

Abstract
Prolonged exposure to high-efficacy agonists results in desensitization of the μ-opioid receptor (MOR). Desensitized receptors are thought to be unable to couple to G-proteins, preventing downstream signaling; however, the changes to the receptor itself are not well characterized. In the current study, confocal imaging was used to determine whether desensitizing conditions cause a change in agonist–receptor interactions. Using rapid solution exchange, the binding kinetics of fluorescently labeled opioid agonist, dermorphin Alexa594 (derm A594), to MORs was measured in live cells. The affinity of derm A594 binding increased after prolonged treatment of cells with multiple agonists that are known to cause receptor desensitization. In contrast, binding of a fluorescent antagonist, naltrexamine Alexa594, was unaffected by similar agonist pretreatment. The increased affinity of derm A594 for the receptor was long-lived and partially reversed after a 45 min wash. Treatment of the cells with pertussis toxin did not alter the increase in affinity of the derm A594 for MOR. Likewise, the affinity of derm A594 for MORs expressed in mouse embryonic fibroblasts derived from arrestin 1 and 2 knock-out animals increased after treatment of the cells with the desensitization protocol. Thus, opioid receptors were “imprinted” with a memory of prior agonist exposure that was independent of G-protein activation or arrestin binding that altered subsequent agonist–receptor interactions. The increased affinity suggests that acute desensitization results in a long-lasting but reversible conformational change in the receptor.
Introduction
Opioid agonists are widely used for the management of pain. The development of tolerance is a major clinical limitation in treating persistent pain in that effective pain relief often requires increasing doses of opioids. Activation of the μ-opioid receptor (MOR) by highly efficacious agonists for as little as 5 min results in a decrease in downstream signaling, characterized as homologous receptor desensitization (Harris and Williams, 1991). Acute receptor desensitization may be the first step in a process leading to the development of tolerance.
Previous studies of receptor desensitization have focused on changes in signaling of downstream effectors to make inferences about changes in the receptor itself. Because of second messenger signal amplification and variability in receptor–effector coupling, it is not clear how receptor desensitization relates to decline in effector readout. A large receptor reserve or efficient receptor–effector coupling can limit the downstream measurement of receptor desensitization. That is, a significant number of receptors can be desensitized while having little effect on effector activity if an efficacious agonist is used at high concentration (Connor et al., 2004). This has resulted in conflicting information about the role of various signaling pathways involved in mediating desensitization. Furthermore, investigations of acute desensitization have used various effectors in many different systems, which has created conflicting information and ideas about the processes governing desensitization (Chuang et al., 1998; Bohn et al., 2000; Tan et al., 2003; Schulz et al., 2004; Bailey et al., 2009; Dang et al., 2009; Arttamangkul et al., 2012).
The current model of desensitization, based on studies of the β2-adrenergic receptor, asserts that agonist binding and G-protein activation induce downstream kinase activation and phosphorylation by G-protein-coupled receptor kinase. This phosphorylation leads to the recruitment of β-arrestin, which binds to receptors with high-affinity preventing receptor association and activation of heterotrimeric G-proteins (Benovic et al., 1987; Dang and Christie, 2012). This model is assumed to hold true for MOR signaling, although studies over the past 15 years have not convincingly elucidated a mechanism.
Real-time measurements of receptor–ligand interactions in living cells have the potential to examine desensitization at the level of the receptor itself without relying on changes in effector function. This study examines a receptor-based model of MOR desensitization in live cells. Fluorescent opioid ligands (an agonist and an antagonist) were imaged to study their binding kinetics to MOR in HEK293 cells that expressed a FLAG-tagged MOR. Prolonged treatment of MOR cells with a protocol known to result in desensitization dramatically slowed the rate of fluorescent agonist unbinding (dissociation) and increased affinity in a long-lived manner. Antagonist unbinding was unaffected. Thus, receptors were imprinted with a signature of previous exposure to agonists. The increase in affinity was agonist dependent but independent of G-protein signaling and β-arrestin proteins. These results suggest that conditions that are known to cause receptor desensitization also induced long-lived changes in agonist–receptor interactions that directly affected agonist affinity.
Materials and Methods
Materials.
The following drugs were used in this study: [Met5] enkephalin, oxycodone, concanavalin A, M1 anti-FLAG, pertussis toxin, naloxone, β-chlornaltrexamine (Sigma-Aldrich) fentanyl, methadone, morphine (National Institute on Drug Abuse, Neuroscience), and Alexa-488 SDP ester and Alexa-594 succinimidyl ester (Invitrogen). M1 anti-FLAG was conjugated with Alexa-488 SDP ester and purified using Bio-Spin 6 Tris columns (Bio-Rad). Plasmids and stable cells lines for expression of FLAG tagged MOR were provided by Mark von Zastrow, University of California San Francisco, and pcDNA 3.1 plasmids encoding G-protein coupled inward rectifying potassium channels 1 and 2 (GIRK 1 and GIRK 2) were gifts from Kevin Wickman, University of Minnesota.
Cell culture.
HEK293 cells stably expressing FLAG tagged MOR (FLAG-MOR) have been described previously (Keith et al., 1996). Cells were cultured in DMEM containing 10% FBS, 0.5% HEPES, 0.8% G418. Mouse embryonic fibroblast cells (MEF), created from WT or β-arrestin 1/2 double knock-out embryonic mice as described previously (Kohout et al., 2001), were a gift from Dr. Robert Lefkowitz, Duke University. MEF cells were efficiently transfected using NeuroMag Magnetofection (OzBiosciences) following the manufacturer's protocol using 5 μg of plasmid DNA and 8.5 μl of NeuroMag solution per well of a 6 well dish.
Fluorescent ligand synthesis.
Naltrexamine Alexa-594 (ntx A594) was synthesized as follows: The 6-β-naltrexamine 2HCl, 0.75 H2O was synthesized as previously described (Sayre and Portoghese, 1980). The compound was dissolved in 0.1 m sodium bicarbonate and then reacted with Alexa-594 succinimidyl ester in DMSO with the molar ratio of 1:7. The crude product was purified by HPLC to at least 98% purity. The molecular mass of the product was further confirmed by electronspray ionization mass spectrometry by the Bioanalytic Shared Resource/Pharmacokinetics Core, Oregon Health & Science University.
The fluorescent peptide dermorphin Alexa-594 (derm A594) was synthesized as previously described (Arttamangkul et al., 2000). Briefly, the peptide (Tyr-D-Ala-Phe-Gly-Tyr-Pro-Lys-Cys-amide) was made using standard Fmoc solid phase peptide protocol. The crude peptide was purified by HPLC before conjugation with the Alexa-594 maleimide dye (Invitrogen). The final product was further purified by HPLC and its molecular mass confirmed by electronspray ionization mass spectrometry (Bioanalytic Shared Resource/Pharmacokinetics Core, Oregon Health & Science University).
Radioligand binding.
HEK293 cells stably expressing FLAG-MOR were grown to confluency and harvested for membrane preparation as previously described (Clark et al., 2003). Cell membranes were thawed, Dounce homogenized, and incubated (5 μg/well) for 60 min at room temperature with 0.2 nm [3H]diprenorphine and 0–10 μm derm A594 or 10 μm naloxone (for nonspecific binding) in Tris buffer (50 mm Tris-HCl, pH 7.4) or G buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2, and 10 μm GTPγS). Samples were filtered through glass-fiber filter mats (Whatman, GF/C) using a Brandel cell harvester and rinsed three times with ice-cold Tris or G buffer. Filters mats were dried, scintillation mixture (EcoLume, ICN) was added, and the filter mats were sealed in a polyethylene bag. Radioactivity retained on the filters was counted in a Wallac 1450 Microβ liquid scintillation counter. Data were fit to a one or two site competition binding curve using GraphPad Prism to determine Ki. After determining that a one site model better fit the data, the Hill slope was fixed to n = 1 to calculate the best fit.
Whole-cell [3H] DAMGO binding.
FLAG-MOR-expressing HEK293 cells grown in 24-well plates were pretreated by replacing the cell media with Krebs buffer containing 600 μg/ml concanavalin A and with or without 30 μm [Met5] enkephalin (ME). After 2 h at 37°C, treatment buffer was replaced 2 times with Krebs buffer alone at 37°C, then with Krebs buffer containing 12 nm [3H] DAMGO with or without 10 μm naloxone. The binding was stopped after 10 min at 37°C by putting the plate on ice and quickly replacing the assay buffer 2 times with ice-cold Krebs buffer followed by replacement with cold 3% perchloric acid (or lysis buffer for protein samples). After 30 min on ice, samples were transferred to vials for liquid scintillation counting.
Imaging solution.
Coverslips were perfused with Krebs buffer containing (in mm) NaCl 126, KCl 2.5, MgCl2 1.2 CaCl2 2.4, NaH2PO4 1.2, NaHCO3 21.4, glucose 11.1, and aerated with 5% CO2/ 95% O2. For fast solution exchange, NaHCO3 and glucose were omitted and HEPES 15 was added, the pH adjusted to 7.4 with HCl, and osmolarity adjusted to 305 mOsm.
Imaging.
Imaging was performed on an Olympus BX51WI inverted microscope and Olympus LUMPlanFL N 60×/1.00 W objective. A Nipkow spinning disk confocal scanner (Solamere Technology) and ICCD camera (XR/MEGA 10, Stanford Photonics) were used to acquire rapid confocal images in a single plane using Piper data acquisition software (Piper Control 2.4.51, Stanford Photonics). Alexa-488 was imaged using a 488 nm Argon ion laser (National Laser Company), whereas Alexa-594 was excited using a 561 nm solid state laser (Cobolt Jive 25). Rapid switching between 488 nm and 561 nm was achieved via AOTF (NEOS). A quadruple bandpass dichroic mirror (Di01 T403/488/568/647, Semrock) and dual bandpass emission filter (FF01–523/610, Semrock) were used.
Data acquisition.
FLAG-tagged MOR was labeled with M1 anti-FLAG antibody conjugated to Alexa-488 to allow for visualization of receptors on the plasma membrane. Solution exchange was achieved using computer-controlled solenoid valves (Warner Instruments) to switch between two solutions that flowed through double-barrel θ glass attached to a piezo bimorph (Piezo Systems) to facilitate exchange. Ratiometric imaging was performed by taking serial images alternating between 488 nm and 561 nm excitation wavelengths to image Alexa-488 and Alexa-594, respectively. A pair of images (100 ms exposure each) was generally taken every 2.5 s before, during, and after application of derm A594 to image baseline fluorescence, binding, and unbinding, respectively.
Steady-state binding.
Cells labeled with M1 A488 were incubated in various concentrations of derm A594 (3–300 nm) and allowed to equilibrate for 5–10 min and subsequently imaged over 10–15 min. Because there was no relationship between equilibration time and the amount of derm A594 bound, it can be concluded that derm A594 was largely equilibrated within 5 min (10 nm derm A594 after ME treatment linear regression of incubation time vs R/G value; r = 0.292, p = 0.45). A 5 s puff of derm A594-free solution was applied during image acquisition to reduce background fluorescence. The images used for quantification were taken within 1 s of derm A594 removal so that unbinding of derm A594 was minimal. Intensity of derm A594 binding relative to M1 488 (R/G) was measured and averaged for each concentration. To allow a direct comparison of binding under different conditions, measurement of steady-state binding in untreated and ME treated cells was interleaved using the same stock solution of M1 A488 antibody to label surface receptors and the same derm A594 to assess binding.
Analysis.
Image quantification was performed using ImageJ software (ImageJ, NIH) and the “Time Series Analyzer” plugin (J. Balaji, Department of Neurobiology, University of California, Los Angeles). Analysis was done using Excel and Origin Laboratory software. Five background ROI surrounding the cell of interest and one signal ROI encompassing the plasma membrane with as little intracellular area as possible were selected, and the fluorescence intensity of each region was measured for each image frame in a time series for both 488 nm and 561 nm excitation wavelengths to measure M1 A488 anti-FLAG and derm A594 intensity, respectively. The average of the 5 background ROI intensities was subtracted from the signal ROI for each frame and the ratio of derm A594: M1 A488 intensity was measured to give an R/G value. The magnitude of the R/G values is dependent upon efficiency of antibody–fluorophore conjugation, exposure time, laser intensity, and efficiency of light collection for each wavelength and do not represent an absolute ratio of ligand bound per receptor. The average R/G value before exposure to derm A594 was taken to represent the baseline fluorescent signal in the absence of any derm A594. This baseline fluorescence was subtracted from all R/G values in the time series. To compare the time course of binding and unbinding between experiments, the R/G intensities were normalized to the intensity of the first image after washout of derm A594 (unbinding) or the final image of derm A594 application (binding). The average fluorescence intensity of the signal ROI was generally ∼5 times that of the background ROIs when measuring M1 A488 fluorescence. derm A594 fluorescence signal was ∼2 times above background when measuring unbinding, but only 5–10% above background ROI intensity when measuring binding at higher derm A594 concentrations.
For steady-state ligand binding and competition assays, relative fluorescence intensity was plotted and fit using the Hill equation with a variable slope. For kinetic binding experiments, apparent association rate or dissociation rates were calculated by fitting data from each experiment with a single exponential decay function. All fitting was done using Origin Laboratory. Apparent association and unbinding rates were averaged among experiments. Apparent association of derm A594 (Kobs) as a function of concentration was plotted and linearly fit (Eq. 1) according to a first-order binding mechanism so that:
 |
with Kon being the slope of the line and Koff being the y-intercept and Kd calculated from
 |
For ntx A594 binding, apparent association (kobs) and unbinding rates (koff) were measured using 100 nm ntx A594, and Equation 1 was used to calculate the rate of association (kon) and Equation 2 used to estimate Kd.
Results
Specificity of binding and unbinding of derm A594 to MORs
The fluorescent opioid agonist derm A594 was rapidly applied to HEK 293 cells stably expressing FLAG-tagged MOR to address whether derm A594 could be used to assay opioid receptor binding kinetics (Fig. 1). Plasma membrane-localized FLAG-MOR receptors were labeled with the M1 anti-FLAG antibody conjugated to Alexa-488 (M1-A488) (Fig. 1A). Binding and unbinding (dissociation) of derm A594 (100 nm, 90 s) were imaged using a spinning disk confocal microscope. By measuring the fluorescence intensity of both derm A594 (red, R) and M1 488 (green, G) bound to the cell membrane, ratiometric quantification (R/G) of derm A594 binding relative to the receptor density was monitored (Fig. 1B). Specificity of binding was confirmed by preincubating cells with the irreversible antagonist, β-chlornaltrexamine (β-CNA; 1 μm, 5 min) and subsequently applying derm A594 (500 nm, 5 min) in the presence of the opioid antagonist naloxone (10 μm, Fig. 1C). A high concentration of naloxone was used to increase the rate of naloxone binding to receptors because derm A594 (500 nm) has a relatively fast apparent rate of association at this concentration. There was no appreciable fluorescence after β-CNA/naloxone treatment. Thus, derm A594 binds specifically to FLAG-MOR receptors in HEK293 cells, and the kinetics of binding can be easily monitored.
Figure 1.

Imaging binding and unbinding of dermorphin A594. A, FLAG-MOR in HEK 293 cells were labeled with M1 anti-FLAG antibody-conjugated Alexa-488 (M1 A488) to visualize receptors localized on the plasma membrane. Dermorphin Alexa-594 (100 nm, derm A594) was applied for 90 s (wash in at t = −90 washout at t = 0 in A). derm A594 binding and unbinding were assessed by imaging M1 A488 and derm A594 every 2.5 s with images from selected time points shown. Scale bar, 20 μm. B, Ratiometric imaging of derm A594: M1 A488 (R/G) intensity was used to quantify binding and unbinding of agonist to receptor from the experiment shown in A. C, Specificity of binding was assessed by determining the relative fluorescence intensity (R/G) of derm A594 bound immediately after application of derm A594 (5 min, 500 nm) under control conditions or on cells pretreated with β-CNA (1 μm, 5 min) and bathed in naloxone (10 μm). Data are mean ± SEM (ctrl, n = 5; β-CNA/naloxone, n = 6).
MOR was largely in low-affinity state in live cells
Agonist binding to MOR in membrane homogenates displays two affinity states, a high agonist affinity state thought to comprise a ternary complex between agonist, MOR, and G-protein, and a low agonist affinity state thought to form between agonist and non-G-protein coupled MOR (Werling et al., 1988). Radioligand competition binding assays were performed to assess the ability of derm A594 to displace [3H]-diprenorphine bound to membrane homogenates from HEK 293 cells stably expressing FLAG-MOR. Displacement assays were performed in 50 mm Tris to observe high-affinity binding and 50 mm Tris in the presence of Na+, Mg2+, and GTPγS to displace G-proteins and observe low-affinity binding. derm A594 displaced [3H]-diprenorphine with a Ki of 2.9 ± 0.8 nm in Tris and 120 ± 40 nm in Tris plus Na+, Mg2+, and GTPγS (Fig. 2A, average of 3 experiments in duplicate; Table 1), revealing high- and low-affinity agonist binding. Under both conditions, the data were best fit to a one site model. The large shift in affinity upon addition of Na+, Mg2+, and GTPγS is indicative of an agonist with generally high efficacy (Lee et al., 1999).
Figure 2.

derm A594 binding to FLAG-MOR in live cells is relatively low affinity. A, Steady-state displacement of [3H]diprenorphine (0.2 nm) with increasing concentrations of derm A594 (0–10 μm) from FLAG-MOR HEK 293 cell membranes was measured in Tris alone (blue) or Tris with Na+, Mg2+, and GTPγS (green) and fit with one site nonlinear regression to define high- and low-affinity binding of derm A594 to FLAG-MOR. The best fit of the data are plotted. Average of 3 experiments performed in duplicate. B, Steady-state binding of derm A594 in live FLAG-MOR HEK 293 cells was relatively low affinity. Cells were incubated in stated concentrations of derm A594, imaged, and relative fluorescence intensity was measured. Data were fit with the Hill equation, and the best fit is plotted (black); n = 5–11. High- and low-affinity binding curves from [3H]diprenorphine competition binding experiments shown in A are overlaid in blue and green, respectively, using the Ki values from competition binding experiments and the slope and R/Gmax values from steady-state whole-cell derm A594 binding to simulate expected results. C, Kinetics of derm A594 binding. derm A594 was applied at 10 (blue), 30, 100 (red), and 200 nm (black), and the apparent association rates were determined by fitting to a single exponential decay function. For display, binding was normalized relative to the amount of derm A594 bound at the end of the 1 min period and averaged results are plotted; n = 4–6. D, Linear fit of the apparent association rate from average data in C plotted as a function of derm A594 concentration. The plotted line has a slope of 612,000 mol−1 s−1 and an intercept of 0.068 s−1, giving a calculated Kd of 110 nm. E, Binding and unbinding kinetics were unaffected by pertussis toxin treatment. Cells were either untreated (black) or treated with pertussis toxin (red) (100 ng/ml, overnight). derm A594 (100 nm) was applied for 90 s at the indicated time and rapidly washed. Data are mean ± SEM (ctrl, n = 6; pertussis toxin, n = 5).
Table 1.
Summary of derm A594 and ntx A594 affinity measurementsa
| Ligand | Assay | Affinity (nm) |
|---|---|---|
| derm A594 | Radioligand competition | 2.9 ± 0.8 (Ki) |
| +Na+, Mg2+, GTPγS | 120 ± 40 nm (Ki) | |
| Association kinetics | 111 (85–149) (Kd) | |
| Association kinetics after ME 2 h | 32 (24–42 (Kd) | |
| Steady-state imaging | 85 (50–142) (EC50) | |
| Steady-state after ME 2 h | 11.6 (8.4–16.2) (EC50) | |
| ntx A594 | Kinetics | 53 (44–65) (Kd) |
a [3H]Diprenorphine competition performed in crude membrane preparations in the presence and absence of Na+, Mg2+, and GTPγS (Ki ± SD; Fig. 2A) revealed low- and high-affinity states of derm A594 binding. Kinetic affinity measurements from live cells were calculated from initial apparent binding kinetics measured for a range of derm A594 concentrations performed in live cells untreated and pretreated with ME 2 h (Kd ± error range; Fig. 2C,D). Steady-state binding of derm A594 to live cells untreated and pretreated with ME 2 h (EC50 ± 95% CI; Figs. 2B, 3B) are shown demonstrating increased affinity after ME treatment. Affinity of ntx A594 was based on measurement of apparent on rate and off rate of ntx A594 (100 nm, 5 min) (Kd ± error range; Fig. 6B). ntx A594 shows similar affinity to derm A594 despite different kinetics.
Next, binding of derm A594 was characterized in live HEK293 cells stably expressing FLAG-MOR to determine whether binding in live cells resembled high- or low-affinity binding seen in the displacement assay. Cells were maintained in HEPES-buffered modified Krebs' solution to mimic physiological ionic conditions as much as possible. M1 A488 was used to visualize surface-localized receptors. derm A594 (3–300 nm) was added to the bath solution, allowed to equilibrate at room temperature, and the ratio of derm A594 bound to cells relative to M1 488 was measured. Half-maximal “steady-state ” binding was estimated to be ∼85 nm derm A594 determined by the best fit of the data (EC50 = 85 nm, 50–142 nm 95% CI, Hill slope = 0.95 ± 0.02, Fig. 2B, n = 5–11 cells for each concentration). This more closely resembled the low agonist affinity binding rather than high agonist affinity binding observed in typical binding assays performed in the absence of sodium. No saturation of whole-cell binding could be observed because derm A594 (300 nm) was the highest concentration possible to image before background fluorescence became unacceptable. Thus, it is possible that the observed binding affinity could appear artificially high, and the actual value may be closer to the 120 nm low-affinity state. It should be noted that the R/G values in Figure 2B are dependent upon the exposure time for each image, relative laser intensity, efficiency of fluorescence excitation, and collection of emitted light. Thus, the maximum R/G value of 4 does not represent an absolute ratio of 4 derm A594 molecules bound per FLAG epitope, and the absolute ratio was not determined in these assays.
Binding kinetics was also used to assess the interaction between derm A594 and MOR in live cells at room temperature. derm A594 was rapidly applied at 10, 30, 100, and 200 nm (Fig. 2C, n = 4–7), and the apparent rate of association was measured. The apparent on rate was plotted as a function of derm A594 concentration (Fig. 2D) and fit linearly according to Equation 1 to yield an association rate of 612,000 ± 114,000 (mol*s)−1, a rate of dissociation of 0.068 ± 0.006 s−1, and a calculated Kd of ∼111 nm (range, 85–149 nm; Eq. 2). Thus, using nonequilibrium binding kinetics, the calculated affinity of derm A594 for MOR was similar to live cell steady-state measurements and radioligand competition results from membrane homogenates in the presence of sodium and GTPγS.
Pertussis toxin is known to eliminate activation of Gi/o G-proteins by MOR and to reduce high-affinity binding of agonists to MOR in the presence of sodium (Kurose et al., 1983; Wüster et al., 1984). If high-affinity binding, which is sodium- and G-protein dependent, makes a significant contribution toward the interaction of agonist and receptor in live cells, one would expect pertussis treatment to decrease the affinity and alter the kinetics of agonist binding to MOR receptor. Treatment with pertussis toxin, however, had no effect on the kinetics of derm A594 binding and unbinding (Fig. 2E). Pertussis toxin treatment did block the activation of GIRK current by ME (1 μm, 5 s) in cells transiently coexpressing FLAG-MOR, GIRK1, and GIRK2 (untreated, 171 ± 61 pA, n = 5; pertussis toxin 100 ng/ml overnight, 3.3 ± 6.8 pA, n = 4) confirming its effectiveness at disrupting Gi/o signaling. This suggests that, although the high-affinity state is likely important for activation of G-proteins and subsequent signaling, most binding and unbinding events in live cells occurred to/from the low-affinity state. Together, these results suggest that binding of derm A594 to receptors in live cells under physiological conditions most closely resembles low-affinity binding seen in traditional radioligand binding assays being both relatively low affinity and pertussis toxin-insensitive. This is consistent with many previous observations that sodium and GTP, present at high concentrations under physiological conditions, lower the affinity of many agonists for GPCRs.
Desensitizing conditions induced a persistent change in agonist–receptor interaction
Classically, desensitization has been defined by a decrease in receptor-mediated signaling in response to prolonged application of high concentrations of efficacious agonists. Full agonists, such as the endogenous opioid agonist ME, cause robust desensitization, whereas partial agonists, such as morphine, are less effective (Dang and Williams, 2005). If the process of desensitization results in a change in receptor conformation state rather than simply a loss of downstream signaling, the kinetics of ligand–receptor interactions may be predicted to change. Therefore, the rate of derm A594 unbinding was used as a reporter for changes in MOR conformation in response to desensitizing agonist exposure. FLAG-MOR-expressing cells were incubated at 37°C and either untreated or treated for 2 min, 20 min, or 2 h in ME 30 μm (a desensitizing concentration) in the presence of concanavalin A (200–600 μg/ml) to reduce MOR internalization. After ME treatment, cells were washed for 1 min and transferred to an imaging chamber during which time cells were perfused with room temperature Krebs' buffer. After 5 min in the imaging chamber, binding and unbinding were measured by imaging FLAG-MOR cells while a 60 or 90 s application of derm A594 (100 nm) was applied. The experimental outline is diagrammed in Figure 3 (top). Surprisingly, pretreatment with ME dramatically slowed the rate of subsequent unbinding of derm A594 (Fig. 3A). The slowed rate of unbinding was quantified by determining the fraction of derm A594 remaining 3 min after washout. The fraction of derm A594 that remained bound after 3 min increased dramatically (Fig. 3B; fraction remaining; untreated, 0.16 ± 0.01, n = 9; ME 30 μm, 2 min, 0.25 ± 0.03, n = 6; ME 30 μm, 20 min, 0.39 ± 0.02, n = 11; ME 30 μm 2 h, 0.52 ± 0.02, n = 7). Thus, although ME was washed from the receptors, MOR pretreated with ME maintained a persistent signature of prior agonist exposure that resulted in a change in the kinetics of subsequent binding of derm A594 to the receptor.
Figure 3.

MOR retained a memory of previous agonist exposure. A, FLAG-MOR-expressing cells were incubated either under control (■) conditions or in the presence of ME 30 μm for 20 min (□) or 2 h (red circles) as indicated. After washing out ME, cells were imaged while derm A594 (100 nm, 60 s) was applied. Individual examples are shown normalized to the relative fluorescence intensity (R/G) immediately after washout of derm A594. B, Summary data quantifying the fraction of derm A594 that remained bound to the receptor 3 min after washout (indicated by arrow in A) in cells that were untreated or treated with ME 30 μm for 2 min, 20 min, or 2 h, and results are shown (n = 6–11). *p < 0.05, compared with untreated (one-way ANOVA, Tukey post hoc). ***p < 0.001, compared with untreated (one-way ANOVA, Tukey post hoc). C, Steady-state binding was performed as described in Figure 2B after pretreatment of cells with ME for 2 h and was fit using the Hill equation and plotted (n = 7–19 cells for each concentration). Data from untreated cells (black) shown in Figure 2B and data from cells incubated with ME (red) are plotted. D, Average observed association of derm A594 (30 nm) in untreated (black) and ME pretreated (30 μm 2 h, red) cells. E, Plot of average observed association rate of derm A594 after ME treatment (red) is plotted as a function of derm A594 concentration. The linear fit has a slope of 353,000 mol−1 s−1 and an intercept of 0.011 s−1 yielding a Kd of 32 nm; n = 4 each. Data from untreated cells (black) shown in Figure 2D are also plotted.
Demonstrating a concentration dependence, a subsaturating concentration of ME (300 nm) applied for 20 min induced a modest change in derm A594 unbinding rate similar to that induced by 2 min of ME 30 μm (fraction derm A594 remaining = 0.27 ± 0.02, n = 4, data not shown). Receptor modulation by agonist exposure persisted after ME washout. Cells that were washed for 45 min at 37°C after a 1 h exposure to ME showed ∼65% reversal of the change in derm A594 unbinding rate (fraction derm A594 remaining: 1 h ME, 0.45 ± 0.01, n = 6; 1 h ME/45 min wash, 0.26 ± 0.01, n = 7; untreated, 0.16 ± 0.01, n = 9; data not shown).Together, there was a change in the kinetics of derm A594 unbinding that was dependent upon both time and concentration of ME preexposure, which reversed slowly over tens of minutes.
Agonist treatment increased agonist affinity
If the rate of unbinding of derm A594 slowed with little or no change in the rate of association, the affinity of the agonist for the receptor should increase. Steady-state binding in live cells was used to estimate the affinity of derm A594 for FLAG MOR after preincubation of cells with ME (30 μm 2 h, Fig. 3C). After ME treatment, the cells were washed, various concentrations of derm A594 were applied, and the amount of derm A594 bound relative to M1 A488 intensity was measured and plotted (Fig. 3C, red). The steady-state binding curve shown in Figure 2B measured under naive conditions is overlaid for reference (black). After pretreatment with ME, there was a leftward shift of the derm A594 binding curve (n = 7–19 cells for each concentration). The EC50 of the best fit of the data increased from ∼85 to 12 nm (Table 1) after incubation in ME (30 μm, 2 h), indicating that desensitizing conditions increased the affinity of agonist for receptor. The Hill slope wash unchanged (1.03 ± 0.3). Consistent with ME treatment increasing the affinity of MOR for derm A594, after ME treatment there was a large increase in derm A594 binding to MOR at low concentrations (derm A594, 10 nm, 275 ± 87% increase after ME treatment) that was not seen at high concentrations (derm A594, 300 nm, 12 ± 6% increase after ME treatment, Fig. 3C). This observation suggests a leftward shift in the binding curve rather than a large change in the number of available binding sites and that 300 nm derm A594 is nearly saturating MOR binding sites.
The apparent association rate was also measured to assess whether prolonged ME treatment increased the affinity of derm A594 for MOR. As was done in untreated cells (Fig. 2C,D), the binding of derm A594 to cells preincubated with ME (30 μm 2 h) was imaged using various concentrations of derm A594 (10–200 nm). Binding of derm A594 (30 nm) to untreated cells (black) and ME treated cells (red) revealed that the average time constant of association increased from 11.4 ± 0.4 s to 51.5 ± 7.8 s (Fig. 3D, n = 4 each). A plot of the apparent association rate as a function of derm A594 concentration (Fig. 3E) was linearly fit in untreated cells (Fig. 2D, black) and cells treated with ME (red). After ME treatment, kon decreased by ∼40% to 353,000 ± 29,000 m−1 s−1, whereas koff decreased ∼6-fold to 0.011 ± 0.002 s−1, yielding a calculated Kd of 32 nm (24–42 nm range). Thus, prolonged exposure of live FLAG-MOR-expressing cells to ME caused an increase in the subsequent affinity of derm A594 for MOR measured using both steady-state and kinetic assays. Because prolonged exposure to ME both slowed the rate of unbinding and increased the affinity of MOR for derm A594, unbinding rate will be used as a proxy for affinity measurements throughout this manuscript.
Increased affinity of nonfluorescent opioid agonists
Two different assays were performed to determine whether an increase in binding affinity after prolonged ME treatment was common to other agonists. First, a subsaturating concentration of [3H] DAMGO (12 nm, 10 min) was applied to live FLAG-MOR HEK293 cells that were naive or pretreated with ME (30 μm, 2 h, + Con A 600 μg/ml) and the amount of specifically bound [3H] DAMGO was quantified. Consistent with steady-state binding of derm A594 (10 nm) shown in Figure 3C, there was a 335% increase in [3H] DAMGO binding after modulation by ME (naive, 91.3 ± 2.3; ME 2 h, 397.7 ± 32.6 fmol of [3H] DAMGO bound/mg protein, n = 3, data not shown). The large increase in binding at subsaturating agonist concentrations is consistent with DAMGO behaving in a similar fashion to derm A594, showing increased affinity for receptors that were previously modulated by ME.
Second, the unbinding rate of ME was inferred from its ability to slow the apparent rate and extent of derm A594 binding. The binding of derm A594 will be occluded as long as ME remains bound to receptors. The pulse-chase-like assay diagrammed in Figure 4A used a prepulse of ME (3 μm, 1 min) instead of buffer immediately preceding application of derm A594 to test whether ME unbinding was slowed after prolonged ME exposure. After prolonged ME treatment (30 μm, 2 h) and wash, followed by either buffer or ME-prepulse (3 μm, 1 min), derm A594 (200 nm, 1 min) was applied and the relative amount of derm A594 bound was measured. Cells subjected to ME-prepulse (3 μm, 1 min) bound approximately one third less derm A594 than cells washed with buffer solution (Figure 4Ai, ME 30 μm, 2 h; then buffer R/G = 1.44 ± 0.10, or ME 3 μm prepulse R/G = 0.95 ± 0.08, n = 6 each). There was no difference in the amount of derm A594 bound after 1 min when naive cells were prepulsed with either ME (3 μm, 1 min) or buffer (Fig. 4Ai, untreated; then buffer R/G = 0.84 ± 0.07, or ME 3 μm prepulse R/G = 0.88 ± 0.07, n = 5 each), suggesting that ME rapidly and completely unbound from MOR in naive cells.
Figure 4.
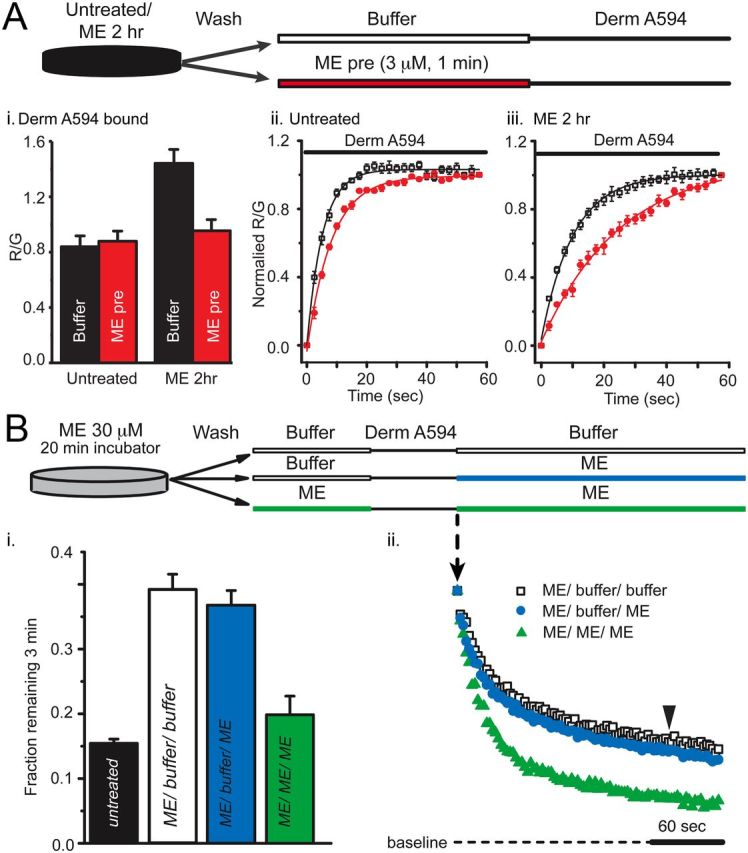
ME unbinds slowly after prolonged ME treatment. A, The amount of derm A594 bound relative to M1 A488 was measured in cells either untreated (Aii) or treated with ME (30 μm, 2 h) (Aiii). After treatment (untreated or ME 2 h), cells were washed with buffer and then subjected to a short pulse of buffer (black) or ME (3 μm, 60 s, red) immediately preceding derm A594 application (200 nm, 60 s) as diagrammed. Ai, Binding of derm A594 was imaged, and the relative amount of derm A594 bound after 60 s (R/G) was measured and plotted the average normalized amount of derm A594 that bound is plotted. Aii, Aiii, Binding of derm A594 (200 nm) is plotted as a function of time in untreated (Aii) or ME (30 μm, 2 h) treated cells (Aiii) and prepulsed with either buffer (black) or ME (3 μm, 60 s, red); n = 5 or 6 each. B, derm A594 (100 nm, 60 s) was applied to cells that had been treated with ME (30 μm, 20 min) and washed with buffer (see diagram). Unbinding of derm A594 was monitored while washing with either buffer (ME/buffer/buffer, □) or ME (3 μm, ME/buffer/ME, blue). ME (3 μm, 1 min) was also pulsed both immediately before and after derm A594 binding to cells that were modulated by ME (30 μm, 20 min) to determine whether residual ME had an effect on derm A594 unbinding kinetics (green, ME/ME/ME). Averaged data in Bi show fraction of derm A594 remaining 3 min after washout (n = 7–11, indicated by arrowhead in Bii), and individual examples of derm A594 unbinding under each condition are shown in Bii.
Binding of derm A594 to untreated cells immediately after a 1 min prepulse of ME (3 μm, 1 min) occurred with an apparent time constant of association of 8.5 ± 0.3 s, only slightly slower than the τ of 5.6 ± 0.2 s observed when there was no ME prepulse (Fig. 4Aii, n = 6 each). This indicates that, under naive conditions, ME rapidly dissociated from the receptor, allowing derm A594 to bind. After the prolonged ME treatment (30 μm 2 h), the basal rate of association of derm A594 slowed down (τ = 12.3 ± 0.1 s) and the ME prepulse further slowed the apparent association (τ = 26.6 ± 2.9 s) (Fig. 4Aiii, n = 7 each), suggesting that ME unbound from the receptor more slowly after the receptor had been desensitized with prolonged ME. Thus, after treatment with ME (30 μm, 2 h), the rate of unbinding of an unlabeled endogenous peptide, ME, was slowed as was derm A594, suggesting that ME affinity increased after extended ME treatment.
In Figure 4A, it was demonstrated that ME could remain bound for an extended period to some receptors that were pretreated with ME (30 μm 2 h). To determine whether this remaining ME had an effect of derm A594 kinetics, the unbinding of derm A594 was measured. Cells were preincubated with ME for a shorter period of time (30 μm, 20 min) to allow determination of whether an ME prepulse increased, decreased, or had no effect on the rate of subsequent derm A594 unbinding. When ME (3 μm) was applied both before (1 min pulse) and after application of derm A594 to cells previously treated with ME (30 μm, 20 min), the rate of derm A594 unbinding was increased relative to cells that were rinsed with buffer (Fig. 4Bii). After a 1 min exposure to ME (1 μm), the fraction of derm A594 remaining after 3 min was not significantly different from untreated cells (Fig. 4Bi, fraction remaining: untreated, 0.15 ± 0.01, n = 9; ME/ME/ME, 0.20 ± 0.03, n = 7). One interpretation of this result is that there were two populations of receptors after incubation with ME (30 μm). One population, having a slow unbinding rate, remained bound to ME, preventing the binding of derm A594. In the second population, ME quickly dissociated, allowing derm A594 to bind. Thus, derm A594 bound only to the rapidly dissociating population of receptors such that derm A594 also dissociated rapidly.
Affinity changes independent of solution exchange
An alternative explanation for the relative speeding of derm A594 unbinding is that ME present during derm A594 washout displaced derm A594 through bulk displacement or negative cooperativity. ME-treated cells (30 μm, 20 min) were washed with buffer, exposed to derm A594, and then washed with either buffer “infinite dilution” (ME/buffer/buffer, Fig. 4Bii, □) or ME (3 μm) “displacement” (ME/buffer/ME, blue) to address these possibilities. Displacing derm A594 with ME had no effect on the rate of unbinding (fraction derm A594 remaining: ME/dilution, 0.39 ± 0.02, n = 11; ME/displace 0.37 ± 0.2, n = 7, Fig. 4Bi), suggesting that rapid agonist rebinding, cooperative binding, or poor solution exchange was insufficient to explain the observed change in agonist affinity induced by prior ME exposure (DeMeyts et al., 1976; Spivak et al., 2006).
Ligand dependence of derm A594 affinity change
FLAG-MOR-expressing cells were next pretreated with a series of opioid ligands, including agonists ME (30 μm), methadone (15 μm), endomorphin 2 (30 μm), fentanyl, morphine, and oxycodone, or the antagonist naloxone (10 μm unless noted otherwise, 2 h) to determine ligand specificity of agonist modulation (Fig. 5B). Slowed unbinding was quantified by determining the fraction of derm A594 bound to MOR 3 min after washout (Fig. 5A,C). Consistent with the efficacy and desensitization profile of each ligand (Virk and Williams, 2008; Rivero et al., 2012), ME, fentanyl, endomorphin 2, and methadone, all efficacious and desensitizing agonists induced robust changes in the rate of unbinding (fraction derm A594 remaining after 3 min: untreated, 0.15 ± 0.01; ME, 0.52 ± 0.02; fentanyl, 0.57 ± 0.01; endomorphin 2, 0.45 ± 0.02; methadone, 0.44 ± 0.01, n = 5–9). Morphine pretreatment resulted in a partial change in unbinding rate relative to ME. Oxycodone pretreatment, which causes even less desensitization than morphine, resulted in less derm A594 remaining after 3 min (morphine, 0.29 ± 0.03 n = 7; oxycodone, 0.23 ± 0.01 n = 6). Pretreatment with naloxone had no significant effect on the fraction of derm A594 bound after 3 min (0.12 ± 0.01, n = 6). In summary, ligand-induced modulation of agonist–receptor interaction was dependent upon both ligand relative efficacy (with the exception of endomorphin 2) and the duration of ligand exposure and correlated with the ability of agonists to induce desensitization.
Figure 5.

Ligand dependence of receptor modulation. A, Images taken either before (pre), immediately after (t = 0), or 3 min after (t = 3 min) application and rapid washout of derm A594 (100 nm, 90 s). Images show derm A594 bound to the plasma membrane of cells expressing FLAG-MOR that were either untreated (ctrl), or treated with ME, fentanyl, or morphine for 2 h. Scale bar, 20 μm. B, Quantification of unbinding of derm A594 as shown in A after 2 h pretreatment with the indicated opioid ligands. Images were taken every 2.5 s imaging both M1 488 and derm A594. Averaged, normalized ratiometric intensity is plotted. Data are mean ± SEM; n = 6–9). Arrow indicates time = 3 min after washout. C, Fraction of derm A594 remaining at t = 3 min relative to t = 0 from data shown in B. Data are mean ± SEM. All treatments except naloxone resulted in a significant change in the fraction bound after 3 min (p < 0.05, ANOVA, Tukey post hoc test).
Antagonist binding not changed
A novel fluorescent antagonist ntx A594 was synthesized and found to bind specifically to MOR in a manner that was inhibited by binding of the irreversible antagonist β-CNA (Fig. 6B). The rate of dissociation (τ = 184 ± 6.1 s) and apparent rate of association (65.8 ± 5.5 s) of ntx A594 (100 nm, Fig. 6A) yielded estimated Kd of 53 nm. In contrast to derm A594, the rate of unbinding of ntx A594 was unaffected by pretreatment of FLAG-MOR cells with ME (30 μm, 2 h; Fig. 6C). Together, the results demonstrate that ME exposure can induce a change in affinity of agonist but not antagonist labeled with the identical fluorophore, suggesting that modulation is specific to agonists.
Figure 6.
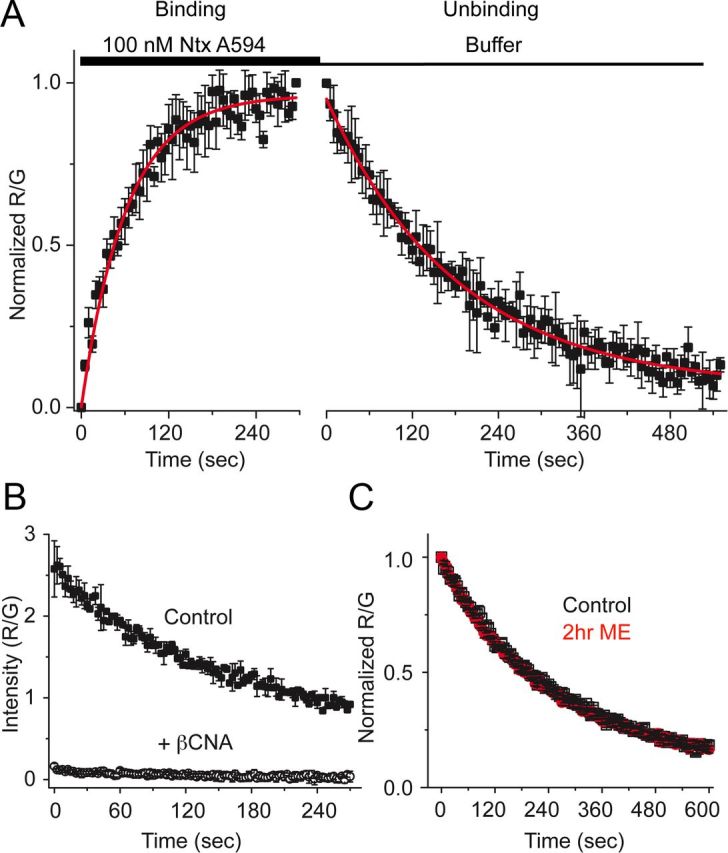
Unbinding of ntx A594 was not affected by previous agonist exposure. A, Binding kinetics of the antagonist ntx A594 were measured by applying ntx A594 (100 nm, 5 min) and imaging binding and washout relative to M1–488 intensity (R/G). Binding and unbinding were plotted and fit with a single exponential decay formula (τ association = 65.8 ± 5.5 s, τ off = 184 ± 6.1 s, n = 4 each, mean ± SEM). B, Preincubation of cells with the irreversible antagonist β-CNA (1 μm, 5 min) prevented all binding when measured by the fluorescence intensity immediately after washout of ntx A594 (300 nm, 90 s), verifying the specificity of binding (n = 2 ctrl, n = 4 β-CNA mean ± SD). C, ntx A594 (300 nm, 90 s) was applied, and unbinding of ntx A594 was imaged from cells either untreated (black) or pretreated with ME (30 μm, 2 h) (red); n = 5 each, mean ± SEM.
Role of G-proteins and β-arrestins
Several possible mechanisms could account for the long-lasting memory of prior agonist exposure. Because the effect of agonist pretreatment persisted long after washout of agonist, GPCR signaling pathways were considered. MOR classically signals through inhibitory G-proteins and nonclassically through β-arrestin. Cells that were treated overnight with pertussis toxin, eliminating inhibitory G-protein signaling, still showed a robust slowing in derm 594 unbinding after treatment with ME (30 μm, 2 h, Fig. 7A, fraction remaining: untreated, ctrl = 0.15 ± 0.01, pertussis toxin = 0.13 ± 0.005; ME 2 h, ctrl = 0.54 ± 0.02, pertussis toxin = 0.51 ± 0.01, n = 3–5, Fig. 7B). This suggested that G-protein signaling and Gi/o-mediated G-protein-coupled receptor kinase phosphorylation were not responsible for this modulation.
Figure 7.

Modulation was independent of G-protein signaling and β-arrestins. A, FLAG-MOR-expressing HEK 293 cells were either untreated (black) or treated with pertussis toxin (100 ng/ml overnight) (red), and the unbinding of derm A594 (100 nm, 90 s) was imaged under control conditions or on cells subjected to agonist pretreatment (ME 30 μm, 2 h). Normalized average data are plotted; n = 3–6. B, The fractional amount of derm A594 that remained bound to cells 3 min after washout was quantified relative to the receptor fluorescence intensity. C, MEFs cultured from β-arrestin 1/2 double knock-out mouse embryos (blue) or their WT controls (black) were either untreated or pretreated with ME (30 μm, 2 h), and unbinding of derm A594 (100 nm, 90 s) was imaged; normalized data are shown. D, Quantification of the fraction of derm A594 remaining 3 min after washout from WT (black) and β-arrestin 1/2 d knock-out MEF cells (blue) shown in C; n = 5 each. Additionally, β-arrestin 1/2 d knock-out MEF cells were treated with pertussis toxin (100 ng/ml overnight), and the fraction of derm A594 remaining in untreated and ME treated cells is shown (purple); n = 4 each. All points are mean ± SEM.
To address the possible role of arrestins in agonist induced imprinting, β-arrestin 1 and 2 double knock-out MEF (β-arrestin 1/2 knock-out) cells and WT controls were transiently transfected with FLAG-MOR and the unbinding of derm A594 was measured. Loss of both β-arrestin 1 and 2 had no effect on the ability of agonist to induce a slowing in the rate of unbinding (Fig. 7C). The rate and extent of unbinding were nearly identical between WT and β-arrestin 1/2 knock-out MEF cells both before and after treatment with ME (30 μm, 2 h) (fraction remaining after 3 min: untreated, WT = 0.09 ± 0.01, β-arrestin knock-out = 0.13 ± 0.02; ME 2 h, WT = 0.50 ± 0.05, β-arrestin knock-out = 0.47 ± 0.04, n = 5, Fig. 7D). Finally, pertussis toxin treatment of β-arrestin 1/2 knock-out MEF cells had no effect on the ability of ME treatment to modulate derm A594 affinity, indicating that there are not parallel pathways of modulation involving both G-protein signaling and arrestin signaling (fraction remaining after 3 min: untreated = 0.12 ± 0.02, ME 30 μm, 2 h = 0.51 ± 0.01 n = 4 each, Fig. 7D).
Discussion
Experiments measuring binding of opioids to receptors on live cells are not often performed, yet they have the potential to better our understanding of opioid pharmacology at the level of binding and the functional effects of opioids on MOR-mediated signaling at the cellular level. One aim of this study was to gain insight into basic features of opioid ligand–receptor interactions in live cells. This was pursued with the goal of determining whether desensitization, which has been defined at the cell signaling level, could also be defined at the receptor level. The results presented here suggest that conditions that are known to desensitize receptors at the functional level also modulate receptor–agonist but not receptor–antagonist interactions, suggesting a long-lived change in receptor conformation states that affect agonist binding and unbinding kinetics. Furthermore, this agonist-induced change in affinity is independent of G-protein and β-arrestin signaling.
The increase in affinity and slowed binding and unbinding kinetics demonstrated here after prolonged ME treatment occurred in receptors that were desensitized. It has not, however, been determined whether the increase in agonist affinity, indicating a distinct conformation state, and acute desensitization of signaling are one and the same; rather, that there is a correlation between desensitizing agonist exposure and agonist–receptor affinity changes. Both phenomena share a similar agonist profile and recover over a similar time scale (Virk and Williams, 2008; Rivero et al., 2012). Although acute desensitization can be induced in as little as 1–2 min (Johnson et al., 2006), the change in agonist affinity occurs over a generally longer time scale, yet this time scale is in line with other measures of cellular desensitization in HEK 293 cells (Koch et al., 1998). It is possible that multiple desensitized states of the receptor exist over different time scales. Thus, it is difficult to directly compare the current results based on direct receptor measurements with previous observations based on downstream signaling.
Receptor desensitization results in a decline in signaling resulting from a functional uncoupling of receptors and G-protein subunits. Although this decline in signaling can be dramatic, prolonged exposure to agonist can never completely abolish signaling, suggesting that there are always some functional receptors present. After 2 h of exposure to ME, fentanyl, endomorphin 2, or methadone, derm A594 unbinding was dramatically slowed. Mirroring functional studies suggesting that both desensitized and functional receptors are always present, the unbinding of derm A594 after agonist imprinting was always poorly fit with a single exponential and was better fit with both a fast and slow unbinding component. Because cells that are not preexposed to desensitizing agonist show predominantly fast unbinding, it is proposed that the fast unbinding receptors are functional and the slow unbinding receptors are desensitized.
The canonical model of GPCR desensitization involves binding of β-arrestin to phosphorylated receptors. The binding of β-arrestin is proposed to prevent binding of the G-protein to the receptor, disrupting signaling (Benovic et al., 1987, Luttrell and Lefkowitz, 2002). High-affinity binding of agonists to GPCRs is thought to be mediated by intimate association of the heterotrimeric G-protein with the opioid receptor (Da Lean et al., 1980). Loss of coupling between receptor and G-protein through desensitization would be expected to result in a loss of high-affinity binding and thus a decrease in affinity. However, our results suggest that the opposite occurred in live cells where the affinity was increased after exposure to desensitizing agonists. Although many ligand gated ion channels, such as the nicotinic acetylcholine receptor (Heidmann et al., 1983), display increased agonist affinity when in a desensitized state, this has not been reported for G-protein-coupled receptors.
The mechanism for the prolonged agonist binding must differ from that of ligand-gated ion channel desensitization whereby desensitized receptors remain bound to agonist while desensitized and upon unbinding of agonist, desensitization is reversed. Because derm A594 binding and unbinding are affected by prior agonist exposure, it is clear that MOR has retained a “memory” of desensitization that persisted after the agonist was removed. Receptor phosphorylation is one mechanism by which information about prior agonist-induced conformational changes could be encoded into a persistent change at the receptor level, and many functional studies of MOR desensitization implicate a role of kinases in the processes of desensitization and recovery from desensitization (Bailey et al., 2009; Dang et al., 2009; Arttamangkul et al., 2012; Dang and Christie, 2012). It has recently been demonstrated that receptor phosphorylation in the C-terminal tail is induced in a ligand-dependent manner similar to agonist treatments described here (Doll et al., 2011; Lau et al., 2011). Although it is recognized that agonists can induce phosphorylation of MOR, it is generally assumed that phosphorylation mechanisms involve downstream signaling for recruitment of kinases to the active receptor, yet not all G-protein-coupled receptor kinases require G-proteins for recruitment to GPCRs. Additionally, tyrosine residues in the intracellular loops and C-terminal tail of MOR undergo phosphorylation in a manner that may be agonist-dependent but G-protein-independent (Pak et al., 1999, McLaughlin and Chavkin, 2001). The present results suggest that neither G-protein nor β-arrestin-mediated signaling was required for agonist modulation of MOR to alter subsequent agonist affinity, and future studies will address whether phosphorylation is involved.
Multiple conformational states are known to exist in MOR and other GPCRs. Opioid receptor dissociation kinetics can be modulated by both sodium and GTP indicating the existence of multiple conformational states (Kurowski et al., 1982). The unbinding kinetics of peptide agonists from both MOR and DOR have been previously reported to be dependent on the time of agonist incubation using radioligand binding assays from hippocampal synaptic membranes, although this time-dependent slowing in unbinding rate was eliminated by the presence of sodium, perhaps suggesting an involvement of G-proteins and stabilization of the ternary complex (Scheibe et al., 1984).
Recent studies have found evidence for dynamic changes in conformation and affinity of other GPCRs. Purified, solubilized β2-adrenergic receptors undergo a fast conformation change upon initial binding of agonists and then a slow conformation change in the continued presence of some but not all agonists (Swaminath et al., 2004). NK2 receptors in intact cells display a time-dependent change in the rate of fluorescent ligand unbinding with a desensitizing fluorescent peptide but not a shorter, nondesensitizing peptide (Palanche et al., 2001). Both of these studies have noted the relationship between these slow changes and desensitizing agonists but have not investigated further. Repetitive activation of the β2-adrenergic receptor by agonist altered the kinetics of subsequent activation, suggesting a transient memory of previous agonist exposure (Ahles et al., 2011). The current results further support and expand upon the existence and ligand modulation of multiple affinity states, suggesting that several G-protein- and β-arrestin-independent conformational states exist for MOR, which are agonist-sensitive yet antagonist-insensitive. Furthermore, this study demonstrates a long-lived modulation of affinity, in which imprinting lasted for tens of minutes after washout of agonist in live cells.
As had been long predicted, the low-affinity binding state may be the most physiologically relevant state of opioid agonist–receptor interactions as judged by binding in live cells (Carroll et al., 1988). This was supported by two observations: (1) derm A594 binding in whole cells had an affinity close to that of the low-affinity radioligand competition binding measured in the presence of Mg2+, Na+, and GTPγS and (2) pertussis toxin had no effect on the kinetics of binding or unbinding of derm A594. It is also notable that, although derm A594 has a relatively high-affinity for MOR even in the low-affinity state (120 nm), dissociation of derm A594 from the receptor was rapid with a time constant on the order of 10 s. In live cells, rapid dissociation is a common property of most opioid agonists (Ingram et al., 1997), perhaps because of the relatively exposed binding pocket as noted in the recent x-ray crystallographic structure of MOR (Manglik et al., 2012).
In conclusion, a novel approach for studying opioid receptor dynamics has demonstrated that prolonged exposure to desensitizing agonists can be imprinted onto receptors. This study suggests that agonist modulation affects the subsequent conformational changes induced by agonists, increasing agonist affinity and stabilizing agonist binding. Although a definitive link between functional agonist-induced desensitization of receptor-mediated signaling and physical agonist-induced modulation of agonist affinity has not been established, both the agonist dependence and time course of action are similar (Virk and Williams, 2008; Rivero et al., 2012). Further, this technique could be adapted for live-cell screening applications, such as screens for allosteric modulators of agonist binding. The ease of manipulation of this system will allow pharmacological and structure–function-based investigation of the mechanism of agonist modulation and determination of how physical changes at the receptor level affect receptor function and signaling.
Footnotes
This work was supported by National Institutes of Health Grants DA08136 and MH083754 to J.R.T. and M.J.C., and in part by the Intramural Research Programs of National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism. We thank Dr. Shane Hentges for feedback during manuscript preparation, Dr. Erica Levitt for manuscript feedback and GraphPad expertise, and Dr. Dale Fortin for technical assistance.
The authors declare no competing financial interests.
References
- Ahles A, Rochais F, Frambach T, Bünemann M, Engelhardt S. A polymorphism-specific “memory” mechanism in the β(2)-adrenergic receptor. Sci Signal. 2011;4:ra53. doi: 10.1126/scisignal.2001681. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Alvarez-Maubecin V, Thomas G, Williams JT, Grandy DK. Binding and internalization of fluorescent opioid peptide conjugates in living cells. Mol Pharmacol. 2000;58:1570–1580. doi: 10.1124/mol.58.6.1570. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Lau EK, Lu HW, Williams JT. Desensitization and trafficking of μ-opioid receptors in locus ceruleus neurons: modulation by kinases. Mol Pharmacol. 2012;81:348–355. doi: 10.1124/mol.111.076208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, Henderson G. Role of protein kinase C and μ-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009;29:307–318. doi: 10.1111/j.1460-9568.2008.06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL, Kühn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc Natl Acad Sci U S A. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Carroll JA, Shaw JS, Wickenden AD. The physiological relevance of low agonist affinity binding at opioid μ-receptors. Br J Pharmacol. 1988;94:625–631. doi: 10.1111/j.1476-5381.1988.tb11569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Yu M, Jan YN, Jan LY. Evidence that the nucleotide exchange and hydrolysis cycle of G proteins causes acute desensitization of G-protein gated inward rectifier K+ channels. Proc Natl Acad Sci U S A. 1998;95:11727–11732. doi: 10.1073/pnas.95.20.11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Harrison C, Zhong H, Neubig RR, Traynor JR. Endogenous RGS protein action modulates μ-opioid signaling through G α o: effects on adenylyl cyclase, extracellular signal-regulated kinases, and intracellular calcium pathways. J Biol Chem. 2003;278:9418–9425. doi: 10.1074/jbc.M208885200. [DOI] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. μ-Opioid receptor desensitization: is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Dang VC, Christie MJ. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br J Pharmacol. 2012;165:1704–1716. doi: 10.1111/j.1476-5381.2011.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VC, Williams JT. Morphine-induced μ-opioid receptor desensitization. Mol Pharmacol. 2005;68:1127–1132. doi: 10.1124/mol.105.013185. [DOI] [PubMed] [Google Scholar]
- Dang VC, Napier IA, Christie MJ. Two distinct mechanisms mediate acute μ-opioid receptor desensitization in native neurons. J Neurosci. 2009;29:3322–3327. doi: 10.1523/JNEUROSCI.4749-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMeyts P, Bainco AR, Roth J. Site-site interactions among insulin receptors: characterization of the negative cooperativity. J Biol Chem. 1976;251:1877–1888. [PubMed] [Google Scholar]
- Doll C, Konietzko J, Pöll F, Koch T, Höllt V, Schulz S. Agonist-selective patterns of μ-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol. 2011;164:298–307. doi: 10.1111/j.1476-5381.2011.01382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Williams JT. Transient homologous μ-opioid receptor desensitization in rat locus coeruleus neurons. J Neurosci. 1991;11:2574–2581. doi: 10.1523/JNEUROSCI.11-08-02574.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidmann T, Bernhardt J, Neumann E, Changeux JP. Rapid kinetics of agonist binding and permeability response analyzed in parallel on acetylcholine receptor rich membranes from Torpedo marmorata. Biochemistry. 1983;22:5452–5459. doi: 10.1021/bi00292a029. [DOI] [PubMed] [Google Scholar]
- Ingram S, Wilding TJ, McCleskey EW, Williams JT. Efficacy and kinetics of opioid action on acutely dissociated neurons. Mol Pharmacol. 1997;52:136–143. doi: 10.1124/mol.52.1.136. [DOI] [PubMed] [Google Scholar]
- Johnson EA, Oldfield S, Braksator E, Gonzalez-Cuello A, Couch D, Hall KJ, Mundell SJ, Bailey CP, Kelly E, Henderson G. Agonist-selective mechanisms of μ-opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. doi: 10.1124/mol.106.022376. [DOI] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Schröder H, Wolf R, Raulf E, Höllt V. Carboxyl-terminal splicing of the rat μ-opioid receptor modulates agonist-mediated internalization and receptor resensitization. J Biol Chem. 1998;232:13652–13657. doi: 10.1074/jbc.273.22.13652. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin F-T, Perry SJ, Conner DA, Lefkowitz RJ. β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci U S A. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurose H, Katada T, Amano T, Ui M. Specific uncoupling by islet-activating protein, pertussis toxin, of negative signal transduction via alpha-adrenergic, cholinergic, and opiate receptors in neuroblastoma × glioma hybrid cells. J Biol Chem. 1983;258:4870–4875. [PubMed] [Google Scholar]
- Kurowski M, Rosenbaum JS, Perry DC, Sadée WE. [3H]-Etorphine and [3H]-diprenorphine receptor binding in vitro and in vivo: differential effect of Na+ and guanylyl imidodiphosphate. Brain Res. 1982;249:345–352. doi: 10.1016/0006-8993(82)90068-3. [DOI] [PubMed] [Google Scholar]
- Lau EK, Trester-Zedlitz M, Trinidad JC, Kotowski SJ, Krutchinsky AN, Burlingame AL, von Zastrow M. Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci Signal. 2011;4:ra52. doi: 10.1126/scisignal.2001748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KO, Akil H, Woods JH, Traynor JR. Differential binding properties of oripavines at cloned μ- and δ-opioid receptors. Eur J Pharmacol. 1999;378:323–330. doi: 10.1016/s0014-2999(99)00460-4. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Chavkin C. Tyrosine phosphorylation of the μ-opioid receptor regulates agonist intrinsic efficacy. Mol Pharmacol. 2001;59:1360–1368. doi: 10.1124/mol.59.6.1360. [DOI] [PubMed] [Google Scholar]
- Pak Y, O'Dowd BF, Wang JB, George SR. Agonist-induced, G protein-dependent and -independent down-regulation of the μ-opioid receptor. J Biol Chem. 1999;274:27610–27616. doi: 10.1074/jbc.274.39.27610. [DOI] [PubMed] [Google Scholar]
- Palanche T, Ilien B, Zoffmann S, Reck M, Bucher B, Edelstein SJ, Galzi J. The neurokinin A receptor activates calcium and cAMP responses through distinct conformational states. Biochemistry. 2001;276:34853–34861. doi: 10.1074/jbc.M104363200. [DOI] [PubMed] [Google Scholar]
- Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA, Rosethorne EM, Charlton SJ, Krasel C, Bailey CP, Henderson G, Kelly E. Endomorphin-2: a biased agonist at the μ-opioid receptor. Mol Pharmacol. 2012;82:178–188. doi: 10.1124/mol.112.078659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Portoghese PS. Stereospecific synthesis of the 6α- and 6β-amino derivatives of naltrexone and oxymorphone. J Org Chem. 1980;45:3366–3368. [Google Scholar]
- Scheibe SD, Bennett DB, Spain JW, Roth BL, Coscia CJ. Kinetic evidence for differential agonist and antagonist binding to bovine hippocampal synaptic membrane opioid receptors. J Biol Chem. 1984;259:13298–13303. [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Höllt V. Morphine induces terminal micro-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak CE, Oz M, Beglan CL, Shrager RI. Diffusion delays and unstirred layer effects at monolayer cultures of Chinese hamster ovary cells: radioligand binding, confocal microscopy, and mathematical simulations. Cell Biochem Biophys. 2006;45:43–58. doi: 10.1385/CBB:45:1:43. [DOI] [PubMed] [Google Scholar]
- Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the β2 adrenoceptor: kinetic evidence for intermediate conformational states. J Biol Chem. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- Tan M, Groszer M, Tan AM, Pandya A, Liu X, Xie CW. Phosphoinositide 3-kinase cascade facilitates μ-opioid desensitization in sensory neurons by altering G-protein-effector interactions. J Neurosci. 2003;23:10292–10301. doi: 10.1523/JNEUROSCI.23-32-10292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virk MS, Williams JT. Agonist-specific regulation of μ-opioid receptor desensitization and recovery from desensitization. Mol Pharmacol. 2008;73:1301–1308. doi: 10.1124/mol.107.042952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling LL, Puttfarcken PS, Cox BM. Multiple agonist-affinity states of opioid receptors: regulation of binding by guanyl nucleotides in guinea pig cortical, NG108–15, and 7315c cell membranes. Mol Pharmacol. 1988;33:423–431. [PubMed] [Google Scholar]
- Wüster M, Costa T, Aktories K, Jakobs KH. Sodium regulation of opioid agonist binding is potentiated by pertussis toxin. Biochem Biophys Res Commun. 1984;123:1107–1115. doi: 10.1016/s0006-291x(84)80247-8. [DOI] [PubMed] [Google Scholar]