

Abstract
Synaptic scaling is a form of homeostatic synaptic plasticity characterized by cell-wide changes in synaptic strength in response to changes in overall levels of neuronal activity. Here we report that bicuculline-induced increase in neuronal activity leads to a decrease in mEPSC amplitude and a decrease in expression of the AMPA receptor subunit GluR2 in rat hippocampal cultures. Bicuculline treatment also leads to an increase in the levels of the transcriptional repressor MeCP2, which binds to the GluR2 promoter along with the corepressors HDAC1 and mSin3A. Downregulation of MeCP2 by shRNA expression or genetic deletion blocks the bicuculline-induced decrease in GluR2 expression and mEPSC amplitude. These observations indicate that MeCP2 mediates activity-dependent synaptic scaling, and suggest that the pathophysiology of Rett syndrome, which is caused by mutations in MeCP2, may involve defects in activity-dependent regulation of synaptic currents.
Introduction
Synaptic scaling is a form of plasticity in which neuronal activity drives cell-wide changes in synaptic strength (Turrigiano et al., 1998; Davis, 2006; Turrigiano, 2007, 2008). In general, an increase in neuronal activity leads to a decrease in quantal amplitude and a decrease in activity leads to an increase in amplitude. It is believed that such a homeostatic regulation of synaptic strength is important for maintaining action potential firing rates in a range suitable for information processing (Davis, 2006; Turrigiano, 2007, 2008).
There is considerable evidence that changes in synaptic strength associated with synaptic scaling are mediated by changes in the levels of AMPA receptors (O'Brien et al., 1998; Turrigiano et al., 1998; Shepherd et al., 2006; Gainey et al., 2009). Most AMPA receptors in the brain consist of GluR1/GluR2 or GluR2/GluR3 heterodimers and scaling up of synaptic currents following silencing of neurons is associated with an increase in synaptic GluR1 and GluR2 receptors (O'Brien et al., 1998; Thiagarajan et al., 2005; Turrigiano, 2008; Gainey et al., 2009). More recently, it has also been shown that GluR2 is required for photostimulation-dependent homeostatic decreases in synaptic strength (Goold and Nicoll, 2010).
The molecular mechanisms by which chronic changes in neuronal activity lead to scaling of synaptic currents are not well understood. In contrast to rapid forms of synapse modification, such as long-term potentiation and long-term depression, changes in synaptic strength associated with synaptic scaling are detected several hours to days after a change in network activity and require transcription (Ibata et al., 2008). Studies investigating synaptic scaling have implicated BDNF, TNFα, β3 integrin, MHC I, CaMKIV, and Arc in scaling up of synaptic currents following silencing of neurons (Turrigiano, 2008). The mechanisms underlying scaling down of synaptic currents in response to elevated activity have not been as extensively studied, but a recent study suggests a role for polo-like kinase 2 (Plk2) in this process (Seeburg et al., 2008). Plk2 is an activity-induced gene and appears to act by regulating the degradation of SPAR, a protein implicated in regulating spine morphology and synaptic function.
Given the observation that synaptic scaling is associated with a change in levels of AMPA receptors, we decided to explore activity-dependent mechanisms that might regulate overall AMPA receptor levels in response to elevated activity. Here we report that the Rett syndrome protein MeCP2 plays a critical role in mediating activity-dependent changes in GluR2 levels and is required for synaptic scaling.
Materials and Methods
Plasmids.
Lentiviral vectors were obtained from Dr. I. Verma's laboratory at the Salk Institute. The LEMPRA strategy for lentiviral shRNA delivery and rescue that we used was described by Zhou et al. (2006). We generated an HA-MeCP2 shRNA-resistant construct for MeCP2 rescue expression. Short hairpin sequence- and rescue shRNA-resistant construct sequences were described by Zhou et al. (2006). The hUbiquitin promoter was taken from the FUGW construct (Addgene).
Molecular biology reagents.
Molecular biology reagents, competent cells, real-time PCR mix and reverse transcription kits were from BioPioneer, Inc.
Antibodies.
Antibodies used were the following: MeCP2 rabbit polyclonal antibody (Abcam, Millipore, Qiu laboratory); HDAC1, mSin3A, ERK1 and control rabbit IgG (Santa Cruz Biotechnology); GluR2 MAB397 (Millipore Bioscience Research Reagents); GFP (Abcam); MeCP2S421 (Abgent AP3693a).
Mice.
Mecp2 conditional knock-out mice made in Rudolf Jaenisch's laboratory were used for dissociated neuronal cultures (Chen et al., 2001). Mecp2 knock-out mice generated in Adrian Bird's laboratory were used for hippocampal slice culture (Guy et al., 2001).
Cell culture.
For biochemical experiments, embryonic day 18 (E18) rat cortical cells were cultured in 12-well plates as previously described (Song and Ghosh, 2004). Cultures were stimulated at 14 DIV with 50 μm bicuculline (in DMSO) to increase synaptic activity. The cultures were pretreated with 10 μm nifedipine for 1 h to selectively drive NMDA receptor-dependent calcium influx. For activity-dependent changes in gene expression, E18 cortical cultures at 16 DIV were stimulated for 6 h with bicuculline and changes in gene expression were assessed using an Affimetrix rat 230A microarray chip. For electrophysiology experiments, postnatal day 0 (P0)–P1 rat hippocampal cells were cultured on Thermanox coverslips (Nunc) in 12 well plates, electroporated at plating or infected with lentivirus at 7/8 DIV with control shRNA or MeCP2 shRNA, stimulated at 12 DIV with 50 μm bicuculline (in DMSO), and recorded at 14 DIV. For immunohistochemistry, P0–P1 mouse hippocampal neurons were electroporated following dissociation and grown on a glial bed cultured from rat cortex on glass coverslips in 12 well plates, then stimulated at 12 DIV with 50 μm bicuculline (in DMSO), and fixed at 14 DIV.
Chromatin immunoprecipitation.
ChIP assays were performed as originally described with minor modifications (Song and Ghosh, 2004). Primers: rat GluR2 forward (F): GCCCAGAGCTCCGACTAAAG, reverse (R): GGCAGTCTCGGGCGCGCGTG, rat GluR1 F: GAGGAGAGAGGCTGCCTGCT, R: CCCTCCCCTCCCTTCGATTC, mouse MeCP2 F: CGCGCGCAACCGATGCCGGGACC, MeCP2 R: CCGCCTCTTTTCCCTGCCTAAACA.
Real-time quantitative reverse transcriptase PCR.
Total RNA was collected from cell lysates for reverse transcription. A first strand cDNA synthesis kit from BioPioneer was used for cDNA synthesis. Primers for rat GluR2 cDNA were F: GAAGCCTTGTGACACCATGA, R: AGCCTTGCCTTGCTCCTCAT. Mouse and rat MeCP2 cDNA were F: ATGGTAGCTGGGATGTTAGGG, R: TGAGCTTTCTGATGTTTCTGCTT. For real-time PCR experiments, SYBR Green PCR Master Mix from Applied Biosystems and 2XqPCR master mix from Biopioneerinc.com were used. An ABI 7000 Q-PCR machine was used to perform the experiments. Datasets are from at least two independent experiments, each of which had triplicate samples. Paired t tests were performed using GraphPad InStat version 3.0a for Macintosh.
Electrophysiology.
Electrophysiological recordings were performed on neurons from hippocampal cell cultures (P0–P1 + 14 DIV), perfused at room temperature in a bicarbonate buffered recording solution containing the following (in mm): 124 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1.5 MgCl2, 2 CaCl2, and 10 glucose and bubbled constantly with 95% O2/5% CO2. Voltage-clamp recordings were made using glass microelectrodes filled with a cesium-substituted intracellular solution containing (in mm) 10 CsCl, 105 CsMeSO3, 8 NaCl, 0.5 ATP, 0.3 GTP, 10 HEPES, 2 MgCl2, and 1 EGTA, pH 7.3. Drug concentrations were (in μm): 0.5 TTX, 50 d-APV, 20 Gabazine. Pipette resistances ranged from 4 to 6 MΩ. Series resistances ranged from ∼8 to 20 MΩ and were monitored for consistency during recording using a −5 mV test pulse. Whole-cell voltage-clamp recordings at −70 mV were made using a Multiclamp 700B amplifier (Molecular Devices). Current signals were filtered at 2 kHz and digitized at 10 kHz with a Digidata 1322A. Gabazine (SR 95531 hydrobromide), d-APV, and TTX were added at the time of recording. mEPSCs were identified and isolated using ClampFit software. Using a control culture, a template was created from a composite average of manually identified mEPSCs and was used with a broad threshold for deviation to ensure all events in subsequent recordings were captured. These events were then manually inspected to discard any non-mEPSC traces.
Immunohistochemistry.
Mouse hippocampal neurons were treated with 50 μm bicuculline or DMSO vehicle at 12 DIV and live labeled with a monoclonal GluR2 antibody (1:200) at 14 DIV. For live labeling, the antibody was added to the culture medium and incubated for 20 min, followed by fixation in 4% paraformaldehyde in 4% sucrose for 5 min. Cultures were then blocked in 5% BSA and appropriate Alexa-secondary antibody (1:1000) in blocking solution was added for 1 h before cell permeabilization in 0.15% Triton X-100. Primary antibodies to GFP, Cre, vGlut or other synaptic markers were then added in blocking buffer for 1 h, followed by incubation in Alexa-secondary antibody (Invitrogen) for 1 h. Images were obtained on a Leica SP-5 scanning confocal microscope under a 63× oil-immersion lens and immunofluorescent puncta were quantified using ImageJ software (NIH).
Results
We performed whole-cell voltage-clamp recordings from P0–P1 hippocampal cultures to examine the effect of a bicuculline-induced increase in neuronal activity on scaling of synaptic currents. As shown in Figure 1A, spontaneous mEPSCs are readily detected in hippocampal cultures at 14 DIV. As previously described (Turrigiano et al., 1998), 48 h bicuculline treatment led to a decrease of ∼20% in average mEPSC amplitudes (Fig. 1A–C). Intrinsic membrane properties and recording parameters were monitored, and control and bicuculline-treated neurons showed no difference in membrane capacitance, input resistance, series resistance, and baseline noise.
Figure 1.
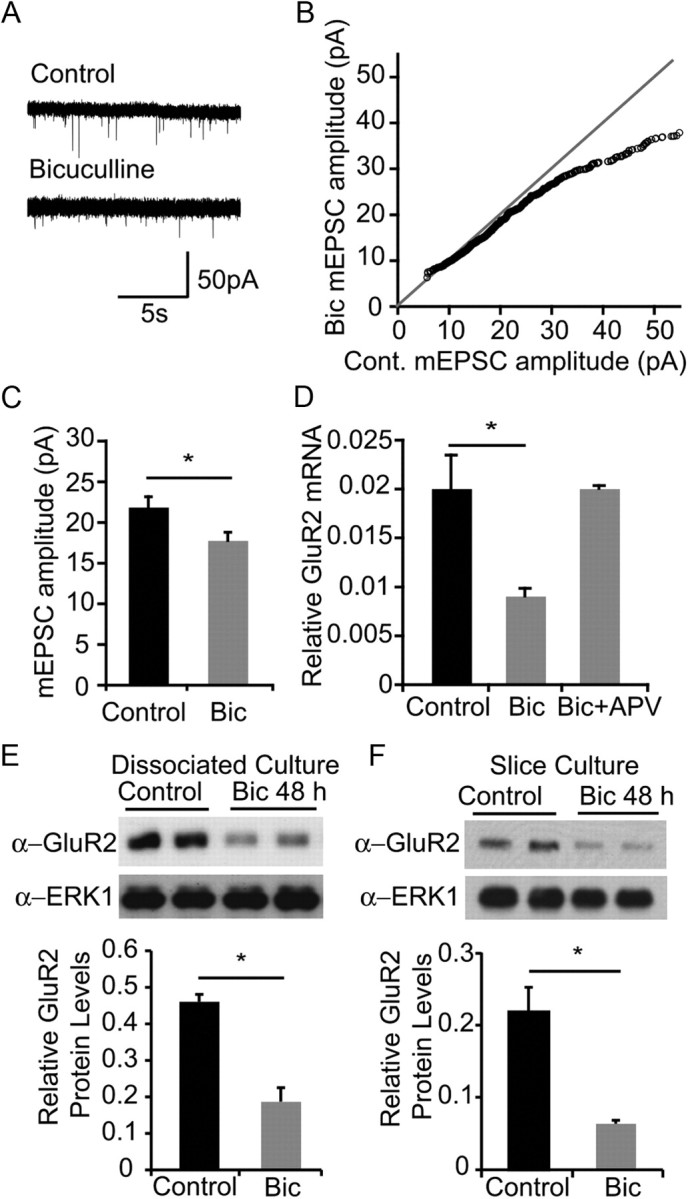
Synaptic scaling is accompanied by downregulation of GluR2 mRNA and protein. A, Representative 15 s mEPSC recordings from hippocampal neurons at P0 + 14 DIV cultured under control conditions or treated with 50 μm bicuculline for 48 h before recording. B, Amplitudes of mEPSC from control cultures or cultures treated with 50 μm bicuculline for 48 h before recording. mEPSCs for each treatment were sorted by amplitude and the resulting distributions were plotted against each other. n = 24 (control), 23 (bicuculline) cells. mEPSCs <55 pA are shown. C, Quantification of average mEPSC amplitude (mean ± SEM). n = 24 (control), 23 (bicuculline). p < 0.05, t test. D, Assessment of GluR2 mRNA levels in rat E18 cortical cultures at 18 DIV, grown under control conditions, or treated with bicuculline for 6 h before cell lysis. Cultures were pretreated with APV for 1 h where indicated. Neurons were lysed for collecting total RNA, and reverse-transcribed for real-time PCR analysis with GluR2-specific primers. E, Assessment of GluR2 protein levels in dissociated neuronal culture after bicuculline treatment. Rat P0 hippocampal neurons were cultured for 14 DIV and stimulated with bicuculline for 48 h. Neurons were lysed, analyzed by SDS-PAGE, and blotted with antibody indicated. F, Assessment of GluR2 protein levels in hippocampal slice culture after bicuculline treatment. Hippocampal slices from rat P7 pups were cultured for 3 DIV and stimulated with bicuculline for 48 h. Neurons were lysed, analyzed by SDS-PAGE, and blotted with the antibody indicated.
The reduction in mEPSC amplitudes is consistent with the report that bicuculline treatment leads to a reduction in postsynaptic response to glutamate (Turrigiano et al., 1998). To determine whether this is accompanied by a reduction in the abundance of AMPA receptors, we performed real-time PCR and Western blot analysis to determine whether bicuculline treatment affects the expression of AMPA receptor subunits. Treatment of cultures with bicuculline for 6 h led to a decrease in GluR2 mRNA levels (Fig. 1D), but not GluR1 mRNA levels (data not shown), indicating that elevated activity leads to a decrease in GluR2 gene expression (O'Brien et al., 1998; Grooms et al., 2006). GluR2 protein levels in both dissociated and slice cultures were also significantly reduced following bicuculline treatment (Fig. 1E,F). The decrease in GluR2 mRNA levels was blocked by the NMDA receptor antagonist APV (Fig. 1D). The inclusion of APV did not grossly affect the stimulation intensity since the synaptic activation in response to bicuculline in control experiments, as measured by charge transfer, did not differ between control and APV-treated cultures (data not shown). These observations suggest that activity-dependent scaling of synaptic currents involves NMDA receptor-dependent downregulation of GluR2 expression.
To explore the possibility that the reduction in GluR2 expression in response to elevated activity is mediated by a transcriptional repressor, we performed a microarray screen for repressor genes that are induced by bicuculline stimulation (data not shown). One gene that was strongly induced by bicuculline stimulation was the methyl CpG-binding protein MeCP2 (Fig. 2A). MeCP2 is a transcriptional repressor that has been extensively studied in the context of Rett syndrome, in which the gene is mutated (Meehan et al., 1992; Amir et al., 1999; Chahrour and Zoghbi, 2007). Given the potential clinical significance of a role for MeCP2 in regulating the response to a global increase in cellular activity, we were motivated to examine whether MeCP2 was involved in regulating GluR2 levels. In support of such a possibility, the GluR2 promoter has been reported to be methylated (Myers et al., 1998), which could facilitate recruitment of MeCP2 (Skene et al., 2010). To determine whether MeCP2 was associated with the GluR2 promoter, we performed chromatin immunoprecipitation (ChIP) experiments on lysates from cortical cultures. Analysis by quantitative PCR indicated that MeCP2 is bound to the GluR2 promoter (Fig. 2B). Additional ChIP experiments revealed that the transcriptional corepressors HDAC1 and mSin3A also bind to the GluR2 promoter, suggesting that GluR2 expression may be regulated by a repressor complex (Fig. 2C). To determine whether occupancy of the GluR2 promoter by MeCP2 was regulated by activity, we performed ChIP experiments from control and bicuculline stimulated cultures and found that bicuculline led to an increase in the binding of MeCP2 to the GluR2 promoter (Fig. 2D), but did not affect occupancy of mSin3a or HDAC to the GluR2 promoter (data not shown). It has been shown that phosphorylation of MeCP2 can be important for transcriptional repression (Zhou et al., 2006). To determine whether the phosphorylation status of MeCP2 changes with activity, we used an antibody specific to MeCP2 protein that has been phosphorylated at Serine 421 (MeCP2S421). We found that cultures stimulated with bicuculline for 30 min show an increase in phosphorylated MeCP2 at S421 (Fig. 2E).
Figure 2.
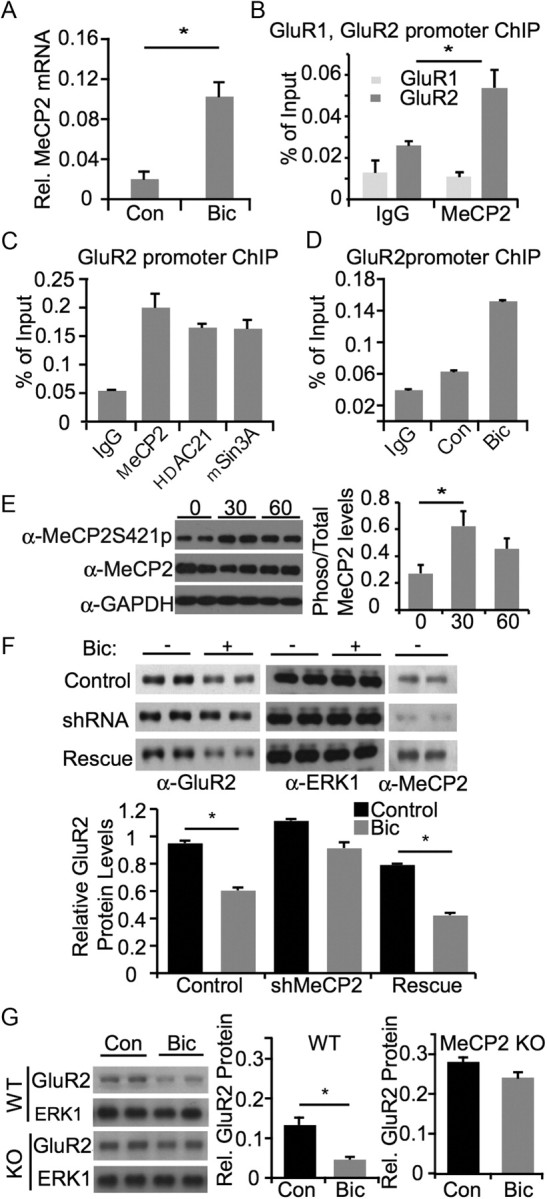
MeCP2 is required for activity-dependent downregulation of GluR2 expression. A, Assessment of MeCP2 mRNA after bicuculline stimulation. Rat E18 cortical neurons were cultured for 16 DIV and stimulated with bicuculline for 6 h. Neurons were lysed for collecting total RNA and reverse-transcribed for real-time PCR analysis with MeCP2-specific primers. B, MeCP2 chromatin immunoprecipitation from E18 + 7 DIV rat cortical cultures. After immunoprecipitation with anti-MeCP2 antibody, PCRs with endogenous GluR1 and GluR2 promoter primers were used to amplify promoter-specific segments. Real-time quantitative PCR was performed and signals were normalized as percentage of input to determine promoter occupancy. C, MeCP2, HDAC1 and mSin3A chromatin immunoprecipitation from rat cortical neurons under basal conditions. After immunoprecipitation with anti-MeCP2, anti-HDAC1 and anti-mSin3a antibody, PCRs with endogenous GluR2 promoter primers were used to amplify GluR2 promoter-specific segments. Real-time quantitative PCR was performed and signals were normalized as percentage of input to determine promoter occupancy. D, Relative binding of MeCP2 to the GluR2 promoter in unstimulated and bicuculline-stimulated neurons examined using chromatin immunoprecipitation. E, Left, Western blot from hippocampal cultures after exposure to bicuculline for 0, 30, or 60 min. Phosphorylated MeCP2 is detected using a Serine 412 phospho-specific antibody. Right, quantification of ratio of total MeCP2 to MeCP2 phosphorylated at Serine 421 at 0, 30, or 60 min bicuculline exposure. Asterisk signifies p < 0.05 by ANOVA. F, Assessment of changes in GluR2 expression following bicuculline stimulation, with and without MeCP2. Rat P0/P1 hippocampal neurons were cultured for 8 DIV and infected with lentivirus containing control shRNA, MeCP2 shRNA, or MeCP2 shRNA with an shRNA-resistant MeCP2 construct (rescue). Cultures were stimulated with bicuculline for 48 h where indicated, and lysed for Western blot analysis with specified antibodies. Asterisk indicates p < 0.05 using t test. G, Assessment of changes in GluR2 protein levels following bicuculline stimulation in wild-type and MeCP2 knock-out neurons. P7 hippocampal slices were prepared from wild-type and MeCP2 knock-out mouse and cultured for 3 d. After stimulation with bicuculline for 48 h, neurons were lysed for Western blot analysis with antibodies as indicated.
We next wanted to determine whether MeCP2 was required for activity-dependent downregulation of GluR2 expression. For these experiments we generated a short hairpin RNA against MeCP2 and cloned it into a lentiviral expression vector. As shown in Figure 2F, this shRNA was very effective in downregulating MeCP2 expression in hippocampal cultures. To test the role of MeCP2 in the bicuculline-induced decrease in GluR2 expression, we examined GluR2 levels in control and MeCP2 shRNA-expressing cultures. As described above for uninfected cultures, bicuculline stimulation of cultures infected with a control shRNA led to a decrease in GluR2 expression (Fig. 2F). This decrease was blocked in cultures expressing the MeCP2 shRNA lentivirus, suggesting that MeCP2 was required for activity-dependent downregulation of MeCP2 (Fig. 2F). In support of the specificity of shRNA experiment, we found that expression of an shRNA-resistant same-sense mutant of MeCP2 rescued the activity-dependent downregulation of GluR2 (Fig. 2F).
To determine whether genetic deletion of MeCP2 also affected activity-dependent downregulation of GluR2 expression, we cultured hippocampal slices from wild-type and Mecp2-null mice and compared the effects of bicuculline stimulation on GluR2 expression. As in the case of rat hippocampal cultures, bicuculline treatment led to a decrease in GluR2 protein levels in wild-type mouse cultures (Fig. 2G). In contrast, bicuculline treatment did not lead to a downregulation of GluR2 levels in Mecp2-null cultures, indicating that endogenous MeCP2 is required for activity-dependent downregulation of GluR2 levels (Fig. 2G). In a separate set of experiments, we directly examined basal GluR2 levels in brain extracts from WT and Mecp2-null animals. Although there was a trend toward reduction in the basal levels of GluR2 in KO tissue, this was not statistically significant (relative GluR2 levels for WT = 0.98 ± 0.27; KO = 0.56 ± 0.19). It may be that additional mechanisms are responsible for setting basal GluR2 levels, but MeCP2 is particularly important for activity-dependent regulation of GluR2 expression.
To further investigate whether cell-wide changes in GluR2 levels were reflected in reduced surface expression of GluR2, we used immunofluorescence to visualize surface GluR2 expression after bicuculline treatment. Hippocampal cultures from MeCP2 floxed mice were transfected at plating with GFP or GFP plus Cre and were stimulated with bicuculline at 12 DIV. At 14 DIV, neurons were live labeled for GluR2 and then fixed and immunostained for GFP and GluR2. We quantified the size of GluR2 puncta on dendrites of control GFP or Mecp2-null neurons. Control neurons show a decrease in GluR2 puncta size following bicuculline stimulation, indicating that the increase in activity triggers the loss of surface GluR2. However, Cre-mediated deletion of Mecp2 in transfected neurons abolished the bicuculline-induced decrease in GluR2 surface expression, suggesting that MeCP2 is necessary for activity-dependent changes in surface GluR2 (Fig. 3A,B).
Figure 3.

MeCP2 is required for synaptic scaling in response to elevated network activity. A, Representative images of dendrites from hippocampal cultures transfected with GFP alone or with GFP + Cre at time of plating then exposed to either vehicle or 50 μm bicuculline for 48 h before live labeling with GluR2 antibodies and subsequent fixation at DIV 14. B, Pooled data of homeostatic changes in surface labeling of the GluR2 subunit measured by puncta size; 10–15 cells per condition. Error bars show SEM. Asterisk indicates p < 0.05 using Kruskal–Wallis nonparametric test. C, Representative 15 s mEPSC recordings from hippocampal neurons at 14 DIV, infected with GFP (control), MeCP2-shRNA, or MeCP2 shRNA plus rescue at 7 DIV. Cultures were treated with vehicle or 50 μm bicuculline for 48 h before recording. D, Distributions of mEPSCs from neurons infected with GFP alone, MeCP2-shRNA, or MeCP2 shRNA + rescue, each grown under control conditions, or treated with bicuculline for 48 h before recordings. For each infection condition, control mEPSCs (x-axis) are sorted by amplitude and plotted against sorted bicuculline mEPSCs (y-axis). mEPSCs <55 pA are shown. The GFP control is the same data as shown in Figure 1B,C and is shown here for ease of comparison. E, Quantification of average mEPSC amplitude (mean ± SEM) under conditions indicated. n = 24, 23, 10, 8, 19, 16 cells. Asterisks indicate p < 0.05, t test. F, Representative 15 s mEPSC recordings from 14 DIV hippocampal neurons from floxed Mecp2 mice electroporated with GFP (control) or Cre. Cultures were treated with 50 μm bicuculline for 48 h before recording. G, Distributions of mEPSCs from floxed Mecp2 neurons electroporated with GFP alone or with GFP + Cre, grown under control conditions, or treated with bicuculline for 48 h before recordings. For each transfection condition, control mEPSCs (x-axis) are sorted by amplitude and plotted against sorted bicuculline mEPSCs (y-axis). mEPSCs <50 pA are shown. H, Quantification of average mEPSC amplitude (mean ± SEM) under conditions indicated. n = 8, 5, 6, and 7 cells. p < 0.05, t test.
To determine whether MeCP2 was required for activity-dependent synaptic scaling, we measured mEPSC amplitude in hippocampal cultures in which MeCP2 levels were downregulated by shRNA expression or gene deletion. In the first set of experiments, we infected hippocampal cultures with lentiviruses encoding GFP or GFP with shRNA targeting MeCP2 at 5 DIV, stimulated with bicuculline at 12 DIV, and recorded from GFP-positive neurons at 14 DIV. GFP-infected neurons showed a decrease in mEPSC amplitude following bicuculline treatment (Fig. 3C–E). Strikingly, this bicuculline-induced scaling of mEPSC amplitudes was absent in neurons expressing MeCP2 shRNA (Fig. 3C–E). This was not due to a failure in adequately increasing activity in knockdown cells, as the bicuculline-induced increase in charge transfer was similar in control and MeCP2 shRNA-expressing cells. Expression of an shRNA-insensitive rescue construct fully restored bicuculline-induced scaling, indicating that activity-dependent scaling down of synaptic currents requires MeCP2 (Fig. 3C–E).
In a complementary set of experiments we asked whether genetic deletion of MeCP2 would also prevent synaptic scaling. For these experiments, we cultured neurons from conditional Mecp2 mutant mice at P0 (Chen et al., 2001). The cultures were electroporated with either GFP alone or GFP and a Cre-expression construct at the time of plating, and the cultures were treated with bicuculline at 13 DIV for 48 h. In contrast to control neurons, which showed bicuculline-induced scaling of mEPSC amplitudes similar to that seen in rat hippocampal cultures, there was no synaptic scaling in Cre-expressing neurons (Fig. 3F–H). In contrast to the shRNA experiments in rat cultures, there was also a decrease in mEPSC amplitudes under control conditions in the mouse cultures (Fig. 3H). This may reflect a more complete loss of MeCP2 function in the mouse cultures where MeCP2 is genetically deleted. These data from the conditional deletion of MeCP2 indicate that endogenous MeCP2 is required for bicuculline-induced synaptic scaling.
Discussion
The observations reported here reveal a role for MeCP2 in activity-dependent scaling of synaptic inputs. We find that increased activity leads to induction of MeCP2 expression. MeCP2 in turn binds to the GluR2 promoter and recruits a repressor complex to inhibit GluR2 expression. Loss of MeCP2 by shRNA expression or genetic deletion blocks the activity-dependent decrease in GluR2 expression and synaptic scaling. Thus MeCP2 plays a critical role in mediating a key adaptive response that allows neurons to adjust synaptic strength following changes in neuronal activity.
The role of MeCP2 in synaptic scaling adds to recent evidence implicating MeCP2 in regulating various aspects of synaptic connectivity and function. While several studies have examined the role of MeCP2 in basal synaptic transmission (Dani et al., 2005; Chao et al., 2007), this is the first study that implicates MeCP2 in activity-dependent scaling of synaptic currents. The fact that MeCP2 regulates GluR2 expression is important since virtually all AMPA receptor complexes in the brain include the GluR2 subunit (Lu et al., 2009) and regulation of GluR2 expression has a direct effect on synaptic AMPA receptor subunit composition (Shi et al., 2001). We also find that increased activity leads to upregulation of MeCP2 levels and increased occupancy of the GluR2 promoter. Although we do not observe activity- or MeCP2-dependent regulation of GluR1 gene expression, in cortical neurons we have found that activity-dependent changes in surface GluR1 level required MeCP2 (DL and AG, unpublished observations). Thus, MeCP2 might regulate AMPA receptor levels by both transcriptional and post-transcriptional mechanisms.
While this study focuses on regulation of AMPA receptors by MeCP2, it is likely that this mechanism works in concert with other mechanisms that mediate activity-dependent changes in synaptic strength. Importantly, a previous study examining transcriptional control of synaptic scaling in response to elevated activity found that activity-dependent induction of Plk2 leads to degradation of SPAR and synaptic weakening (Seeburg et al., 2008). Also, activity regulates the expression of Arc, which has been implicated in AMPA receptor internalization (Shepherd et al., 2006). Thus increased activity might engage multiple pathways to regulate AMPA receptor expression and trafficking to regulate synaptic currents.
The identification of MeCP2 as a key regulator of synaptic scaling is noteworthy in terms of our understanding of Rett syndrome (RTT). RTT is a childhood neurological disorder caused by mutation in the MECP2 gene (Amir et al., 1999; Chahrour and Zoghbi, 2007). The relationship between MECP2 mutation and loss of cognitive and motor function in RTT patients is not well understood, but the associated mental retardation as well as the relatively subtle neuropathology has focused attention on the potential role of MeCP2 in the development of functional neural circuits. Regulating synaptic strength during the process of circuit formation may be particularly important to prevent uncontrolled recurrent excitation. Based on our observations we suggest that MeCP2 may play a key role in this process by mediating activity-dependent regulation of synaptic strength. Loss of this pathway due to mutations in MECP2 may lead to a pathophysiological increase in neuronal excitability, resulting in aberrant network activity and seizures, which are common in RTT patients.
Footnotes
This work was supported by NIH Grant MH068578 (A.G.) and Rett Syndrome Research Foundation/International Rett Syndrome Foundation postdoctoral fellowship (Z.Q.). Z.Q. and X.-Y.L. are also supported by the Hundred Talents Program from the Chinese Academy of Sciences, China. We thank Dr. Inder Verma for lentiviral vectors and Drs. Roberto Malinow and Charles Stevens for comments on the manuscript.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29:6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grooms SY, Noh KM, Regis R, Bassell GJ, Bryan MK, Carroll RC, Zukin RS. Activity bidirectionally regulates AMPA receptor mRNA abundance in dendrites of hippocampal neurons. J Neurosci. 2006;26:8339–8351. doi: 10.1523/JNEUROSCI.0472-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SJ, Peters J, Huang Y, Comer MB, Barthel F, Dingledine R. Transcriptional regulation of the GluR2 gene: neural-specific expression, multiple promoters, and regulatory elements. J Neurosci. 1998;18:6723–6739. doi: 10.1523/JNEUROSCI.18-17-06723.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MR, Ghosh A. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci. 2004;7:229–235. doi: 10.1038/nn1192. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]