

Abstract
The polo-box domain (PBD) has critical roles in the mitotic functions of PLK1. The REPLACE strategy to develop inhibitors of protein-protein interactions has identified alternatives for the N-terminal tripeptide of a Cdc25C substrate. In addition, a peptide structure activity relationship described key determinants and novel information useful for drug design. Fragment ligated inhibitory peptides (FLIPs) were generated with comparable affinity to peptide PBD inhibitors and possessed anti-proliferative phenotypes in cells consistent with the observed decrease in PLK1 centrosomal localization. These FLIPs demonstrated evidence of enhanced PLK1 inhibition in cells relative to peptides and induced monopolar and multipolar spindles, which stands in contrast to previously reported small molecule PBD inhibitors that display phenotypes only partially representative of PLK1 knockdown. Progress obtained applying REPLACE validates this approach for identifying fragment alternatives for determinants of the Cdc25C binding motif and extends its applicability of the strategy for discovering protein-protein interaction inhibitors. In addition, the described PBD inhibitors retain high specificity for PLK1 over PLK3 and therefore show promise as isotype selective, non-ATP competitive kinase inhibitors that provide new impetus for the development of PLK1 selective anti-tumor therapeutics.
Keywords: Phosphorylation and proteolysis in cell cycle control, Protein serine-threonine kinases, Molecular modelling, In silico evaluation of targets and design of libraries, Protein/protein interactions, Cell cycle mechanisms of anticancer drug action, Kinase and phosphatase inhibitors, Novel antitumor agents
INTRODUCTION
The polo-like kinases are central players in regulating entry into and progression through mitosis (1). A significant body of literature has determined that profound anti-proliferative activity is achieved through selective inhibition of PLK1 functions (2). The four known human PLKs have non-redundant and non-overlapping functions. Overexpression of PLK1 is frequently observed and PLK1 expression is a prognostic indicator for outcome of patients suffering from various tumors (3–5). More than half of prostate cancers overexpress PLK1 and this expression is positively correlated with tumor grade (6). PLK1 is extensively overexpressed in colorectal cancer (7) and recently was demonstrated to be a potential therapeutic target in such tumors with inactivated p53 (8). Moreover, it has been recently reported that p53 transcriptionally regulates PLK1 expression, providing direct evidence that PLK1 is oncogenic when p53 is mutated (9). Thus, there is a strong rationale for pursuing PLK1 as an anti-tumor drug target. Indeed, the therapeutic rationale for PLK inhibition has been validated through several studies and shown to profoundly inhibit cancer cell proliferation both in vitro and in vivo (10, 11). Numerous inhibitors of ATP binding site of PLKs have been identified, with some entering clinical trials after showing significant anti-tumor activity in preclinical models. At least two compounds have been evaluated in phase I clinical trials. Results from two compounds suggest acceptable toxicity profiles warranting further investigation in phase II trials (12, 13). A significant potential drawback of compounds targeting the ATP cleft, however, is that at least 3 of the 4 known members of the mammalian PLKs are inhibited by BI2536, currently the most advanced PLK inhibitor to date (14). Since PLK3 has been reported to have opposing functions to PLK1, PLK3 inhibition may lead to diminution of the anti-tumor effect mediated by blocking PLK1 (15–18). These issues were revealed after the initial clinical development of ATP-competitive PLK inhibitors and strongly suggest that inhibiting PLK3 would not a desirable feature of a clinical candidate. In addition, ATP competitive inhibitors will only block the enzymatic functions of PLK1 and will not necessarily affect its other critical functions in mitosis. Therefore, alternative approaches to develop potent and highly selective PLK1 inhibitors are required. Studies with peptides provide evidence that the substrate and sub-cellular targeting binding site in the Polo Box Domain (PBD) forms a compact and druggable interface amenable to small molecule inhibitor development (19–22). Although high-throughput screening approaches have identified small molecule inhibitors of the PBD-peptide interaction, these either are weakly binding or non-drug-like in nature (23, 24). In addition, these inhibitors display a contrasting phenotype to PLK1 knockdown and to cellular treatment with inhibitors of catalytic activity (14, 25, 26). Recently progress has been reported in the generation of derivatized peptides that occupy a novel site in the PBD binding groove (27, 28).
Peptides, while also non-drug-like, can discriminate between the PLK family members and can therefore provide structural basis for the development of selective PLK1 inhibitory compounds. Herein, REPLACE, a validated strategy for the iterative discovery of non-peptidic protein-protein interaction inhibitors, has been applied to discover fragment alternatives for the N-terminal hydrophobic motif in a Cdc25C PBD substrate peptide. Furthermore, using transfected PBD binding peptides and fragment ligated inhibitory peptides (FLIPs), a workflow for phenotypic and PLK1 specific cellular effects has been established. Results demonstrate that PBD-targeted inhibitors replicate a PLK1 phenotype, in contrast to the partial phenotype obtained with PBD dominant negative and small molecule inhibitors, suggesting that they inhibit both subcellular localization and substrate phosphorylation.
MATERIALS & METHODS
Peptide & FLIP Synthesis
Phospho-Peptides were synthesized and purified using standard Fmoc chemistry by GenScript (Piscataway, NJ) and unless stated otherwise, all peptides were synthesized with an N-terminal amino group and a C-terminal carboxyl group. HPLC and MS were used to confirm the purity and structure of each peptide (see Supplementary Information Table 1). R group definitions for FLIPs are given in Supplementary Table 2.
Fluorescent Polarization Binding Assay
FLIPs and peptides to be tested were dissolved in DMSO (10 mM), and diluted from 10 nM to 600 µM. The PLK1 PBD (367–603) and PLK3 PBD (335–646) proteins were obtained from BPS Bioscience Inc. (San Diego, CA) and 250 ng was used per reaction. The fluorescein-tracer phospho-peptides (MAGPMQS[pT]PLNGAKK for PLK1, and GPLATS[pT]PKNG for PLK3) were used at a final concentration of 100 nM. Incubation was carried out at room temperature for 45 minutes. Fluorescence was measured using a DTX 880 plate reader and Multimode Analysis software (Beckman Coulter, Brea, CA). The polarization values in millipolarization (mP) units were measured at an excitation wavelength of 488 nm and an emission wavelength of 535 nm. Each data point was performed in triplicate for every experiment, and experiments were performed at least three times. An IC50 value for each compound was calculated from linear regression analysis of the plots (Supplementary Figure 1).
Cell Culture
HeLa cervical cancer cells were obtained from ATCC (Manassas, VA) and were not authenticated by the authors for this study. Histone H2B GFP-labeled HeLa cells (HeLa-H2B-GFP) were kindly provided by Dr. Geoffrey Wahl (Gene Expression Laboratory, Salk Institute) [29], and were confirmed as >95% GFP positive by FACS (data not shown) but were not otherwise authenticated. Cells were maintained in DMEM (Invitrogen, Carlsebad, CA) supplemented with 10% Nu-serum (BD Bioscience, Franklin Lakes, NJ) and 1% penicillin/streptomycin (Invitrogen) in a humidified incubator and 5% CO2 at 37°C.
Peptide Transfection
Peptides and FLIPs were transfected into cells using as the QQ reagent, (29). The following modifications were made to the protocol: a 10 mg/mL stock solution of PEI was made by diluting PEI (50 wt. % in H2O) with 50 mM phosphate buffer and using 6 M HCl (Sigma Aldrich) to adjust the pH to 7.4. MG132 was resuspended in 0.1 M DMSO to make a 5 µg/mL stock solution of MG132. The final concentration of PEI and MG132 was 0.25 mg/mL and 5 ng/mL, respectively.
Immunofluorescence, PLK1 localization, and aberrant prometaphase/metaphase analysis
HeLa-H2B-GFP cells were plated at a density of 10,000 cells per coverslip 24 h prior to thymidine block. Cells were synchronized with 2 mM thymidine for 18 h and followed by two PBS washes. Fresh medium was added and the cells incubated at 37°C with or without peptide or FLIP. After the indicated treatment time, coverslips were washed with PBS. The cells were fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100. Fixed cells were incubated with PLK1 antibody (Cell Signaling Technology, Danvers, MA) overnight at 4° C and Alexa Fluor 488–conjugated anti-rabbit secondary IgG (Molecular Probes) for 1 h at room temperature to visualize PLK1. A minimum of 20 centrosomes were analyzed per slide. Cells were scored as normal metaphase when two clearly defined centrosomes were observed while those with a single or multiple centrosomes were scored aberrant. Metaphase cells with misaligned chromosomes were scored as aberrant. Experiments were performed in triplicate and the results averaged.
RESULTS
Structure-activity relationship of peptidic inhibitors of the polo-box domain
While the binding of various synthetic peptides based on sequences that interact with the PBD have been studied, there has been little systematic analysis of the residues that comprise the motif and their contributions. PBD inhibitory peptides thus far reported consist of variants of both the native Cdc25C PBD binding sequence, those from non-native ligands (20, 21) and from the PBIP also known as CENP-Q (19). From the latter, Yun et al. identified minimized PBD binding sequences consisting of 5 and 6-mer peptides that possessed an increased level of selectivity for PLK1 over longer sequences previously studied (19). Specifically, the minimal recognition motif for the PBD is PLHS(pT), a peptide with a Kd of 0.45 µM, which was slightly less potent than the 6-mer with a C-terminal alanine extension (0.26 µM). The LHSpTAI sequence of similar potency possessed a Kd of 0.25 µM.
To increase the body of data available for structural determinants of the PBD motif, in this present study peptide analogs were synthesized and tested in a fluorescence polarization (FP) binding assay for their ability to compete off the fluorescein labelled “non-natural” PBD sequence (MAGPMQS[pT]PLNGAKK, IC50 = 0.85 µM for the unlabeled competitor) (20). Analysis of the SAR of LLCS[pT]PNGL, the Cdc25C PBD binding sequence, was undertaken through analog synthesis and computational design. The phosphorylated motif LLCS[pT]PNGL comprises the PBD interacting sequence of Cdc25C, thereby recruiting it as a PLK1 substrate. Since non-phosphothreonine containing peptides have been shown to bind to the PBD (22), a number of peptide analogs of the Cdc25C sequence were synthesized in this laboratory. These included natural and non-natural amino acid residues, exploring substitution of the LLC hydrophobic tripeptide motif on the N-terminus and replacement of the phosphorylated threonine residue. No detectable binding was observed for non-phosphorylated peptides incorporating various hydrophobic substitutions, despite previous observations that such peptides retain activity (22). This possibly reflects the use of different PBD domain proteins in prior studies, which include a shorter version produced for crystallography (residues 367–603) (22) and a longer construct (326–603) used in binding studies (21). To probe this further, PBD sequences were examined for binding to both constructs. No evidence for affinity of non-phosphorylated peptides was detected despite robust binding of the native phospho-Cdc25C sequence, LLCS[pT]PNGL (Table 1, 292). This discrepancy is most likely a consequence of the competitive binding assay since the previous study measured direct binding through intrinsic tryptophan fluorescence (22). It was observed, however, that phospho-Cdc25C binds with significantly higher affinity (0.42 µM) for the shorter PBD construct (1.3 µM IC50 for competition of fluorescent peptide binding to PLK1 326-603), perhaps indicating that the multiple conformational states previously described for PLK1 affect its sub-cellular localization (30, 31).
Table 1.
IC50 values for PBD peptides derived from CDC25c and PBIP
| SCCP ID | Sequence | PLK1 PBD IC50 |
|---|---|---|
| 292 | LLCS[pT]PNGL | 0.42 |
| 5821 | Ac-LLCS[pT]PNGL | 0.5 |
| 345 | LLCSEPNGL | 350 |
| 5795 | LDCS[pT]PNGL | 3 |
| 5783 | LLAAAPNGL | >600 |
| 5714 | S[pT]PNGL | >600 |
| 5649 | LHS[pT]AI | 200 |
| 5781 | Ac-LHS[pT]AI | 5 |
| 5782 | Ac-AHS[pT]AI | 15 |
| 5743 | Ac-PLHS[pT]A | 2 |
| 5744 | Ac-PLHSEA | 500 |
Synthesis and testing of a peptide library incorporating semi-conservative replacements and phosphothreonine isosteres (data not shown) revealed that only the glutamic acid containing sequence (Table 1, 345) possessed detectable binding to the PBD. While almost a thousand fold less potent, the carboxylate side chain nonetheless partially mimics the phosphate, thereby suggesting useful SAR information that could be exploited. Additional peptide analogs were synthesized based upon the PBIP sequence and where the N-terminal end was truncated and modified by replacing Leu-Leu-Cys with Leu-His and where the residues C-terminal to the pThr (Pro-Asp-Gly-Leu) were replaced with Ala-Ile. A serendipitous discovery resulted from generating peptides without the N-terminal acetyl group previously incorporated (19). Interestingly, the des-acetyl peptide exhibited dramatically reduced potency relative to the expected value (Table 1, 5649). After resynthesis with the acetyl group on the N-terminus, the potency was found to be similar to the reported compound (Table 1, 5781). The acetylated peptide (IC50 of 5 µM) is 40 fold more potent than the non-acetylated version (IC50 of 200 µM) and is just six-fold less potent than the parent peptide containing the N-terminal LLC. The essential contribution of this acetyl group to binding was not previously apparent despite evidence of H-bonding in crystal structures of Ac-LHS[pT]AI (5781) in complex with the PBD (19). The decreased potency of the non-actetylated peptide suggests that the H-bond from the carbonyl of the acetyl group to the guanidinum side chain of Arg516 is a critical determinant for binding to the PBD since no interactions of the amide nitrogen or methyl group are observed. In addition, a partial contribution to the potency increase derives from the fact that repulsion of the positively charged N-terminal amino group with the basic side chain of Arg516 is mitigated in the acetylated compound.
Furthermore, other acetylated sequences were generated and tested in the PBD binding assay to probe the SAR of truncated molecules. To determine the contributions of the Leu side chain this context, Ac-AHS[pT]AI (5782) was synthesized. As observed from the available crystal structures (5781), the branched aliphatic side chain has no significant non-bonded interactions with the PBD groove suggesting that the alanine replacement should display similar potency. After testing, the resulting IC50 was 3 fold higher (Table 1, 5782), which indicates that the side chain contributes entropically to peptide binding through conformational effects.
An acetylated peptide, truncated at the C-terminus with respect to the native Cdc25C sequence, Ac-PLHS[pT]A (Pro-Leu-His replacing Leu-Leu-Cys), was synthesized, tested in the FP assay and shown to have comparable activity to Ac-LHs[pT]AI (Table 1, compare 5743 to 5781). This observation suggests that the N-terminal proline compensates for removal of the C-terminal isoleucine and the relative affinities are consistent with results previously obtained (19). Peptide 5743 also reveals that high affinity PLK1 binding can be obtained through interaction of the PBD subsite occupied by the N-terminal tripeptide of Cdc25C. A glutamic acid was substituted for phospho-threonine in the PBIP sequence (5744). In agreement with results seen for the corresponding substitution in the Cdc25C context (345), 5744 weakly but detectably bound to the PBD (Table 1). To assess the contribution of the N-terminal tripeptide to binding of the Cdc25C native sequence, the peptide S[pT]PNGL was synthesized. In vitro testing confirmed the critical interactions of the LLC trimer as no detectable binding was observed for the truncated peptide (Table 1, 5714). A negative control in which three residues were mutated to alanines was devoid of binding (Table 1, 5783).
The SAR information generated suggests that focusing on the PBD subsite occupied by the LLC tripeptide in conjunction with the incorporation of phosphothreonine mimetics should yield non-peptides with sufficient potency.
Design and synthesis of fragment alternatives of the N-terminal tripeptide of the Cdc25C PBD binding sequence
Using REPLACE, low molecular weight fragments are computationally docked into the volume of a binding site known to interact with key peptidic determinants. Through use of generated SAR data, REPLACE was applied to identify fragment alternatives to the N-terminal tripeptide (32). A requirement for docked fragments is that they have the potential for incorporation of appropriate functionality to allow ligation onto a truncated peptide (i.e. minus the determinant requiring replacement). For example, in the previous CDK2/cyclin A context, a critical arginine residue was replaced with a phenyltriazole, N-terminally appended to a tetrapeptide via a carboxylate group (32). After using REPLACE to identify capping group alternatives to one region of a peptidic inhibitor, this approach can then be used iteratively to find fragment alternatives to the remaining determinants. Applying this strategy to the PBD groove involved docking of 1800 carboxylate containing fragments using LigandFit into the volume of the peptide binding groove occupied by the LLC tripeptide. Since S[pT]PNGL (5714) possesses no measurable affinity, capped peptides are considered hits if they have measurable binding in the FP assay. Based on predictions of high scoring fragment alternatives for the tripeptide, 34 Fragment Ligated Inhibitory Peptides (FLIPs) were synthesized.
After initial screening in the FP assay, 7 FLIPs were identified as having measurable activity (IC50 values ranging between 200 and 400 µM; 1G1 -1G3 are 1st iteration examples shown in Table 2) with a further five FLIPs having an IC50 between 400 and 600 µM. These results indicated a total hit rate of 30% and thereby confirmed the success of the strategy in the initial application to the PBD. While initial partial ligand alternative (PLA) capped peptides were of considerably lower potency than the native Cdc25C sequence, an increase relative to the truncated molecule (S[pT]PNGL, 5714) was apparent. The most potent substructures observed in the first generation were a benzo[1,4]oxazin-3-one pharmacophore and a similar 5-phenylpyrazinone core (Table 2, 1G1 and 1G2). Based on the assumption that interactions of the predicted docked fragment structure and the truncated peptide will be preserved in the FLIP, it is straightforward to obtain a protein-ligand complex for the capped molecule through molecular modeling (i.e. binding mode of 5-phenylpyrazinone group in Figure 1). The SAR obtained here suggests that appropriate modification of the 5-phenyl group with H-bond acceptors should lead to binding improvements on order of the increases observed with addition of the acetyl group to the LHS[pT]AI (5781).
Table 2.
PLK PBD In vitro binding and cellular activity for FLIP compounds (Ncap-S[pT]PNGL)
| SCCP ID | Abbreviation | N Capping Group | PLK1 PBD FP IC50 [µM] |
PLK3 PBD FP IC50 [µM] |
Apoptosis @ 24hr, 30 µM |
Aberrant mitoses, @ 24hr, 30 µM |
|---|---|---|---|---|---|---|
| 5594 | 1G1-S[pT]PNGL | 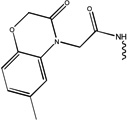 |
350 | ND | ND | ND |
| 5598 | 1G2-S[pT]PNGL | 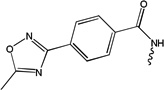 |
320 | ND | ND | ND |
| 5603 | 1G3-S[pT]PNGL | 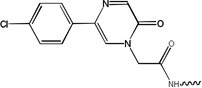 |
250 | ND | ND | ND |
| 5756 | 2G1-S[pT]PNGL | 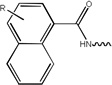 |
99 | ND | ND | ND |
| 5788 | 3G1-S[pT]PNGL | 16.5 | >600 | 36.2% | 47.9% | |
| 5827 | 3G2-S[pT]PNGL | 8.6 | >600 | 55.5% | 53.8% | |
| 5743 | Ac-PLHS[pT]A | - | 2 | >600 | 34.0% | 40.3% |
Figure 1.

Binding mode of the SCCP5594, a benzo[1,4]oxazin-3-one fragment ligated to S[pT]PNGL (generated using Discovery Studio 3.0). H-bonds to the fragment and other non-bonded contacts to the PBD are indicated by black dashed and solid green lines respectively.
As follow up to these initial hits, it was proposed that incorporation of appropriately substituted napthoic and benzoic acid derivatives onto the N-terminus of the hexapeptide would uniquely take advantage of features of the PBD groove. Second generation derivatives were predicted to be simpler pharmacophores of the initial hits and were incorporated as capping groups through synthesis of additional FLIP molecules (Table 2, 2G1, 5756). After in vitro testing of second generation FLIPS, substantial potency enhancements were observed relative to the initial hits. The best of these compounds, 3G1-S[pT]PNGL (Table 2, 5788) had an IC50 of 16.5 µM and therefore was the most successful thus far in recapitulating the potency represented in the Cdc25C peptide. Among several analogs obtained, it is apparent that these compounds exploit novel features of the PBD groove and that a clear SAR is evident from the range of activities observed. Indeed, synthesis of third generation derivatives exploiting this novel interaction resulted in compound 3G2-S[pT]PNGL (Table 2, 5827) with an IC50 of 8.6 µM. The benzoic acid PLA core structure is suitable for further derivatization and allows for the incorporation of appropriate functionality to mimic the H-bond interactions of the acetyl group observed in the peptide context. Several studies indicate that both Cdc25C and PBIP derived peptide sequences have a high degree of specificity for the PBD of PLK1 compared to those of PLK2 and PLK3. To determine if the benzoic acid derived FLIPs retain this selectivity, a similar FP competitive binding assay was developed using the PBD of PLK3 (residues 335-646). Using this assay, a robust signal was obtained for the fluorescein labeled PLK3 ligand in complex with the PLK3 PBD as previously described (23). Furthermore, an unlabeled version of this peptide successfully competed the tracer from the PLK3 construct with an IC50 value of 6.5 µM. Dose-response testing of the two benzoic acid capped FLIPS, 3G1-S[pT]PNGL (5788) and 3G2-S[pT]PNGL (5827) showed insignificant levels of inhibition even at the highest ligand concentrations (Table 2). The peptide PBD inhibitor (5743) evaluated showed minor inhibition at 600 µM (31%) therefore suggesting that the small molecule peptide hybrid compounds have greater specificity towards the PBD of PLK1.
PBD binding compounds reduce PLK1 sub-cellular localization
To uncover evidence of PLK1 targeting in living cells, studies were conducted using the more promising PBD inhibitory compounds identified by the FP binding assay. To date, a limited body of data is available on cellularly administered peptides due to their lack of permeability and stability. The recently developed QQ reagent (29) was utilized in order to successfully transfect peptides into cells. To demonstrate successful transfection, HeLa cells were treated with the QQ modified, native Cdc25C phosphopeptide with an N-terminal fluorescein label. Extensive green fluorescence was observed intracellularly, which indicated nuclear localization of the peptide (data not shown).
PLK1 is recruited to the centrosomes and kinetochores; interference with this step and its catalytic activity causes improper centrosome duplication and spindle defects (2, 33, 34). To determine if QQ transfected peptides were able to generate phenotypes consistent with blocking of recruitment through the PBD, localization of PLK1 at the centrosomes in metaphase was visualized and quantified through immunofluorescence in HeLa cells expressing GFP fused to Histone H2B (used to visualize chromosomes during mitosis) (35). The average relative fluorescence intensity of PLK1 at the centrosome was 51.2 and 49.2 (arbitrary fluorescence units) for the PBS treated control and QQ controls respectively (Figure 2). Three peptides and one FLIP (Ncapped peptide) were tested for their ability to interfere with PLK1 centrosomal localization. These included the Cdc25C parent peptide LLCS[pT]PNGL (292), two truncated and acetylated peptides, Ac-PLHS[pT]A (5743) and Ac-LHS[pT]AI (5781) and one of the most promising FLIP generated thus far, 3G1-S[pT]PNGL (5788). Treatment with the acetylated peptides 5743 and 5781 (30 μM) reduced the fluorescence intensity of PLK1 staining at centrosomes to 33 and 35, respectively, while treatment with 5788 resulted in even more marked effects (32 at 10 μM) suggesting that the peptides and FLIP 5788 were specifically targeting PLK1 (Figure 2). Treatment with 292 only slightly reduced the fluorescent intensity to 45, which was not a statistically significant difference. Although 292 provided the lowest IC50 value in the in vitro binding assay, the reduced potency is not surprising given the instability of unmodified peptides in cells and contrasted with more effective results for the acetylated and fragment-ligated peptides.
Figure 2.

Treatment of HeLa cells with PBD inhibitors leads to reduced localization of PLK1 to the centrosomes. The Y-axis represents arbitrary fluorescent intensity units. Control represents fluorescence intensity data from untreated cells. QQ represents the data from cells treated with transfection reagent alone. The remainder represent data from cells transfected with the following peptides: 292 (LLCS[pT]PNGL), 5743 (Ac-PLHS[pT]A), 5781 (Ac-PLHS[pT]A), 5788 (3G1-S[pT]PNGL). Statistically significant decreases in fluorescent intensity were observed for 5743, 5781 and 5788.
Anti- mitotic effects of PBD binding compounds
In addition, cells were examined for mitotic phenotypes consistent with PLK1 inhibitory effects following treatment with the PBD inhibitors. Two such phenotypes include a prometaphase/metaphase arrest, and monopolar or multipolar spindles in metaphase (Figure 3). The GFP-H2B HeLa cells (35) were utilized to visualize chromosomes during mitosis after treatment with PBD inhibitors transfected using QQ reagent.
Figure 3.

Representative images from HeLa cells expressing GFP-H2B and immunostained for PLK1. (a) normal metaphase from an untreated cell, (b) aberrant prometaphase from a cell transfected with 5743, (c) aberrant, quadripolar metaphase from a cell transfected with 5743, (c) aberrant, tripolar metaphase from a cell transfected with 5788.
The large majority of mitotic cells mock or QQ-alone treated were observed to be in normal prometaphase or metaphase (<10% of the cells were scored as aberrant, Figure 4). Treatment of cells with the negative control peptide (5783) and the Cdc25C sequence (292) resulted in 14% and 25% aberrant prometaphase/ metaphase features (30 µM dose, Figure 4) respectively. Peptide 5743 (Ac-PLHS[pT]A) resulted in a potent and dose dependent induction of cells with aberrant prometaphase or metaphase since at a 10 µM dose, 32% of cells showed mitotic abnormalities and at 30 µM, the fraction rose to 40.3%. Treatment with 5781, a weaker in vitro PBD inhibitor, resulted in a less pronounced cellular effect with approximately 23% demonstrating aberrant metaphase at 30 µM. Interestingly, of all the compounds tested, the two FLIPs identified using REPLACE, namely 3G1-S[pT]PNGL (5788) and 3G2-S[pT]PNGL (5827), led to the most pronounced effects in the induction of aberrant mitosis, with values of 48 and 54% respectively at 30 µM.
Figure 4.

Treatment with PBD inhibitors induced aberrant mitoses. The y-axis represents the percentage of aberrant mitoses seen in mitotic cells. Control represents data from untreated cells. QQ represents data following treatment with QQ transfection reagent alone. The remainder represent data from the following treatments: 292 (LLCS[pT]PNGL), 5743 (Ac-PLHS[pT]A), 5781 (Ac-LHS[pT]AI), 5788 (3G1-S[pT]PNGL), or 5827 (3G2-S[pT]PNGL).
Treatment of HeLa cells with PBD inhibitors induces apoptosis and necrosis
An Annexin V (AV) – Propidium Iodide (PI) assay was used to measure cell death induced by the PBD inhibitors. Using flow cytometry, cells were scored as viable, early apoptotic and late apoptotic/necrotic. The mock and QQ treatments did not significantly induce cell death although a slight increase in apoptosis was observed with QQ alone (Scatter Plots are shown in Supplementary Figure 2). Parental HeLa cells were transfected with PBD inhibitors 292, 5821, 5743 and 5781 at a 30 µM dose. Both 6-mer peptides 5743 and 5781 behaved similarly after 24 hours post-treatment with the percentage of viable cells decreasing by 30% and similar levels of early and late apoptosis with each transfected inhibitor (Figure 5). Interestingly, the most potent PBD inhibitory peptide 292, when transfected did not induce significant levels of apoptosis relative to the controls. An acetylated version of this compound (5821), however, was more potent despite having a similar IC50 in the FP assay. After 24 hours, it reduced the fraction of viable cells to 57%, therefore indicating a stabilizing effect of the acetyl group on its cellular activity. Treatment of HeLa cells with the two PBD FLIPs, 5788 (3G1) and 5827 (3G2) resulted in the most profound effects in apoptotic induction at 30 µM. Relative to the peptide inhibitors, and despite having weaker in vitro affinity, these two compounds had the lowest proportion of viable cells with 64 and 45% respectively after 24 hours. In particular, the most potent FLIP in the in vitro assay, 5827, had a similar proportion of viable and early apoptotic cells and a significant fraction of late apoptotic cells. These results suggest that the capping group provides protection from cellular degradation of the peptides and leads to increased cellular potency. Taken together, these results confirm that PBD inhibitors induced significant levels of cell death through apoptosis and that our two most potent FLIPs are the most effective in this cellular assay.
Figure 5.

Induction of apoptosis by PBD inhibitory compounds. The y-axis represents the percentage of cells. PBS represents cells treated with PBS alone. QQ represents cells treated with QQ transfection reagent alone. The remainder represent cells transfected with: 292 (LLCS[pT]PNGL), 5821 (Ac-LLCS[pT]PNGL), 5743 (Ac-PLHS[pT]A), 5781 (Ac-LHS[pT]AI), 5788 (3G1-S[pT]PNGL), or 5827 (3G2-S[pT]PNGL).
DISCUSSION
In this study, a systematic approach was undertaken to examine the structure-activity relationship of PBD inhibitory peptides, to apply REPLACE to discover new fragment alternatives for the N-terminal tripeptide establishing proof of concept, and to characterize the cellular mechanism of action for these compounds. The peptide analogues synthesized were derived from the Cdc25C (LLCS[pT]PNGL) and PBIP1 (PLHS[pT]AI) proteins that bind to the PLK1 PBD (19, 22). The importance of the phosphothreonine for PBD binding was confirmed in both peptide contexts, although the weak activity of Glu replacements suggests that incorporation of other functionalities is tolerated and is useful for future inhibitor design. SAR information from the peptides containing the core PBIP1 sequence agreed with previously reported data (19). Furthermore, this present study uncovered a novel observation that N-terminal acetylation is crucial for truncated peptides to bind with high affinity to the PBD. This result suggests that incorporation of appropriate functional groups into a PBD inhibitor to facilitate H-bonding to Arg516 would result in a substantive potency increase. In a different context, addition of an acetyl group to the Cdc25C nine residue peptide resulted in no effect on its in vitro binding (compare 292 to 5821), an observation consistent with the minimal interactions of the N-terminal Leucine residue. The acetyl group did, however, significantly increase the cellular potency relative to 292 as measured by induction of apoptosis. The capped N-terminus likely protects the peptide from proteolytic degradation thereby improving half life within the cell. It was also determined that replacing the Leu residue of the PBIP1 sequence with Ala led to a measurable decrease in binding (3-fold), an observation inconsistent with the lack of non-bonded interactions in published crystal structures. This suggests that the conformational requirements imposed by the larger Leu side-chain make a significant contribution to binding. The criticality of the N-terminal tripeptide of the Cdc25C sequence was established by testing a truncated peptide (5714) as a prelude to the design of non-peptidic inhibitory compounds.
Having established novel SAR data for PBD inhibition, the REPLACE was applied to the critical N-terminal region. Three iterations of REPLACE enabled substitution of the Leu-Leu-Cys tripeptide with derivatized benzoic acids approximately 1/3 the size, which were within one log of the activity of the endogenous Cdc25C peptide and almost equivalent in potency to the truncated PBIP1 peptides. A clear structure-activity relationship was observed, therefore providing a scaffold for further optimization. Addition of H-bond acceptor groups to the 3rd generation scaffold in order to mimic the acetyl group in the PBIP peptides should result in increased potency. Future iterations of REPLACE will exploit SAR information suggesting that when synthesized in the C-terminally truncated contexts (i.e. NCap-S[pT]A), FLIP potencies will be similar.
Further to the development of PBD inhibitors as anti-tumor therapeutics, data obtained from experiments in cancer cells demonstrate that PBD FLIPs indeed directly interfere with the functions of PLK1. Specifically, treatment resulted in reduced PLK1 localization to centrosomes, aberrant mitoses as visualized by mono and multipolar spindles, abnormal chromosome alignment during metaphase, and apoptotic cell death. Consistent data from each cellular endpoint for the 3rd generation FLIPs suggests that despite weaker in vitro binding, the small-molecule-peptide hybrids have increased cellular activity relative to peptide inhibitors. Addition of the N-terminal capping group may stabilize these compounds against cellular proteolysis, therefore improving their drug-likeness. As a whole, this outcome validates the REPLACE strategy for conversion of peptides into more pharmaceutically appropriate molecules, in the context of the PLK1 polo-box domain.
It has been reported for ATP competitive PLK1 inhibitors that treated cells have spindle defects including monopolar spindles and improper attachment of the spindle to the kinetochores (14, 26). These defects result from the failure of PLK1 to localize to the centrosomes and kinetochores and to act on substrates such as BubR1, PBIP and CENP-E. While these defects are predominant with ATP competitive compounds and after depletion of PLK1 (25, 36), they have not been observed with the two small molecule PBD compounds identified to date nor with a dominant negative PBD construct expressed in cells (37). Although treatment with purpurogalin (24) (PPG) or poloxin (23) resulted in profound effects on chromosome condensation (at relatively high inhibitor concentrations), a bipolar spindle was found in contrast to the monopolar spindles induced using inhibitors of PLK1 catalytic activity. Indeed it has recently been shown that neither PPG nor Poloxin inhibit PLK1 catalytic activity in vitro (38). In the assays utilized in this study, PPG treatment induced very few aberrant mitoses occurred and little evidence of monopolar or multipolar spindles (data not shown). Results with peptidic and FLIP PBD inhibitors in this study clearly demonstrate a phenotype more consistent with blocking the enzymatic and localization functions of PLK1. As this phenotype was observed with both peptide and small molecule hybrid compounds, it is unlikely to be the result of an off target effect. These data imply that small molecule inhibitors such as PPG and Poloxin, while interesting biological probes, do not recapitulate a complete PLK1 phenotype and therefore may not possess adequate anti-tumor activity. In addition, this raises interesting mechanistic questions concerning PLK1 regulation in that some compounds can apparently block sub-cellular localization while not affecting substrate recruitment. Further implications of this observation require clarification since it is possible that blocking catalytic and sub-cellular localization of PLK1 might be required for sensitization of p53 deficient and KRas mutant tumors to PLK1 inhibition as has previously been described (8, 39). Although some computational studies have been carried out (38), crystal structures of PPG or poloxin in complex with the PBD have not yet been solved. It is unknown how these compounds compete off PBD peptides in binding assays yet not inhibit other functions of PLK1.
The results of this study demonstrate that fragment modifications to the Cdc25C PBD binding sequence through application of REPLACE result in non-ATP competitive inhibitors with significant effects in blocking PLK1 cellular functions, inducing mitotic defects and triggering apoptosis. These data were enabled through the use of a novel peptide transfection agent and indicate that FLIPs possess enhanced cellular stability and therefore potency relative to peptidic PBD inhibitors despite having similar or lower in vitro binding activity. Results also suggest that peptide-small molecule hybrid PBD inhibitors recapitulate a PLK1 phenotype not observed with other small molecule PBD binding compounds. Furthermore, compounds obtained using REPLACE retain PLK1 (versus PLK3) specificity, thus further demonstrating their suitability for anti-cancer drug development. Taken together, this study provides new validation for the PBD of PLK1 as an approach to generate non-ATP competitive anti-tumor therapeutics while presenting further confirmation of the REPLACE strategy for developing protein-protein interaction inhibitors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Douglas Pittman for helpful discussions, Dr Joshua Bolger for laboratory assistance and Dr. Preeti Rajesh for assistance with immunofluorescence experiments.
GRANT SUPPORT
This work was funded by an American Cancer Society Institutional Research Grant award (#IRG – 97-219-08) to the Hollings Cancer Center at the Medical University of South Carolina and by a South Carolina Translational Research Institute award (CTSA, NIH/NCRR Grant Numbers UL1RR029882 and UL1 TR000062), to C. McInnes.
Abbreviations
- PLK
polo-like kinase
- PBD
polo box domain
- FLIP
fragment ligated inhibitory peptide
- REPLACE
Replacement with partial ligand alternative through computational enrichment
- PLA
partial ligand alternative
- SAR
structure-activity relationship
Footnotes
The authors of this manuscript have no conflict of interest to disclose.
REFERENCES
- 1.Xie S, Xie B, Lee MY, Dai W. Regulation of cell cycle checkpoints by polo-like kinases. Oncogene. 2005;24:277–286. doi: 10.1038/sj.onc.1208218. [DOI] [PubMed] [Google Scholar]
- 2.McInnes C, Wyatt MD. PLK1 as an oncology target: current status and future potential. Drug Discov Today. 2011;16:619–625. doi: 10.1016/j.drudis.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Yuan J, Hoerlin A, Hock B, Stutte HJ, Ruebsamen-Waigmann H, Strebhardt K. Polo-like kinase, a novel marker for cellular proliferation. American Journal of Pathology. 1997;150:1165–1172. [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, et al. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14:543–549. doi: 10.1038/sj.onc.1200862. [DOI] [PubMed] [Google Scholar]
- 5.Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. International Journal of Oncology. 1999;15:687–692. doi: 10.3892/ijo.15.4.687. [DOI] [PubMed] [Google Scholar]
- 6.Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, et al. Polo-like kinase I is overexpressed in prostate cancer and linked to higher tumor grades. The Prostate. 2004;60:240–245. doi: 10.1002/pros.20050. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Science. 2003;94:148–152. doi: 10.1111/j.1349-7006.2003.tb01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr, Kinzler KW, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106:3964–3649. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, et al. p53-dependent repression of polo-like kinase-1 (PLK1) Cell cycle (Georgetown, Tex. 2010;9:4200–4212. doi: 10.4161/cc.9.20.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elez R, Piiper A, Kronenberger B, Kock M, Brendel M, Hermann E, et al. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene. 2003;22:69–80. doi: 10.1038/sj.onc.1206038. [DOI] [PubMed] [Google Scholar]
- 11.Spankuch-Schmitt B, Wolf G, Solbach C, Loibl S, Knecht R, Stegmuller M, et al. Downregulation of human polo-like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene. 2002;21:3162–3171. doi: 10.1038/sj.onc.1205412. [DOI] [PubMed] [Google Scholar]
- 12.Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, et al. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J Clin Oncol. 2008;26:5504–5510. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, et al. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2008;26:5511–5517. doi: 10.1200/JCO.2008.16.1547. [DOI] [PubMed] [Google Scholar]
- 14.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z, Waldman AS, Wyatt MD. DNA damage and homologous recombination signaling induced by thymidylate deprivation. Biochem Pharmacol. 2008;76:987–996. doi: 10.1016/j.bcp.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Xie S, Chen J, Fukasawa K, Naik U, Traganos F, et al. Cell cycle arrest and apoptosis induced by human Polo-like kinase 3 is mediated through perturbation of microtubule integrity. Molecular and cellular biology. 2002;22:3450–3459. doi: 10.1128/MCB.22.10.3450-3459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Bai J, Shen R, Brown SA, Komissarova E, Huang Y, et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008;68:4077–4085. doi: 10.1158/0008-5472.CAN-07-6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu D, Yao Y, Jiang X, Lu L, Dai W. Regulation of PTEN stability and activity by PLK3. J Biol Chem. doi: 10.1074/jbc.M110.166462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nature structural & molecular biology. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 21.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, et al. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Alvarez B, de Carcer G, Ibanez S, Bragado-Nilsson E, Montoya G. Molecular and structural basis of polo-like kinase 1 substrate recognition: Implications in centrosomal localization. Proc Natl Acad Sci U S A. 2007;104:3107–3112. doi: 10.1073/pnas.0609131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reindl W, Yuan J, Kramer A, Strebhardt K, Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chemistry & biology. 2008;15:459–466. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe N, Sekine T, Takagi M, Iwasaki J, Imamoto N, Kawasaki H, et al. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem. 2009;284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 25.van Vugt MATM, van de Weerdt BCM, Vader G, Janssen H, Calafat J, Klompmaker R, et al. Polo-like Kinase-1 Is Required for Bipolar Spindle Formation but Is Dispensable for Anaphase Promoting Complex/Cdc20 Activation and Initiation of Cytokinesis. J Biol Chem. 2004;279:36841–36854. doi: 10.1074/jbc.M313681200. [DOI] [PubMed] [Google Scholar]
- 26.Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 27.Liu F, Park JE, Qian WJ, Lim D, Scharow A, Berg T, et al. Identification of High Affinity Polo-like Kinase 1 (Plk1) Polo-box Domain Binding Peptides Using Oxime-Based Diversification. ACS Chem Biol. 2012 doi: 10.1021/cb200469a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu F, Park JE, Qian WJ, Lim D, Graber M, Berg T, et al. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nature chemical biology. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Huang Y, Xiao N, Murray V, Chen J, Wang J. Real time investigation of protein folding, structure, and dynamics in living cells. Methods Cell Biol. 2008;90:287–325. doi: 10.1016/S0091-679X(08)00814-5. [DOI] [PubMed] [Google Scholar]
- 30.Jang Y-J, Lin C-Y, Ma S, Erikson RL. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proceedings of the National Academy of Sciences of the USA. 2002;99:1984–1989. doi: 10.1073/pnas.042689299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lowery DM, Lim D, Yaffe MB. Structure and function of Polo-like kinases. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 32.Andrews MJ, Kontopidis G, McInnes C, Plater A, Innes L, Cowan A, et al. REPLACE: a strategy for iterative design of cyclin-binding groove inhibitors. Chembiochem. 2006;7:1909–1915. doi: 10.1002/cbic.200600189. [DOI] [PubMed] [Google Scholar]
- 33.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature reviews. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 34.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 35.Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 36.Qian Y-W, Erikson E, Li C, Maller JL. Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis. Molecular and Cellular Biology. 1998;18:4262–4271. doi: 10.1128/mcb.18.7.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seong YS, Kamijo K, Lee JS, Fernandez E, Kuriyama R, Miki T, et al. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem. 2002;277:32282–32293. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- 38.Liao C, Park JE, Bang JK, Nicklaus MC, Lee KS. Exploring Potential Binding Modes of Small Drug-like Molecules to the Polo-Box Domain of Human Polo-like Kinase 1. ACS medicinal chemistry letters. 2010;1:110–114. doi: 10.1021/ml100020e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.