

Abstract
Dysfunction of basal forebrain cholinergic neurons (BFCNs) and γ-aminobutyric acid (GABA) interneurons, derived from medial ganglionic eminence (MGE), is implicated in disorders of learning and memory. Here we present a method for differentiating human embryonic stem cells (hESCs) to a nearly uniform population of NKX2.1+ MGE-like progenitor cells. After transplantation into the hippocampus of mice in which BFCNs and some GABA neurons in the medial septum had been destroyed by mu P75-saporin, human MGE-like progenitors, but not ventral spinal progenitors, produced BFCNs that synaptically connected with endogenous neurons, whereas both progenitors generated similar populations of GABA neurons. Mice transplanted with MGE-like but not spinal progenitors showed improvements in learning and memory deficits. These results suggest that progeny of the MGE-like progenitors, particularly BFCNs, contributed to learning and memory. Our findings support the prospect of using human stem cell–derived MGE-like progenitors in developing therapies for neurological disorders of learning and memory.
During development, basal forebrain neurons, including BFCNs and GABA interneurons, originate in the MGE and preoptic area (POa) in the most ventral part of the telencephalon1. The MGE and POa form from the developing neural tube in response to high concentrations of sonic hedgehog (SHH)2,3; cells in these areas express the ventral forebrain homeodomain transcription factor NKX2.1 (refs. 1,4,5). During neurogenesis, NKX2.1+ progenitors differentiate to BFCNs and GABA interneurons; the mechanism that determines the choice between these two cell fates has not yet been elucidated. BFCNs mainly project to the cerebral cortex and hippocampus6 and are associated with learning, memory and spatial recognition7,8. GABA interneurons migrate into the cortex and hippocampus and may also be involved in learning and memory9,10. Hence, degeneration or dysfunction of MGE progeny is often associated with learning and memory disorders. The availability of large quantities of human MGE and POa progenitors would aid the study of the ontogeny and degeneration of BFCNs and GABA interneurons and may facilitate the development of cell therapies for diseases that affect learning and memory.
In principle, MGE and POa progenitors could be generated from hESCs or human induced pluripotent stem cells (iPSCs)11,12. We have identified a primitive neuroepithelial cell that arises during hESC differentiation13,14; in response to specific sets of morphogens, this cell generates progenitors of cerebral glutamatergic neurons15,16, striatal GABA neurons17, midbrain dopaminergic neurons18–21 and spinal motoneurons13,22. Neurons produced in this manner have shown promise in improving behavioral deficits in animal disease models, including a rat model of Parkinson’s disease treated with dopamine neurons23–26 and a mouse model of Huntington’s disease treated with GABA neurons17. Here we demonstrate the conversion of hESCs without cell sorting to a nearly homogeneous population of NKX2.1+ MGE-like progenitors, achieved by patterning the primitive neuroepithelia with high concentrations of SHH. After transplantation into mice with a medial septum lesion, the MGE-like progenitors produced GABA interneurons and BFCNs, which formed synapses with host hippocampal neurons and corrected learning and memory deficits.
RESULTS
MGE progenitors are efficiently induced from hESCs by SHH
hESCs can be differentiated in vitro by around day 8–15 to primitive neuroepithelial cells14, which predominantly generate cerebral neurons in the absence of exogenous morphogens15. We hypothesized that treatment with SHH, a ventralizing morphogen, would pattern the primitive neuroepithelia to MGE progenitors that would subsequently give rise to BFCNs and GABA interneurons. First, we differentiated hESCs (lines H9 and H1) to PAX6+ neuroepithelial cells in a chemically defined medium, as previously described13,27. Application of SHH for a week starting on either day 10, 13 or 17 after hESC differentiation (Supplementary Fig. 1a) indicated that treatment beginning on day 10 resulted in the most efficient induction of NKX2.1+ ventral telencephalic progenitors at day 25 (Supplementary Fig. 1b).
We next determined the most effective dosage of SHH by applying 0 ng ml−1, 100 ng ml−1, 200 ng ml−1, 500 ng ml−1 or 1,000 ng ml−1 SHH to the cultures at day 10 and assessing gene expression using qRT-PCR at day 17 and immunocytochemistry at day 25 (Fig. 1a). NKX2-1 mRNA levels (n = 3 cultures, two from different passages of H9 and one from H1) increased in response to SHH in a dose-dependent manner (Fig. 1b). Expression of ISL1, OLIG2 and ASCL1, all of which are expressed in both lateral ganglionic eminence (LGE) and MGE cells in vivo28, also increased, although to a lesser extent at the highest SHH concentration (1,000 ng ml−1; Fig. 1b). In contrast, expression of the dorsal telencephalic transcription factor genes PAX6 and EMX1 decreased (Fig. 1b). Immunostaining and western blotting analyses confirmed that the NKX2.1+ cell population increased over time, reaching 93% ± 4.1% (±s.e.m.) by day 25 (n = 5 cultures, four cultures from line H9 and one culture from line H1), whereas the population of PAX6+ dorsal progenitors decreased to <0.1% with 1,000 ng ml−1 SHH (Fig. 1c–e). Additional ventral transcriptional factors, including OLIG2 (40 ± 3.4%) and ISL1 (21% ± 3.1%), were expressed (Supplementary Fig. 1). The MGE-like progenitors exhibited a forebrain identity, with expression of the telencephalic marker FOXG1 (89% ± 1.9%) at day 25 but not the midbrain marker EN1 or the hindbrain and spinal marker HOXB4 (Supplementary Fig. 1c). MEIS2, a transcription factor expressed by LGE progenitors, was highly expressed in the presence of a low concentration of SHH (100 ng ml−1) but was nearly completely blocked by a high concentration of SHH (500 ng ml−1; Supplementary Fig. 1d, e), supporting the conclusion that the differentiation protocol generated MGE rather than LGE progenitors. In addition, expression of LHX8 and LHX6, encoding transcription factors involved in BFCN and GABA interneuron specification4,28,29, increased substantially in response to higher concentrations of SHH, mainly at day 25 (Fig. 1b), suggesting that the NKX2.1+ ventral progenitors have cholinergic and GABAergic potential.
Figure 1.
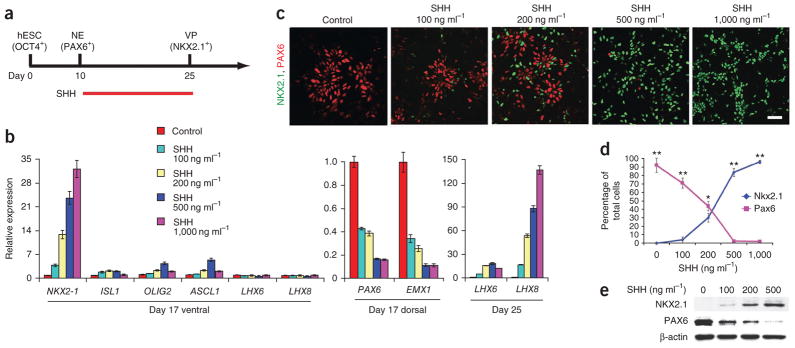
SHH-dependent specification of basal forebrain progenitors from hESCs. (a) Schematic showing specification of NKX2.1+ ventral progenitors (VP) from neuroepithelia (NE) by SHH for 2 weeks starting from day 10. (b) Quantitative RT-PCR analysis of various markers (expression relative to control: 0 ng ml−1 SHH treatment group) on indicated days of differentiation. Dorsal and ventral refer to dorsal and ventral forebrain markers. Error bars, s.e.m. (c) Immunocytochemistry analysis of hESC-derived neural progenitors for NKX2.1 and PAX6 expression in response to SHH on day 25 of differentiation. Scale bar, 50 μm. (d) Quantification of data in c. Error bars, s.e.m.; *P < 0.05 and **P < 0.01. (e) Western blot analysis of hESC-derived neural progenitor lysates with antibodies to indicated proteins in response to SHH treatment on day 25 after differentiation.
MGE progenitors generate BFCNs and GABA neurons in vitro
To differentiate NKX2.1+ progenitors, we dissociated the cells by treatment with accutase on day 28 and plated them on hESC-derived astroglia30, followed by withdrawal of SHH and addition of nerve growth factor31, a survival factor for cholinergic neurons (Fig. 2a). Four days after plating (day 32), the majority of cells (90% ± 2.1%, n = 5 cultures, two cultures from line H1 and three from line H9) were process-bearing neurons, as indicated by immunostaining for βIII-tubulin (data not shown). Among the βIII-tubulin+ cells, 38% ± 2.3% were ChAT+ neurons (Fig. 2b, c) by day 40. Most of the ChAT+ neurons expressed ISL1 (data not shown), NKX2.1 and FOXG1 (Fig. 2c), transcription factors that are associated with BFCNs. By day 45, nearly all the ChAT+ cells coexpressed the neurotrophin receptor P75 (data not shown) and vesicular acetylcholine transporter (VAChT), which are associated with maturing cholinergic neurons, and the synaptic marker synapsin (Fig. 2c). These data indicated that the cholinergic neurons generated in our culture system had a mature, ventral forebrain neuronal identity.
Figure 2.

Differentiation of basal forebrain progenitors to cholinergic neurons and GABAergic interneurons. (a) Schematic of neuronal differentiation from basal forebrain progenitors. NE, neuroepithelia. (b) Quantification of the percentage of ChAT+ and GABA+ neurons in βIII-tubulin+ (βIII-T+) cells. Error bars, s.e.m. (c) Immunofluorescence images of ChAT+ cholinergic neurons immunostained for βIII-tubulin (βIII-T), VAChT, NKX2.1 and FOXG1. (d) Immunofluorescence images of GABA neurons immunostained for βIII-tubulin (βIII-T), NKX2.1 and GAD65. TO, topo3; HO, Hoechst staining. Scale bars, 50 μm.
The βIII-tubulin+ population also included a cell population expressing GABA that did not coexpress ChAT; 45% ± 3.1% expressed this marker at day 40 (Fig. 2b, d). Of these GABA+ cells, <1% expressed DARPP32, a marker of GABA projection neurons, at day 40 (not shown). The GABAergic neurons coexpressed βIII-tubulin, NKX2.1 and GAD65 (another marker of GABA neurons; Fig. 2d). These data suggested that the GABA+ neurons were mostly interneurons.
Whole-cell patch-clamp recording on neurons differentiated for 8–10 weeks (n = 12 neurons, from three different H9 cultures) showed that the cells fired action potentials with inward Na+ and outward K+ currents, in addition to having normal membrane properties (resting membrane potential, input resistance and capacitance; Supplementary Fig. 2a–c). We observed spontaneous synaptic currents in all of the tested cells; these currents were blocked partially by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), a competitive AMPA receptor antagonist, and almost completely by a combination of CNQX and bicuculline (Supplementary Fig. 2d), revealing both inhibitory and excitatory inputs.
Grafted neural progenitors generate neurons and glia
To study the in vivo function of our in vitro–produced human MGE-like progenitors, we used a previously described mouse model (n = 47 mice) of learning and memory loss, which is generated by intraparenchymal injection of anti–mouse P75-saporin (p75-SAP), an immunotoxin that binds to low-affinity neurotrophin receptors on BFCNs, into the medial septum of severe combined immunodeficiency (SCID) mice32–35 (Fig. 3a). Cholinergic neurons in the medial septum, examined by immunostaining for ChAT 2 weeks after immunotoxin injection, were nearly eliminated by p75-SAP injection, whereas aCSF injection produced no visible damage to the neurons (n = 10 mice) (Fig. 3a, b). GABAergic interneurons, imaged by parvalbumin staining, in the medial septum were also decreased (Fig. 3b).
Figure 3.

Survival, proliferation and differentiation of grafted cells in lesioned hippocampi. (a) Schematics showing medial septal lesion and hippocampal transplantation sites. (b) Immunofluorescence images showing the expression of ChAT and parvalbumin (PV) in the p75-SAP–injected medial septum and the CSF injection control (sham). Dashed outlines show medial septum area. (c) Total grafted cell numbers (human nuclear protein–immunopositive; hN+) in MGE and VSP groups. (d) Immunofluorescence images of human cells showing hN and human neural fibers (hTau) in MGE and VSP grafts. Hoechst (HO)-stained cell nuclei reveal hippocampal layer structures. Scale bars, 50 μm (b, d). (e) Proportion of KI67+, NESTIN+ and SOX2+ cells among total human (hN+) cells 1 week, 1 month and 6 months after transplantation in MGE as well as Sox2+ cells in adult SCID mouse hippocampus (HIP). Values for KI67 and NESTIN at 6 months are too small to be shown with bars and are labeled with values. (f) Quantification of neural subtypes among total grafted human (hN+) cells by 6 months. **P < 0.01. Error bars, s.e.m.
The medial septum lesion results in cholinergic and GABAergic denervation in the hippocampus, which partially underlies memory loss6–10. We transplanted to the hippocampi on both sides MGE-like progenitors that had been differentiated from hESCs for 5 weeks in suspension (Fig. 3a). Ventral spinal progenitors (VSPs) generate similar proportions of ChAT+ neurons and GABAergic neurons in vitro13 and were used as a control for functional specificity of neural progenitors and for possible trophic contributions. The control groups were (i) p75-SAP-injected mice transplanted in the hippocampi with VSPs differentiated from the same hESCs for 5 weeks; (ii) p75-SAP-injected mice injected in the hippocampi with aCSF; and (iii) mice injected with CSF but not p75-SAP (sham). These four groups had 7–13 mice each (Supplementary Table 1). In addition, we transplanted MGE progenitors into six mice and VSP progenitors into another six mice for short-term (1 week and 1 month) histological analysis. Finally, six mice received MGE progenitors labeled with mCherry under the synapsin promoter for in vivo analysis of neuronal electrophysiological activities.
One week, one month and six months after transplantation, the grafted brains exhibited no obvious difference in gross morphology from those that received only aCSF injection (septal lesion but no cell transplantation) or sham surgery (aCSF injection to the medial septum but no transplantation surgery). Stereological measurement 6 months after transplantation produced estimated total human nuclear protein–immunopositive cell numbers of 150,167 ± 40,010 in an average MGE graft and 186,894 ± 40,919 in an average VSP graft (Fig. 3c). Thus, there was no significant difference in graft cell numbers between the two groups. Immunostaining for human nuclear protein and human Tau (hTau) indicated that human cells were present in all grafted brains and distributed to the hippocampus proper, with a higher concentration in the dentate gyrus, leading to an enlarged dentate gyrus in some of the brains, especially in the plane with the injection site, thus displacing the CA1 region upward (Fig. 3d). Staining of cross-brain sections with a DNA dye (Hoechst) revealed that the cell-layer structures in the grafted hippocampus were maintained, with the exception of injection sites (Fig. 3d).
Analysis of cell-proliferation and stem cell-markers in the MGE group at 1 week indicated that 13% ± 1.8% of human cells (human nuclear protein–immunopositive) were KI67+, 52% ± 3.3% were NESTIN+ and 39% ± 2.6% were SOX2+ (Fig. 3e and Supplementary Fig. 3). In the MGE and the VSP groups, no cells were positive for the pluripotency factors NANOG or OCT3/4 (data not shown) at 1 week and 1 month after transplantation (n = 3 mice each). In the MGE group one month after transplantation, only 3.8% ± 0.7% of the cells were KI67+, 21% ± 2.7% were NESTIN+ and 21% ± 3.4% were SOX2+ (Fig. 3e and Supplementary Fig. 3c; n = 3 mice); 6 months after transplantation, KI67+ cells (<0.01%) were rarely found in the grafts, <1% of the cells expressed NESTIN, and 14% ± 1.9% of the cells were SOX2+ (Fig. 3e and Supplementary Fig. 3c; n = 7 mice), similar to the expression of these proteins in adult mouse hippocampus36. In the VSP group, the proportions of cell-proliferation and stem-cell markers were similar to those in the MGE group (Supplementary Fig. 3d). In both groups, the vast majority of grafted cells at 6 months after transplantation were post-mitotic.
The cells in the MGE and VSP grafts at 6 months were primarily neurons (Fig. 3f). In the MGE group, 74.3% ± 1.5% of the total grafted (human nuclear protein–immunopositive) cells were βIII-tubulin+ (neurons), 11.7 ± 1.0% were GFAP+ (astrocytes), 4% were MBP+ (oligodendrocytes) and 1% were NESTIN+ (neural progenitor cells) (Fig. 3f and Supplementary Fig. 3a). With regard to expression of markers of neuronal subtypes, 45% were GABA+, 8% were ChAT+ (Fig. 3f), 4% were TH+ and 16% were calbindin+. Confocal analysis of grafted sections that were also immunostained for a mouse-specific antibody M2/M6 as well as a human-specific nuclear antigen, human nuclei and a cytoplasmic marker (STEM121) indicated that the grafted human cells did not fuse with mouse cells (Supplementary Fig. 3e).
MGE and VSP cells produce different proportions of BFCNs
The major neuronal population in both grafts was GABAergic interneurons (Fig. 3f). We observed human nuclear protein–immunopositive GABA+ neurons in the first week after transplantation, and these neurons constituted ~30% of all human cells. The proportion of GABA+ neurons increased over time, and by 6 months, ~45% of the grafted cells were GABA+ in both the MGE and VSP grafts (Fig. 4). The MGE group, however, had more neurons expressing subtype markers: 31.9 ± 3.4% for somatostatin, 16.7% ± 3.4% for calbindin, 13.6% ± 2.9% for parvabumin, and less than 1% neuropeptide Y (NPY) in the MGE group compared with 6.2% ± 2.7% for somatostatin, 5.9% ± 1.8% for calbindin, 5.3% ± 2.1% for parvabumin and 1% for NPY in the VSP group (Supplementary Fig. 4a). Another marker, calretinin, was not observed in either group.
Figure 4.

Differentiation and integration of grafted GABA+ neurons. (a) Immunofluorescence images of GABA+ neurons among human (human nuclear protein–immunopositive; hN+) cells over 1 week, 1 month and 6 months after transplantation. Insets, higher magnification of GABA+ and human nuclear protein–immunopositive (hN+) neurons in grafts 6 months after transplantation. (b) Immunostaining images showing GABA, human synaptophysin (Syn) and βIII-tubulin (βIII-T) (top) and VGLU punctae, GABA and STEM121 (S121) (bottom) for MGE and VSP groups. Insets, magnified views of boxed regions (arrows indicate colocalization). Scale bars, 50 μm. (c) Quantification of GABAergic neurons among total grafted cells in the MGE and VSP groups at indicated times. Error bars, s.e.m.
Analysis of synapses revealed that human (STEM121+) GABA+ neurons in both types of graft were surrounded by vGLU+ punctae in the cell bodies and fibers (Fig. 4b), suggesting potential glutamatergic inputs into grafted GABA+ neurons. Human (synaptophysin+) GABA+ neuronal fibers apposed mouse hippocampal pyramidal dendritic trees (Fig. 4b). These results suggested that human GABA+ neurons from both the ventral forebrain and spinal progenitors were integrated into local neural circuitry.
We rarely observed another major neuronal type, ChAT+ cholinergic neurons, in either group in the first week after transplantation (Fig. 5a). Over time, these neurons increased to large numbers in the MGE group and to a much lesser extent in the VSP group (Fig. 5). By 6 months after transplantation, there were 11,779 ± 2,783 ChAT+ cells on average in MGE grafts but only 1,457 ± 277 ChAT+ cells on average in VSP grafts (Fig. 5d). The ChAT+ cells in the MGE group also expressed the neuronal marker βIII-tubulin, the cholinergic markers VAChT and p75, and human-specific synaptophysin (Supplementary Fig. 3a). The size of individual ChAT+ cells in the MGE grafts increased substantially from 1 month after graft; by 6 months after graft, the average cell size, as measured by cell body area, was 289.5 μm2 ± 17.1 μm2, compared with 126.6 μm2 ± 10.1 μm2 for ChAT+ mouse cells in the medial septum (Supplementary Fig. 3b). Thus, human BFCNs were substantially larger than their mouse counterparts.
Figure 5.

Differentiation and integration of cholinergic neurons. (a) Immunostaining of ChAT+ neurons over time in MGE and VSP groups. hN, human nuclear protein. (b) Immunostaining images of ChAT, human synaptophysin (Syn) and βIII-tubulin (βIII-T) (top) and of ChAT and VGLU (bottom) for MGE and VSP groups. Insets, magnified views of boxed regions (arrows indicate colocalization). Scale bars, 50 μm. (c) Percentage of human ChAT+ neurons among total grafted human cells 1 week, 1 month and 6 months after transplantation. hN, human nuclear protein. (d) Quantification of total ChAT+ cell numbers in MGE and VSP groups. Error bars, s.e.m; **P < 0.01.
Although MGE progenitors tended to concentrate in the dentate gyrus, human cholinergic neurons (ChAT+ and human nuclear protein+) in the MGE-grafted mice were mainly distributed along the pyramidal cell layer (pyramidal stratum) in the CA3 region and were also scattered in other areas of the hippocampus. Large numbers of fibers projected along the molecular layer in the hippocampus and dentate gyrus, thus crossing the dendritic trees as well as cell bodies of the pyramidal neurons. Confocal microscopy analysis revealed that the ChAT+ processes in MGE grafts, but not in VSP grafts, closely apposed the processes of mouse pyramidal neurons and also expressed human-specific synaptophysin (Fig. 5b), suggesting formation of synaptic contact between human cholinergic neurons and mouse pyramidal neurons, most of which are glutamatergic neurons37. Furthermore, cholinergic neurons in MGE grafts (Fig. 5b) were surrounded and overlapped with punctae of vGLU, suggesting glutamatergic inputs from endogenous brain areas such as the hippocampus and thalamus. MGE grafts themselves were devoid of glutamatergic neurons (data not shown). The human cholinergic neurons in the mouse hippocampus expressed nicotinic and muscarinic acetylcholinereceptors (Supplementary Fig. 4b).
We have recently shown that human ESC-derived neurons can functionally integrate into an existing neural circuitry in vitro and after transplantation into the mouse brain38. To assess whether MGE-derived neurons formed functional synapses in the denervated hippocampus, we transplanted into another group of septum-lesioned mice (n = 6 mice) MGE progenitors expressing mCherry under the synapsin promoter39. Whole-cell patch-clamp recordings on human MGE-derived neurons in brain slices 2 months after transplantation revealed no action potentials in the grafted neurons (n = 3 mice). However, by 6 months after graft, the human MGE-derived neurons showed inward Na+ and outward K+ currents and action potentials (n = 3 mice; Supplementary Fig. 4c). We also observed spontaneous postsynaptic currents (n = 12 neurons, Supplementary Fig. 4c), indicating that the human cells had synaptically integrated into neural circuits in the mouse brain. The mCherry-expressing cells expressed human nuclear protein, confirming that they were human cells (Supplementary Fig. 4c).
MGE-progenitor transplants correct behavioral deficits
The medial septum lesion results in cholinergic and possibly GABAergic denervation in the hippocampus, which is associated with functional deficits in learning and memory7–10. We evaluated all mice in each of the four groups, MGE, VSP, aCSF and sham (Supplementary Table 1), before and after cell transplantation in learning and memory tests (Morris water maze and passive avoidance) and in an anxiety test (open field) (Fig. 6a). The behavioral tests were performed by a person who was blinded to the experimental groups, and statistical data were analyzed by another person who had no knowledge about the experimental groups. We first conducted a Morris water-maze test. We trained mice to locate a hidden platform for four consecutive days (with eight 60-s trials each day) and recorded the latency (time mice spent locating the hidden platform). After the training was finished, we recorded the swimming pattern in the absence of a platform (a probe test). Before training, the four groups after septum lesion and before transplantation exhibited a similar swimming pattern (trace of mice swimming recorded by a monitor; Supplementary Fig. 5a). After training, the lesioned mice took much longer (P = 0.0006) to reach a hidden platform compared with the sham-treated group 2 weeks after lesion (Fig. 6b), showing that the lesion diminished learning ability. Two months after transplantation, the MGE-transplanted mice began to exhibit a decreased latency in identifying the hidden platform compared with the VSP- and aCSF-injected mice (Fig. 6b). The trend was statistically significant by 4 and 6 months after transplantation (P < 0.05 and P < 0.01, respectively; Fig. 6b, c).
Figure 6.

Transplantation of hESC-derived MGE progenitors contributes to functional recovery. (a) Timeline of surgery and behavioral analyses. (b) Morris water-maze test–based analysis of latency in finding a hidden platform before transplantation in mice treated as indicated. Sham: aCSF injection in medial septum. After transplantation, the MGE group, but not the VSP or CSF group, progressively shortened the latency in finding the platform: −0.5 months, F = 19.434 and P = 0.0006; 2 months, F = 5.671 and P = 0.01; 4 months, F = 12.14 and P = 0.0002; 6 months, F = 9.966 and P = 0.0007. (c) Summary of the latency in finding the hidden platform for the MGE group over time. (d) Times crossing the removed platform in the MGE-transplanted group compared to controls 6 months after transplantation. F = 3.359, P = 0.048. (e) Differential crossing and time spent in each of the quadrants by the mice. MGE crossings, F = 3.034 and P = 0.046; time, MGE time spent in quadrant, F = 12.093 and P < 0.001. (f) Latency in landing on the visible platform. (g) Latency in passive avoidance tests. F = 5.257 and P = 0.0159. Error bars, s.e.m.; *P < 0.05, **P < 0.01.
In the probe test, we measured the search time and crossing times in each of the four quadrants after the platform had been removed, a measure of memory. The MGE group crossed the removed platform area more often than did the two control groups, especially at month 6 (P < 0.05) (Fig. 6d). In particular, at six months, the MGE group exhibited preferential crossings and spent significantly (P < 0.05) more time in the quadrant with the hidden platform than in other quadrants, whereas the control transplant groups did not exhibit quadrant preference (Fig. 6e and Supplementary Fig. 5a).
To ensure that the changes in latency and in the probe test were not due to physical impairment (motor or vision), we performed a visible-platform water-maze test. It showed that although the sham-treated mice (no lesion) reached the platform faster, all transplanted mice (with lesion) had a similar latency in landing on the platform (Fig. 6f), validating that the different water-maze behaviors were not caused by differences in motor- or vision-based search.
Another test for short-term memory, passive avoidance, in which the animal learns to avoid an aversive stimulus previously experienced, indicated that MGE-grafted mice had a similarly increased latency as did sham mice to enter a dark compartment where they had received a shock 24 h earlier, whereas control mice (aCSF and VSP groups) exhibited a significantly shorter latency (P < 0.05; Fig. 6g).
We investigated whether the behavioral changes described above were due to changes in anxiety rather than in learning and memory. Analysis of swimming traces at 6 months showed no difference between the groups in the tendency to swim near the wall of the maze (Supplementary Fig. 5a), a sign of anxiety. An open field test, another measure of anxiety, at the same time point revealed that all mice exhibited a similar center ratio (ratio of the distance traveled in the center versus the outside of the area) and proportion of time spent along the side grids of the chamber 6 months after graft (Supplementary Fig. 5b). Together, these results indicated that transplantation of human MGE-like cells, but not VSPs, into mice with a septal lesion ameliorated not anxiety but deficits in learning, memory and spatial cognition.
DISCUSSION
This study presents a method for efficiently differentiating hESCs in vitro to NKX2.1+ MGE-like progenitors (93% ± 4.1% of all cells without cell sorting) and differentiating these progenitors into high proportions of BFCNs and GABAergic interneurons. During development, MGE progenitors are specified by ventrally derived SHH signals2,3, whereas hESC-derived neural progenitors have dorsal telencephalic (cortical) phenotypes because Wnt signals override SHH signals15. We therefore patterned the primitive neuroepithelia14 with high concentrations (500–1,000 ng ml−1) of SHH, which produced a nearly uniform population of NKX2.1+ MGE-like progenitors. This is similar to mouse ESC differentiation in that Nkx2.1+ cells were generated only in the presence of high concentrations of Shh, although only 30% of the sorted Foxg1+ cells became Nkx2.1+ (ref. 40). The human MGE-like progenitors differentiated to the expected neuronal types, BFCNs and GABA interneurons, upon removal of SHH in vitro. Notably, after transplantation into mice with septohippocampal lesions, the MGE-like cells survived and produced GABA interneurons and BFCNs that formed synaptic connections with the host neurons and corrected short-term learning and memory deficits.
A recent report described differentiation of hESCs to BFCNs by treating hESCs with 10 μM of retinoic acid for 7 d followed by expansion of neurospheres and treatment with bone morphogenetic protein 9 (BMP9) for 3 d in a dissociated culture41. However, the identity of the BFCNs produced was not clear as the cells did not express fore-brain markers such as FOXG1 (some of the cells expressed FOXG1 in the cytoplasm instead of in the nuclei), and some of the cholinergic markers (for example, VAChT) also stained MAP2-negative cells. A side-by-side comparison between this approach and ours is provided in Supplementary Table 2. In repeating the protocol of ref. 41, we found that, similar to previous reports25,42, early treatment of hESCs with 10 μM retinoic acid resulted in <1% of the cells becoming neurospheres, and most of these neural progenitors expressed hindbrain and spinal marker HoxB4 but not the forebrain marker FoxG1, indicating that cells differentiated under high concentrations of retinoic acid for 7 d are not forebrain cells. The basis for the differences in the findings of the two laboratories is at present not clear. Nevertheless, the cholinergic neurons described here have known features of BFCNs in vitro and in vivo. Our protocol also allows the generation of NKX2.1+ progenitors from human and monkey iPSCs (data not shown), suggesting that it could be applied to the derivation of BFCNs and GABA interneurons from disease-affected iPSCs for pathology analysis and drug discovery.
GABA interneurons and BFCNs are implicated in locomotion regulation as well as in learning, memory and cognition7–10. Transplantation of human fetal MGE cells has improved motor ability in Parkinson’s rats43, and transplantation of fetal medial septum tissues has rescued learning and memory deficits in animal models32,33,44. Here we showed that hippocampal transplantation of hESC-derived MGE progenitors into septohippocampal lesioned mice34,35 corrected memory loss, suggesting that in vitro–produced MGE progenitors behave like their in vivo counterparts. The lack of functional contribution by hESC-derived ventral neural progenitors having hindbrain and spinal characteristics indicates the requirement of subtype-specific progenitors or neurons to achieve functional replacement. Similarly, we found that only striatal GABA neurons but not spinal GABA interneurons corrected locomotion deficit in a Huntington’s disease model17.
The mechanism underlying the improvements in learning and memory is not fully delineated in the present study and could involve synaptic integration and/or trophic support. Grafted neural progenitors can release trophic factors that promote functional and/or structural recovery45,46. The behavioral recovery we observed occurred mainly at later time points, when the grafted cells had become mature neurons and glia (Fig. 5), but mature neurons and glia may also provide trophic support. However, the MGE and VSP groups produced similar proportions of neurons and glia, suggesting that the differences in functional outcomes were not primarily due to trophic factors.
The model we used involves injection of p75-SAP specifically into the septum rather than into the commonly used target, the lateral ventricles. This consistently destroys nearly all cholinergic neurons and many GABA neurons, whereas ventricle injection results in variable extent of neuronal loss (Fig. 3b). Both cholinergic neurons and GABA neurons are involved in learning and memory, although there is debate about their precise roles9,10,34,35,47. We observed large numbers of GABA neurons from grafts of both forebrain and spinal progenitors, suggesting that GABA neurons may have contributed to the functional improvements. However, the numbers of GABA neurons and their synaptic density were similar in the two groups and thus do not likely explain the differences in behavioral outcomes. Other possible explanations are differences in GABA interneuron subtypes and differences in the numbers and localization of cholinergic neurons. In particular, the MGE-derived cholinergic neurons were preferentially localized to the dentate gyrus and to the CA3 region, a target of septal cholinergic neurons. They not only projected their fibers to pyramidal neurons and their dendritic trees in the hippocampus and expressed human synaptophysin but also received synaptic inputs from glutamatergic neurons, presumably from thalamus and/or cortex, as many vGlu punctae were present on cell bodies and dendrites of the ChAT+ cells. Additionally, cholinergic fibers from the MGE graft but not the VSP group projected to cerebral cortex (data not shown). Thus, the BFCNs may have contributed more directly to improvements in learning and memory in our grafted mice, whereas the GABA interneurons, which participate in local network formation, may have formed a substrate or a modifier that aided BFCN function.
Neural transplantation of hESC derivatives, especially neural progenitors, is often associated with tumor formation or overgrowth23,24,48. Notably, our neural progenitors distributed throughout the hippocampus quite evenly, mitotic progenitors decreased over time, and no tumors were detected (Fig. 3 and Supplementary Fig. 3). We speculate that this was due to the purity of the transplanted cells and their developmental stage. In our differentiation system, hESCs are converted to nearly pure populations of NKX2.1+ ventral progenitors by SHH or to Olig2+ ventral spinal progenitors by retinoic acid and SHH49. There were no remaining hESCs and very few PAX6+ primitive neuroepithelial cells; the latter are the main contributors to overgrowth as human cortical progenitors proliferate rapidly for a long period23. Furthermore, we identified an optimal differentiation stage of MGE progenitors, day 35, at which to transplant the cells so as to generate functional BFCNs and GABA neurons in vivo without producing displacing grafts in the relatively small and neurogenic mouse hippocampus over 6 months. Grafted cells from both groups migrated to the dentate gyrus and proliferated, presumably in response to the local neurogenic milieu, thus enlarging the dentate gyrus. Nevertheless, the grafted cells stopped dividing at later stages, and the GABA interneurons and BFCNs formed connections with host neurons.
In summary, we developed a method for differentiating human pluripotent stem cells to MGE-like progenitors. The progenitors generated expected BFCNs and GABA interneurons in vitro and after transplantation into the mouse brain corrected learning and memory deficits in lesioned mice. These results support the possibility of using human stem cell–derived MGE progenitors to develop therapeutics for neurological diseases that affect learning and memory.
ONLINE METHODS
hESC culture and neural differentiation
hESCs (line H9, passages 18–35; line H1 passages 30–36) were maintained on a feeder layer of irradiated embryonic mouse fibroblasts, as previously described13. For neuroepithelial cell induction, hESCs were first differentiated to primitive neuroepithelia in a chemically defined medium13,27. To induce MGE-like progenitors, the primitive neuroepithelial cells were treated with SHH (100–1,000 ng/ml; 1845-SH; R&D Systems). The neuroepithelial cells formed neural tube-like rosettes at day 14–17 which were gently blown off by a 1-ml pipette and suspended in the same medium for 1 week.
For neuronal differentiation, from day 28, the neural progenitors were cultured together with hESC-derived astrocytes30 that were previously plated onto laminin at a density of 5,000 cells/cm2. The plated cells were grown in a neuronal differentiation medium consisting of Neurobasal medium (Gibco), N2 supplement (Invitrogen), nerve growth factor (NGF; 50–100 ng/ml; R&D), cAMP (1 μM; Sigma), brain-derived neurotrophic factor (BDNF), insulin-like growth factor I (IGFI), bone morphogenetic protein 9 (BMP9) and SHH (50 ng/ml; R&D). Differentiation of ventral spinal progenitors (VSP), which contain cholinergic spinal motor neurons, was performed as previously described50.
Immunocytochemistry and cellular quantification
Immunocytochemistry staining on coverslip cultures was performed as previously described13,23. The primary antibodies used in this study are listed in Supplementary Table 3. Images were visualized using a Nikon TE600 fluorescence microscope (Nikon Instruments) or a Nikon C1 laser-scanning confocal microscope. To quantify the differentiation efficiency of ventral forebrain progenitors, neural progenitor spheres were dissociated into single cells, plated onto coverslips and stained for NKX2.1. For quantifying differentiated cholinergic neurons and GABAergic neurons, the neural progenitor spheres were digested with accutase (Innovation Cell Technology) into small clusters and plated on coverslips or astrocytes for immunostaining, as previous described13,50. Fields were randomly selected, and the neuronal cells and total cells (Hoechst-stained) were counted using ImageJ software. All the experiments were performed in triplicate, and the results were reproduced in two different cell lines (H1 and H9). A two-tailed Student’s t-test was used for statistical analysis.
Quantitative reverse-transcription PCR (qRT-PCR)
Total RNA was extracted using Trizol reagent (Invitrogen), and cDNA was reverse-transcribed using the SuperScript III First-Strand (Invitrogen). RT-PCR was performed using the Bio-Rad MyiQ real-time PCR detection system, as described51. Primers used are listed in Supplementary Table 4.
Whole-cell patch-clamp recording
Whole-cell patch-clamp recordings were made from hESC-derived cholinergic neurons at 8–10 weeks of age, as previously reported52. Biocytin (1%, Sigma) was injected into cells after recording to determine the neuronal identity13. Data were analyzed with pClamp 9.0 (Axon instruments) and are presented as mean ± s.e.m.
Western blot
Protein samples were collected on day 25 of differentiation. Western blot analysis was performed as described53. Proteins (25 μg) were separated and blotted with antibodies to NKX2.1 (1:2,000, mouse IgG, Chemicon & Millipore, MAB5460), PAX6 (1:4,000, rabbit IgG, Covance, prb 278p), and β-actin (1:4,000, mouse IgG, Sigma, A1978).
Medial septum immunolesion and cell transplantation
The animal experiments were carried out following the protocols approved by the University of Wisconsin–Madison Animal Care and Use Committee. The medial septum immunolesion model was created in a total of 57 male SCID mice, 8–10 weeks old (47 mice with medial septum lesions and 10 sham-treated mice). Most of the p75-saporin lesion models were created by injecting the toxin into the lateral ventricles33,34. Such a model results in variable cellular damage to tissues surrounding the ventricles as well as the cerebellum and hence variable behavioral outcomes. To avoid the variability, we titrated the dosage and targeted to the medial septum so that the lesion will destroy most cells in but will be limited to the septum area, thus leading to a reproducible and consistent deficit in learning. The mice were anesthetized with 1% isoflurane mixed with oxygen, and 0.8 μl of undiluted anti–mouse-p75-SAP (Advanced Targeting System, IT-16) was injected into the medial septum over a 5-min period with a 15° angle toward the midline at the following stereotaxic coordinates: anterior-posterior = +0.38 mm; left lateral = +1 mm; and dorsoventral = −4.12 mm. Two weeks after surgery, mice were subjected to Morris water maze (Nodule) tests. Six weeks after lesion, the affected mice were subdivided randomly into three groups, one group received transplantation with hESC-derived MGE-like progenitors, one with hESC-derived VSP, and the other with aCSF. Cells or CSF were injected into the hippocampus at the following stereotaxic coordinates: anterior-posterior = −2.46 mm; left-right lateral = ±2 mm; and dorsoventral = −2.25 mm. About 100,000 cells were injected into each side of the hippocampus in 2 μl over a 5-min period. All the 29 transplanted mice and 10 sham-operated mice, which were injected with aCSF (Harvard Apparatus), underwent behavioral tests until we killed them (Supplementary Table 1). Morris water maze tests were performed 2 months, 4 months and 6 months after transplantation, and other behavioral tests, including visible platform Morris water maze and passive avoidance, were carried out 6 months after transplantation.
For short-term histological analysis, another 12 lesioned mice were transplanted with VSP or MGE progenitors separately (6 with MGE and 6 with VSP). For electrophysiology measurement of grafted cells, we transplanted fluorescently labeled MGE progenitors (mCherry under the control of the synapsin promoter) into the hippocampus of 6 septal lesioned mice and examined the brain slices 2 months and 6 months after transplantation, as described38.
Behavioral tests
Morris water maze tests were performed 2 weeks after lesion, and 2 months, 4 months and 6 months after cell transplantation, and passive avoidance and open field were performed 6 months after transplantation.
Morris water maze
The swimming tank (130 cm diameter) was divided into quarters with an escape platform hidden 0.5–1 cm under opaque water (22 °C ± 2 °C). The hidden platform was placed in a different quadrant at the start of each time point, so that each of the four tests (−0.5, 2, 4 and 6 months) used a different platform location. The swimming behavior of mice was monitored by an automated video system. Mice were trained to locate the platform for four consecutive days. There were two ‘blocks’ of four 60-s trials (8 total) each day, with 30 min of interval between the blocks. For the training trials, the latency to find the platform was recorded. After completion of the 32 trials over 4 d, the hidden platform was removed, and one 60-s probe trial was conducted to record the number of ‘platform’ crosses and the time spent in each quadrant.
To ensure that the water maze behaviors were not caused by differences in vision, a visible water maze test was performed for two consecutive days (four blocks total) after the 6th-month water maze. The visible platform, 5 cm above the water surface with a black cube (5 cm × 5 cm × 5 cm), was placed randomly in a different quadrant in every single trial. The latency in finding the visible platform was recorded. Two-way ANOVAs with repeated measures were performed to analyze regular water maze, and one-way ANOVA was performed to analyze visible platform water maze.
Passive avoidance
A box was divided into light and dark sides by a door which was closed initially. A mouse was placed into the bright side, and the door was opened after 10 s. After the mouse entered the dark side, the door was closed, and the mouse received a 2-s 0.5 mA foot shock. To test whether the mice learned and remembered the shock, the mouse was placed into the bright side again 24 h later, and the latency to enter the dark side was measured. The latency was compared between groups by one-way ANOVA.
Open field
By 6 months after transplantation, all the mice were tested by open field, as described previously17.
Brain-slice electrophysiological recording
Brain tissue at months 2 and 6 after transplantation were sectioned at 400 μm using LEICA vibrotome (VT 1000S). Whole-cell patch-clamp recordings using Axon Multiclamp 700B and pClamp software (v.9.2) were performed as described previously38. Human MGE derived neurons were identified by mCherry expression under control of a synapsin promoter, which was introduced into the progenitors at day 25 (10 d before transplantation) using a lentivirus.
Histological analysis of transplants
The mice were perfused with 4% paraformaldehyde. After 4 h of post-fixation, the brain was serially sectioned coronaly to 30 μm in thickness, and free-floating sections were stained. Grafted cells were counted on every six sections as described23 using a Stereo Investigator software (MicroBrightField, Inc). The graft area was outlined according to the presence of human nuclear protein–positive cells under a ×10 objective of a fluorescent scope. Cell counting was performed under a ×40 objective of a Zeiss fluorescence scope in fields chosen by the software. The total cells in each graft were estimated by Stereo Investigator software54.
For measuring the cell body size, fields were randomly taken under ×40 by confocal z-stack. Cells were measured according to the cytoplasm edge, and the body area was calculated by ImageJ software. Data were presented as mean ± s.e.m.
Supplementary Material
Acknowledgments
We thank M.E. Andrzejewski and H. Mitchell for help in analyzing animal behavioral data. This study was supported in part by the US National Institute of Neurological Disorders and Stroke (NS045926) and the National Institute of Child Health and Human Development (P30 HD03352).
Footnotes
Note: Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS
Y.L. and S.-C.Z. designed the experiments and wrote the manuscript. Y.L., J.P.W., H.L., R.K., X.Z., L.M., G.Z. and M.A. performed the experiments. Y.L., J.P.W., H.L., R.K., X.Z., L.M., M.A. and S.-C.Z. analyzed the data.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 2.Rubenstein JL, Shimamura K, Martinez S, Puelles L. Regionalization of the prosencephalic neural plate. Annu Rev Neurosci. 1998;21:445–477. doi: 10.1146/annurev.neuro.21.1.445. [DOI] [PubMed] [Google Scholar]
- 3.Wilson SW, Rubenstein JL. Induction and dorsoventral patterning of the telencephalon. Neuron. 2000;28:641–651. doi: 10.1016/s0896-6273(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Y, et al. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA. 2003;100:9005–9010. doi: 10.1073/pnas.1537759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell K. Dorsal-ventral patterning in the mammalian telencephalon. Curr Opin Neurobiol. 2003;13:50–56. doi: 10.1016/s0959-4388(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 6.Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- 7.Oliveira AA, Jr, Hodges HM. Alzheimer’s disease and neural transplantation as prospective cell therapy. Curr Alzheimer Res. 2005;2:79–95. doi: 10.2174/1567205052772759. [DOI] [PubMed] [Google Scholar]
- 8.Whitehouse PJ, et al. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 9.Pang KC, Jiao X, Sinha S, Beck KD, Servatius RJ. Damage of GABAergic neurons in the medial septum impairs spatial working memory and extinction of active avoidance: effects on proactive interference. Hippocampus. 2011;21:835–846. doi: 10.1002/hipo.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dwyer TA, Servatius RJ, Pang KC. Noncholinergic lesions of the medial septum impair sequential learning of different spatial locations. J Neurosci. 2007;27:299–303. doi: 10.1523/JNEUROSCI.4189-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang SC. Neural subtype specification from embryonic stem cells. Brain Pathol. 2006;16:132–142. doi: 10.1111/j.1750-3639.2006.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamanaka S. A fresh look at iPS cells. Cell. 2009;137:13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Li XJ, et al. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- 14.Pankratz MT, et al. Directed neural differentiation of human embryonic stem cells via an obligated primitive anterior stage. Stem Cells. 2007;25:1511–1520. doi: 10.1634/stemcells.2006-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XJ, et al. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development. 2009;136:4055–4063. doi: 10.1242/dev.036624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eiraku M, et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Ma L, et al. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10:455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol. 2000;18:675–679. doi: 10.1038/76536. [DOI] [PubMed] [Google Scholar]
- 19.Xi J, et al. Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells. 2012;30:1655–1663. doi: 10.1002/stem.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirkeby A, et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012;1:703–714. doi: 10.1016/j.celrep.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Kriks S, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh Roy N, et al. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp Neurol. 2005;196:224–234. doi: 10.1016/j.expneurol.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 23.Yang D, Zhang ZJ, Oldenburg M, Ayala M, Zhang SC. Human embryonic stem cell-derived dopaminergic neurons reverse functional deficit in parkinsonian rats. Stem Cells. 2008;26:55–63. doi: 10.1634/stemcells.2007-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy NS, et al. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med. 2006;12:1259–1268. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- 25.Cooper O, et al. Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci. 2010;45:258–266. doi: 10.1016/j.mcn.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hargus G, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc Natl Acad Sci USA. 2010;107:15921–15926. doi: 10.1073/pnas.1010209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 28.Flames N, et al. Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci. 2007;27:9682–9695. doi: 10.1523/JNEUROSCI.2750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manabe T, et al. L3/Lhx8 is involved in the determination of cholinergic or GABAergic cell fate. J Neurochem. 2005;94:723–730. doi: 10.1111/j.1471-4159.2005.03261.x. [DOI] [PubMed] [Google Scholar]
- 30.Krencik R, Weick JP, Liu Y, Zhang ZJ, Zhang SC. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat Biotechnol. 2011;29:528–534. doi: 10.1038/nbt.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reilly JO, Karavanova ID, Williams KP, Mahanthappa NK, Allendoerfer KL. Cooperative effects of Sonic Hedgehog and NGF on basal forebrain cholinergic neurons. Mol Cell Neurosci. 2002;19:88–96. doi: 10.1006/mcne.2001.1063. [DOI] [PubMed] [Google Scholar]
- 32.Cassel JC, et al. Grafts of fetal septal cells after cholinergic immunotoxic denervation of the hippocampus: a functional dissociation between dorsal and ventral implantation sites. Neuroscience. 2002;113:871–882. doi: 10.1016/s0306-4522(02)00226-9. [DOI] [PubMed] [Google Scholar]
- 33.Leanza G, Martinez-Serrano A, Bjorklund A. Amelioration of spatial navigation and short-term memory deficits by grafts of foetal basal forebrain tissue placed into the hippocampus and cortex of rats with selective cholinergic lesions. Eur J Neurosci. 1998;10:2353–2370. doi: 10.1046/j.1460-9568.1998.00247.x. [DOI] [PubMed] [Google Scholar]
- 34.Berger-Sweeney J, et al. Selective immunolesions of cholinergic neurons in mice: effects on neuroanatomy, neurochemistry, and behavior. J Neurosci. 2001;21:8164–8173. doi: 10.1523/JNEUROSCI.21-20-08164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh TJ, Herzog CD, Gandhi C, Stackman RW, Wiley RG. Injection of IgG 192-saporin into the medial septum produces cholinergic hypofunction and dose-dependent working memory deficits. Brain Res. 1996;726:69–79. [PubMed] [Google Scholar]
- 36.Jinno S. Regional and laminar differences in antigen profiles and spatial distributions of astrocytes in the mouse hippocampus, with reference to aging. Neuroscience. 2011;180:41–52. doi: 10.1016/j.neuroscience.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 37.Francis PT. Glutamatergic systems in Alzheimer’s disease. Int J Geriatr Psychiatry. 2003;18:S15–S21. doi: 10.1002/gps.934. [DOI] [PubMed] [Google Scholar]
- 38.Weick JP, Liu Y, Zhang SC. Human embryonic stem cell-derived neurons adopt and regulate the activity of an established neural network. Proc Natl Acad Sci USA. 2011;108:20189–20194. doi: 10.1073/pnas.1108487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weick JP, et al. Functional control of transplantable human ESC-derived neurons via optogenetic targeting. Stem Cells. 2010;28:2008–2016. doi: 10.1002/stem.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danjo T, et al. Subregional specification of embryonic stem cell–derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J Neurosci. 2011;31:1919–1933. doi: 10.1523/JNEUROSCI.5128-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bissonnette CJ, et al. The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells. 2011;29:802–811. doi: 10.1002/stem.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pankratz MT, et al. Directed neural differentiation of human embryonic stem cells via an obligated primitive anterior stage. Stem Cells. 2007;25:1511–1520. doi: 10.1634/stemcells.2006-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez-Cerdeno V, et al. Embryonic MGE precursor cells grafted into adult rat striatum integrate and ameliorate motor symptoms in 6-OHDA-lesioned rats. Cell Stem Cell. 2010;6:238–250. doi: 10.1016/j.stem.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gage FH, Bjorklund A, Stenevi U, Dunnett SB, Kelly PA. Intrahippocampal septal grafts ameliorate learning impairments in aged rats. Science. 1984;225:533–536. doi: 10.1126/science.6539949. [DOI] [PubMed] [Google Scholar]
- 45.Emborg ME, et al. GDNF-secreting human neural progenitor cells increase tyrosine hydroxylase and VMAT2 expression in MPTP-treated cynomolgus monkeys. Cell Transplant. 2008;17:383–395. [PubMed] [Google Scholar]
- 46.Gage FH, Bjorklund A. Trophic and growth-regulating mechanisms in the central nervous system monitored by intracerebral neural transplants. Ciba Found Symp. 1987;126:143–159. doi: 10.1002/9780470513422.ch9. [DOI] [PubMed] [Google Scholar]
- 47.Lecourtier L, et al. Septohippocampal pathways contribute to system consolidation of a spatial memory: sequential implication of gabaergic and cholinergic neurons. Hippocampus. 2011;21:1277–1289. doi: 10.1002/hipo.20837. [DOI] [PubMed] [Google Scholar]
- 48.Aubry L, et al. Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci USA. 2008;105:16707–16712. doi: 10.1073/pnas.0808488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li XJ, et al. Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells. 2008;26:886–893. doi: 10.1634/stemcells.2007-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu BY, Zhang SC. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat Protoc. 2009;4:1295–1304. doi: 10.1038/nprot.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du ZW, Hu BY, Ayala M, Sauer B, Zhang SC. Cre recombination-mediated cassette exchange for building versatile transgenic human embryonic stem cells lines. Stem Cells. 2009;27:1032–1041. doi: 10.1002/stem.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson MA, Weick JP, Pearce RA, Zhang SC. Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci. 2007;27:3069–3077. doi: 10.1523/JNEUROSCI.4562-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LaVaute TM, et al. Regulation of neural specification from human embryonic stem cells by BMP and FGF. Stem Cells. 2009;27:1741–1749. doi: 10.1002/stem.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peterson DA. Quantitative histology using confocal microscopy: implementation of unbiased stereology procedures. Methods. 1999;18:493–507. doi: 10.1006/meth.1999.0818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.