNEUROSCI.3794-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15:158–163. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol. 2002;12:1157–1163. doi: 10.1016/s0960-9822(02)00926-0. [DOI] [PubMed] [Google Scholar]
- Traka M, Wollmann RL, Cerda SR, Dugas J, Barres BA, Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci. 2008;28:11537–11549. doi: 10.1523/JNEUROSCI.1490-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Imura T, Sofroniew MV, Fushiki S. Loss of adenomatous polyposis coli in Bergmann glia disrupts their unique architecture and leads to cell nonautonomous neurodegeneration of cerebellar Purkinje neurons. Glia. 2011;59:857–868. doi: 10.1002/glia.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 2004;6:820–830. doi: 10.1038/ncb1160. [DOI] [PubMed] [Google Scholar]
- Wickline ED, Awuah PK, Behari J, Ross M, Stolz DB, Monga SP. Hepatocyte gamma-catenin compensates for conditionally deleted beta-catenin at adherens junctions. J Hepatol. 2011;55:1256–1262. doi: 10.1016/j.jhep.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y, Kim WY, Chen Y, Wang X, Stanco A, Komuro Y, Snider W, Anton ES. The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron. 2009;61:42–56. doi: 10.1016/j.neuron.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Zhou J, Qu J, Yi XP, Graber K, Huber L, Wang X, Gerdes AM, Li F. Upregulation of gamma-catenin compensates for the loss of beta-catenin in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;292:H270–H276. doi: 10.1152/ajpheart.00576.2006. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]