

Abstract
Ototoxicity is a main dose-limiting factor in the clinical application of aminoglycoside antibiotics. Despite longstanding research efforts, our understanding of the mechanisms underlying aminoglycoside ototoxicity remains limited. Here we report the discovery of a novel stress pathway that contributes to aminoglycoside-induced hair cell degeneration. Modifying the previously developed bioorthogonal noncanonical amino acid tagging method, we used click chemistry to study the role of protein synthesis activity in aminoglycoside-induced hair cell stress. We demonstrate that aminoglycosides inhibit protein synthesis in hair cells and activate a signaling pathway similar to ribotoxic stress response, contributing to hair cell degeneration. The ability of a particular aminoglycoside to inhibit protein synthesis and to activate the c-Jun N-terminal kinase (JNK) pathway correlated well with its ototoxic potential. Finally, we report that a Food and Drug Administration-approved drug known to inhibit ribotoxic stress response also prevents JNK activation and improves hair cell survival, opening up novel strategies to prevent and treat aminoglycoside ototoxicity.
Introduction
Protein homeostasis, the composite of synthesis and degradation of cellular proteins, is tightly regulated by various cellular mechanisms, ensuring adaptation to different growth, nutrient, and stress conditions (Gebauer and Hentze, 2004; Sonenberg and Hinnebusch, 2009). Dysregulation of protein homeostasis contributes to neurodegenerative diseases, cancer and diabetes (Keller, 2006; Kasinath et al., 2009). Especially the regulation of protein synthesis in response to stress is highly conserved evolutionarily, producing a general translational arrest, accompanied by a selective upregulation of protective proteins (Holcik and Sonenberg, 2005). However, protein synthesis inhibition is not only a mere response to cellular stress, but affects the cell's fate under stress in a reciprocal manner. This is best exemplified by the fact that most protein synthesis inhibitors such as cycloheximide and anisomycin are also well-established inducers of apoptosis (Kochi and Collier, 1993; Kageyama et al., 2002), reflecting an intimate relationship between protein synthesis inhibition and apoptosis. Further complicating the matter, it is also well established that apoptosis depends on protein synthesis (Lockshin and Zakeri, 1992, Mesner et al., 1992). Depending on cell type and cellular state, protein synthesis inhibitors can therefore elicit or prevent apoptosis (Rehen et al., 1996). Such seemingly contradictory findings suggest that apoptosis is governed by a fine balance between proapoptotic and antiapoptotic gene products, so that inhibition of de novo protein synthesis can tip the balance in either direction, depending on the cellular context.
Sensory hair cells in the inner ear are the primary receptors of auditory and vestibular information (Hudspeth, 1989; Gillespie and Walker, 2001). In most cases of sensory hearing loss, degeneration of hair cells is the primary event; however, despite its recognized relevance in many degenerative disorders, protein synthesis activity and regulation have not been studied as a factor in ototoxicity. The goal of the present study, therefore, was to elucidate the relationship between protein synthesis activity and stress response in sensory hair cells.
To visualize protein synthesis activity in a manner that allows correlation with markers for pathway activation and cellular health, we adopted the bioorthogonal noncanonical amino acid tagging (BONCAT) method developed by Dieterich et al. (2006, 2010), based on the so-called “click-chemistry” reaction pioneered by Kolb and Sharpless (2003). Using this method, we described protein synthesis activity in vestibular and auditory organ explant cultures from chick and mouse. Furthermore, we tested the effect of the ototoxic aminoglycoside antibiotics on protein synthesis activity in hair cells. We report for the first time that aminoglycosides inhibit protein synthesis in hair cells, and that protein synthesis inhibition is correlated with the activation of the c-Jun N-terminal kinase (JNK) pathway. We postulate that aminoglycosides inhibit protein synthesis by binding to ribosomal RNA (rRNA), causing a cellular stress response similar to ribotoxic stress. Finally, we show that the anti-cancer drug sorafenib, known to inhibit ribotoxic stress response, also inhibits aminoglycoside-induced JNK activation and, to a limited degree, hair cell apoptosis. Together, these observations reveal a novel candidate strategy for the prevention of ototoxic hair cell injury and related hearing loss.
Materials and Methods
Animal care and handling
The protocol for care and use of animals was approved by the University of Virginia Animal Care and Use Committee. The University of Virginia is accredited by the American Association for the Accreditation of Laboratory Animal Care. Chick embryos before hatching are exempt from animal care guidelines, but we made sure to follow guidelines established for vertebrates when euthanizing chick embryos (by rapid decapitation using sharp scissors). All mouse experiments were performed using the CBA/J inbred mouse strain. Neonatal mouse pups [postnatal day 3 (P3)–P4] were killed by rapid decapitation, and mature mice were killed by CO2 asphyxiation followed by cervical dislocation.
Organotypic explant cultures
Chick basilar papillae (BPs) and utricles were dissected out from chick embryo heads. The otoconial mass was removed, and organs were cultured free floating in complete high-glucose DMEM (Invitrogen) containing 1% FBS supplemented with ampicillin (Fisher Scientific) and ciprofloxacin (LKT Laboratories) antibiotics. Mouse cochleae and utricles were dissected in HBSS (Invitrogen) containing 25 mm HEPES, pH 7.5. The organ of Corti was separated from the spiral lamina and the spiral ligament using fine forceps and stuck down onto the bottom of sterile 35 mm Petri dishes (BD Falcon), the hair bundle side facing up. The dissection medium was then replaced by two exchanges with culture medium (complete high-glucose DMEM containing 1% FBS, supplemented with ampicillin and ciprofloxacin). Gentamicin sulfate (Fisher Scientific), kanamycin sulfate (USB), and apramycin sulfate (Sigma) were dissolved in water. Chloramphenicol was dissolved in 100% ethanol, and sorafenib (Selleckchem) was dissolved in DMSO (1 mm stock solution).
BONCAT and click-chemistry reaction
We used both azidohomoalanine (AHA; bearing an azide residue) and the homopropargylglycine (HPG; bearing an alkyne residue) as methionine analogues in the BONCAT experiments. Detection via click-chemistry reaction needs to be performed with the respective complementary reagent: to detect incorporated AHA, an alkyne-conjugated hapten (biotin in our case) or fluorophore is used. Incorporated HPG is detected using an azide-conjugated biotin or fluorophore. Biotin was then detected using streptavidin (SA)-horseradish peroxidase (HRP) for immunoblots and SA-fluorophore for fluorescence microscopy.
For click-chemistry reaction in cell lysates (for immunoblots), we followed the procedure described in the Invitrogen Click-iT Protein Reaction buffer kit (catalog #C10276). In short, organotypic explants were cultured for various times in methionine-free medium containing AHA (Invitrogen, catalog #C10102, or Anaspec) at a final concentration of 400 μm. After the desired culture time, organs were washed in HBSS for 15 min at 37°C, after which protein lysates were prepared (10 mg/ml) in protein extraction buffer. Twenty-five microliters of the protein lysate was mixed with 50 μl of 2× Click-it reaction buffer and vortexed. In subsequent steps, copper sulfate solution and other additives were added, each addition followed by vortexing. After addition of the final additive, the entire reaction was vortexed at room temperature for 20 min. Proteins were precipitated using methanol/chloroform protein extraction. The protein pellet was resolubilized into 100 μl Laemmli buffer, and 10 μl was run on the gels.
For click reaction in fixed whole-mount organs to be imaged by fluorescence microscopy, Invitrogen provides the Click-iT Cell Reaction buffer kit (catalog #C10269), which has the caveat that it is not compatible with phalloidin staining, a helpful counterstain for imaging hair cells. We found, however, that the Invitrogen Click-iT Protein Reaction buffer kit (catalog #C10276) we used for cell lysates can be modified for fixed tissue, and that this procedure was compatible with phalloidin staining. Organotypic explant cultures were established as described above, precultured in methionine-free medium at 37°C for 15 min (to deplete cellular methionine), and cultured in methionine-free medium containing AHA or HPG, followed by a wash step at 37°C for 15 min in HBSS. Organs were then fixed in 4% paraformaldehyde, permeabilized in 0.2% Triton X-100, and washed in 50 mm Tris/HCl, pH 7.5. Up to three organs were then transferred into 50 μl of 50 mm Tris/HCl buffer (in a 96-well plate), and 50 μl of 2× reaction buffer from the kit was added and mixed (containing alkyne or azide-biotin or fluorophore, depending on whether AHA or HPG was detected). Subsequently, 5 μl of the copper sulfate (II) solution, additive 1, and additive 2 were added (separately), each time followed by brief mixing using a pipette. After the addition of the last solution (additive 2), the solution turned bright orange, after which the reaction was incubated at room temperature for 30 min, accompanied by mixing every 5 min. In the AHA/HPG dual-labeling protocol, HPG was reacted with azide-biotin (followed by conjugation with streptavidin Alexa 548), and AHA was reacted directly to Alexa Fluor 647-Alkyne (Invitrogen).
Alternatively, AHA can be detected using a copper-free version of the click-chemistry reaction (strain-promoted click chemistry) (Agard et al., 2004), which requires significantly less experimental manipulation. The copper-free version can only be used to detect AHA, and it showed similar sensitivity compared to the copper-mediated reaction described above. The azide group on AHA is reacted with a dibenzylcyclooctyne (DBCO)-conjugated biotin (instead of alkyne-biotin) in aqueous solution that does not require copper catalysis. For the copper-free version, organs that have incorporated AHA were fixed, permeabilized, and washed in 50 mm Tris/HCl, pH 7.5. Organs were then simply incubated in 50 mm Tris/HCl, pH 7.5, containing 10 μm sulfo-DBCO-biotin conjugate (Click Chemistry Tools, catalog #A115-10) for 1 h and washed three times in PBS. Biotin was then detected using fluorophore-conjugated SA.
Quantification of AHA and HPG signal
For a relative quantification of AHA incorporation, the fluorescence intensity of the AHA-biotin-Alexa 647 conjugate was normalized to parvalbumin 3 (PV3) immunoreactivity in chick, and to myosin 7a (Myo7a) immunoreactivity in mouse organs. For hair cells, the AHA signal was normalized to PV3 (or Myo7a for mouse) of the same cell, and for supporting cells, AHA immunofluorescence was normalized to PV3 (or Myo7a for mouse) of the adjacent hair cell. Staining procedures and confocal microscopy settings (gain, offset, laser power, magnification, and z-stack numbers) were kept identical for all comparative experiments.
Antibodies
The following antibodies were used: rabbit polyclonal anti-gentamicin antibody [G1015; Sigma; 1:100 for immunocytochemistry (ICC)]; mouse monoclonal anti-gentamicin antibody (QED Biosciences; 1:100 for ICC), mouse monoclonal Y10B antibody (Santa Cruz Biotechnology; 1:100 for ICC); mouse monoclonal anti-Myosin VIIa antibody (Developmental Studies Hybridoma Bank; concentrate; 1:200 in ICC); rabbit anti-GRP78 (Abcam; 1:1000 in immunoblots); mouse IgM anti-β-actin JLA20 (Developmental Studies Hybridoma Bank; concentrate; 1:1000 in immunoblots); rabbit anti-MLTK (MLK7) (Sydlabs; 1:100 in ICC); rabbit anti-PV3 (GenScript; 1:100 in ICC). All following antibodies were purchased from Cell Signaling Technology and used at a concentration of 1:100 for ICC and 1:1000 for immunoblots: rabbit anti-phospho-eukaryotic elongation factor 2 (eEF2; Thr56; catalog #2331); rabbit anti-eEF2 (catalog #2332); rabbit anti-phospho-ribosomal protein S6 (p-rpS6; Ser235/236; catalog #2211); rabbit anti-phospho-eukaryotic initiation factor 4E-binding protein 1 (p-4EBP1; Thr36/42; catalog #2855); rabbit anti-phospho-c-Jun (Ser73; catalog #3270); mouse anti-phospho-JNK (Thr183/Tyr185; catalog #9255); rabbit anti-activated caspase-3 (Asp175; catalog #9661).
Immunoblots
Organs were homogenized in reducing SDS-PAGE sample buffer, heated to 70°C for 5 min, and microcentrifuged for 5 min to remove insoluble debris. Proteins were resolved using Bis-Tris SDS PAGE gel (Novex 4–12%, Invitrogen, and TGX gels from Bio-Rad), transferred to PVDF membranes, and stained with India Ink. Blots were then blocked in blocking buffer (ECL prime blocking reagent; GE Healthcare) for 1 h and probed with primary antibodies overnight at 4°C. After three 5 min washings in PBS/0.3% Tween 20, blots were incubated with HRP-conjugated goat anti-rabbit secondary antibody (Cell Signaling Technology) for 1 h, and bands were visualized by ECL reagent (Pierce Biotechnology ECL Western blotting substrate and GE Healthcare GE ECL prime Western blotting reagent). Chemiluminescence was detected using an ImageQuant LAS4000 mini imager (GE Healthcare).
Immunocytochemistry
Tissues were fixed for 25 min in 3% formaldehyde and washed three times for 5 min each in PBS. After blocking for 1 h with 1% bovine serum albumin, 3% normal donkey serum, and 0.2% saponin in PBS, organs were incubated with primary antibodies overnight at room temperature in blocking buffer. After incubation in primary antibody, organs were washed three times for 5 min each with PBS and incubated with secondary antibodies (fluorophore-conjugated IgGs at 1:100; Invitrogen) and 0.25 μm phalloidin-Alexa 488 (Invitrogen) in the blocking solution for 1–3 h. Finally, organs were washed five times in PBS and mounted in Vectashield (Vector Laboratories). Samples were imaged using Zeiss LSM510 and LSM700 confocal microscopes.
rRNA UV-monitored thermal stability analysis
RNA corresponding to nucleosides 3722–3740 of the stem and terminal loop of human 28S rRNA Helix 69 (H69) was chemically synthesized (Dharmacon RNA Technologies) without pseudouridine modifications. The RNA was deprotected per the manufacturer's instructions and dialyzed extensively. Experiments were performed as described previously with the following changes (Scheunemann et al., 2010). H69 (1 μm) was prepared in 20 mm sodium phosphate, pH 6.8, and titrated with final concentrations of gentamicin from 0 to 9 μm. Thermal denaturations and renaturations were monitored by UV absorbance at 260 nm using a Varian Cary 100 Bio UV-visible spectrophotometer. Data were collected at the rate of 1°C/min from 20 to 90°C, averaged over six cycles, and performed in triplicate. The binding constant was determined by plotting the change in melting temperature (ΔTm) versus gentamicin concentration, and the data were fitted to a one-site nonlinear regression using Prism (GraphPad).
Systemic drug administration
Methods for inducing rapid cochlear hair cell death and hearing loss in mice have been established, using aminoglycosides in combination with loop diuretics such as furosemide and bumetanide (Taylor et al., 2008; Oesterle and Campbell, 2009; Hirose and Sato, 2011). Gentamicin sulfate (Fisher Scientific), apramycin sulfate (Sigma), and kanamycin sulfate (USB) were dissolved in 0.9% sterile saline to a concentration of 50 mg/ml. Adult CBA/J mice received one subcutaneous injection of (1) 60 mg/kg gentamicin, (2) 100 mg/kg gentamicin, (3) 600 mg/kg apramycin, (4) 1000 mg/kg apramycin, (5) 600 mg/kg kanamycin, or (6) 1000 mg/kg kanamycin or an equivalent volume of saline. This was followed by a single intraperitoneal injection of furosemide (10 mg/ml; Hospira) at a dosage of 400 mg/kg 30 min later. Mice were allowed to recover for 48 h before final auditory brainstem response (ABR) analysis.
Auditory brainstem response
ABRs of adult CBA/J mice were recorded before and after receiving injections of gentamicin, apramycin, or kanamycin. Mice were anesthetized with a single intraperitoneal injection of 100 mg/kg ketamine hydrochloride (Fort Dodge Animal Health) and 10 mg/kg xylazine hydrochloride (Lloyd Laboratories). All ABRs were performed in a sound-attenuating cubicle (Med Associates), and mice were kept on a Deltaphase isothermal heating pad (Braintree Scientific) to maintain body temperature. ABR recording equipment was purchased from Intelligent Hearing Systems. Recordings were captured by subdermal needle electrodes (FE-7; Grass Technologies). The noninverting electrode was placed at the vertex of the midline, the inverting electrode over the mastoid of the right ear, and the ground electrode on the upper thigh. Stimulus tones (pure tones) were presented at a rate of 21.1 per second through a high-frequency transducer (Intelligent Hearing Systems). Responses were filtered at 300–3000 Hz, and threshold levels were determined from 1024 stimulus presentations at 8, 11.3, 16, 22.4, and 32 kHz. Stimulus intensity was decreased in 5–10 dB steps until a response waveform could no longer be identified. Stimulus intensity was then increased in 5 dB steps until a waveform could again be identified. If a waveform could not be identified at the maximum output of the transducer, a value of 5 dB was added to the maximum output as the threshold.
Hair cell counts
Mouse utricle.
Organs were imaged using confocal microscopy, and hair cells counted based on their Myo7a staining. Three random fields of 50 × 50 μm were counted in both the striolar and extrastriolar (ES) regions, and hair cells numbers were averaged for each organ.
Mouse organ of Corti.
Hair cells were counted based on Myo7a staining over a length of 200 or 100 μm of the basal turn of the cochlea, omitting the last 100 μm of the basal tip. Activated caspase-3 immunoreactivity was counted over a stretch of 700 μm of the basal turn.
Chick basilar papilla.
Hair cells were identified based on their PV3 staining. We counted hair cells of the entire basilar papilla in Figure 4.
Figure 4.

Aminoglycoside antibiotic gentamicin induces translational arrest in sensory hair cells. A, AHA incorporation (24 h) in chick and mouse auditory and vestibular explant cultures, maintained in AHA-supplemented culture medium and exposed to 100 μm gentamicin or vehicle. When exposed to gentamicin, hair cells in all organs showed significant decreases in protein synthesis, while supporting cell protein synthesis was unchanged. Scale bars: 20 μm. B, Quantification of AHA incorporation (relative to PV3 immunoreactivity) in chick BP and utricle hair cells (HC) and supporting cells (SC) exposed to gentamicin or vehicle (n = 6–8; top). Quantification of AHA incorporation (relative to Myo7a immunoreactivity) in mouse inner (IHCs) and outer hair cells (OHCs) and utricle hair cells exposed to gentamicin or vehicle (n = 5–11; bottom). C, AHA incorporation (AHA immunoreactivity in hair cells, normalized to PV3 in chick or Myo7a in mouse, in percentage relative to vehicle control) as a function of gentamicin concentration in growth medium. Organs were cultured for 24 h. D, Hair cell numbers (phalloidin positive) in chick and mouse auditory and vestibular explants as a function of gentamicin concentration in growth medium. Organs were cultured for 24 h. *p < 0.01; **p < 0.001; ***p < 0.0001.
Chick utricle.
PV3-positive cells were counted in an area of 320 × 320 μm, covering an extrastriolar region adjacent to the striolar region.
Statistical analysis
For statistical analysis, GraphPad Prism was used. Two-tailed Mann–Whitney tests were used for comparing two conditions. p values smaller than 0.05 were considered significant. All n in statistical analysis refer to number of organs per experimental condition. All error bars indicate SEM.
Results
Click chemistry-mediated analysis of protein synthesis in sensory hair cells
To visualize protein synthesis activity, we adopted the BONCAT method (Dieterich et al., 2006). Methionine analogues that are metabolically incorporated into newly synthesized proteins are functionalized using the so-called click-chemistry reaction, a cyclic, copper-mediated reaction that is highly specific due to the virtual absence of alkynes in nature, hence the “bioorthogonalilty” of this detection method (Kolb and Sharpless, 2003). Figure 1A depicts the chemical structure of AHA and HPG, the two methionine analogues used in the present study. Figure 1B illustrates the principles of the two applications of BONCAT used in this study. Unless noted otherwise, explant cultures were precultured for 24 h, switched to methionine-free AHA-containing (or HPG-containing) medium, and maintained in culture for various time periods. After the culture period, the click-chemistry reaction was performed on either cell lysates or formaldehyde-fixed tissues, attaching a biotin (or a fluorophore) to all newly synthesized proteins. The immunoblot in Figure 1C illustrates the time-dependent increase of AHA incorporation into the BP, the chick auditory organ. We then used BONCAT to visualize cell-specific protein synthesis activity using light microscopy. BP explant cultures were metabolically labeled with AHA followed by the click-chemistry reaction in the fixed tissue. Figure 1D shows the time-dependent increase of AHA-biotin-fluorophore signal in the BP. The uptake of AHA was mediated by de novo protein synthesis, as shown by inhibition of AHA incorporation by the protein synthesis inhibitor anisomycin (see Fig. 7).
Figure 1.

Click chemistry can be used to monitor protein synthesis in inner ear sensory epithelia. A, Chemical structures of methionine analogs AHA and HPG. B, Diagram illustrating the experimental procedure of BONCAT: Cultures maintained in methionine-free medium were incubated in AHA (or HPG, not shown here) and further processed for click-chemistry reaction followed by immunoblot or immunolabeling. C, Immunoblot analysis of AHA incorporation shows a time-dependent increase of AHA in newly synthesized proteins in the chick BP. D, Click-chemistry allows for analysis of protein synthesis on a cell-to-cell basis. BP treated with AHA for various periods of time and analyzed by confocal microscopy confirm time-dependent increases in AHA (green) incorporation in hair cells (red). Scale bar: 50 μm.
Figure 7.

The ribotoxin anisomycin causes protein synthesis inhibition, mTOR activation, and cell death in hair cells. A, Mouse cochleae and utricles were exposed to the ribotoxic compound anisomycin at various concentrations for 24 h. Analysis of AHA incorporation showed a significant dose-dependent decrease of protein synthesis in hair cells (n = 5). AHA intensity was normalized to Myo7a immunoreactivity and plotted as the percentage relative to vehicle control. B, Mouse cochleae and utricles were exposed to the same concentrations of anisomycin as in A, and hair cells were counted. Anisomycin induced significant dose-dependent loss of hair cells (n = 5). C, Immunoblot analysis of mTOR substrate phosphorylation status indicated that 1 μm anisomycin (Aniso) activates the mTOR pathway in mouse P4 cochlea cultures (reduction of p-eEF2 and increase of p-rpS6 and p-4EBP1). Con, Control. Comparable results were obtained with mouse utricle cultures.
Variations in protein synthesis activity in hair cells and supporting cells
Next, we examined protein synthesis activity in various hair cell-bearing sensory epithelia from the mouse and chick (Fig. 2). Protein synthesis activity in hair cells and supporting cells in the vestibular and auditory organs displayed striking differences. While chick BP hair cells exhibited robust protein synthesis activity that was stronger than in surrounding supporting cells (Fig. 2A), AHA incorporation in mouse cochlea hair cells was weaker compared to supporting cells (Fig. 2C). In the vestibular system, both in mouse and chick, most hair cells showed lower protein synthesis compared to supporting cells (Fig. 2B,D), with the exception of a few smaller hair cells that exhibited robust protein synthesis (Fig. 2D, arrows). We hypothesize that the smaller hair cells are developing hair cells. Accordingly, such hair cells with high translational activity were missing in the utricles of mature mice (Fig. 2E).
Figure 2.
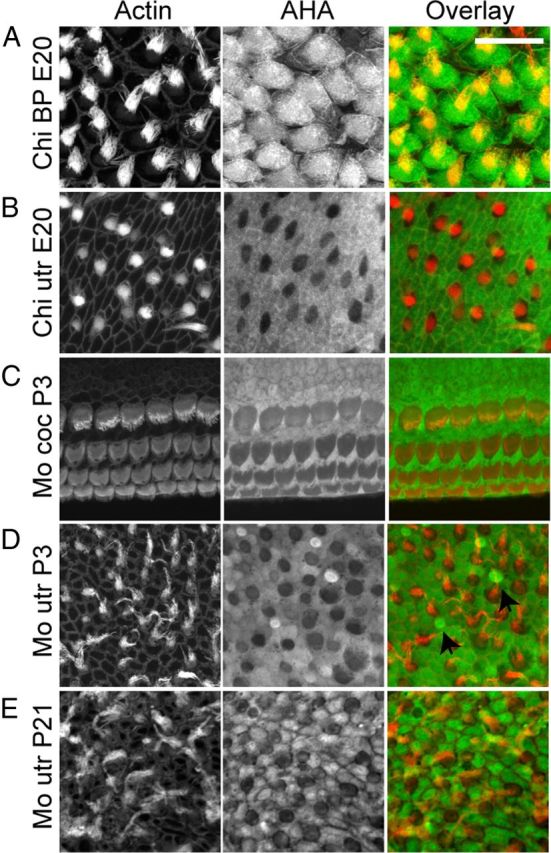
Protein synthesis in inner ear sensory epithelia varies between species and organs. A–E, Immunolabeling for AHA (24 h) was performed in auditory and vestibular organs from chick (Chi) and mouse (Mo). A, AHA incorporation in chick BP hair cells (red) was higher when compared to adjacent supporting cells. However, hair cells from neonatal mouse cochlear explants (C) displayed lower protein synthesis when compared with neighboring supporting cells. In vestibular organs of both species (B, chick E20; D, mouse P4; E, mouse P21), protein synthesis activity was higher in supporting cells compared to hair cells, except for some small, presumably immature hair cells in neonatal mouse utricle where AHA incorporation was high (D, arrows). Scale bar: 20 μm.
Effect of “culture shock” on protein synthesis activity
The experiments above (Fig. 2) reflect protein synthesis activity in explant cultures that were allowed to acclimatize to culture conditions for 24 h, common practice justified by the assumption that initial cultivation represents a stress impact that requires a recovery period. Since many forms of cellular stress cause protein synthesis inhibition (Holcik and Sonenberg, 2005), we wondered whether protein synthesis activity was affected in freshly cultured organs. To test this, organs were cultured without a preculture period in AHA medium for 12 h. As illustrated in Figure 3A, protein synthesis activity in the mouse cochlea and utricle, as well as chick utricle cultures, displayed no difference compared to organs precultured for 24 h (Fig. 2). The chick BP, however, displayed a significant difference (Fig. 3B): Without preculture, protein synthesis activity varied dramatically among adjacent BP hair cells. While some hair cells displayed robust AHA incorporation, other cells showed little AHA incorporation in the 12 h culture period. Such heterogeneous appearance of protein synthesis activity in adjacent cells was seldom observed in BPs that were precultured for at least 12 h (Fig. 2A).
Figure 3.

The effect of culture shock on protein synthesis in explanted sensory epithelium. A, AHA incorporation in mouse cochlea and utricle, and chick utricle explants without preculture period appeared similar to precultured explants. B, AHA incorporation (12 h; green) in chick BP that were not precultured. Most hair cells (red) displayed robust AHA immunoreactivity, but a few had little to no AHA incorporation (inset). C, Dual labeling with AHA and HPG was used to monitor temporal changes in protein synthesis activity after establishment of the explant culture. Explants were pulse labeled with AHA (green) for 24 h and subsequently with HPG (blue) for an additional 24 h. The majority of hair cells displaying initial translational arrest were found to have significant HPG incorporation in the subsequent 24 h. We occasionally identified hair cells that remained in a state of translational arrest (arrowhead). Scale bars: A, B, 20 μm; C, 10 μm.
To test whether BP hair cells recover their protein synthesis activity after a period of quiescence, we used a dual-labeling paradigm in which we pulse labeled in the first 24 h with AHA, and in the subsequent 24 h with HPG, as used previously for temporal resolution of protein synthesis activity in neurons (Beatty and Tirrell, 2008; Dieterich et al., 2010). Each methionine analog can be detected by separate click-chemistry reactions and visualized by different fluorophores. In our case, the AHA signal (Fig. 3C, overlay, green) represented protein synthesis activity in the first 24 h, while the HPG signal (blue) was a measure for protein synthesis activity in the subsequent 24 h. As illustrated in Figure 3C, most cells with low protein synthesis recovered to normal activity, with the exception of only one cell that displayed continued low AHA incorporation (black arrowhead). This finding indicates that inhibition of protein synthesis immediately after start of the explant culture in chick BP hair cells is a reversible and regulated phenomenon.
Protein synthesis inhibition in organs exposed to the aminoglycoside gentamicin
We next investigated whether inhibition of protein synthesis might play a role in drug-induced hair cell damage and death. As an example, we chose to test the effects of the ototoxic aminoglycoside gentamicin on hair cell and supporting cell protein synthesis. Aminoglycosides comprise a highly potent class of antibiotics, but their clinical use is limited due to nephrotoxicity and ototoxicity (Xie et al., 2011). The mechanisms underlying aminoglycoside ototoxicity are not fully understood, and therapeutic and preventative measures are still elusive (Rybak and Whitworth, 2005). Strikingly, when chick and mouse auditory and vestibular hair cells were exposed to gentamicin, the incorporation of AHA over a time period of 24 h was reduced by 30–60% compared to control conditions (Fig. 4A,B). Gentamicin inhibited AHA uptake (and therefore protein synthesis) at lower concentrations than required to cause hair cell loss (Fig. 4C,D), indicating that protein synthesis inhibition is not a mere side effect of hair cell death. In agreement with the hair cell specific toxicity of aminoglycosides, protein synthesis inhibition was confined to hair cells, while supporting cell protein synthesis was not altered (Fig. 4A,B). We therefore conclude that gentamicin inhibits protein synthesis specifically in hair cells.
Mechanisms underlying protein synthesis inhibition
Next, we investigated the mechanisms underlying protein synthesis inhibition in gentamicin-exposed hair cells. To avoid redundancy, we focus on mouse organs for the remainder of this paper. It should be noted that in all experiments performed, we obtained similar results with chick organs. Protein synthesis inhibition could be caused by an activation of the unfolded protein response (UPR) in response to ER stress (Schröder and Kaufman, 2005) and/or by an inhibition of the mammalian target of rapamycin (mTOR) pathway (Ma and Blenis, 2009). GRP78, a marker for ER stress (Bertolotti et al., 2000), did not change in gentamicin-treated hair cells (Fig. 5B). We also tested multiple other markers for ER stress and UPR, but did not detect any changes (data not shown), indicating that ER stress is not involved in gentamicin-induced protein synthesis inhibition. We then tested the role of the mTOR pathway by monitoring phosphorylation states of its downstream substrates, 4EBP1, rpS6, and eEF2 (Wang and Proud, 2007). Activation of the mTOR pathway results in the phosphorylation of 4EBP1 and rpS6, but in dephosphorylation of eEF2. Surprisingly, mTOR activity increased in hair cells treated with gentamicin (Fig. 5). In immunocytochemistry experiments, p-rpS6 immunoreactivity in hair cells increased, while p-eEF2 immunoreactivity decreased after gentamicin treatment, changes consistent with mTOR activation (Fig. 5A). Findings were further confirmed in the utricle using a double-labeling experiment: P3 mouse utricles were incubated in 100 μm gentamicin for 4 h and double labeled with an anti-gentamicin antibody and antibodies for p-rpS6 or p-eEF2. As shown previously (Lyford-Pike et al., 2007), type I hair cells in the striolar region accumulated more gentamicin than the ES hair cells. Consistent with gentamicin-induced mTOR activation, gentamicin immunoreactivity was positively correlated with p-rpS6 (Pearson's r = 0.77; p < 0.05), and inversely correlated with p-eEF2 immunoreactivity (Pearson's r = −0.64; p < 0.005; Fig. 5C). We were unable to detect significant changes in p-4EBP1 immunoreactivity (data not shown), but differential regulation of mTOR substrates has been reported in other systems (Choo et al., 2008). Immunoblot analysis using whole organ of Corti confirmed the changes seen in immunocytochemistry experiments (Fig. 5B). Activation of the mTOR pathway must therefore be considered the hair cell's compensatory response to protein synthesis inhibition. As to the cause for the protein synthesis inhibition in gentamicin-exposed hair cells, we conclude that it is mediated by an alternative mechanism.
Figure 5.
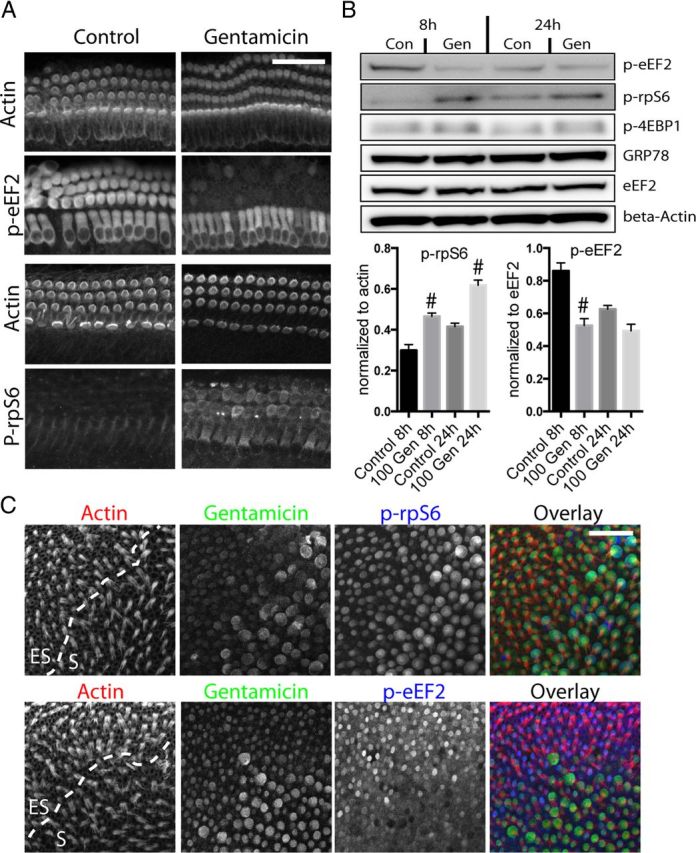
Gentamicin (Gen) exposure activates the mTOR pathway in hair cells. A, Immunocytochemistry for mTOR substrates indicated that exposure to gentamicin (100 μm) results in decreased phosphorylation of eEF2 and increased phosphorylation of rpS6 (most evident between 4–8 h after start of gentamicin exposure) in mouse cochlea hair cells, consistent with mTOR activation. B, Immunoblot analysis and quantification of mTOR substrate phosphorylation status also shows a significant increase in p-rpS6 and reduction of p-eEF2 in cochlea explants treated with 100 μm gentamicin (8 and 24 h; n = 4 experiments). Changes in p-4EBP1 were inconsistent and not significant. Levels of GRP78 (marker for ER stress), unphosphorylated eEF2, and β-actin remained unchanged. #p < 0.05 compared to control conditions. C, P3 mouse utricles were treated with 100 μm gentamicin for 4 h, then double labeled with a gentamicin antibody (green) and antibodies against p-rpS6 (blue, top) or p-eEF2 (blue, bottom). Hair cells (red) with stronger gentamicin immunoreactivity display stronger p-rpS6 (Pearson's r = 0.77; p < 0.05) and lower p-eEF2 immunoreactivity (Pearson's r = −0.64; p < 0.005). More than 100 cells from three different organs were analyzed.
Gentamicin binds to ribosomal RNA
We considered the possibility that gentamicin inhibits protein synthesis by interfering directly with the ribosomal translational machinery. According to established paradigms, aminoglycosides kill bacteria by binding to the bacterial 16S rRNA of the small (30S) ribosomal subunit, thereby causing mistranslation and fatal translational arrest (Davies et al., 1965; Moazed and Noller, 1987). Reduced affinities to the corresponding eukaryotic rRNA provides the rationale for bacterial specificity of aminoglycosides (Recht et al., 1999). However, aminoglycosides also bind to other rRNA sites, most notably to H69 of the 23S rRNA on the large subunit, a site involved in a crucial step of translation called ribosome recycling (Borovinskaya et al., 2007). We showed previously that the aminoglycoside neomycin binds to the human H69 19 nt hairpin, corresponding to nucleotides 3722–3740 of 28S rRNA with relatively high affinity (Kd ≈ 1.5 μm) (Scheunemann et al., 2010). We therefore investigated whether gentamicin also binds to this construct similarly. Using UV-monitored thermal denaturation, the effect of gentamicin binding on the thermal stability of the chemically synthesized 19 nt human H69 was analyzed. The changes in the Tm of H69 induced by gentamicin binding demonstrated an interaction with a Kd of ∼1.7 μm (Fig. 6A). Next, we investigated whether gentamicin colocalizes with rRNA in hair cells: mouse cochleae were incubated for 30 min with 100 μm gentamicin and double stained with an anti-gentamicin antibody and the mouse monoclonal Y10B antibody that specifically recognizes rRNA (Garden et al., 1994). As seen in Figure 6B, gentamicin and Y10B immunoreactivity colocalized in a limited fashion (Pearson correlation coefficient of 0.25; analysis of 50 outer hair cells). We therefore conclude that gentamicin binds, among other targets, to cytosolic rRNA.
Figure 6.

Gentamicin binds to and partially colocalizes with eukaryotic rRNA. A, UV-monitored thermal stability analysis demonstrates gentamicin binding to the H69 19 nt hairpin of human 28S rRNA. Unmodified H69 (1 μm) was titrated with increasing concentrations of gentamicin (0–9 μm), and the rRNA/gentamicin complex was subjected to repeated thermal denaturations and renaturations. Each curve is the result of point-by-point averages of six transitions for each concentration. The differences in melting temperatures (ΔTm) of H69 as a function of the gentamicin concentration provided the binding curve (bottom). The Kd of gentamicin to H69 was determined as 1.7 μm. B, Partial colocalization of gentamicin and rRNA immunoreactivity. Mouse cochleae were incubated for 30 min with 100 μm gentamicin and double labeled with a rabbit anti-gentamicin antibody and the mouse monoclonal Y10B antibody that specifically recognizes rRNA (Garden et al., 1994). Gentamicin (red) and Y10B (green) immunoreactivity colocalized in a limited fashion (Pearson correlation coefficient of 0.25, analysis of 50 outer hair cells). Scale bar, 20 μm.
Hair cells are susceptible to ribotoxic stress
Binding to rRNA and subsequent translational inhibition is reminiscent of a class of toxins that causes the so-called ribotoxic stress response. Ribotoxic stress can be elicited by toxins of various structural origins, ranging from plant-derived toxins such as ricin to antibiotics such as anisomycin (Iordanov et al. 1997; Pestka 2010). Common to ribotoxins is their property to bind or otherwise compromise rRNA, thereby causing translational arrest. The binding of the ribotoxin to rRNA also activates the JNK pathway and triggers apoptosis (Iordanov et al. 1997). First, we tested whether known ribotoxins cause hair cell death, and whether the death profile is similar to that elicited by aminoglycosides. Treatment with anisomycin, a well-established ribotoxic antibiotic, caused a dose-dependent protein synthesis arrest and hair cell death in mouse cochlea and utricle cultures (Fig. 7A,B). As with aminoglycosides, anisomycin also activated the mTOR pathway, as evident in an increase of 4EBP1, rpS6 phosphorylation, and decrease of eEF2 phosphorylation (Fig. 7C). We therefore conclude that anisomycin, a well-established ribotoxin, elicits a stress response in hair cells that is similar to the effects seen after aminoglycoside exposure.
JNK activation by gentamicin is correlated with protein synthesis inhibition
It is well established that aminoglycosides cause activation of the JNK pathway, and that this pathway is at least partially responsible for hair cell apoptosis. For example, inhibitors of JNK activation and upstream MAP3Ks partially prevent hair cell death in various in vitro and in vivo settings (Pirvola et al., 2000; Wang et al., 2003). JNK activation is also a hallmark feature of ribotoxic stress response (Iordanov et al., 1997). If gentamicin indeed causes ribotoxic stress response in hair cells, we should be able to detect a correlation between protein synthesis inhibition and JNK activation in hair cells. We tested this by combining the BONCAT method with immunocytochemistry experiments for phosphorylated c-Jun. Figure 8A–J shows mouse utricles treated for 24 h with 100 μm gentamicin, while the organs were maintained in methionine-free medium containing AHA. Protein synthesis activity in hair cells was lower than in surrounding supporting cells, but in gentamicin-treated utricles, we detected a further reduction of hair cell protein synthesis, while supporting cells remained unchanged in their protein synthesis activity. At the same time, we observed a robust activation of c-Jun phosphorylation. As seen in Figure 8H–J (quantified in S), activation of JNK correlated with low AHA signal. Hair cells with p-c-Jun immunoreactivity exhibited low protein synthesis, while cells lacking JNK activity displayed stronger AHA signals (Fig. 8J′, asterisks). We indicated one cell that exhibited neither JNK activity nor strong AHA signal (J′, white arrow). Accordingly, the cell-by-cell correlation was significant, but not perfect (Pearson's r = −0.7), probably due to the fact that the AHA signal represents an integration of protein synthesis activity over 24 h, while JNK activation in each cell is transient and not synchronized among the cells in a given organ. Similar results were obtained in the mouse cochlea, as shown in Figure 8K–R, with the difference that c-Jun phosphorylation was detectable only briefly (∼4 h after beginning of gentamicin exposure, and not detectable after 8 h).
Figure 8.

Aminoglycoside-induced JNK activation is correlated with translational arrest. A–J, P4 mouse utricles were incubated in AHA-supplemented medium under control conditions (A–E) or treated with 100 μm gentamicin for 24 h (F–J). A′–J′ are representative profile views (using reslice function in ImageJ). Phalloidin and Myo7a (red) staining indicate hair cells (A, B, F, G), AHA staining indicates protein synthesis activity (C, H, green), and p-c-Jun immunoreactivity indicates JNK activation (D, I, blue). AHA incorporation was lower in hair cells compared to adjacent supporting cells (C), but exposure to gentamicin resulted in a further decrease of hair cell protein synthesis (H). p-c-Jun immunoreactivity was strongly induced by gentamicin compared to control (D, I). The majority of hair cells without p-c-Jun immunoreactivity (J′, asterisks) exhibited higher AHA incorporation than those positive for p-c-Jun. In some cases, hair cells had neither strong p-c-Jun immunoreactivity nor high AHA incorporation (J′, arrow). K–R, JNK activity (p-c-Jun, blue) and AHA incorporation (green) are also correlated (negatively) in mouse cochlea hair cells incubated with 100 μm gentamicin (24 h). K′–R′ are representative profile views. Dotted circles in R′ indicate the locations of p-c-Jun-positive nuclei in outer and inner hair cells. Scale bars: E (for A–J), E′ (for A′–J′), N (for K–R), 25 μm. S, Correlation analysis of JNK activity and AHA incorporation in mouse utricles exposed to 100 μm gentamicin shows significant negative correlation (Pearson's r = −0.7; p < 0.0001).
MLK7 is expressed in hair cells and downregulated after gentamicin exposure
It was shown previously that the MAP3K MLK7 (also known as ZAK, MLTK) mediates ribotoxic stress response caused by doxorubicin in a keratinocyte cell line (Jandhyala et al., 2008; Sauter et al. 2010). Figure 9 shows that MLK7 is strongly expressed in mouse cochlea hair cells. Exposure to 100 μm gentamicin for 24 h caused weakening and condensation of MLK7 immunoreactivity in hair cells, consistent with the well described negative feedback mechanism by which activated MAP kinases are degraded through the ubiquitin–proteasome pathway (for review, see Lu and Hunter, 2009). Similar results were obtained in the mouse utricle (data not shown).
Figure 9.

MLK7 is expressed in sensory hair cells. MLK7 (green) is expressed in both inner hair cells and outer hair cells (red) of the neonatal mouse cochlea (P4). Exposure to 100 μm gentamicin (Gen; 24 h) resulted in sporadic loss of outer hair cells and a reduced or condensed pattern of MLK7 immunoreactivity in outer hair cells. Con, Control. Scale bar, 20 μm.
Sorafenib prevents JNK activation
Sorafenib is a multikinase inhibitor used to treat various types of cancer (Wilhelm et al., 2006). Besides its inhibitory effect on Raf kinase, proangiogenic vascular endothelial growth factor receptor, and other kinases relevant for tumor growth, sorafenib was also shown to have high, “off-target” affinity to the MAP3K MLK7 (Kd = 6.3 nm) (Karaman et al., 2008). In a subsequent study, sorafenib was shown to inhibit doxorubicin-induced ribotoxic stress response (Sauter et al., 2010). We therefore tested the involvement of MLK7 in gentamicin-induced hair cell toxicity by testing whether sorafenib inhibits gentamicin-induced JNK activation and apoptosis in mouse hair cells. To illustrate the time dependency of JNK activation, mouse utricle and cochlea cultures were incubated for different time periods with 200 μm gentamicin, with or without a preincubation with 500 nm sorafenib for 1 h (sorafenib was present during entire culture period). Figure 10A illustrates the time-dependent activation of the JNK pathway in mouse utricles, as evident in the phosphorylation of JNK and its substrate c-Jun. In agreement with an involvement of MLK7, sorafenib nearly completely prevented JNK and c-Jun phosphorylation (Fig. 10B, quantified in D). The comparative dose–response curve is illustrated in Figure 10E. Similar results were obtained with mature utricles (P21) (data not shown). Sorafenib-mediated inhibition of JNK activation did not prevent protein synthesis inhibition (data not shown), indicating that protein synthesis inhibition is upstream of JNK activation, in agreement with the proposed mechanism of ribotoxic stress. Similar results were obtained in mouse cochlea cultures (Fig. 10C, quantified in F).
Figure 10.

Sorafenib inhibits gentamicin-induced JNK activation. A, P4 mouse utricles were exposed to 200 μm gentamicin for various time periods. Immunoreactivity for phosphorylated JNK (green) and c-Jun (blue) in hair cells increased in a time-dependent manner. B, Hair cells from explants pretreated with 500 nm sorafenib displayed a near complete inhibition of JNK activation at all time points analyzed. C, Sorafenib (Sora) also prevents gentamicin (Gen)-induced JNK activation in mouse cochlea cultures. D, Quantification of A and B (JNK immunoreactivity) indicated a near complete suppression of JNK activation in sorafenib treated hair cells at all time points examined (n = 4). E, Sorafenib prevents gentamicin-induced JNK activation across the entire gentamicin dose–response curve (n = 4). F, Quantification of C showing that sorafenib prevents gentamicin-induced JNK activation in mouse cochlea hair cells (n = 4). Asterisks illustrate the significance of difference between gentamicin-only and corresponding gentamicin plus sorafenib samples. p-c-Jun-positive cells were counted in a 320 × 320 μm area of the utricle and in a 200 μm stretch of the basal end of the cochlea. Scale bars: A, B, 100 μm; C, 20 μm. ***p < 0.0001.
Sorafenib partially protects hair cells from caspase-mediated apoptosis
Next, we tested whether sorafenib also prevents gentamicin-induced hair cell death. Mouse utricle (P4 and P21) and cochlea cultures (P4) were pretreated for 1 h with 500 nm sorafenib and exposed to different concentrations of gentamicin. Hair cell numbers and caspase-3 activation were quantified in cochlea cultures after 24 h in gentamicin, and in utricle cultures, hair cell numbers were counted after a 72 h culture period. We found that sorafenib protects vestibular hair cells in a limited fashion; in the utricle, we saw moderate protection at all tested concentrations, in both the striolar and extrastriolar regions (Fig. 11A, C, top). For example, at 500 μm gentamicin concentration, percentage of hair cell survival in the striolar region improved from ∼20 to ∼60% compared to the DMSO-treated control. Significant protection was also observed in mature (P21) mouse utricles at 200 and 500 μm gentamicin concentrations (Fig. 11C, bottom). In the cochlea, however, sorafenib protects only at lower gentamicin concentrations (Fig. 11B). At higher concentrations, even the partial protective effect seen at 50 μm gentamicin concentration disappeared. Loss of Myo7a immunoreactivity coincided well with cleaved caspase-3 immunoreactivity, suggesting that most hair cell death is mediated by caspase-mediated apoptosis (Fig. 11B). Accordingly, we observed a reduction in caspase-3-positive cells after sorafenib cotreatment at 50 μm gentamicin concentration (Fig. 11D, right). In summary, although sorafenib nearly completely prevented JNK activation in response to gentamicin exposure in hair cells, it did not translate into a complete protection from hair cell death, suggesting the existence of alternative and parallel pathways leading to apoptosis and other cell death pathways.
Figure 11.

Sorafenib partially protects against gentamicin (Gen)-induced hair cell death. A, Representative images of mouse utricles exposed to vehicle (DMSO) only and 500 μm gentamicin (24 h), with and without sorafenib (Sora) pretreatment. Hair cells, immunolabeled for Myo7a, were partially protected from gentamicin-induced cell death when pretreated with sorafenib (500 nm). B, P4 mouse cochlea cultures were exposed to 50 μm gentamicin (24 h), with and without sorafenib pretreatment, and immunolabeled for Myo7a (red) and activated caspase-3 (green). Gentamicin induced sporadic loss of OHCs and caspase-3 activation, which was reduced by pretreatment with sorafenib. C, Quantification of striolar and extrastriolar hair cell counts from neonatal (P4) and mature (P21) mouse utricles exposed to concentrations of gentamicin across the dose–response curve. Sorafenib provided significant inhibition of hair cell death in both neonatal and mature utricles at a range of gentamicin concentrations (n > 5). D, Quantification of caspase-3 activation and outer hair cell counts in neonatal mouse cochlea also indicated partial protective effect of sorafenib at 50 μm gentamicin concentration (n > 5). Asterisks illustrate significance of difference between gentamicin-only and corresponding gentamicin plus sorafenib samples. Scale bars: A, 200 μm; B, 50 μm. #p < 0.05; *p < 0.01; **p < 0.001.
Differential ototoxicity of apramycin, kanamycin, and gentamicin correlates with their differential ability to inhibit cytosolic protein synthesis and to activate JNK
A previous study reported that another aminoglycoside, apramycin, exhibits reduced ototoxicity without losing bactericidal effects (Matt et al., 2012). The reduced ototoxicity was attributed to the fact that apramycin binds less strongly to mitochondrial rRNA and therefore inhibits mitochondrial translation less than other aminoglycosides such as gentamicin. Matt et al. (2012) therefore concluded that the block of mitochondrial translation is the main factor determining the ototoxic potential of aminoglycosides. Since our findings emphasized the importance of cytosolic (as opposed to mitochondrial) protein synthesis inhibition in gentamicin ototoxicity, we compared the effects of different aminoglycosides (apramycin, kanamycin, and gentamicin) on cytosolic protein synthesis and JNK activation in relation to their respective ototoxicities. As illustrated in Figure 12A, apramycin's ability to inhibit protein synthesis and activate JNK was much reduced compared to the same concentration of gentamicin. For example, 200 μm apramycin caused protein synthesis inhibition and JNK activation only in the striolar region, while the same concentration of gentamicin caused widespread protein synthesis inhibition and JNK activation throughout the entire utricle. A concentration of 1000–2000 μm apramycin was required to elicit effects comparable to 200 μm gentamicin. Kanamycin's ability to activate JNK and inhibit cytosolic protein synthesis was similar to apramycin's. Notably, apramycin and kanamycin were also ∼5–10 times less effective in killing hair cells: As seen in Figure 12B, apramycin or kanamycin at concentrations higher than 1000 μm produced utricular and cochlear hair cell loss similar to 200 μm gentamicin, a reduction in ototoxicity that resembled the reduction in JNK activation (and translational inhibition) seen in Figure 12A. The comparative dose–response curves for JNK activation (counts of p-c-Jun-positive cells; Fig. 12C) illustrate that both the kanamycin and apramycin curves are shifted to 5- to 10-fold higher concentrations compared to the gentamicin curve. A similar shift is also seen in the dose–survival curves for utricle and cochlea hair cells in Figure 12, D and E.
Figure 12.

Hair cell loss correlates with the ability of aminoglycosides to inhibit cytosolic protein synthesis and to activate the JNK pathway. A, P4 mouse utricle cultures were incubated for 24 h in 200 μm gentamicin (Gen), 200 μm apramycin, (Apra), 1000 μm apramycin, 1000 μm kanamycin (Kan), or 100 μm chloramphenicol (Chlora) in AHA-supplemented medium. Hair cells (red) exposed to gentamicin or apramycin exhibited decreased AHA incorporation (green) and increased p-c-Jun immunoreactivity (blue); gentamicin was ∼5- to 10-fold more effective in causing protein synthesis inhibition and JNK activation compared to apramycin (200 μm gentamicin corresponded to >1000 μm apramycin). Kanamycin's ability to inhibit protein synthesis and activate JNK was virtually identical to apramycin's. Exposure to chloramphenicol, an inhibitor of mitochondrial translation, did not result in JNK activation or reduction of protein synthesis (bottom). B, P4 mouse utricle (left) and cochlea (right) cultures were incubated for 72 h (24 h for cochlea) under same conditions as described in A. A concentration of 200 μm gentamicin caused a degree of hair cell loss similar to 1000 μm apramycin and kanamycin in both utricle and cochlea cultures. A concentration of 100 μm chloramphenicol failed to cause any hair cell loss. C, Quantification of A. p-c-Jun-positive hair cells were counted in an area of 300 × 300 μm. Gentamicin is ∼5–10 times more potent in activating JNK, compared to equal concentrations of kanamycin and apramycin. D, E, Quantification of B. Utricle hair cells were counted in three random areas (50 × 50 μm) of the extrastriolar region (n = 4), and cochlea outer hair cells were counted in a 100 μm stretch of the basal end (n = 4). Similar to the JNK activation dose–response curve in C, gentamicin also caused ∼5–10 times more hair cell loss in both utricle and cochlea cultures, compared to equal concentrations of kanamycin and apramycin. Scale bars: 50 μm.
To elucidate the role of mitochondrial protein synthesis, utricle explants were treated with chloramphenicol, an inhibitor of mitochondrial protein synthesis (Ramachandran et al., 2002). Mouse utricles cultured in 100 μm chloramphenicol, a concentration well above the range that causes maximal inhibition of mitochondrial protein synthesis in endothelial cells (50 μm) (Ramachandran et al., 2002), failed to produce significant hair cell loss (Fig. 12B, bottom). As expected, cytosolic protein synthesis (AHA incorporation) and JNK activity were not affected by chloramphenicol (Fig. 12A, bottom).
Differences in the ototoxicity of apramycin, kanamycin, and gentamicin were further corroborated in vivo, using functional hearing tests in mice. Adult CBA/J mice were injected with apramycin, kanamycin, or gentamicin (each at two different concentrations, using a coinjection protocol with furosemide), and the shifts in ABR thresholds were measured. Figure 13 shows that apramycin and kanamycin (both injected at 1000 mg/kg body weight) caused a similar shift in ABR threshold (∼50 dB), while 10 times lower concentrations of gentamicin (100 mg/kg) sufficed to produce a comparable threshold shift. This toxicity relationship was preserved at lower concentrations (600 mg/kg for kanamycin and apramycin, and 60 mg/kg for gentamicin). We therefore conclude that both the in vitro and in vivo ototoxicities of gentamicin, apramycin, and kanamycin correlate well with their ability to inhibit cytosolic protein synthesis and to activate the JNK pathway.
Figure 13.
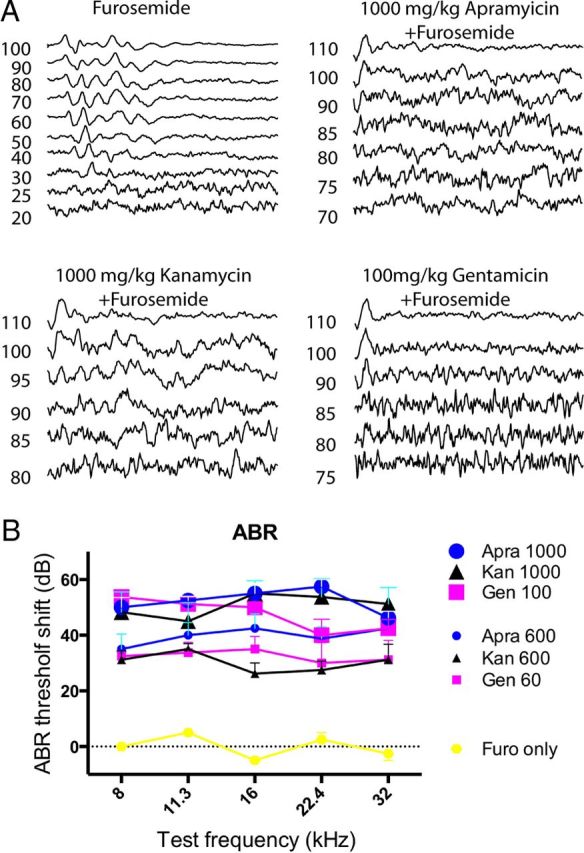
In vivo ABR threshold measurements. A, Representative ABR traces of mice systemically treated with furosemide only, 1000 mg/kg apramycin, 1000 mg/kg kanamycin, and 100 mg/kg gentamicin (coinjected with furosemide). B, Summary of ABR measurements. Injection of 1000 mg/kg apramycin (n = 4 mice) or 1000 mg/kg kanamycin (n = 4 mice) produced ABR threshold shifts comparable to the injection of 100 mg/kg gentamicin (n = 4; ∼50 dB) at all measured frequencies. Reduction of injection dose by 40% (600 mg/kg for kanamycin and apramycin, 60 mg/kg for gentamicin) resulted in similar reduction of ABR threshold shift. Furosemide injections alone did not cause a threshold shift.
Discussion
Using the BONCAT method, we performed an initial description of protein synthesis activity in cultured sensory hair cells and studied the role of protein synthesis inhibition in aminoglycoside ototoxicity. Previous studies have demonstrated that the BONCAT method does not disturb cellular function (Dieterich et al., 2006). We therefore conclude that this method allows a true representation of protein synthesis activity in hair cells, within the biological constraints of organotypic explant cultures. Analogous pulse-labeling experiments using HPG in live mice and frogs were unsuccessful, probably due to the high preference of the translational machinery for endogenous methionine as opposed to HPG (∼500-fold difference) (Kiick et al., 2002). Similar studies in living animals have so far been limited to zebrafish larvae in which nearly complete substitution of endogenous methionine with AHA can be achieved (Hinz et al., 2012).
A possible explanation for the relatively high protein synthesis activity in supporting cells compared to hair cells might be related to metabolic activity and/or the proliferative potential of supporting cells (Burns et al., 2008). For unknown reasons, protein synthesis activity in chick BP is relatively high and initially sensitive to culture conditions, with the ability to recover.
We found that gentamicin causes a significant inhibition of protein synthesis in all hair cells examined. Protein synthesis in supporting cells was not affected, in agreement with the hair cell specific toxicity of aminoglycosides. The mTOR pathway and unfolded protein response, which usually mediate translational regulation in response to stress, were not responsible for the translational block. On the contrary, the mTOR pathway was upregulated, likely a compensatory response to the protein synthesis inhibition. Previous studies have shown that the mTOR pathway is activated by an increase in free amino acids as a result of translational arrest (Beugnet et al., 2003). This induces the translation of proteins needed for translation itself, such as ribosomal proteins, which are encoded by mRNA characterized by a 5′ tract of pyrimidine nucleosides (5′ TOP mRNA) (Hara et al., 1998; Meyuhas, 2000).
We suggest that gentamicin inhibits protein synthesis in hair cells by a mechanism similar to the ribotoxic stress response (Iordanov et al., 1997; Pestka 2010). Ribotoxic stressors bind and/or inactivate a specific site of the rRNA, thereby causing translational arrest. We recognize that our hypothesis that aminoglycosides bind to eukaryotic rRNA and inhibit eukaryotic cytosolic protein synthesis contradicts established paradigms: according to a well-established model, aminoglycoside binding to the A site on the 16S rRNA of the small (30S) ribosomal subunit, and the much reduced binding to the eukaryotic counterpart, is the main rationale for bacterial specificity of aminoglycosides (Moazed and Noller, 1987; Magnet and Blanchard, 2005; Böttger, 2007). However, because hair cells accumulate aminoglycosides through the mechanotransduction channel (Richardson et al., 1997; Marcotti et al., 2005), concentrations reached may be much higher than in other cells. Although the reported binding affinities of aminoglycosides for the eukaryotic version of the A site on the 16S rRNA are low [∼40 μm for gentamicin according to Matt et al. (2012)], additional binding sites for aminoglycosides exist. Indeed, our in vitro binding assay showed that gentamicin binds to the 19 nt hairpin Helix 69 of human 28S rRNA involved in ribosome recycling (Borovinskaya et al., 2007). The Kd was 1.7 μm, a concentration that is likely to be reached in hair cells in vivo at clinically relevant concentrations (Tran Ba Huy et al., 1986; Marcotti et al., 2005). Interestingly, the 28S rRNA has also been implicated in ribotoxic stress response (Iordanov et al., 1997). We therefore hypothesize that any cell type that has the unfortunate ability to accumulate aminoglycosides, be it through the “one-way valve” that is the transduction channel in hair cells (Marcotti et al., 2005) or through the megalin receptor in renal proximal tubule cells (Schmitz et al., 2002), will expose its ribosomes to aminoglycoside binding.
Colocalization of intracellular gentamicin and rRNA (Y10B immunoreactivity) in immunocytochemistry experiments was limited, which is consistent with previous studies that demonstrated colocalization of gentamicin with the ribosome-enriched endoplasmic reticulum, in addition to mitochondria, lysosomes, and Golgi bodies (Steyger et al., 2003).
Although this report is the first to show that gentamicin inhibits protein synthesis in hair cells, it is not a surprising finding. Gentamicin's ability to bind to eukaryotic cytosolic rRNA and to cause faulty translation is a phenomenon used to suppress premature stop codons in gentamicin-based therapy of cystic fibrosis and other genetic disorders (Burke and Mogg, 1985; Bedwell et al., 1997). Furthermore, it was reported that rats treated with gentamicin in vivo showed a reduction of protein synthesis in kidney cortices and brush border membrane preparations (Sundin et al., 2001).
It is well established that aminoglycosides activate the JNK pathway, and the protective effect of inhibitors of the JNK pathway is consistent with a causal involvement of JNK in hair cell apoptosis in response to aminoglycosides (Pirvola et al., 2000; Wang et al., 2003). The activation of JNK is also a hallmark of ribotoxic stress response, and BONCAT-enabled visualization of protein synthesis activity allowed us to show for the first time a cell-by-cell correlation of translational arrest with JNK activation (Fig. 8). Furthermore, the MAP3K MLK7, implicated previously in ribotoxic stress (Sauter et al., 2010), was strongly expressed in sensory hair cells. Last, we show that the multikinase inhibitor sorafenib, which displays high affinity for MLK7 and inhibits ribotoxic stress (Sauter et al., 2010), nearly completely ameliorates JNK activation, further pointing to the similarity between the two stress responses. Interestingly, near complete inhibition of the JNK pathway failed to translate into a similarly strong inhibition of hair cell apoptosis. One reason might be that residual JNK activity (after sorafenib block) was sufficient to permit proapoptotic signals. Alternatively, sorafenib, befitting its role as an anti-cancer drug, might trigger proapoptotic signals that might override the prosurvival effects of JNK inhibition. We believe this scenario to be less likely because sorafenib by itself did not cause any hair cell death. Our results therefore suggest that a mechanism similar to ribotoxic stress response accounts nearly fully for gentamicin-induced protein synthesis inhibition and JNK activation, but only partially contributes to overall extent of apoptosis.
One model for aminoglycoside ototoxicity suggests that aminoglycosides exert their toxicity by inhibiting mitochondrial protein synthesis (Lu et al., 2010; Guan, 2011). This is supported by a previous finding that apramycin, an aminoglycoside with reduced ability to block mitochondrial protein synthesis, exhibits reduced ototoxicity (Matt et al., 2012). We show, however, that apramycin's reduced ototoxicity correlates equally well with a reduced block of cytosolic protein synthesis and JNK activity in hair cells. Apramycin's dose–response curve was shifted to an equal extent (to 5- to 10-fold higher concentrations) for JNK activation, hair cell loss, and hearing loss compared to gentamicin, suggesting a mechanistic connection between protein synthesis inhibition, JNK activation, and hair cell death. We have also performed UV-monitored thermal stability analysis to determine whether apramycin binds to human H69, but the binding was too weak to assess (data not shown), consistent with apramycin's reduced effect on protein synthesis and ototoxicity. This is in agreement with our hypothesis that binding to H69 of the 28S rRNA contributes to protein synthesis inhibition and toxicity caused by aminoglycosides.
According to Matt et al. (2012), kanamycin is approximately fourfold more potent in blocking mitochondrial protein synthesis compared to apramycin, but their abilities to block cytosolic protein synthesis in a reticulocyte extract assay were virtually identical (Matt et al., 2012). Our data not only confirm that kanamycin and apramycin are indeed equally efficient in inhibiting cytosolic protein synthesis, but also show that kanamycin and apramycin are similarly ototoxic (both in culture and in vivo), suggesting that ototoxicity correlates more closely with cytosolic rather than mitochondrial protein synthesis inhibition. Furthermore, inhibiting mitochondrial protein synthesis using chloramphenicol failed to elicit hair cell loss in our system, similar to the finding in a previous study that chloramphenicol by itself is not ototoxic, but exacerbates the ototoxic effects of aminoglycosides (Hyde and Rubel, 1995). It needs to be noted, however, that chloramphenicol has occasionally been shown to cause hearing loss (Iqbal and Srivatsav, 1984) and also cause hair cell death in zebrafish (Coffin et al., 2010). More detailed studies are needed to evaluate the contribution of mitochondrial and cytosolic translational inhibition in aminoglycoside ototoxicity. Nevertheless, a consistent framework for the aforementioned findings is that inhibition of mitochondrial as well as cytosolic protein synthesis, with the resulting ribotoxic stress, contribute to aminoglycoside ototoxicity.
The role of translational arrest in aminoglycoside ototoxicity is likely a complicated one. For example, even in the course of ribotoxic stress response, protein synthesis inhibition per se is not necessarily apoptotic. In fact, the execution of the ribotoxic stress response program depends on residual protein synthesis (Iordanov et al., 1997). This is consistent with the finding that cycloheximide-induced translational block protects from aminoglycoside-induced hair cell death (Matsui et al., 2002). On the other hand, protein synthesis inhibitors consistently elicit apoptosis in other systems (Kochi and Collier, 1993; Kageyama et al., 2002). Identifying the cellular factors that can tip the impact of translational arrest toward the death or survival pathway will contribute to a deeper understanding of the role of protein synthesis activity and inhibition in hair cell degeneration.
Footnotes
This work was funded by National Institute for Deafness and Communication Disorders Grant K99/R00 DC009412 (J.-B.S.) and the National Science Foundation Grant MCB 1101859 (P.F.A.).We thank Drs Peter Gillespie, Kevin Lee and Jeff Corwin for reading this manuscript.
The authors declare no competing financial interests.
References
- Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Beatty KE, Tirrell DA. Two-color labeling of temporally defined protein populations in mammalian cells. Bioorg Med Chem Lett. 2008;18:5995–5999. doi: 10.1016/j.bmcl.2008.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J. 2003;372:555–566. doi: 10.1042/BJ20021266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- Böttger EC. Antimicrobial agents targeting the ribosome: the issue of selectivity and toxicity—lessons to be learned. Cell Mol Life Sci. 2007;64:791–795. doi: 10.1007/s00018-007-6431-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985;13:6265–6272. doi: 10.1093/nar/13.17.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J, Christophel JJ, Collado MSOL, Magnus C, Carfrae M, Corwin JT. Reinforcement of cell junctions correlates with the absence of hair cell regeneration in mammals and its occurrence in birds. J Comp Neurol. 2008;414:396–414. doi: 10.1002/cne.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin AB, Ou H, Owens KN, Santos F, Simon JA, Rubel EW, Raible DW. Chemical screening for hair cell loss and protection in the zebrafish lateral line. Zebrafish. 2010;7:3–11. doi: 10.1089/zeb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Gorini L, Davis BD. Misreading of RNA aminoglycoside codewords induced by antibiotics. Mol Pharmacol. 1965;1:93–106. [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci U S A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Canady KS, Lurie DI, Bothwell M, Rubel EW. A biphasic change in ribosomal conformation during transneuronal degeneration is altered by inhibition of mitochondrial, but not cytoplasic protein synthesis. J Neurosci. 1994;14:1994–2008. doi: 10.1523/JNEUROSCI.14-04-01994.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PG, Walker RG. Molecular basis of mechanosensory transduction. Nature. 2001;413:194–202. doi: 10.1038/35093011. [DOI] [PubMed] [Google Scholar]
- Guan MX. Mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Mitochondrion. 2011;11:237–245. doi: 10.1016/j.mito.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Hinz FI, Dieterich DC, Tirrell DA, Schuman EM. Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci. 2012;3:40–49. doi: 10.1021/cn2000876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Sato E. Comparative analysis of combination kanamycin-furosemide versus kanamycin alone in the mouse cochlea. Hearing Res. 2011;272:108–116. doi: 10.1016/j.heares.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Hudspeth AJ. How the ear's works work. Nature. 1989;341:397–404. doi: 10.1038/341397a0. [DOI] [PubMed] [Google Scholar]
- Hyde GE, Rubel EW. Mitochondrial role in hair cell survival after injury. Otolaryngol Head Neck Surg. 1995;113:530–540. doi: 10.1177/019459989511300503. [DOI] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, Magun BE. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal SM, Srivatsav CB. Chloramphenicol ototoxicity. A case report. J Laryngol Otol. 1984;98:523–525. [PubMed] [Google Scholar]
- Jandhyala DM, Ahluwalia A, Obrig T, Thorpe CM. ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol. 2008;10:1468–1477. doi: 10.1111/j.1462-5822.2008.01139.x. [DOI] [PubMed] [Google Scholar]
- Kageyama A, Kusano I, Tamura T, Oda T, Muramatzu T. Comparison of the apoptosis-inducing abilities of various protein synthesis inhibitors in U937 Cells. Biosci Biotech Biochem. 2002;66:835–839. doi: 10.1271/bbb.66.835. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Kasinath BS, Feliers D, Sataranatarajan K, Ghosh Choudhury G, Lee MJ, Mariappan MM. Regulation of mRNA translation in renal physiology and disease. Am J Physiol Renal Physiol. 2009;297:1153–1165. doi: 10.1152/ajprenal.90748.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN. Interplay between oxidative damage, protein synthesis, and protein degradation in Alzheimer's disease. J Biomed Biotechnol. 2006;2006:12129. doi: 10.1155/JBB/2006/12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc Natl Acad Sci U S A. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochi SK, Collier RJ. DNA fragmentation and cytolysis in U937 cells treated with diphteria toxin or other inhibitors of protein synthesis. Exp Cell Res. 1993;208:296–302. doi: 10.1006/excr.1993.1249. [DOI] [PubMed] [Google Scholar]
- Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- Lockshin RA, Zakeri ZF. Physiology and protein synthesis in programmed cell death: Early Synthesis and DNA degradation. Ann N Y Acad Sci. 1992;663:234–249. doi: 10.1111/j.1749-6632.1992.tb38667.x. [DOI] [PubMed] [Google Scholar]
- Lu J, Li Z, Zhu Y, Yang A, Li R, Zheng J, Cai Q, Peng G, Zheng W, Tang X, Chen B, Chen J, Liao Z, Yang L, Li Y, You J, Ding Y, Yu H, Wang J, Sun D, et al. Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss. Mitochondrion. 2010;10:380–390. doi: 10.1016/j.mito.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitination. Annu Rev Biochem. 2009;78:435–475. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyford-Pike S, Vogelheim C, Chu E, Della Santina CC, Carey JP. Gentamicin is primarily localized in vestibular type I hair cells after intratympanic administration. J Assoc Res Otolaryngol. 2007;4:497–508. doi: 10.1007/s10162-007-0093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- Marcotti W, van Netten SM, Kros CJ. The aminoglycoside antibiotic dihydrostreptomycin rapidly enters hair cells through the mechano-electrical transducer channels. J Physiol. 2005;567:505–521. doi: 10.1113/jphysiol.2005.085951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui JI, Ogilvie JM, Warchol ME. Inhibition of caspases prevents ototoxic and ongoing hair cell death. J Neurosci. 2002;22:1218–1227. doi: 10.1523/JNEUROSCI.22-04-01218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt T, Ng CL, Lang K, Sha SH, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Böttger EC. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc Natl Acad Sci U S A. 2012;14:2012. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem. 2000;267:6321–6330. doi: 10.1046/j.1432-1327.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- Oesterle EC, Campbell S. Supporting cell characteristics in long-deafened aged mouse ears. J Assoc Res Otolaryngol. 2009;10:525–544. doi: 10.1007/s10162-009-0183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka JJ. Deoxynivalenol: mechanisms of action, human exposure, and toxicological relevance. Arch Toxicol. 2010;84:663–679. doi: 10.1007/s00204-010-0579-8. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Moellering DR, Ceaser E, Shiva S, Xu J, Darley-Usmar V. Inhibition of mitochondrial protein synthesis results in increased endothelial cell susceptibility to nitric oxide-induced apoptosis. Proc Natl Acad Sci U S A. 2002;99:6643–6648. doi: 10.1073/pnas.102019899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht MI, Douthwaite S, Puglisi JD. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999;18:3133–3138. doi: 10.1093/emboj/18.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen SK, Varella MH, Freitas FG, Moraes MO, Linden R. Contrasting effects of protein synthesis inhibition and of cyclic AMP on apoptosis in the developing retina. Development. 1996;122:1439–1448. doi: 10.1242/dev.122.5.1439. [DOI] [PubMed] [Google Scholar]
- Richardson GP, Forge A, Kros CJ, Fleming J, Brown SD, Steel KP. Myosin VIIA is required for aminoglycoside accumulation in cochlear hair cells. J Neurosci. 1997;17:9506–9519. doi: 10.1523/JNEUROSCI.17-24-09506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak LP, Whitworth CA. Ototoxicity: therapeutic opportunities. Drug Discov Today. 2005;10:1313–1321. doi: 10.1016/S1359-6446(05)03552-X. [DOI] [PubMed] [Google Scholar]
- Sauter KA, Magun EA, Iordanov MS, Magun BE. ZAK is required for doxorubicin, a novel ribotoxic stressor, to induce SAPK activation and apoptosis in HaCaT cells. Cancer Biol Ther. 2010;10:258–266. doi: 10.4161/cbt.10.3.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheunemann AE, Graham WD, Vendeix FA, Agris PF. Binding of aminoglycoside antibiotics to helix 69 of 23S rRNA. Nucleic Acids Res. 2010;38:3094–3105. doi: 10.1093/nar/gkp1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hilpert J, Jacobsen C, Boensch C, Christensen EI, Luft FC, Willnow TE. Megalin deficiency offers protection from renal aminoglycoside accumulation. J Biol Chem. 2002;277:618–622. doi: 10.1074/jbc.M109959200. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyger PS, Peters SL, Rehling J, Hordichok A, Dai CF. Uptake of gentamicin by bullfrog saccular hair cells in vitro. J Assoc Res Otolaryngol. 2003;4:565–578. doi: 10.1007/s10162-003-4002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundin DP, Sandoval R, Molitoris BA. Gentamicin inhibits renal protein and phospholipid metabolism in rats: implications involving intracellular trafficking. J Am Soc Nephrol. 2001;12:114–123. doi: 10.1681/ASN.V121114. [DOI] [PubMed] [Google Scholar]
- Taylor RR, Nevill G, Forge A. Rapid hair cell loss: a mouse model for cochlear lesions. J Assoc Res Otolaryngol. 2008;9:44–64. doi: 10.1007/s10162-007-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Ba Huy P, Bernard P, Schacht J. Kinetics of gentamicin uptake and release in the rat. Comparison of inner ear tissues and fluids with other organs. J Clin Invest. 1986;77:1492–1500. doi: 10.1172/JCI112463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Proud CG. Methods for studying signal-dependent regulation of translation factor activity. Methods Enzymol. 2007;431:113–142. doi: 10.1016/S0076-6879(07)31007-0. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Xie J, Talaska AE, Schacht J. New developments in aminoglycoside therapy and ototoxicity. Hearing research. 2011;281:28–37. doi: 10.1016/j.heares.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]