

Abstract
Myelin is a biologically active membrane receiving and processing signals from axons. Although much is known about its structure and molecular composition, the intracellular signal transduction pathways, active during specific phases of myelinogenesis for regulating myelin formation, remain poorly understood. Recent genetic loss-of-function studies have suggested a key role of extracelluar signal-regulated kinases-1 and -2 (ERK1/2), downstream mediators of mitogen-activated protein kinases (MAPKs), in promoting CNS and PNS myelination. In contrast, other studies, largely in vitro, have suggested that activation of ERK1/2 pathway can be detrimental for glial cell function and myelination. Given these conflicting reports, we investigated the effects of cell-autonomous activation of ERK1/2 in glial cells during developmental myelination in the intact CNS and PNS. Two lines of transgenic mice with sustained activation of ERK1/2 in oligodendrocyte progenitors (OPCs), oligodendrocytes, and Schwann cells were generated. Consistent with our loss-of-function studies, gain of ERK1/2 function in oligodendrocyte-lineage cells significantly increased myelin thickness, independent of oligodendrocyte differentiation or initiation of myelination. Additionally, increased activation of ERK1/2 in OPCs during early development resulted in transient hyperproliferation and overproduction of OPCs but generation of normal numbers of myelinating oligodendrocytes. Thus, these in vivo studies suggest a beneficial biphasic requirement of ERK1/2 during developmental myelination in the CNS, deployed first during early stages of the oligodendrocyte lineage for promoting OPC expansion and then redeployed later in myelinating oligodendrocytes for promoting myelin growth. Furthermore, Schwann cells with activated ERK1/2 hypermyelinate PNS axons, suggesting that ERK1/2 signaling is a conserved mechanism that promotes both CNS and PNS developmental myelination.
Introduction
Oligodendrocytes in the CNS and Schwann cells in the PNS progress through multiple stages of development and finally wrap axons with myelin sheaths by elaboration of their plasma membranes, resulting in dramatic increases in nerve conduction rate and efficiency (Baumann and Pham-Dinh, 2001; Jessen and Mirsky, 2005; Fancy et al., 2011). Inadequate myelination, or damage to the myelin sheaths as in multiple sclerosis, leads to severe neurological deficits. Limited remyelination of demyelinated lesions is characterized by the formation of myelin sheaths that are thinner than normal (Ludwin and Maitland, 1984; Franklin, 2002). Therefore, identifying signals that regulate myelin sheath thickness is critically important for therapeutic intervention to promote efficient remyelination. Several studies have implicated the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway as an important regulator of myelin growth (Flores et al., 2008; Goebbels et al., 2010; Harrington et al., 2010; Sherman et al., 2012). We recently showed a prominent role of ERK1/2-MAPK (mitogen-activated protein kinase) (Rubinfeld and Seger, 2005), in the regulation of myelin thickness, independent of oligodendrocyte differentiation, and initiation of myelination (Ishii et al., 2012). In the PNS, ERK1/2 also play a significant role because ablation of ERK1/2 in Schwann cells arrests their differentiation, resulting in dramatic hypomyelination (Newbern et al., 2011; Ishii and Bansal, unpublished).
These genetic loss-of-function studies strongly suggest a previously unrecognized function of ERK1/2 signaling as a positive regulator of myelination both in the CNS and PNS. Whether a converse genetic gain-of-function approach would support this hypothesis was uncertain because several studies have suggested that increased levels of ERK1/2 activity could indeed have detrimental effects on Schwann cells and oligodendrocytes. For example, overactivation of components of the Ras/Raf/Mek/Erk pathway in isolated Schwann cells or in cocultures prevents their differentiation, induced dedifferentiation, and demyelination (Harrisingh et al., 2004; Ogata et al., 2004), and in transgenic mice, it leads to dedifferentiation of Schwann cells and demyelination (Napoli et al., 2012). Stimulation of mature oligodendrocytes by high doses of soluble neuregulin-1 or fibroblast growth factor-2 (FGF-2) in vitro led to strong ERK1/2 activation, concomitant with downregulation of myelin proteins and aberrant cell-cycle reentry (Fressinaud et al., 1995; Bansal and Pfeiffer, 1997; Canoll et al., 1999). In contrast, constitutive activation of Mek1 in oligodendrocyte-lineage cells in vitro increased myelin protein expression (Du et al., 2006) and enhanced myelination in oligodendrocyte-neuron cocultures (Xiao et al., 2012), and constitutive activation of Ras in Schwann cells increased P0 protein expression (Rosenbaum et al., 1999), together suggesting a positive outcome of stimulating this pathway. Given these conflicting reports, it was important to investigate the effect of elevated ERK1/2 signaling in oligodendrocytes and Schwann cells in the complex environment of the intact CNS and PNS.
We therefore generated two lines of conditional transgenic mice, which expressed constitutively active Mek1, upstream activator of ERK1/2, in oligodendrocyte progenitors (OPCs), oligodendrocytes, and Schwann cells. Sustained activation of ERK1/2 in either oligodendrocytes or Schwann cells significantly enhanced myelin growth, resulting in hypermyelinated axons, independent of glial cell differentiation or initiation of myelination. Activation of ERK1/2 early during development in Olig1+ cells resulted in transient hyperproliferation and overproduction of OPCs, but normal numbers of oligodendrocytes were generated. Together, these studies suggest that, in the context of the in vivo environment, cell-autonomous activation of ERK1/2 is beneficial for developmental myelination in the CNS and PNS.
Materials and Methods
Generation of transgenic mice.
Rosa26StopFlMek1DD,EGFP mice (generated and described: Srinivasan et al., 2009) were appropriately crossed with CnpCre/+ (Lappe-Siefke et al., 2003; 2′-3′ cyclic nucleotide 3′-phosphodiesterase) or Olig1Cre/+ mice (Lu et al., 2002) to generate heterozygous CnpCre/+;Rosa26StopFlMek1DD,EGFP/+ and Olig1Cre/+;Rosa26StopFlMek1DD,EGFP/+ mice (referred to as CnpCre;MekDD and Olig1Cre;MekDD, respectively). In these transgenic mice, Cre-mediated excision of floxed STOP cassette leads to the expression of constitutively active Mek1 and EGFP. This results in sustained activation of ERK1/2 in CNP-expressing oligodendrocyte-lineage cells and Schwann cells (Gravel et al., 1998; Toma et al., 2007) or in Olig1-expressing OPCs earlier in the lineage (Lu et al., 2002). Littermate mice with no Cre were used as controls. The CnpCre;MekDD or Olig1Cre;MekDD transgenic mice did not display any obvious loss of coordination and remained generally healthy. For ablation of ERK1/2 in Olig1+ cells, ERK1−/−;ERK2flox/flox mice (obtained from Dr. J. Richards, Baylor College, Houston) were appropriately crossed with Olig1Cre/+ mice to generate Olig1Cre/+;ERK1−/−;ERK2flox/flox (referred to as Olig1Cre;ERK1/2 dKO). These mice die at birth. Genotyping of different lines of mice of both sexes was performed by PCR analysis using appropriate primers as described previously (Furusho et al., 2011, Ishii et al., 2012).
Immunolabeling.
As described previously (Kaga et al., 2006, Furusho et al., 2012; Ishii et al., 2012), cryostat transverse sections (15 μm) of cervical spinal cord and sciatic nerves from unperfused or perfused (4% paraformaldehyde/PBS) mice of both sexes were cut after overnight fixation in 4% paraformaldehyde/PBS and overnight cryoprotection in 20% sucrose/PBS. For MBP immunolabeling, sections were delipidated with 100% ethanol for 10 min; washed with PBS (3 times, 10 min); blocked (1 h) in 10% normal goat serum (Invitrogen), 5% BSA, and 0.1% fish gelatin; and incubated overnight (4°C) in polyclonal anti-MBP (1:3000; Dr. E. Barbarese, University of Connecticut) and monoclonal anti-neurofilament-m (1:200; Millipore Bioscience Research Reagents). Before immunolabeling with anti-Ki67 (1:250; BD Biosciences PharMingen), anti-Olig2 (1:100; Immuno-Biological Laboratories Co., Ltd.), anti-pan-ERK1/2 (1:200; Promega), and CC1 antibody (1:40; Millipore), spinal cord sections were subjected to antigen retrieval by 5 min of incubation at 95°C in citrate buffer, pH 6.0, and 10 min on the bench. Anti-GFP (1:200; Invitrogen) labeling was performed before antigen retrieval and double-labeling with CC1 or Olig2. Sections were incubated (1 h) with appropriate secondary antibodies conjugated to Alexa 488 (1:500, Invitrogen) or Cy3 (1:500, Jackson ImmunoResearch), and nuclei were counterstained with Hoechst blue dye 3342 (1 mg/ml; Sigma). For phospho-ERK1/2 immunolabeling, sections were incubated in 5% methanol/3% H2O2 for 10 min, followed by 10% Triton X-100 for 30 min and blocked in 10% NGS for 1 h. Incubation in phospho-ERK1/2 antibody (1:500; Sigma) in 3% BSA/0.02% Triton X-100, was performed for 72 h at 4°C. Tris-buffered saline containing 100 μm sodium fluoride, 100 μm o-vanadate was used or all dilutions and washes. Sections were incubated overnight in secondary antibody conjugated to Cy3 (1:1000; Jackson ImmunoResearch) at 4°C.
Measurements of the white matter area of spinal cords was done by Photoshop CS4. Images from matched sections of spinal cord from control and transgenic mice of both sexes were captured. Before measurement of the area, the optional plug-in “PHSPCS4_Cont_LS1” was installed, and the scale of magnification was set. The lateroventral regions of the MBP-stained white matter were selected by the Quick selection tool and applied to the area measurement.
In situ hybridization.
Transverse sections of cervical spinal cord were prepared as described for immunohistochemistry, and in situ hybridization was performed as previously described (Furusho et al., 2011, Furusho et al., 2012; Ishii et al., 2012) using riboprobes specific for proteolipid protein (PLP) mRNA (Dr. W.B. Macklin, University of Colorado School of Medicine), MBP mRNA (Dr. M. Qiu, University of Louisville), or platelet-derived growth factor receptor-α (PDGFRα) mRNA (Dr. W.D. Richardson, University College London). Briefly, after incubation in 1 μg/ml proteinase K at 37°C for 30 min, sections were hybridized overnight at 65°C with digoxigenin-labeled antisense cRNA probe and washed in 50% formamide, 2×SSC, and 1% SDS at 65°C for 2–3 h, followed by rinses in 2×SSC and 0.2×SSC at room temperature and 0.1×SSC at 60°C. After blocking in 1% Tween 20 and 1% normal goat serum (1 h), sections were incubated (2 h) in alkaline phosphatase-conjugated antidigoxigenin antibody (1:5000; Roche Diagnostics). Color was developed with 4-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate and nuclei-counterstained with Hoechst blue dye 3342.
Electron microscopy.
Transgenic and littermate control mice of both sexes were perfused with 4% paraformaldehyde, 2% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4 (Electron Microscopy Sciences). Cervical spinal cords, optic nerves, and sciatic nerves of transgenic and littermate control mice were postfixed in 1% OsO4. Samples were dehydrated through graded ethanol, stained en bloc with uranyl acetate, and embedded in Poly/Bed812 resin (Polysciences). Thin (1 μm) sections were stained with toluidine blue, and ultrathin (0.1 μm) sections from matching areas of experimental and control tissue blocks were cut and visualized using an electron microscope (JEOL 1200CX) at 80 kV. For g-ratio analysis, between 100 and 400 axons were measured per genotype from matched regions of the ventral cervical spinal cord, optic nerves, or sciatic nerves. Statistical analysis was performed using Student's t test.
Immunoblotting.
Immunoblotting was performed as described previously (Fortin et al., 2005). Briefly, equal amounts of total proteins from spinal cord and sciatic nerve homogenates were loaded onto SDS-PAGE, transferred to PVDF membranes, and immunolabeled for phosphorylated ERK1/2 (1:10,000; Sigma) and pan-ERK1/2 (1:5000; Promega).
Results
Expression and activation of ERK1/2 in myelinating oligodendrocytes correlates with the period of active myelination in the spinal cords of normal mice
Conditional loss of ERK1/2 function in oligodendrocyte-lineage cells suggested that ERK1/2 signaling might significantly influence the myelinogenic potential of mature oligodendrocytes during the period of active myelination (Ishii et al., 2012). To determine whether the normal temporal and cellular pattern of ERK1/2 expression in vivo is consistent with a role of ERK1/2 signaling during this period, normal mice were harvested at P6, P9, P15, P30, and P60, and transverse sections of cervical spinal cord were double-labeled for pan-ERK1/2 and CC1, a marker of oligodendrocytes (Fig. 1A). We found that, at all the ages examined, the majority of mature CCI+ oligodendrocytes strongly colabeled for pan-ERK1/2 (P6 = 60%, P9 = 68%, P15 = 73%, P60 = 62%) and represented the predominant ERK1/2-expressing population of cells in the white matter. A recent study showed that phospho-ERK1/2 also predominantly colocalizes with the vast majority of CC1+ oligodendrocytes but is rarely detected in PDGFRα+ OPCs in the spinal cords and brains of normal mice (Xiao et al., 2012). When we labeled spinal cord sections for phospho-ERK1/2, we found that consistent with this study, the highest intensity of phospho-ERK1/2 signal in the white matter was detected in oligodendrocytes between P9 and P30, a period of active myelination. Importantly, analysis of active ERK1/2 expression over longer periods (up to 5 months) showed that, subsequent to the period of active myelination, phospho-ERK1/2 expression progressively declined with age and was barely detectable at P60 (Fig. 1C; compare phospho-ERK1/2 intensity in “Controls” at P30 and P60). We conclude that ERK1/2 activation in oligodendrocytes correlates temporally with the period of active myelination, which is consistent with the notion that ERK1/2 signaling in myelinating oligodendrocytes plays a significant role in myelination during this period.
Figure 1.

Expression and activation of ERK1/2 in myelinating oligodendrocytes correlate with the period of active myelination in the spinal cords of normal mice, and ERK1/2 activity is elevated in oligodendrocytes of CnpCre;MekDD mice. A, Transverse sections of cervical spinal cord from normal mice at P6, P9, P15, P30, and P60, double-immunolabeled for CC1 (red), a marker of oligodendrocytes, and pan-ERK1/2 (green), show a comparable overlap (yellow) of ERK1/2 expression with CC1+ in oligodendrocytes (P6 = 60%, P9 = 68%, P15 = 73%, P60 = 62%; N = 3). B, Schematic representation of the Rosa26StopFlMek1DD,EGFP transgene. Sections of spinal cords from control and CnpCre;MekDD mice, double-labeled for EGFP and CC1, show that almost all CC1+ cells are colabeled with EGFP in the white matter, indicating efficient excision of the floxed STOP cassette by CnpCre recombinase and expression of constitutive active Mek-1 and EGFP in oligodendrocytes of transgenic mice. C, Sections double-labeled with anti-phospho-ERK1/2 and EGFP show that, in the control mice, there is an easily detectable level of phospho-ERK1/2 expression in the white matter of the spinal cord at P30, which becomes almost undetectable by P60. In the CnpCre;MekDD mice, phospho-ERK1/2 expression remains elevated, even at P60, and colocalizes with 86% of EGFP+ oligodendrocytes. Representative images of 3 mice per genotype are shown. D, Immunoblot analysis of P45 spinal cord lysates shows significantly higher levels of phospho-ERK1/2 expression in the CnpCre;MekDD (MekDD) mice compared with controls (normalized to actin levels). Pan-ERK1/2 levels remain unchanged. Scale bars: A, 50 μm; B, 25 μm; C, top, 10 μm; C, bottom, 25 μm. Error bars indicate SEM. *p < 0.05. N = 3. Arrowheads show examples of double-labeled cells.
Elevation of ERK1/2 signaling in oligodendrocytes increases myelin sheath thickness, but the initiation of myelination is unaltered
Analysis of mice lacking ERK1/2 signaling in oligodendrocytes has shown that, after normal axonal ensheathment and initiation of myelination, myelin growth is arrested, resulting in reduced thickness of the myelin sheath in these mice (Ishii et al., 2012). This raises the important question of whether elevating ERK1/2 activity in oligodendrocytes will conversely stimulate myelin growth (a question that has obvious therapeutic implications for stimulating remyelination). Therefore, we generated mice that conditionally expressed constitutively active Mek1 (and EGFP), an upstream activator of ERK1/2, in CnpCre-expressing oligodendrocytes (CnpCre;MekDD; Fig. 1B, see Materials and Methods). Double-immunolabeling of spinal cord sections from CnpCre;MekDD mice for EGFP and CC1 showed a complete overlap (99%) of CC1 with EGFP expression (Fig. 1B), indicative of transgenic expression of Mek1-EGFP in oligodendrocytes. To validate that the ERK1/2 activity was elevated in oligodendrocytes of CnpCre;MekDD mice relative to controls, we double-labeled spinal cord sections for phospho-ERK1/2 and EGFP (Fig. 1C). At P30, phospho-ERK1/2 expression appeared quite strong in both control and transgenic mice; therefore, it was difficult to identify obvious differences in the intensities at this age. But at P60, when phospho-ERK1/2 expression was normally downregulated in the controls, the elevated intensity of phospho-ERK1/2 labeling was easily detectable in 86% of EGFP+ oligodendrocytes of CnpCre;MekDD mice. Furthermore, immunoblot analysis for the expression of phospho-ERK1/2 in lysates from control and transgenic spinal cords showed that, at P45, there was a moderate but significant increase of phospho-ERK1/2 (∼1.6-fold) in heterozygous CnpCre;MekDD compared with littermate control mice (Fig. 1D).
To investigate whether onset of axon ensheathment and the initial wrapping of the myelin sheath were affected by elevation of ERK1/2 activity in the CnpCre;MekDD mice, we performed EM analysis and examined the ventral spinal cords from CnpCre;MekDD mice and littermate controls at P4 (Fig. 2A). This is a good time to examine alterations in the onset and extent of myelin formation because, in the mouse spinal cord, axonal fibers are first myelinated in the ventral spinal cord around the time of birth and then gradually increase in numbers with age. We observed that transgenic and control mice showed a similar pattern of axonal ensheathment by thin myelin sheaths. Quantification of the percentages of myelinated and unmyelinated axons from EM micrographs showed that there was no difference between the transgenic and control mice (Fig. 2A). Furthermore, as shown later (Fig. 4A, P4), immunolabeling of P4 spinal cord sections from CnpCre;MekDD and control mice with anti-MBP and neurofilament-m also showed similar patterns of MBP+ myelinated fibers in the controls and transgenic mice, together indicating that elevation of ERK1/2 activity in oligodendrocytes did not alter the initiation of myelin formation in the spinal cords of transgenic mice.
Figure 2.

Myelin sheath thickness is increased in the spinal cords of CnpCre;MekDD mice, but initiation of myelination is unaltered. EM micrographs of ventral cervical spinal cords from CnpCre;MekDD and littermate controls are shown at P4, P15, P30, and P60. A, At P4, ensheathment of axons by thin myelin sheaths appears similar in controls and transgenic mice. Quantification of the percentages of myelinated and unmyelinated axons shows no difference between CnpCre;MekDD and littermate control mice. B, At P15, myelin thickness appears similar, but by P30 and P60, axons are wrapped by thicker myelin sheaths in the CnpCre;MekDD mice compared with littermate controls. C, High-magnification images of myelin wrapped around axons of similar diameter show that the ultrastructure and periodicity of myelin wraps appear similar in the control and transgenic mice. D, Quantification of g-ratios of individual fibers in relation to respective axon diameters (presented as scatter plots) at P15, P30, and P60 confirms that myelin thickness is significantly increased in CnpCre;MekDD at P30 and P60 compared with littermate controls (P30: control = 0.78 ± 0.003, transgenic = 0.72 ± 0.003, p = 0.19 × 10−37; P60: control = 0.76 ± 0.004, transgenic = 0.69 ± 0.004, p = 0.3 × 10−39). E, Quantification, presented as myelin thickness (μm) in relation to respective axon diameters, shows that, in the CnpCre;MekDD mice, disproportionately thicker myelin sheaths wrap the majority of axons. For g-ratio measurements, at each time point, ∼300–500 axons were measured from 3 mice of each genotype per time point. Quantification of percentages of myelinated or unmyelinated axons was done from ∼1000 axons from 3000× EM images of 3 mice per genotype. Scale bars: A, B, 1 μm; C, 50 nm
Figure 4.
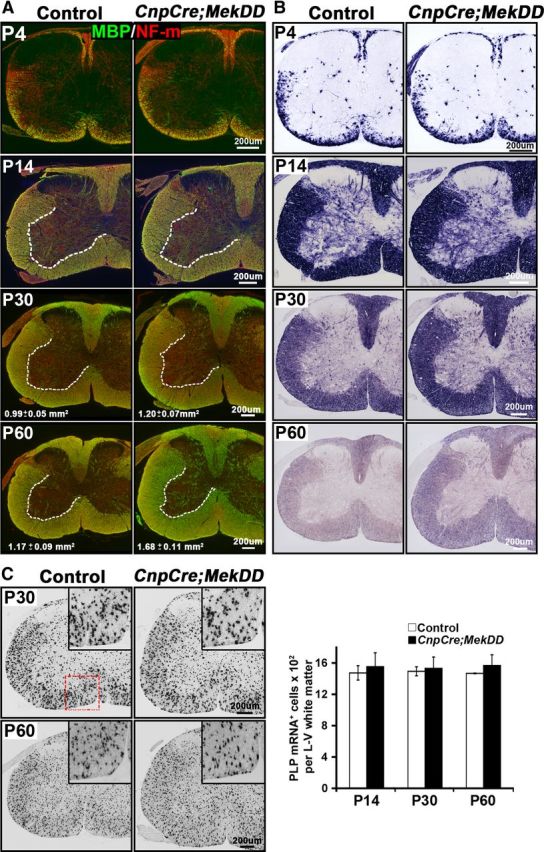
The size of the myelinated white matter in the spinal cord is increased, but the total numbers of oligodendrocytes remain unchanged in CnpCre;MekDD mice. A, B, Transverse sections of cervical spinal cord from CnpCre;MekDD and littermate control mice were analyzed at P4, P15, P30, and P60 to examine the progression of myelination by immunolabeling for the myelin marker MBP (green) and the axonal marker neurofilament-m (red) (A) and by in situ hybridization for MBP mRNA (B). At P4 and P15, the pattern of MBP-immunolabeled myelinated fibers and the size of white matter appear similar in the CnpCre;MekDD and littermate control mice. At P30 and P60, measurement of MBP-labeled lateroventral white matter area shows that it is significantly larger in the transgenic mice compared with their littermate controls (P30: control = 0.99 ± 0.05 mm2, transgenic = 1.20 ± 0.07 mm2, *p < 0.05, SEM, N = 3; P60: control = 1.17 ± 0.09 mm2, transgenic = 1.68 ± 0.11 mm2, *p < 0.05, SEM, N = 3). Similar increases in white matter sizes at P30 and P60 are shown by MBP in situ hybridization. Representative images from 3 mice of each group are shown. C, Spinal cord sections were analyzed at P14, P30, and P60 by in situ hybridization for PLP mRNA expression, as a marker of mature oligodendrocytes. Quantification of the total numbers of PLP mRNA+ oligodendrocytes in the whole lateroventral (L–V) white matter of the cervical spinal cords shows no differences in the total numbers of oligodendrocytes between control and transgenic mice at any of the ages. Three sections each from 3 mice of each genotype were analyzed at each age. Error bars indicate SEM. N = 3. Scale bars, 200 μm.
To determine the effect of ERK1/2 elevation on myelin thickness and ultrastructure of myelinated tracks, ventral cervical spinal cord sections from control and CnpCre;MekDD mice were examined at P15, P30, and P60 by EM analysis. The micrographs showed that the thickness of myelin was comparable in control and CnpCre;MekDD mice at P15, but by P30 and P60, it appeared to be progressively thicker (Fig. 2B). High-magnification images showed that the periodicity of compact myelin appeared similar in the transgenic and control mice (Fig. 2C). Morphometric quantification of myelin thickness by g-ratio analysis (ratio of individual axon diameters to myelinated fiber diameters) in matched regions of the ventral cervical spinal cord confirmed the relative increase in myelin thickness (lower g-ratios) in the CnpCre;MekDD mice compared with littermate controls at both P30 and P60 (Fig. 2D). At P30, the most striking difference in g-ratios was detected in axons of diameter <4 μm (P30: control = 0.78 ± 0.003, transgenic = 0.72 ± 0.003; p = 0.19 × 10−37). By P60, the difference became even more pronounced in this group (P60: control = 0.76 ± 0.004, transgenic = 0.69 ± 0.004; p = 0.3 × 10−39). Similarly, when myelin thickness (μm) was plotted as a function of axon diameter (Fig. 2E), it showed that, in the transgenic mice, the majority of axons were wrapped by disproportionately thicker myelin sheaths.
To examine whether elevation of ERK1/2 signaling has a general effect on myelin thickness in different regions of the CNS or is specific to the spinal cord, we analyzed myelin thickness in the optic nerves of CnpCre;MekDD and littermate control mice at P60 (Fig. 3A, top). As for the spinal cord, EM analysis showed that the myelin thickness was also increased in the optic nerves of the transgenic compared with control mice. The ultrastructure and periodicity of compact myelin in the transgenic mice appeared to be similar to controls (Fig. 3A, bottom). Quantification of myelin thickness by g-ratio analysis (Fig. 3B) confirmed the increase of myelin thickness in the optic nerves of CnpCre;MekDD mice (average g-ratios: controls = 0.82 ± 0.003; transgenic = 0.77 ± 0.003, p = 6.7 × 10−28, N = 264,245).
Figure 3.
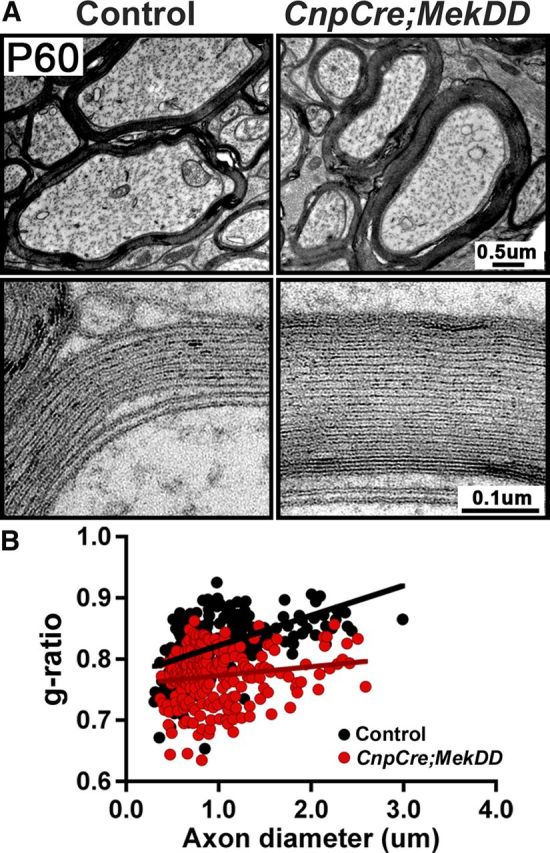
Increase in myelin sheath thickness also occurs in other regions of the CNS of CnpCre;MekDD mice. A, EM micrographs of P60 optic nerves show that, in the CnpCre;MekDD mice, axons are wrapped by thicker myelin sheaths, compared with littermate controls. High-magnification images of myelin wrapped around axons of similar diameter show that the ultrastructure and periodicity of myelin wraps appear similar in the control and transgenic mice. B, Scatter plots of g-ratios measurements of ∼250 axons from 3 mice of each genotype show a statistically significant increase in myelin thickness in the optic nerves of CnpCre;MekDD mice compared with littermate controls (average g-ratios: controls, 0.82 ± 0.003; transgenic, 0.77 ± 0.003; p = 6.7 × 10−28). Scale bars: A, top, 0.5 μm; A, bottom, 0.1 μm.
We conclude that the overall increase of myelin thickness results from increased number of myelin wraps.
The size of myelinated white matter in the spinal cord is increased, but the total numbers of oligodendrocytes remain unchanged in the CnpCre;MekDD mice
Variations in the thickness of myelin sheath are often reflected as increased or decreased area of the myelinated white matter tracks (Flores et al., 2008; Goebbels et al., 2010; Harrington et al., 2010; Furusho et al., 2012; Ishii et al., 2012). Therefore, we examined transverse sections of cervical spinal cord from CnpCre;MekDD mice and littermate control mice at P4, P14, P30, and P60 by immunolabeling for the myelin marker MBP and the axonal marker neurofilament-m (Fig. 4A) or by MBP in situ hybridization (Fig. 4B). Although the pattern of myelination and the area of myelinated white matter appeared to be comparable in the control and transgenic mice at P4 and P14, it progressively became larger with age in the CnpCre;MekDD mice. This was confirmed by measurement of the MBP-labeled lateroventral white matter area, which showed significant increases at P30 and P60 in the transgenic compared with control mice (P30: control = 0.99 ± 0.05 mm2, transgenic = 1.20 ± 0.07 mm2, N = 3, p = 0.04; P60: control = 1.17 ± 0.09 mm2, transgenic = 1.68 ± 0.11 mm2, N = 3, p = 0.02). Similar differences in the sizes of the white matter between control and transgenic spinal cords were apparent by MBP in situ hybridization. In addition, the intensity of MBP mRNA expression appeared to be somewhat higher in the transgenic compared with control mice at P30 and P60 (Fig. 4B).
To examine whether elevation of ERK1/2 in CnpCre;MekDD affected the numbers of differentiated oligodendrocytes, we performed in situ hybridization for PLP mRNA to identify and quantify mature oligodendrocytes in the whole lateroventral white matter of the spinal cords at P14, P30, and P60 (Fig. 4C). We found that the total numbers of oligodendrocytes remained unchanged between controls and transgenic mice at each of the ages examined. However, the density of oligodendrocytes was reduced at P30 and P60, as expected, because the size of the lateroventral white matter had increased in the transgenic mice at these ages (P30: control = 1353 ± 22/mm2, transgenic = 1146 ± 12/mm2; P60: control = 971 ± 34/mm2, transgenic = 802 ± 24/mm2). We conclude that hypermyelination in the CnpCre;MekDD is not caused by increases in the numbers of differentiated oligodendrocytes.
Conditional activation of ERK1/2 in Olig1Cre;MekDD mice results in transient hyperproliferation and overproduction of OPCs, but the generation of myelinating oligodendrocytes remains largely unaffected
Several in vitro studies have implicated elevation of ERK1/2 activity in OPCs (usually by growth factor stimulation) with stimulation of OPC proliferation. We asked whether genetically elevating ERK1/2 activity in OPCs in vivo would also stimulate their proliferation and increase their numbers. Because our initial analysis showed that in the spinal cords of the CnpCre;MekDD line, transgene expression in OPCs was not very efficient (determined by EGFP expression), we generated a second line of conditional transgenic mice using the Olig1Cre driver, which is known to recombine with higher efficiency in OPCs and the recombination occurs earlier during development. To validate the efficiency of transgene expression in OPCs in the Olig1Cre;MekDD mice, we double-labeled spinal cord sections at P2 with EGFP and Olig2 (pan-marker for oligodendrocyte lineage cells that primarily marks white matter OPCs at P2). As expected, in the heterozygous Olig1Cre;MekDD line, the vast majority of Olig2+ cells expressed EGFP (Fig. 5A), indicating that the transgene is expressed in OPCs with a high level of Olig1Cre-mediated recombination efficiency (99%). Therefore, this line of mice was used subsequently to investigate the effects of activating ERK1/2 on OPC proliferation and differentiation during the first 2 postnatal weeks in the spinal cords.
Figure 5.
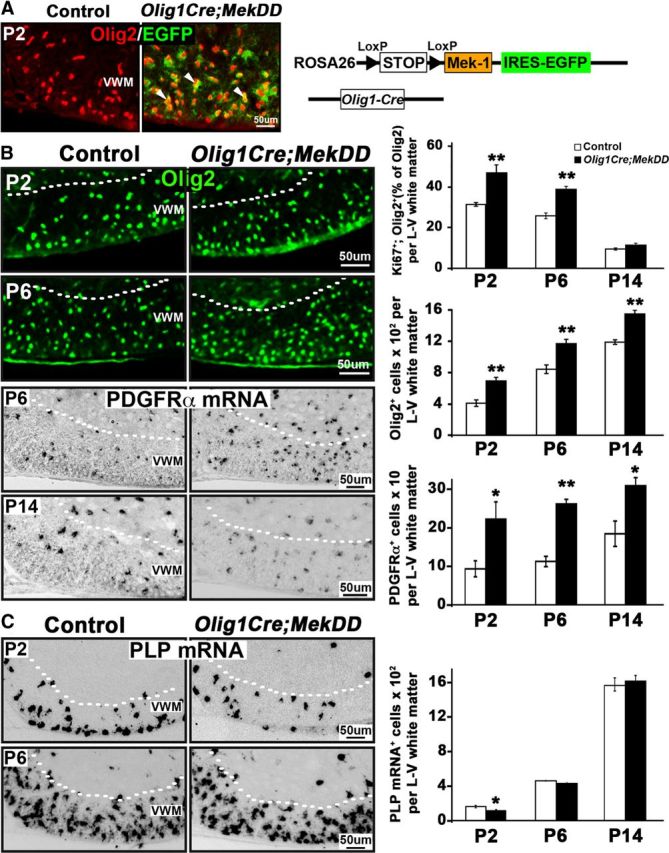
Conditional activation of ERK1/2 in Olig1Cre;MekDD mice results in transient hyperproliferation and overproduction of OPCs, but the generation of myelinating oligodendrocytes remains largely unaffected. A, Transverse sections of P2 spinal cords from control and Olig1Cre;MekDD mice, double-labeled for EGFP and Olig2 (which primarily marks OPCs at P2), show an almost complete overlap (yellow) in the white matter of the cervical spinal cord, indicating efficient excision of the floxed STOP cassette by Olig1Cre recombinase and expression of Mek1-EGFP in OPCs of transgenic mice. B, Transverse sections of P2, P6, and P14 spinal cords from Olig1Cre;MekDD and littermate controls were analyzed by immunolabeling for the proliferation marker Ki67 and Olig2 and by in situ hybridization for the OPC marker PDGFRα mRNA. Quantification of Ki67+; Olig2+ double-labeled cells (expressed as percentage of Olig2+ cells) and the total numbers of Olig2+ or PDGFRα mRNA+ cells quantified from the whole lateroventral (L–V) region of the spinal cord show that, at P2 and P6, there is a significant increase in both the percentages of proliferating cells (Ki67+/Olig2+) and total numbers of PDGFRα mRNA+ and Olig2+ cells in the Olig1Cre;MekDD mice compared with littermate controls. At P14, the numbers of PDGFRα mRNA+ and Olig2+ cells remain elevated in the Olig1Cre;MekDD mice, but the percentage of proliferating cells (Ki67+/Olig2+) is normalized to similar levels as in the controls. C, Transverse sections of cervical spinal cords from P2, P6, and P14 Olig1Cre;MekDD and littermate control mice were analyzed by in situ hybridization for PLP mRNA to identify differentiated oligodendrocytes. Quantification of the total numbers of PLP mRNA+ oligodendrocytes in the whole lateroventral white matter of the spinal cords shows that, at P2, there is a transient decrease in oligodendrocyte numbers in the transgenic mice, which is readily normalized, and the numbers of oligodendrocytes become comparable at P6 and P14 in transgenic and control mice. Two or 3 sections each from 3 mice of each genotype were analyzed at each time point. Error bars indicate SEM. *p < 0.05. **p < 0.01. N = 3. Scale bars, 50 μm. VWM, Ventral white matter.
To examine potential alterations in the proliferation capacity of OPCs and determine the effects of ERK1/2 elevation on the numbers of OPC, we analyzed spinal cords from Olig1Cre;MekDD and littermate control mice at P2, P6, and P14. Spinal cord sections were double-labeled for Olig2 and the proliferation marker Ki67 to quantify proliferating cells. To determine the total numbers of OPCs at each time point, we used in situ hybridization to identify OPCs by the expression of PDGFRα mRNA. These cells were quantified from the whole lateroventral white matter area of the spinal cords from controls and transgenic mice. Total numbers of Olig2+ cells were also counted similarly at each time point. We found that, at P2 and P6, the percentage of proliferating Olig2+ cells (%Ki67+/Olig2+/total Olig2+ cells) and the total numbers of Olig2+ and PDGFRα mRNA+ cells were all significantly increased in the Olig1Cre;MekDD mice compared with controls. However, by P14, although the total numbers of PDGFRα mRNA+ and Olig2+ cells still remained elevated in the Olig1Cre;MekDD mice, the percentage of Ki67+/Olig2+ cells normalized to control levels.
We next investigated the effect of ERK1/2 elevation on OPC differentiation in the Olig1Cre;MekDD mice (Fig. 5C). The numbers of oligodendrocytes, identified by in situ hybridization for PLP mRNA expression, were quantified at P2, P6, and P14 in equivalent areas of the whole lateroventral white matter of spinal cords from the Olig1Cre;MekDD and littermate control mice. At P2, there was a transient delay in the appearance of differentiated oligodendrocytes (perhaps resulting from temporary delay in exiting from the cell cycle); however, it was readily normalized within a few days, and similar numbers of oligodendrocytes appeared at P6 and P14 in the Olig1Cre;MekDD and control mice.
Together, we conclude that conditional activation of ERK1/2 in OPCs over the normal threshold level results in hyperproliferation and overproduction of OPCs in the spinal cords of Olig1Cre;MekDD mice. However, despite increased numbers of OPCs and a transient delay, overall OPC differentiation into myelinating oligodendrocytes was not affected in the Olig1Cre;MekDD line, and normal numbers of myelinating oligodendrocytes were generated during the period of active myelination, as was observed in the CnpCre;MekDD line.
Loss of ERK1/2 function at Olig1+ stage in the Olig1Cre;ERK1/2 dKOs reduces OPC proliferation and numbers but does not directly reduce their capacity to differentiate into oligodendrocytes
Although elevation of ERK1/2 activity in Olig1+ cells (Fig. 5) resulted in hyperproliferation and expansion of the OPC population, our recent studies showed that loss of ERK1/2 signaling in NG2+ cells did not conversely decrease OPC numbers or their proliferative capacity (Ishii et al., 2012). Because the NG2+ stage is slightly later than the Olig1+ stage of OPC maturation (Ligon et al., 2006; Whitman et al., 2012), one probable reason for this difference might be that signaling by ERK1/2 is significant at the Olig1+ stage but becomes dispensable once the cells have reached the more mature NG2+ stage. To test this possibility, we generated mice where ERK1/2 were ablated using the Olig1Cre driver line (Olig1cre/+; ERK1−/−;ERK2flox/flox, referred as Olig1Cre;ERK1/2 dKO). These mice die soon after birth; so, analysis was performed at P0 only (Fig. 6). Proliferation and numbers of OPCs were determined by double-labeling and quantification of Olig2+ and Ki67+ cells in equivalent areas of the whole lateroventral regions of spinal cords from Olig1Cre;ERK1/2 dKO and littermate control mice (Fig. 6A). We found that the percentage of proliferating OPCs (%Ki67+/Olig2+) was significantly decreased in the spinal cords of Olig1Cre;ERK1/2 dKO compared with their littermate controls. The total numbers of Olig2+ cells, quantified similarly and expressed as percentage of controls, were also significantly decreased in the Olig1Cre;ERK1/2 dKO compared with controls (Fig. 6A). Because proliferation was reduced and cell viability (TUNEL assay) remained unaffected (data not shown), we conclude that reduced proliferation is at least in part responsible for the reduction in OPC numbers in the Olig1Cre;Erk1/2 dKO mice. Whether alterations in lineage decisions additionally contribute toward the reduction in OPC numbers remains to be determined in future studies by the analysis of OPC generation in the embryonic spinal cords and brains of these mutant mice.
Figure 6.

Loss of ERK1/2 function at Olig1+ stage in the Olig1Cre;ERK1/2 dKOs reduces OPC proliferation and numbers but does not directly reduce their capacity to differentiate into oligodendrocytes. A, To identify proliferating OPCs, transverse spinal cord sections from Olig1Cre;ERK1/2 dKO and their littermate controls were double-labeled for Ki67 and Olig2 at P0. Quantification of the percentages of Ki67+;Olig2+ double-labeled cells of total Olig2+ cells in the whole lateroventral white matter area of the spinal cord shows a significant decrease in both the percentage of proliferating OPCs and the total numbers of Olig2+ OPCs in the Olig1Cre;ERK1/2 dKOs compared with their littermate controls. B, Numbers of mature PLP mRNA+ oligodendrocytes counted similarly also show a significant reduction in the Olig1Cre;ERK1/2 dKOs compared with controls (expressed as percentage of controls). Quantification of cells double-immunolabeled for CC1 and Olig2 shows that the percentages of Olig2+ cells that differentiate into CC1+ oligodendrocytes are similar in controls and Olig1Cre;ERK1/2 dKOs, indicating that the reduction in oligodendrocytes is a reflection of reduced OPC generation. Three sections each from 3 mice of each genotype were analyzed at each time point. Error bars indicate SEM. *p < 0.05. **p < 0.01. N = 3. Arrowheads show examples of double-labeled cells. VWM, Ventral white matter region. Scale bars, 50 μm.
To examine the effects of ERK1/2 ablation from Olig1+ cells on OPC differentiation, we first performed in situ hybridization for PLP mRNA expression on spinal cord sections from Olig1Cre;ERK1/2 dKOs and littermate control mice and counted the total numbers of PLP mRNA+ oligodendrocytes in equivalent areas of the whole lateroventral regions of spinal cords (Fig. 6B). We found that the numbers of oligodendrocytes were also significantly reduced in the Olig1Cre;ERK1/2 dKOs. Because the relative reduction of OPCs and oligodendrocytes in Olig1Cre;ERK1/2 dKOs was similar (∼25–35% of controls), it suggested that the reduction of oligodendrocytes was most generation and not the result of reduced probability of OPC differentiation into oligodendrocytes. To investigate this further, we double-immunolabeled sections for CC1 and Olig2. Quantification of labeled cells confirmed that the proportion of Olig2+ cells that differentiated into CC1+ oligodendrocytes at P0 was similar in both Olig1Cre;ERK1/2 dKOs and controls.
Together, we conclude that, although loss of ERK1/2 function at the NG2+ stage does not affect OPC proliferation or differentiation (Ishii et al., 2012), the loss of ERK1/2 function at the earlier Olig1+ stage of OPC maturation does reduce the expansion of the OPC population and in turn reduces the appearance of oligodendrocytes. However, regardless of whether ERK1/2 were ablated at the NG2+ or Olig1+ stage, its loss does not directly reduce the capacity of OPCs to differentiate into oligodendrocytes in vivo.
Hypermyelination also occurs in the PNS by elevation of ERK1/2 activity in Schwann cells
Because Cnp-Cre is also expressed by Schwann cells in addition to oligodendrocytes (Gravel et al., 1998; Toma et al., 2007), the Schwann cells of the CnpCre;MekDD mice should also have constitutive activation of Mek1 and sustained elevation of ERK1/2 activity. This was validated by immunoblotting and quantification of phosphorylated ERK1/2 levels, which showed a 1.5-fold increase in P14 sciatic nerves. Further, transgene expression in the PNS was confirmed by demonstration of EGFP expression in the sciatic nerves of CnpCre;MekDD mice (Fig. 7A).
Figure 7.

Hypermyelination also occurs in the PNS by sustained activation of ERK1/2 in Schwann cells of CnpCre;MekDD mice. A, Longitudinal sections of sciatic nerves from control and CnpCre;MekDD mice at P14 show EGFP expression in the transgenic mice, indicative of transgene expression. Elevation of ERK1/2 activity in sciatic nerves of these mice is shown by immunoblotting for phospho-ERK1/2 in lysates of P14 sciatic nerves. Error bars indicate SEM. *p < 0.05. N = 3. B, Toludine blue-stained semithin sections of sciatic nerves show that, at P4, Schwann cells from CnpCre;MekDD mice appear to ensheath and wrap axons in a similar manner as in the control mice. C, EM micrographs of sciatic nerves from CnpCre;MekDD and littermate controls at P15 and P30 show that axons of transgenic mice are wrapped by myelin sheaths that are thicker compared with littermate controls. Morphometric quantification of g-ratios of individual fibers in relation to respective axon diameters (presented as scatter plots) shows significantly increased myelin sheath thickness in the CnpCre;MekDD mice compared with littermate controls at both the ages examined (P15: p = 3.6 × 10−53; P30: p = 7.2 × 10−34). Similarly, myelin thickness (μm), plotted as a function of axon diameter, shows that myelin thickness is disproportionately increased relative to axon diameter in the transgenic mice. Approximately 100–300 axons each were measured from 3 mice per genotype at each age. Scale bars: A, 50 μm; B, 5 μm; C, 2 μm.
To determine the effect of sustained activation of ERK1/2 in Schwann cells on developmental myelination in the PNS, sciatic nerve cross-sections from control and CnpCre;MekDD mice were examined at P4, P15, and P30. Toluidine blue-stained semithin sections of sciatic nerves showed that at P4, Schwann cells in transgenic mice appeared to ensheath and wrap axons in a similar manner as in the control mice (Fig. 7B). Examination of EM micrographs at P15 and P30 showed that the thickness of the myelin sheath was clearly increased in the transgenic mice compared with controls (Fig. 7C). Morphometric quantification of myelin thickness by g-ratio analysis confirmed this and showed a significant increase in myelin thickness (lower g-ratios) in the CnpCre;MekDD mice compared with littermate controls at P15 and P30 (P15: p = 3.6 × 10−53; P30: p = 7.2 × 10−34). Similarly, when myelin thickness (μm) was plotted as a function of axon diameter, it showed that, in transgenic mice, myelin thickness was disproportionately increased relative to axon diameter.
Discussion
Here, we demonstrate that elevation of ERK1/2 activity in CNP+ oligodendrocyte-lineage cells stimulates myelin growth and increases myelin thickness without affecting oligodendrocyte differentiation or ensheathment of axons, whereas overstimulation or, conversely, loss of ERK1/2 function early during development in Olig1+ OPCs, leads to defects in the production of normal numbers of OPCs. Thus, these studies reveal a critical requirement of ERK1/2 signaling first during very early stages for OPC expansion and then again during later stages of myelinogenesis for promoting myelin growth. In the PNS, activation of ERK1/2 in Schwann cells also leads to hypermyelination, suggesting a conserved mechanism underlying myelin wrapping between the CNS and PNS. Interestingly, this regulatory mechanism is also evolutionarily conserved in myelinating glia of Drosophila (Franzdóttir et al., 2009).
It has been suggested that an important determinant of the biological consequence of ERK1/2 activation is the strength and duration of ERK-MAPK activation; e.g., sustained elevation of ERK1/2 activity led to neurite extension in PC12 cells (Marshall, 1995; Ramos, 2008). Thus, enhanced extension of the myelin sheath resulting from sustained elevation of ERK1/2 activity in oligodendrocytes in the CnpCre;MekDD mice is consistent with this notion. In normal mice, the fact that ERK1/2 activity remains elevated in oligodendrocytes during the period of active myelination is also consistent with its role in myelin growth. In addition, in vitro studies have linked elevated ERK1/2 activation with enhanced myelin protein expression in oligodendrocytes (Du et al., 2006) and with increased myelination inoligodendrocyte-neuron cocultures (Xiao et al., 2012).
Although these studies suggest that activation of ERK1/2 is beneficial for promoting myelination, there is also strong evidence that suggests a negative effect of increased activation of this pathway. For example, in vitro activation of ERK1/2 signaling or overexpression of Ras or Raf, upstream of ERK1/2, prevents Schwann cell differentiation in response to cAMP; induces downregulation of myelin proteins in already differentiated Schwann cells; and causes demyelination in Schwann cell-DRG cocultures (Harrisingh et al., 2004; Ogata et al., 2004), and in vivo activation of ERK1/2 in differentiated Schwann cells of the adult peripheral nerves leads to their dedifferentiation and demyelination (Napoli et al., 2012). In the CNS, there is also correlative evidence that predicts a detrimental role for ERK1/2 activation in oligodendrocytes. Specifically, exposure of differentiated oligodendrocytes in vitro, to high doses of glial growth factor, an isoform of neuregulin-1, or to FGF-2, results in ERK1/2 activation along with phenotypic reversion of oligodendrocytes, downregulation of myelin proteins, and aberrant reentry into the cell cycle (Fressinaud et al., 1995; Bansal and Pfeiffer, 1997; Canoll et al., 1999). One possible explanation for the difference between our study and recent in vivo study (Napoli et al., 2012) is that, when ERK1/2 is continuously activated during developmental myelination, as in our study, it promotes myelination, whereas when it is terminated and then reactivated in differentiated myelinating cells it triggers demyelination. Thus, ERK1/2 activation in myelinating glia can be either promyelinating or demyelinating, depending on the context of activation. Further, in the present study, ERK1/2 were only moderately elevated (<2-fold), which may simply represent a potentiation of the normal function of ERK1/2 in promoting myelination. It remains to be investigated whether continued long-term elevation of ERK1/2 or increasing the threshold of ERK1/2 activation in oligodendrocytes or Schwann cells would result in continued myelin growth and/or would become detrimental in the long run.
Several growth factors, such as neuregulin-1 type III, IGF-1, FGFs, and brain-derived neurotrophic factor (BDNF), which are implicated in the regulation of myelination, activate the Ras/Mek/ERK-MAPK pathway. Although neuregulin-1 and IGF1 are thought to signal primarily via the P13K/Akt pathway, activation of ERK1/2 downstream of these growth factors may also (directly or indirectly) play a role in promoting myelination. The link of FGF and BDNF with ERK1/2 signaling in the regulation of myelination is better understood. Specifically, mice lacking FGF receptors-1 and -2 in oligodendrocytes show reduced ERK1/2 phosphorylation and arrest of myelin growth, similar to that observed in the ERK1/2 knock-out mice (Furusho et al., 2012). Although such a cell-autonomous connection of BDNF/TrkB with ERK1/2 activity for myelin growth has not yet been established in vivo, BDNF is reported to increase myelin protein expression in isolated oligodendrocytes and enhance myelination in oligodendrocyte-neuron cocultures through the ERK-MAPK pathway (Du et al., 2006; Xiao et al., 2012). Thus, ERK1/2 may serve to integrate signals for myelin growth from multiple growth factors. Additionally, because ERK1/2 are activated by several other extracellular signals that play a role in myelination (Rubinfeld and Seger, 2005; Piaton et al., 2010), ERK1/2 activation in oligodendrocytes may have wider implications for myelination.
The molecular mechanisms by which ERK1/2-MAPK promote myelin growth are challenging to fully predict at this point. ERK1/2 are known to integrate multiple upstream signals into numerous downstream substrates, including nuclear transcription factors, cytoskeletal proteins, signaling proteins, and receptors (Roux and Blenis, 2004). Regulation of the transcription of myelin genes by downstream effectors of MAPK pathways, such as transcription factors, Elk1, AP2-complex, or CREB, is a potential mechanism. For example, CREB-dependent myelin protein expression in oligodendrocytes has been shown to require MAPK activation (Palacios et al., 2005). Because ERK1/2 are well-known transcriptional activator by themselves, they may directly phosphorylate key myelination-specific transcription factors; e.g., YY1 phosphorylation in Schwann cells by ERK1/2 is important for neuregulin-induced activation of peripheral myelination (He et al., 2010). Similarly, phosphorylation of the transcription factor Olig2 at its putative tyrosine phosphorylation sites by ERK1/2 could potentially regulate its function, as shown recently by phosphorylation of its threonine phosphorylation sites (Li et al., 2011; Sun et al., 2011). Moreover, it is plausible that cross talk between the PI3K/Akt/mTOR or Mek1/ERK1/2-MAPK pathways may exist in oligodendrocytes and Schwann cells, perhaps converging at central regulators of the translation machinery, driving myelin growth. One possible point of interaction of ERKs with the PI3K/Akt/mTOR pathway is at the TSC1/2 suppressor complex. Both ERK-RSK and Akt negatively regulate the TSC1/2 complex, which in turn regulates mTORC1, a key regulator of both CNS and PNS myelination (Wullschleger et al., 2006; Narayanan et al., 2009, Sherman et al., 2012). In addition, ERK-RSK are also known to directly interact with other proteins involved in protein translation (Shahbazian et al., 2006; Steelman et al., 2011). Future studies are required to determine whether P13K/Akt/mTOR and Mek/ERK1/2-MAPK are independent parallel pathways or whether they cooperate with each other to facilitate myelin growth.
Several in vitro studies, including ours (Bhat and Zhang, 1996; Canoll et al., 1999; Baron et al., 2000; Bansal et al., 2003; Du et al., 2006; Cui and Almazan, 2007; Van't Veer et al., 2009), have shown that one of the most robust responses of isolated OPCs upon exposure to several growth factors is the activation of ERK1/2 and proliferation. Our studies show that, in the in vivo context of the intact CNS, cell-autonomous elevation or loss of ERK1/2 activity in Olig1+ cells can increase or decrease OPC proliferation and numbers, respectively. This suggests that a strict control of ERK1/2 levels is critically important to produce normal numbers of OPCs during the early stages of development in the spinal cord, as shown recently for the production of astrocytes (Tien et al., 2012). Interestingly, the analysis of mice with conditional loss of ERK1/2 signaling in NG2+ or CNP+ cells did show a significant effect on OPC proliferation (Ishii et al., 2012). It is possible that the Olig1+ stage is more proliferative and regulated by more than one mitogenic intracellular signal (ERK1/2 being one major signal), whereas the NG2+ stage is less proliferative, requiring fewer mitogenic signals. Thus, loss of ERK1/2 at the Olig1+ stage has a bigger impact than at the NG2+ stage where OPCs are in the process of exiting the cell cycle and entering terminal differentiation. Thus, it appears that, in vivo, the requirement of ERK1/2 signaling at the Olig1+ stage becomes largely dispensable as cells progress to more mature NG2+ and CNP+ stages of development. Whether elevation of ERK1/2 activity in NG2+ cells will show increased proliferation, as observed by its elevation in Olig1+ cells, remains to be established. However, regardless of the stage of maturation, modulation of ERK1/2 in OPCs does not directly alter their capacity to differentiate and generate myelinating oligodendrocytes. Collectively, these studies have revealed a critically important, stage-specific, “early” function of ERK1/2 for OPC expansion in addition to its “later” function in the regulation of myelin growth.
In summary, one of the main conclusions drawn from the present genetic gain-of-function studies, which are consistent with our previous loss-of-function studies (Ishii et al., 2012), is that ERK1/2 signaling plays a central role in the regulation of myelin growth and that this is a common mechanism used by both oligodendrocytes and Schwann cells. In the future, it will be important to define the molecular mechanisms by which intracellular signaling by ERKs in turn influences the expression and activity of downstream effectors, such as transcriptional regulators, epigenetic regulators, and microRNAs, to efficiently orchestrate the complex process of developmental myelination. Further, because a fine balance of ERK1/2 levels may be critically important for normal function, a better understanding of the regulation (or dysregulation) of this exquisitely controlled signaling system is required before manipulation of ERK1/2 activity for therapeutic intervention in demyelinating diseases or peripheral nerve injury will be possible.
Footnotes
This work was supported by NIH Grant NS38878, and in part by Grant RG 4087-A03 from the National Multiple Sclerosis Society and NIH Grant NS41078. We thank Dr. K.-A. Nave (Max Plank Institute, Gottingen, Germany) for providing the CNP-Cre mice, Dr. D.H. Rowitch (University of California at San Francisco, CA) for Olig1-Cre mice, Dr. D.J. Richards (Baylor College of Medicine, Houston, TX) for the Erk1−/−;Erk2 floxed mice, and G. Wark for technical assistance.
References
- Bansal R, Pfeiffer SE. FGF-2 converts mature oligodendrocytes to a novel phenotype. J Neurosci Res. 1997;50:215–228. doi: 10.1002/(SICI)1097-4547(19971015)50:2<215::AID-JNR10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Bansal R, Magge S, Winkler S. Specific inhibitor of FGF receptor signaling: FGF-2-mediated effects on proliferation, differentiation, and MAPK activation are inhibited by PD173074 in oligodendrocyte-lineage cells. J Neurosci Res. 2003;74:486–493. doi: 10.1002/jnr.10773. [DOI] [PubMed] [Google Scholar]
- Baron W, Metz B, Bansal R, Hoekstra D, de Vries H. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol Cell Neurosci. 2000;15:314–329. doi: 10.1006/mcne.1999.0827. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Canoll PD, Kraemer R, Teng KK, Marchionni MA, Salzer JL. GGF/neuregulin induces a phenotypic reversion of oligodendrocytes. Mol Cell Neurosci. 1999;13:79–94. doi: 10.1006/mcne.1998.0733. [DOI] [PubMed] [Google Scholar]
- Cui QL, Almazan G. IGF-I-induced oligodendrocyte progenitor proliferation requires PI3K/Akt, MEK/ERK, and Src-like tyrosine kinases. J Neurochem. 2007;100:1480–1493. doi: 10.1111/j.1471-4159.2006.04329.x. [DOI] [PubMed] [Google Scholar]
- Du Y, Lercher LD, Zhou R, Dreyfus CF. Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J Neurosci Res. 2006;84:1692–1702. doi: 10.1002/jnr.21080. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Franzdóttir SR, Engelen D, Yuva-Aydemir Y, Schmidt I, Aho A, Klämbt C. Switch in FGF signaling initiates glial differentiation in the Drosophila eye. Nature. 2009;460:758–761. doi: 10.1038/nature08167. [DOI] [PubMed] [Google Scholar]
- Fressinaud C, Vallat JM, Labourdette G. Basic fibroblast growth factor down-regulates myelin basic protein gene expression and alters myelin compaction of mature oligodendrocytes in vitro. J Neurosci Res. 1995;40:285–293. doi: 10.1002/jnr.490400302. [DOI] [PubMed] [Google Scholar]
- Furusho M, Kaga Y, Ishii A, Hébert JM, Bansal R. Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J Neurosci. 2011;30:5055–5066. doi: 10.1523/JNEUROSCI.4800-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Nave KA, Bansal R. Fibroblast Growth Factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci. 2012;32:6631–6641. doi: 10.1523/JNEUROSCI.6005-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Möbius W, Liu X, Lappe-Siefke C, Rossner MJ, Groszer M, Suter U, Frahm J, Boretius S, Nave KA. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci. 2010;30:8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel M, Di Polo A, Valera PB, Braun PE. Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J Neurosci Res. 1998;53:393–404. doi: 10.1002/(SICI)1097-4547(19980815)53:4<393::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Harrington EP, Zhao C, Fancy SP, Kaing S, Franklin RJ, Rowitch DH. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol. 2010;68:703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisingh MC, Perez-Nadales E, Parkinson DB, Malcolm DS, Mudge AW, Lloyd AC. The Ras/Raf/ERK signaling pathway drives Schwann cell dedifferentiation. EMBO J. 2004;23:3061–3071. doi: 10.1038/sj.emboj.7600309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Kim JY, Dupree J, Tewari A, Melendez-Vasquez C, Svaren J, Casaccia P. Yy1 as a molecular link between neuregulin and transcriptional modulation of peripheral myelination. Nat Neurosci. 2010;13:1472–1480. doi: 10.1038/nn.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci. 2012;32:8855–8864. doi: 10.1523/JNEUROSCI.0137-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Kaga Y, Shoemaker WJ, Furusho M, Bryant M, Rosenbluth J, Pfeiffer SE, Oh L, Rasband M, Lappe-Siefke C, Yu K, Ornitz DM, Nave KA, Bansal R. Mice with conditional inactivation of fibroblast growth factor receptor-2 signaling in oligodendrocytes have normal myelin but display dramatic hyperactivity when combined with Cnp1 inactivation. J Neurosci. 2006;26:12339–12350. doi: 10.1523/JNEUROSCI.3573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Li H, de Faria JP, Andrew P, Nitarska J, Richardson WD. Phosphorylation regulates OLIG2 cofactor choice and the motor neuron oligodendrocyte fate switch. Neuron. 2011;69:918–929. doi: 10.1016/j.neuron.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Kesari S, Kitada M, Sun T, Arnett HA, Alberta JA, Anderson DJ, Stiles CD, Rowitch DH. Development of NG2 neural progenitor cells requires Olig gene function. Proc Natl Acad Sci U S A. 2006;103:7853–7858. doi: 10.1073/pnas.0511001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Ludwin SK, Maitland M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J Neurol Sci. 1984;64:193–198. doi: 10.1016/0022-510x(84)90037-6. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh MC, White IJ, Woodhoo A, Lloyd AC. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron. 2012;73:729–742. doi: 10.1016/j.neuron.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern JM, Li X, Shoemaker SE, Zhou J, Zhong J, Wu Y, Bonder D, Hollenback S, Coppola G, Geschwind DH, Landreth GE, Snider WD. Specific functions for ERK/MAPK signaling during PNS development. Neuron. 2011;69:91–105. doi: 10.1016/j.neuron.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Iijima S, Hoshikawa S, Miura T, Yamamoto S, Oda H, Nakamura K, Tanaka S. Opposing extracellular signal regulated kinase and Akt pathways control Schwann cell myelination. J Neurosci. 2004;24:6724–6732. doi: 10.1523/JNEUROSCI.5520-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios N, Sánchez-Franco F, Fernández M, Sánchez I, Cacicedo L. Intracellular events mediating insulin-like growth factor I-induced oligodendrocyte development: modulation by cyclic AMP. J Neurochem. 2005;95:1091–1107. doi: 10.1111/j.1471-4159.2005.03419.x. [DOI] [PubMed] [Google Scholar]
- Piaton G, Gould RM, Lubetzki C. Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair. J Neurochem. 2010;114:1243–1260. doi: 10.1111/j.1471-4159.2010.06831.x. [DOI] [PubMed] [Google Scholar]
- Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40:2707–2719. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T, Kim HA, Boissy YL, Ling B, Ratner N. Neurofibromin, the neurofibromatosis type 1 Ras-GAP, is required for appropriate P0 expression and myelination. Ann NY Acad Sci. 1999;883:203–214. [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006;25:2781–2791. doi: 10.1038/sj.emboj.7601166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Krols M, Wu LM, Grove M, Nave KA, Gangloff YG, Brophy PJ. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J Neurosci. 2012;32:1817–1825. doi: 10.1523/JNEUROSCI.4814-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011;3:192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien AC, Tsai HH, Molofsky AV, McMahon M, Foo LC, Kaul A, Dougherty JD, Heintz N, Gutmann DH, Barres BA, Rowitch DH. Regulated temporal-spatial astrocyte precursor cell proliferation involves BRAF signaling in mammalian spinal cord. Development. 2012;139:2477–2487. doi: 10.1242/dev.077214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma JS, McPhail LT, Ramer MS. Differential RIP antigen (CNPase) expression in peripheral ensheathing glia. Brain Res. 2007;1137:1–10. doi: 10.1016/j.brainres.2006.12.053. [DOI] [PubMed] [Google Scholar]
- Van't Veer A, Du Y, Fischer TZ, Boetig DR, Wood MR, Dreyfus CF. Brain-derived neurotrophic factor effects on oligodendrocyte progenitors of the basal forebrain are mediated through TrkB and the MAP kinase pathway. J Neurosci Res. 2009;87:69–78. doi: 10.1002/jnr.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman LM, Blanc CA, Schaumburg CS, Rowitch DH, Lane TE. Olig1 function is required for remyelination potential of transplanted neural progenitor cells in a model of viral-induced demyelination. Exp Neurol. 2012;235:380–387. doi: 10.1016/j.expneurol.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xiao J, Ferner AH, Wong AW, Denham M, Kilpatrick TJ, Murray SS. Extracellular signal-regulated kinase 1/2 signaling promotes oligodendrocyte myelination in vitro. J Neurochem. 2012;122:1167–1180. doi: 10.1111/j.1471-4159.2012.07871.x. [DOI] [PubMed] [Google Scholar]