

Abstract
Dominant mutations in the human insulin gene can lead to pancreatic β-cell dysfunction and diabetes mellitus due to toxic folding of a mutant proinsulin. Analogous to a classical mouse model (the Akita mouse), this monogenic syndrome highlights the susceptibility of human β-cells to endoreticular stress due to protein misfolding and aberrant aggregation. The clinical mutations directly or indirectly perturb native disulfide pairing. Whereas the majority of mutations introduce or remove a cysteine (leading in either case to an unpaired residue), non-cysteine-related mutations identify key determinants of folding efficiency. Studies of such mutations suggest that the evolution of insulin has been constrained not only by its structure and function, but also by the susceptibility of its single-chain precursor to impaired foldability.
Keywords: hormone, human genetics, protein folding, folding efficiency, metabolism
Insulin plays a central role in the regulation of vertebrate metabolism. The mature hormone, the post-translational product of a single-chain precursor, is a globular protein containing two chains, A (21 residues) and B (30 residues). Advances in human genetics over the past five years have identified dominant mutations in the insulin gene associated with diabetes mellitus (DM) (for reviews, see [1–3]). This syndrome arises from the toxic misfolding: the clinical mutations impair the folding of proinsulin in the endoplasmic reticulum (ER) of pancreatic β-cells [4,5]. Originally described in relation to neonatal-onset DM [6–9], DM may also present in childhood [10] or early adulthood [11]. Age of onset is proposed to reflect mutational differences in the degree of impaired folding and its cellular consequences [5,12]. Phenotypic variation may also arise due to polygenic differences in β-cell susceptibility to ER stress [13].
Patients with the mutant proinsulin syndrome are heterozygous. Although expression of one wild-type insulin allele would in other circumstances be sufficient to maintain metabolic homeostasis, studies of a corresponding mouse model (the Akita mouse [14–16]) have demonstrated that misfolding of the variant proinsulin perturbs the biosynthesis of wild-type insulin [17,18]. In a survey of human mutations similar perturbations in trans has been demonstrated in β-cell lines [4,5,12]. Impaired β-cell secretion is associated with ER stress, distorted organelle architecture, and eventual cell death [19,20]. These findings have stimulated renewed interest in the biosynthesis of insulin [21–23] and the structural basis of disulfide pairing [24–29]. The foundational importance of this problem and its clinical relevance have motivated construction of novel molecular reagents and animal models [30–33]. Such studies have led in parallel to an enhanced understanding of structure-functional relationships [13] and cellular mechanisms of proinsulin folding and insulin biosynthesis [1].
Clinical mutations in the insulin gene provide probes into evolutionary constraints shared by a family of single-chain factors (such as mammalian insulin like growth factors) and in part by a superfamily of invertebrate insulin-like proteins [34–36]. Such relationships have motivated studies of the mitogenic properties of proinsulin, revealing preferential signaling through the A isoform of the insulin receptor (IR) [35]. Although beyond the scope of this review, these activities may be relevant to carcinogenesis [37]. Therapeutic use of proinsulin in the treatment of human DM has been investigated but would have unclear clinical benefits [38].
Biosynthesis of Insulin
The insulin gene encodes a single-chain precursor polypeptide, designated preproinsulin (Fig. 1A, top). The signal peptide (gray bar in Fig. 1A) is cleaved on translocation into the ER to yield proinsulin. Reduced and unfolded, the translocated polypeptide contains a connecting domain (black in Fig. 1A) between the C-terminal residue of the B domain (blue) and N-terminus of the A domain (red) [39]. Its folding in the ER is coupled to the specific pairing of three cystines (Fig. 1A, center). The disulfide bridges (A6-A11, A7-B7, and A20-B19; shown in gold in Fig. 1B) contribute to protein stability and biological activity [27,40–48]. The solution structure of proinsulin as an engineered monomer consists of a folded insulin-like moiety and a flexible C domain (black segment in Fig. 1B) [49]. The overall structure is in accordance with prior biochemical evidence [50–55]. The structure, stability, and receptor-binding activity of insulin (Fig. 1A, bottom) require maintenance of each disulfide bridge [27,40–42,44–48]. Conserved among insulin-related ligands, the cystines provide interior struts (A19-B20 and A6-A11) and an external staple between chains (A7-B7). Insulin disulfide isomers exhibit molten structures of marginal stability and low activity [56–58].
Figure 1. Biosynthesis of proinsulin.

(A) Pathway begins with preproinsulin (top): signal peptide (gray), B-domain (blue), dibasic BC junction (green), C-domain (red), dibasic CA junction (green), and A-domain (red). Specific disulfide pairing in the ER yields native proinsulin (middle panels). BC- and CA cleavage (mediated by prohormone convertases PC1 and PC2) releases insulin and C-peptide (bottom). (B) Solution structure of proinsulin: insulin-like moiety and disordered connecting peptide (dashed line). A- and B-domains are shown in red and blue, respectively; C-domain contains a nascent α-helical turn near the CA junction [49]. Cystines are labeled in yellow boxes. The solution structure exploited an engineered monomer (DKP-proinsulin) as characterized by multi-dimensional 1H-13C-15N NMR. Panel A is adapted from Ref 13 panel B depicts a representative member of an ensemble of solution structures (Protein Databank entry 2KQP).
Proinsulin binds only weakly to the insulin receptor; proteolytic processing liberates the mature hormone [22,38]. On transit through the Golgi apparatus (GA) and immature secretory granules [59], the C-peptide is excised by prohormone convertases [60], which cleave conserved dibasic sites flanking the C domain (BC and CA junctions; green in Fig. 1A, B). Insulin is stored as Zn2+-stabilized hexamers as microcrystalline arrays [61–63] within specialized secretory granules [64]. The hexamers dissociate on secretion into the portal circulation, enabling the circulating hormone to function as a Zn2+-free monomer. Because the insulin monomer is exquisitely susceptible to fibrillation at millimolar concentration and body temperature [65], its zinc-mediated assembly within β-cells may represent a defense against toxic misfolding [66] during storage in the secretory granule [23]. Evidence that such assembly contributes to insulin storage and efficient secretion has recently been obtained through studies of a mouse model lacking the β-cell zinc transporter [67].
Although insulin biosynthesis occurs via a single-chain precursor, its chemical synthesis has traditionally employed isolated A- and B-chain peptides (for review see [68]). The success of insulin chain combination implies that chemical information required for folding is contained within the sequences of the A- and B domains [69,70]. A variety of analogs have been prepared by this protocol, facilitating their pharmaceutical development for treatment of DM [71,72]. Despite the general robustness of insulin chain combination, synthesis of certain analogs has been confounded by low yields [26,73–79]. Such synthetic failures provide models of impaired folding, providing insight into structural mechanisms that underlie the mutant proinsulin syndrome.
Mechanism of Disulfide Pairing
Oxidative folding of globular proteins has traditionally been investigated by chemical trapping of populated disulfide intermediates [80]. Such studies of proinsulin and related polypeptides – foreshortened single-chain analogs (mini-proinsulin [81]) and insulin-like growth factors – are notable for the transient accumulation of one- and two-disulfide intermediates [24,25,82]. Successive steps of partial folding define a series of trajectories on a sequence of free-energy landscapes (Fig. 2A). Each landscape underlies the dynamics of an ensemble of accessible polypeptide conformations in the presence of a specific subset of disulfide bridges. The landscapes proceed from shallow to steep, enabling the folding chain to acquire structure stepwise on successive disulfide pairing. The preferred sequence of disulfide intermediates, as defined by chemical trapping, thus corresponds to a progression of multiple folding trajectories on funnel-shaped landscapes. This perspective extends the classical disulfide-centered view of protein-folding intermediates [83] to encompass general biophysical paradigms of protein folding [84–86].
Figure 2. Energy landscape paradigm.

(A) Successive disulfide pairing enables a sequence of folding trajectories on ever-steeper funnel-shaped free-energy landscapes. (B) Preferred pathway of disulfide pairing. Initial formation of cystine A20-B19 (left) is directed by a nascent hydrophobic core comprising the central B-domain α-helix (residues B9-B19), part of the C-terminal B-chain β-strand (B24-B26), and part of the C-terminal A-domain α-helix (A16-A20). Alternative pathways mediate successive disulfide pairing (middle panel) leading in turn to the native state (right). The mechanism of disulfide pairing is perturbed by clinical mutations associated with a monogenic syndrome of DM due to toxic misfolding of the variant proinsulin in the ER. Figure is adapted from Ref 49; panel A is adapted from an image kindly provided by J. Williamson.
Although refolding studies of proinsulin are limited by aggregation (imposing a requirement for pH conditions > 9, confounding interpretation [87]), the disulfide pathways of mini-proinsulin and IGF-I are well characterized near neutral pH [24,25,40,43,82,88,89]. A structural pathway is suggested by spectroscopic studies of equilibrium models (Fig. 2B) [27,40–42,44–48,90]. In this pathway a key role is played by initial formation of cystine A20-B19. In the native state this disulfide bridge packs within a cluster of conserved aliphatic and aromatic side chains in the hydrophobic core to connect the C-terminal α-helix of the A domain to the central α-helix of the B domain [57,58,75]. Because cystine A20-B19 is the only one-disulfide species to accumulate in the refolding of mini-proinsulin and IGF-I [24,25,89], its initial pairing proposed to contribute to the stabilization of a specific folding nucleus [27,90,91].
The structural role of cystine A20-B19 in populated one- and two-disulfide intermediates motivated construction of analogs (of insulin, mini-proinsulin, and IGF-I) containing pairwise substitution of the other cysteines by Ala or Ser [27,40–48]. Such analogs exhibit attenuated α-helix content. Nucleation of native-like structure was observed in a disulfide-linked peptide fragment of IGF-I containing cystine A20-B19: at low temperature the disulfide bridge native tethers α-helices within a cluster of nonpolar side chains resembling the native core [27]. Mutations within this A20-B19-associated subdomain impair disulfide pairing in insulin chain combination and the biosynthesis of single-chain precursors in Saccharomyces cerevisciae [44,77,91–93].
1H-NMR spectra of the above equilibrium models exhibit a progressive increase in chemical-shift dispersion with successive disulfide pairing, suggesting stepwise stabilization of structure. These findings are in accordance with the landscape perspective [40,90]. After A20-B19 pairing, folding can proceed through multiple alternative channels. Folding of mini-proinsulin near neutral pH exhibits subsequent rapid formation of cystine A7-B7 (lower pathway in Fig. 2B) or slow pairing of A6-A11 (upper pathway). Although it is not intuitive why pairing of cysteines distant in the sequence (A7 and B7) is favored relative to pairing of proximate cysteines (A6 and A11), spectroscopic studies have demonstrated that removal of cystine A7-B7 (by pairwise substitution) destabilizes the structure of insulin more markedly than removal of A6-A11 [91]. These findings suggest that nascent structure in the one-disulfide [A20-B19] intermediate either more effectively aligns CysA7 and CysB7 or more significantly impairs pairing of CysA6 and CysA11. Either on-pathway two-disulfide intermediate may interconvert with non-native disulfide isomers as off-pathway kinetic traps (central panel of Fig. 2B).
The hazard of competing traps in the refolding of proinsulin is suggested by the refolding of a homologous polypeptide (IGF-I) to form native and non-native disulfide isomers as alternative ground states (native IGF-I and IGF-swap) [24,82]. Containing respective pairings [A20-B19, A7-B7, A6-A11] (native) and [A20-B19, A6-B7, A7-A11] (swap), the isomers exhibit similar core structures near the shared A20-B19 disulfide bridge [24,94]. The relative stabilities of these isomers in a mini-IGF model is controlled by the N-terminal segment of the B domain [95,96]. Although proinsulin has a unique disulfide-specific ground state, this and a second isomer (pairings [A20-B19, A11-B7, A6-A7]) readily form following disulfide exchange in the presence of a chemical denaturant [57]. Whereas the stability of IGF-swap is similar to that of native IGF-I, the accessible non-native isomers of insulin and proinsulin are molten globules of marginal stability [58].
Evidence for the pathological relevance of non-native disulfide isomers of proinsulin and related polypeptides has been obtained in eukaryotic cell culture [20,26,97–99]. An assay for non-native isomers has been developed based on native polyacrylamide gel electrophoresis. This assay exploits the slower mobilities of misfolded proteins, presumably reflecting their less compact structure. Formation of non-native proinsulin isomers has thus been observed on transfection of expression constructs in both S. cerevisciae and rodent neurosecretory cell lines. Although non-native isomers are generally not secreted, amino-acid substitutions in human proinsulin can enhance the fraction of mispairing in the ER [98,99]. Because propensity to misfold in this assay does not correlate with effects of the substitutions on thermodynamic stability in vitro, its mechanism is not well understood.
Studies of proinsulin variants containing substitutions or deletions in the N-terminal segment of the B chain suggest that the contribution of specific side chains to foldability may not be apparent in the native state [100]. We imagine that the substituted side chains perturb the relative stabilities or kinetic accessibility of disulfide intermediates disproportionately to their effects on the native state, once achieved. Alternatively, it is possible that these residues contribute to interactions of the nascent polypeptide with ER chaperones and the oxidative machinery of disulfide pairing [101]. The ER of β-cells may contain a lineage-specific set of chaperones and foldases required in combination for insulin biosynthesis. Defining such an “ER proteome” of β-cell-specific foldases represents a major goal of current studies. Altered ER proteomes in non-neurosecretory cell lines is likely to underlie their impaired ability to support the efficient folding and secretion of proinsulin [102].
Remarkably, mini-proinsulin (which refolds in vitro more efficiently than proinsulin [103]) misfolds in S. cerevisciae to form a metastable disulfide isomer as the predominant product. This non-native protein is secreted, indicating that its globular structure can resemble a native state sufficiently to fulfill quality-control checkpoints. Quantitative misfolding of mini-proinsulin in yeast demonstrates that the ER folding machinery of a eukaryotic cell can selectively target folding into a non-ground-state conformation. It would be of future interest to characterize this alternative pairing scheme and determine the extent of structural resemblance to the native fold. Because the aberrant protein is not degraded prior to exit through the ER and GA, mini-proinsulin provides an example of “stealth” misfolding, leading to secretion of a protein caught in a kinetic trap.
Diabetes-Associated Mutations
The majority of mutations in the insulin gene lead to permanent neonatal-onset DM [8]. Impaired β-cell function develops prior to maturation of the immune system and so presents as an auto-antigen negative form of apparent Type 1 DM. This presentation may be due to mutations in genes other than the insulin gene [104]. The most common cause is a heterozygous activating mutation in a subunit of the β-cell voltage-gated potassium channel, either KCNJ11 (encoding the Kr6.2 subunit) or ABCC8 (encoding the Sur1 subunit) [105,106]. The resulting diabetic phenotype may be transient or permanent. In favorable cases recognition of this syndrome enables patients to be treated with oral agents that inhibit the channel (sulfonylureas) rather than insulin [104].
Dominant mutations in the insulin gene define the second most common genetic cause of permanent neonatal DM [6–8,10]. Such mutations occur in each region of preproinsulin: its signal peptide, A-, B- and C domains (Fig. 1A). The majority of such mutations result in the addition or removal of a cysteine, leading in either case to an odd number of potential pairing sites. This imbalance leads in general to misfolding and aggregation [4,5,12]. Remarkably, the catalogue of human mutations encodes the same “Akita” substitution (CysA7→Tyr) as in the Ins2 gene of the Mody4 mouse [14–16]; this dominant murine substitution thus provides an attractive model of progressive postnatal β-cell failure [18–20]. The variant murine proinsulin in vitro undergoes partial unfolding with increased aggregation [107]. Analogous perturbations were characterized in human insulin- and proinsulin analogs lacking cystine A7-B7 [47,91]. Heterozygous expression of a variant Ins2 allele encoding CysA6→Ser (identified in an N-ethyl-N-nitrosourea screen) likewise induces DM [108].
The identification of identical human and murine mutations at position A7 suggests that the pathogenesis of neonatal DM in these patients is likewise similar to that of the Akita mouse, which has been extensively characterized [14–16,18–20]. Although the mechanism of β-cell degeneration in the Akita mouse remains incompletely understood, its β-cells exhibit an early defect in the folding and trafficking of both wild-type and variant proinsulins. Such impaired biosynthesis is accompanied by elevated markers of ER stress, electron-dense deposits in abnormal ER and GA, mitochondria swelling, and progressive loss of β-cell mass [18–20]. Extension of such findings to human variants has been aided by the construction of innovative fluorescent proinsulin fusion proteins and their use in cell lines and transgenic mice as probes of subcellular localization, and aggregation [30–33].
Biochemical studies of clinical variants in β-cells or neurosecretory cell lines have revealed perturbations in disulfide pairing, which range from severe or mild depending on the site of mutation and the properties of the substituted side chain [1–3]. Whereas an odd number of cysteines presumably induces a severe block to folding, better-tolerated mutations present later in life as auto-antibody-negative presumed Type 1 DM or Type 2 DM. One such mutation, presenting in the second decade as maturity-onset diabetes of the young (MODY), is due to substitution of ArgB22 by Gln. This mutation alters a solvent-exposed site in the B21-B24 β-turn not required for receptor binding [6–8,10]. DM presenting in the third decade of life is associated with substitution of PheB24 by Ser [109]. This mutation causes only a mild impairing of receptor binding [110] but like GlnB22 imposes ER stress at a level intermediate between over-expression of wild-type proinsulin and neonatal variants [5]. Chronic elevation of moderate ER stress in β-cells presumably leads to a slow but progressive loss of β-cell mass.
Evidence is accumulating that subtle perturbations of insulin biosynthesis (due either to variant insulin genes or mutations in the ER folding machinery) may contribute to the pathogenesis of non-syndromic Type 2 DM in humans [111,112] as in the Akita mouse [17,113]. A newly recognized biophysical contribution to such β-cell dysfunction is “molecular crowding” in the ER due to over-expression of nascent proinsulin in the face of peripheral insulin resistance [114], a concept that may have therapeutic implications [33].
Mutations in the insulin gene unassociated with the defective folding of proinsulin can also be associated with adult-onset phenotypes of variable penetrance [115]. The diversity of genetic syndromes reflects a range of molecular mechanisms in accord with Murphy’s Law of genetics. Classical insulinopathies (LeuA3 and LeuB25), for example, markedly perturb receptor binding [109], leading to reduced clearance and hence mutant hyperinsulinemia [110]. Substitution of HisB10 by Asp by contrast would enhance the receptor-binding activity of the variant insulin if secreted [116], but within β-cells leads to aberrant sorting of the mutant proinsulin to a constitutive (i.e., non-glucose-regulated) granule [117,118]. Because constitutive granules lack prohormone convertases, the patients exhibit mutant hyperproinsulinemia. Yet another syndrome is caused by a mutation at the CA junction [119]. Impaired prohormone processing leads to the secretion of a split proinsulin, which has reduced biological activity. Secretion of mutant insulins or proinsulins in the above patients does not exclude concomitant induction of chronic ER stress or perturbation of other organelles as a contributing pathobiological mechanism. Indeed, we speculate that the variable genetic penetrance of the above syndromes may be the hallmark of a genetic variant that modifies long-term survival of β-cells as one component of a multi-genic trait.
Structural Implications
Mutations in proinsulin associated with neonatal DM can encode amino-acid substitutions not involving cysteine (for review, see [13]). Within the insulin moiety such mutations have been found only in the B domain. These substitutions are of structural interest in relation to the mechanism of protein folding. The mutations occur within the N-terminal segment of the B-chain (residues B1-B8), central α-helix (B9-B19), and adjoining β-turn (B20-B23). An overview of key positions in these elements in provided in Figure 3B. We discuss these structural elements in turn.
Figure 3. Critical sites governing the foldability of proinsulin are widely distributed in insulin.
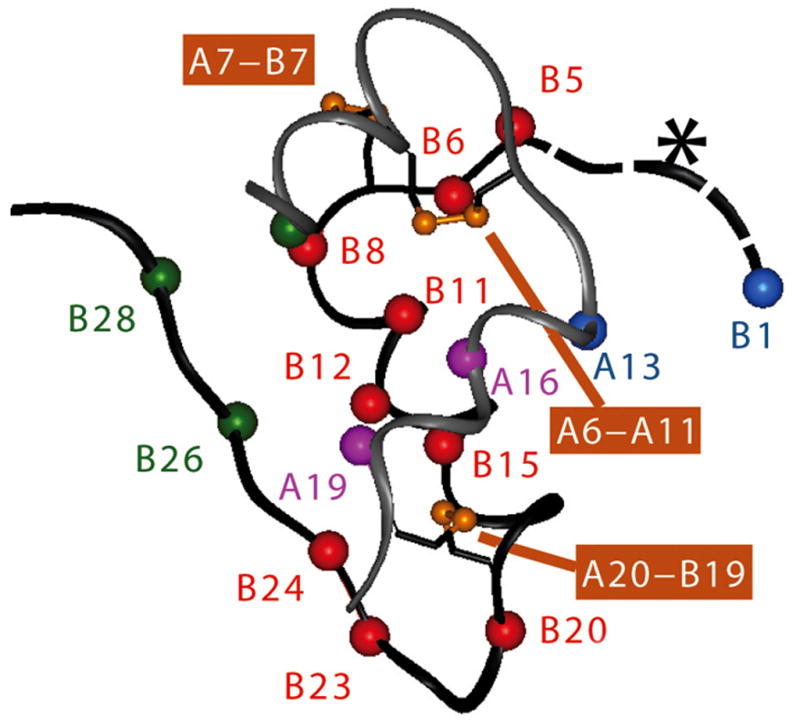
Asterisk indicates N-terminal segment of the B chain, which promotes foldability but is dispensable in the mature hormone [100]. Conserved side chains in or adjoining the C-terminal α-helix of the A chain (LeuA16 and TyrA19; Cα purple spheres) and at multiple sites in the B chain (Cα red spheres) impair insulin chain combination in accord with studies of mutant proinsulins in mammalian cell lines and the distribution of clinical non-cysteine mutations in the insulin gene. Contacts between the side chains PheB1 and IleA13 (Cα blue spheres), although not well ordered in the native state, contribute to the cellular foldability of proinsulin. Residues IleA2, TyrB26, and ProB28 (Cα green spheres) contribute to the structure and stability of the native state but are not required for efficient disulfide pairing in chain combination. Disulfide bridges are as indicated (orange). Coordinates were obtained from Protein Databank file 4INS and correspond to molecule 1 of the classical 2-Zn insulin hexamer [123].
N-terminal segment
In both the solution structure of insulin [41,120,121] and its crystallographic T-state [122,123] residues B1-B6 are extended (asterisk in Fig. 3) whereas B7-B10 comprise a β-turn adjoining the central α-helix. N-terminal residues contribute to the specification of the A7-B7 disulfide bridge in vitro [95,96] and to the overall efficiency of proinsulin folding in cell lines [100]. Sites of clinical mutation (HisB5 and GlyB8; broadly conserved among vertebrate insulins) have been well characterized [26,76,78,79]. (i) In the native state HisB5 packs within an inter-chain crevice, making one or more hydrogen bonds to carbonyl oxygens in the A chain. Diverse substitutions impair chain combination [26]; ArgB5 (which is observed in some non-mammalian insulins) is by contrast well tolerated, presumably through formation of analogous inter-chain hydrogen bonds as visualized in its crystal structure [79]. In mammalian cell culture substitution of HisB5 by Asp blocks the folding and secretion of human proinsulin [26]. (ii) GlyB8 ordinarily exhibits a positive φ dihedral angle and so occupies a position in the Ramachandran plane ordinarily forbidden to L-amino acids. Any L-substitution at this site impairs chain combination whereas yield is enhanced by D-substitutions [76]. Such stereospecificity implies that the sign of the B8 φ angle contributes to the alignment or misalignment of neighboring CysB7 for disulfide pairing.
The solution structure of AlaB5 and SerB8 insulin analogs retain native-like features and exhibit substantial biological activity despite their reduced foldabilities [26,78]. Decreased thermodynamic stabilities are observed, presumably due to altered inter-chain packing (AlaB5) or an unfavorable main-chain conformation (SerB8). Whereas decreased native-state stability can in principle contribute to misfolding, it is likely that such perturbations also impose a kinetic block to disulfide pairing and concomitantly decrease the stability of populated folding intermediates. Evidence for such kinetic blocks has been provided by cellular studies in which these and related mutations lead to formation of non-native disulfide isomers [26,78]. Interestingly, the substantial biological activities of such “non-foldable” analogs [26,78,100] indicates that residues required for efficient folding may be dispensable in the native state [124,125].
Central α-helix
Nascent folding of the B-chain α-helix is observed in equilibrium models of the key [A20-B19] folding intermediate [27,40] and subsequent two-disulfide intermediates [44,47,48,91]. In these models LeuB11 and LeuB15 each contribute to the nascent clustering of nonpolar residues. It is thus plausible that clinical mutations at these sites would impede initial disulfide pairing and, should native disulfide pairing be achieved, perturb native structure and assembly. Ala substitutions at B11 and B15 (although helicogenic in model peptides) impair the expression and secretion of mini-proinsulin in S. cerevisciae [93]. Similarly, insulin chain combination is markedly impaired by interchange of LeuB11 and ValB12, which would be predicted to perturb tertiary structure without net change in intrinsic helical potential [73]. It is not clear why substitution of TyrB16 by His, a clinical mutation that is predicted to project at the surface of this α-helix [7], may impair folding.
B20-B23 β-Turn
B-chain super-secondary structure requires a β-turn between the central α-helix and the neighboring β-strand (residues B24-B28). This turn contains conserved glycines at B20 and B23, each of which exhibits a positive φ dihedral angle. Whereas Ala substitutions impair the expression of mini-proinsulin in S. cerevisciae and impede chain combination, efficient disulfide pairing (like that at B8 above) can be rescued by D-amino acid substitutions. A clinical mutation has been found at B23 and is expected to perturb pairing of cystine A20-B19. The mechanism by which a mutation at B22 (Arg→Gln; see above) causes MODY is not apparent.
The absence of clinical mutations in the A domain may reflect incomplete sampling of patients, or alternatively, may indicate that non-cysteine residues in the A domain plays a less critical role in disulfide pairing than the B domain. We believe that both explanations have partial validity. On the one hand, studies of the mechanism of chain combination have shown that the N-terminal A-chain α-helix (residues A1-A8) is not required for disulfide pairing [75]. Such dispensability is in accord with a putative structural pathway in which segmental folding of this α-helix is a late event (Fig. 2B). On the other hand, the efficiency of chain combination is exquisitely sensitive to substitutions of core residue in the C-terminal A-chain α-helix (LeuA16 and TyrA19; purple spheres in Fig. 3) [74,126]. Accordingly, we anticipate that continuing genetic screening of infants and children with antibody-negative DM will uncover analogous A-domain mutations.
Protein Folding and Induced Fit
The evolution of insulin is enjoined by multiple biological constraints, including biosynthesis, structure, and function. We imagine that conserved residues may contribute to one or more essential requirements of viability: foldability in the β-cell, protection from intra- or extracellular toxic misfolding, self-assembly within the secretory granule, and receptor binding. The overlapping nature of these constraints may account for the limited sequence diversity among vertebrate insulins [123].
Evolutionary constraints may be coincident at some positions but opposing at others. Studies of stereospecific B8 analogs, for example, suggest that at this site kinetic determinants of foldability are at odds with conformational requirements of receptor binding [76,78]. Whereas a positive φ dihedral angle (like that of a D-amino acid) facilitates disulfide pairing, a negative dihedral angle (like that of an L-amino acid) is required for receptor binding [76,78]. These opposing requirements presumably underlie the invariance of glycine – the only achiral amino acid – at a site of conformational change. Indeed, a classical model for the induced fit of insulin on receptor binding is provided by the TR transition among zinc insulin hexamers [127]. Although the choreography of conformational changes seen in the TR transition may pertain only to hexamers [79], the transition is remarkable for a change in the sign of the B8 dihedral angle from positive (the foldable state) to negative (the active state). That B8 provides a hot spot for mutations causing neonatal DM in turn suggests that induced fit provides protective mechanism: a premature setting of the B8 dihedral angle in its active conformation within the β-cell may lead to toxic misfolding.
The recent co-crystal structure of insulin bound to a fragment of the IR ectodomain has provided evidence for induced fit within the C-terminal segment B-chain [128]. As predicted based on studies of anomalous insulin analogs [120,129,130], detachment of the C-terminal β-strand (residues B24-B28) enables docking of a receptor α-helix (denoted αCT) against the N-terminal A-chain α-helix. Such induced fit of the B chain may likewise represent an evolutionary response to the danger of proteotoxicity. Indeed, pre-detachment of the B-chain β-strand by a chiral perturbation (substitution by PheB24 by D-Ala) enhances receptor binding at the expense of increased susceptibility to fibrillation [130]. The aromatic ring of PheB24 anchors the native β-strand, stabilizes the hydrophobic core, contributes to dimerization and hexamer formation [123], enables induced fit on receptor binding [130], and even further, is likely to contact the receptor itself [131]. Much remains to be learned. How induced fit of the B chain is triggered on IR binding, the nature of the switch at (or near) residue B24, and the contacts subsequently made by PheB24, PheB25, and TyrB26 were not addressed in the recent co-crystal structure of a “micro-receptor” model [128].
Understanding the co-evolution of insulin and the IR will require further structural analysis of their binding interfaces. A framework is provided by the elegant classification of hormone-binding surfaces in the receptor as Site 1 and Site 2 [132,133]. The cognate IR-binding surfaces of insulin correspond in large part to its respective dimerization and hexamer-forming surfaces (Fig. 4) (for review, see [134]). Because the recent co-crystal structure was restricted to Site 1, it will be of particular interest to extend such analysis to larger fragments of the ectodomain containing Site 2 surfaces. Given the ancestral status of insulin signaling in both vertebrates and invertebrates [34–36], an intriguing possibility exists that distinct evolutionary constraints and histories are associated with these complementary interfaces. Such an historical understanding would highlight the relationship between proinsulin and single-chain homologues (including mammalian IGF-I and IGF-II), which may have translational implications in cancer biology [37,135].
Figure 4. Proposed binding surfaces of insulin.

(A) Front view and (B) back view. Whereas the classical receptor-binding surface of insulin engages IR Site 1 (blue; Ref 132,133, its Site 2-related surface includes hexamer contacts HisB10, ValB18, SerA12, LeuA13 and GluA17 (red). The A- and B chains are otherwise shown in light gray and dark gray, respectively. The structures shown are based on an R crystallographic protomer, which contains a receptor-active positive phi angle at GlyB24 (in accordance with the results of Ref 76) but is otherwise unlikely to represent the receptor-bound conformation of insulin [128].
We imagine in general that conserved side chains in insulin play multiple distinct roles at each stage of its conformational “life cycle,” representing a remarkable compression of information within a short protein sequence. It would be of future interest to probe the range of structures within this life cycle through crystal structures of insulin-holoreceptor complexes and through solid-state NMR-based models of non-native insulin aggregates and fibrils.
Concluding Remarks
The mutant proinsulin syndrome provides an opportunity to investigate principles of protein folding at the intersection of cell biology and human genetics. Consistent with classical diffusion-collision and framework models [136], folding of a globular protein represents the coalescence of discrete subdomains [137,138]. Whereas funnel-like energy landscapes highlight the role of parallel events in folding [84], evidence for preferred trajectories [139] has been provided by disulfide trapping studies of insulin-related polypeptides. Characterization of predominant intermediates promises to enable structural interpretation of many of the clinical mutations. Sites of mutation reflect mechanisms of folding or misfolding that may not be apparent in the native state [13].
Foldability of proteins is an evolved property of polypeptide sequences [140]. The general threat of toxic misfolding provides a hidden constraint underlying such evolution. Application of these principles to insulin promises to connect bench to bedside. An intrinsic tension between folding-competent and active conformations of insulin, only partially resolved in the wild-type hormone by induced fit, may underlie the role of chronic ER stress in the progression of β-cell dysfunction in Type 2 DM. Molecular dissection of this progression promises to provide novel therapeutic targets.
Acknowledgments
This work was supported by grants from the American Diabetes Association and National Institutes of Health (DK040949 and DK0697674) to M.A.W. The author thanks Q.-X. Hua and Y. Yang for assistance with figures, N. F. Phillips for advice, and P. Arvan, S. B. Kent, M. C. Lawrence, M. Liu, D. F. Steiner, and J. Whittaker for discussion. This article is a contribution from the Cleveland Center for Membrane and Structural Biology and is dedicated to the memory of the late Prof. G. G. Dodson.
Abbreviations
- DM
diabetes mellitus
- ER
endoplasmic reticulum
- GA
Golgi apparatus
- MODY
maturity-onset diabetes of the young
- and NMR
nuclear magnetic resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, Arvan P. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21:652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greeley SA, Tucker SE, Naylor RN, Bell GI, Philipson LH. Neonatal diabetes mellitus: a model for personalized medicine. Trends Endocrinol Metab. 2010;21:464–472. doi: 10.1016/j.tem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greeley SA, Naylor RN, Philipson LH, Bell GI. Neonatal diabetes: an expanding list of genes allows for improved diagnosis and treatment. Curr Diab Rep. 2011;11:519–532. doi: 10.1007/s11892-011-0234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SY, Ye H, Steiner DF, Bell GI. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2010;391:1449–1454. doi: 10.1016/j.bbrc.2009.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu M, Haataja L, Wright J, Wickramasinghe DN, Hua QX, Phillips NB, Barbetti F, Weiss MA, Arvan P. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on nonmutant proinsulin transport. PLos-One. 2010;5:e13333. doi: 10.1371/journal.pone.0013333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, Beccaria L, Monciotti C, Toni S, Pedersen O, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J Clin Invest. 2008;118:2148–2156. doi: 10.1172/JCI33777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, et al. Insulin mutation screening in 1044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polak M, Dechaume A, Cave H, Rivtal N, Crosnier H, Sulmont V, De Kerdanet M, Scharfmann R, Lebenthal U, Froguel P, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy. Diabetes. 2008;57:1115–1119. doi: 10.2337/db07-1358. [DOI] [PubMed] [Google Scholar]
- 10.Molven A, Ringdal M, Nordbo AM, Raeder H, Stoy J, Lipkind GM, Steiner DF, Philipson LH, Bergmann I, Aarskog D, et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes. 2008;57:1131–1135. doi: 10.2337/db07-1467. [DOI] [PubMed] [Google Scholar]
- 11.Boesgaard TW, Pruhova S, Andersson EA, Cinek O, Obermannova B, Lauenborg J, Damm P, Bergholdt R, Pociot F, Pisinger C, et al. Further evidence that mutations in INS can be a rare cause of Maturity-Onset Diabetes of the Young (MODY) BMC Med Genet. 2010;12:42. doi: 10.1186/1471-2350-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, Fetita S, Tarasov AI, Guillausseau PJ, Boesgaard TW, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. 2010;59:653–661. doi: 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss MA. Proinsulin and the genetics of diabetes mellitus. J Biol Chem. 2009;284:19159–19163. doi: 10.1074/jbc.R109.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic β-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest. 2002;109:443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, Takeuchi T. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52:409–416. doi: 10.2337/diabetes.52.2.409. [DOI] [PubMed] [Google Scholar]
- 19.Zuber C, Fan JY, Guhl B, Roth J. Misfolded proinsulin accumulates in expanded pre-Golgi intermediates and endoplasmic reticulum subdomains in pancreatic β cells of Akita mice. FASEB J. 2004;18:917–919. doi: 10.1096/fj.03-1210fje. [DOI] [PubMed] [Google Scholar]
- 20.Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci U S A. 2007;104:15841–15846. doi: 10.1073/pnas.0702697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967;157:697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- 22.Steiner DF, Clark JL, Nolan C, Rubenstein AH, Margoliash E, Aten B, Oyer PE. Proinsulin and the biosynthesis of insulin. Recent Prog Horm Res. 1969;25:207–282. doi: 10.1016/b978-0-12-571125-8.50008-9. [DOI] [PubMed] [Google Scholar]
- 23.Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol. 1998;8:189–194. doi: 10.1016/s0959-440x(98)80037-7. [DOI] [PubMed] [Google Scholar]
- 24.Miller JA, Narhi LO, Hua QX, Rosenfeld R, Arakawa T, Rohde M, Prestrelski S, Lauren S, Stoney KS, Tsai L, et al. Oxidative refolding of insulin-like growth factor 1 yields two products of similar thermodynamic stability: a bifurcating protein-folding pathway. Biochemistry. 1993;32:5203–5213. doi: 10.1021/bi00070a032. [DOI] [PubMed] [Google Scholar]
- 25.Qiao ZS, Guo ZY, Feng YM. Putative disulfide-forming pathway of porcine insulin precursor during its refolding in vitro. Biochemistry. 2001;40:2662–2668. doi: 10.1021/bi001613r. [DOI] [PubMed] [Google Scholar]
- 26.Hua QX, Liu M, Hu SQ, Jia W, Arvan P, Weiss MA. A conserved histidine in insulin is required for the foldability of human proinsulin. Structure and function of an AlaB5 analog. J Biol Chem. 2006;281:24889–24899. doi: 10.1074/jbc.M602617200. [DOI] [PubMed] [Google Scholar]
- 27.Hua QX, Mayer J, Jia W, Zhang J, Weiss MA. The folding nucleus of the insulin superfamily: a flexible peptide model foreshadows the native state. J Biol Chem. 2006;281:28131–28142. doi: 10.1074/jbc.M602616200. [DOI] [PubMed] [Google Scholar]
- 28.Qiao ZS, Guo ZY, Feng YM. In vitro folding/unfolding of insulin/single-chain insulin. Protein Pept Lett. 2006;13:423–429. doi: 10.2174/092986606776819583. [DOI] [PubMed] [Google Scholar]
- 29.Guo ZY, Qiao ZS, Feng YM. The in vitro oxidative folding of the insulin superfamily. Antioxid Redox Signal. 2008;10:127–140. doi: 10.1089/ars.2007.1860. [DOI] [PubMed] [Google Scholar]
- 30.Hodish I, Liu M, Rajpal G, Larkin D, Holz RW, Adams A, Liu L, Arvan P. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem. 2010;285:685–694. doi: 10.1074/jbc.M109.038042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodish I, Absood A, Liu L, Liu M, Haataja L, Larkin D, Al-Khafaji A, Zaki A, Arvan P. In vivo misfolding of proinsulin below the threshold of frank diabetes. Diabetes. 2011;60:2092–2101. doi: 10.2337/db10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haataja L, Snapp E, Wright J, Liu M, Hardy AB, Wheeler MB, Markwardt ML, Rizzo M, Arvan P. Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J Biol Chem. 2013;288:1896–1906. doi: 10.1074/jbc.M112.420018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Absood A, Gandomani B, Zaki A, Nasta V, Michail A, Habib PM, Hodish I. Insulin therapy for pre-hyperglycemic β-cell endoplasmic reticulum crowding. PLoS One. 2013;8:e54351. doi: 10.1371/journal.pone.0054351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, Ferguson KC, Heller J, Platt DM, Pasquinelli AA, et al. Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 2001;15:672–686. doi: 10.1101/gad.867301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hua QX, Nakagawa SH, Wilken J, Ramos RR, Jia W, Bass J, Weiss MA. A divergent INS protein in Caenorhabditis elegans structurally resembles human insulin and activates the human insulin receptor. Genes Dev. 2003;17:826–831. doi: 10.1101/gad.1058003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sajid W, Kulahin N, Schluckebier G, Ribel U, Henderson HR, Tatar M, Hansen BF, Svendsen AM, Kiselyov VV, Norgaard P, et al. Structural and biological properties of the Drosophila insulin-like peptide 5 show evolutionary conservation. J Biol Chem. 2011;286:661–673. doi: 10.1074/jbc.M110.156018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A. Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology. 2012;153:2152–2163. doi: 10.1210/en.2011-1843. [DOI] [PubMed] [Google Scholar]
- 38.Galloway JA, Hooper SA, Spradlin CT, Howey DC, Frank BH, Bowsher RR, Anderson JH. Biosynthetic human proinsulin. Review of chemistry, in vitro and in vivo receptor binding, animal and human pharmacology studies, and clinical trial experience. Diabetes Care. 1992;15:666–692. doi: 10.2337/diacare.15.5.666. [DOI] [PubMed] [Google Scholar]
- 39.Steiner DF. Evidence for a precursor in the biosynthesis of insulin. Trans N Y Acad Sci. 1967;30:60–68. doi: 10.1111/j.2164-0947.1967.tb02452.x. [DOI] [PubMed] [Google Scholar]
- 40.Narhi LO, Hua QX, Arakawa T, Fox GM, Tsai L, Rosenfeld R, Holst P, Miller JA, Weiss MA. Role of native disulfide bonds in the structure and activity of insulin-like growth factor 1: genetic models of protein-folding intermediates. Biochemistry. 1993;32:5214–5221. doi: 10.1021/bi00070a033. [DOI] [PubMed] [Google Scholar]
- 41.Hua QX, Hu SQ, Frank BH, Jia W, Chu YC, Wang SH, Burke GT, Katsoyannis PG, Weiss MA. Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J Mol Biol. 1996;264:390–403. doi: 10.1006/jmbi.1996.0648. [DOI] [PubMed] [Google Scholar]
- 42.Dai Y, Tang JG. Characteristic, activity and conformational studies of [A6-Ser, A11-Ser]-insulin. Biochim Biophys Acta. 1996;1296:63–68. doi: 10.1016/0167-4838(96)00054-4. [DOI] [PubMed] [Google Scholar]
- 43.Hober S, Uhlen M, Nilsson B. Disulfide exchange folding of disulfide mutants of insulin-like growth factor I in vitro. Biochemistry. 1997;36:4616–4622. doi: 10.1021/bi9611265. [DOI] [PubMed] [Google Scholar]
- 44.Weiss MA, Hua QX, Jia W, Chu YC, Wang RY, Katsoyannis PG. Hierarchiacal protein “un-design”: insulin’s intrachain disulfide bridge tethers a recognition α-helix. Biochemistry. 2000;39:15429–15440. doi: 10.1021/bi001905s. [DOI] [PubMed] [Google Scholar]
- 45.Guo ZY, Feng YM. Effects of cysteine to serine substitutions in the two intra-A-chain disulfide bonds of insulin. Biol Chem. 2001;382:443–448. doi: 10.1515/BC.2001.054. [DOI] [PubMed] [Google Scholar]
- 46.Feng Y, Liu D, Wang J. Native-like partially folded conformations and folding process revealed in the N-terminal large fragments of staphylococcal nuclease: a study by NMR spectroscopy. J Mol Biol. 2003;330:821–837. doi: 10.1016/s0022-2836(03)00660-0. [DOI] [PubMed] [Google Scholar]
- 47.Jia XY, Guo ZY, Wang Y, Xu Y, Duan SS, Feng YM. Peptide models of four possible insulin folding intermediates with two disulfides. Protein Sci. 2003;12:2412–2419. doi: 10.1110/ps.0389303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan H, Guo ZY, Gong XW, Xi D, Feng YM. A peptide model of insulin folding intermediate with one disulfide. Protein Sci. 2003;12:768–775. doi: 10.1110/ps.0237203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Hua QX, Liu J, Shimizu EH, Choquette MH, Mackin RB, Weiss MA. Solution structure of proinsulin: connecting domain flexibility and prohormone processing. J Biol Chem. 2010;285:7847–7851. doi: 10.1074/jbc.C109.084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frank BH, Veros AJ. Physical studies on proinsulin-association behavior and conformation in solution. Biochem Biophys Res Commun. 1968;32:155–160. doi: 10.1016/0006-291x(68)90362-8. [DOI] [PubMed] [Google Scholar]
- 51.Pekar AH, Frank BH. Conformation of proinsulin. A comparison of insulin and proinsulin self-association at neutral pH. Biochemistry. 1972;11:4013–4016. doi: 10.1021/bi00772a001. [DOI] [PubMed] [Google Scholar]
- 52.Frank BH, Pekar AH, Veros AJ. Insulin and proinsulin conformation in solution. Diabetes. 1972;21:486–491. doi: 10.2337/diab.21.2.s486. [DOI] [PubMed] [Google Scholar]
- 53.Snell CR, Smyth DG. Proinsulin: a proposed three-dimensional structure. J Biol Chem. 1975;250:6291–6295. [PubMed] [Google Scholar]
- 54.Brems DN, Brown PL, Heckenlaible LA, Frank BH. Equilibrium denaturation of insulin and proinsulin. Biochemistry. 1990;29:9289–9293. doi: 10.1021/bi00491a026. [DOI] [PubMed] [Google Scholar]
- 55.Weiss MA, Frank BH, Khait I, Pekar A, Heiney R, Shoelson SE, Neuringer LJ. NMR and photo-CIDNP studies of human proinsulin and prohormone processing intermediates with application to endopeptidase recognition. Biochemistry. 1990;29:8389–8401. doi: 10.1021/bi00488a028. [DOI] [PubMed] [Google Scholar]
- 56.Sieber PS, Eisler K, Kamber B, Riniker B, Rittel W, Marki F, deGasparo M. Synthesis and biological activity of two disulphide bond isomers of human insulin: [A7-A11, A6-B7-cystine]- and [A6-A7, A11-B7-cystine] insulin (human) Hoppe Seylers Z Physiol Chem. 1978;359:113–123. doi: 10.1515/bchm.1978.359.1.113. [DOI] [PubMed] [Google Scholar]
- 57.Hua QX, Gozani SN, Chance RE, Hoffmann JA, Frank BH, Weiss MA. Structure of a protein in a kinetic trap. Nat Struct Biol. 1995;2:129–138. doi: 10.1038/nsb0295-129. [DOI] [PubMed] [Google Scholar]
- 58.Hua QX, Jia W, Frank BH, Phillips NB, Weiss MA. A protein caught in a kinetic trap: structures and stabilities of insulin disulfide isomers. Biochemistry. 2002;41:14700–14715. doi: 10.1021/bi0202981. [DOI] [PubMed] [Google Scholar]
- 59.Huang XF, Arvan P. Formation of the insulin-containing secretory granule core occurs within immature β-granules. J Biol Chem. 1994;269:20838–20844. [PubMed] [Google Scholar]
- 60.Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 61.Greider MH, Howell SL, Lacy PE. Isolation and properties of secretory granules from rat islets of Langerhans. II Ultrastructure of the β granule. J Cell Biol. 1969;41:162–166. doi: 10.1083/jcb.41.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lange RH. Crystalline islet B-granules in the grass snake (Natrix natrix (L.)): tilting experiments in the electron microscope. J Ultrastruct Res. 1974;46:301–307. doi: 10.1016/s0022-5320(74)80064-x. [DOI] [PubMed] [Google Scholar]
- 63.Michael J, Carroll R, Swift HH, Steiner DF. Studies on the molecular organization of rat insulin secretory granules. J Biol Chem. 1987;262:16531–16535. [PubMed] [Google Scholar]
- 64.Huang XF, Arvan P. Intracellular transport of proinsulin in pancreatic β-cells. Structural maturation probed by disulfide accessibility. J Biol Chem. 1995;270:20417–20423. doi: 10.1074/jbc.270.35.20417. [DOI] [PubMed] [Google Scholar]
- 65.Brange J, Langkjaer L. Insulin formation and delivery. In: Sanders LM, Hendren RW, editors. Protein Delivery: Physical Systems. Plenum Press; New York, New York, NY: 1997. pp. 343–410. [Google Scholar]
- 66.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 67.Lemaire K, Ravier MA, Schraenen A, Creemers JW, Van de Plas R, Granvik M, Van Lommel L, Waelkens E, Chimienti F, Rutter GA, et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc Natl Acad Sci U S A. 2009;106:14872–14877. doi: 10.1073/pnas.0906587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katsoyannis PG. Synthesis of insulin. Science. 1966;154:1509–1514. doi: 10.1126/science.154.3756.1509. [DOI] [PubMed] [Google Scholar]
- 69.Tang JG, Tsou CL. The insulin A and B chains contain structural information for the formation of the native molecule. Studies with protein disulphide- isomerase. Biochem J. 1990;268:429–435. doi: 10.1042/bj2680429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang CC, Tsou CL. The insulin A and B chains contain sufficient structural information to form the native molecule. Trends Biochem Sci. 1991;16:279–281. doi: 10.1016/0968-0004(91)90114-b. [DOI] [PubMed] [Google Scholar]
- 71.Brange J. The new era of biotech insulin analogues. Diabetologia. 1997;40:S48–S53. doi: 10.1007/s001250051400. [DOI] [PubMed] [Google Scholar]
- 72.Hirsch IB. Insulin analogues. N Engl J Med. 2005;352:174–183. doi: 10.1056/NEJMra040832. [DOI] [PubMed] [Google Scholar]
- 73.Hu SQ, Burke GT, Schwartz GP, Ferderigos N, Ross JB, Katsoyannis PG. Steric requirements at position B12 for high biological activity in insulin. Biochemistry. 1993;32:2631–2635. doi: 10.1021/bi00061a022. [DOI] [PubMed] [Google Scholar]
- 74.Weiss MA, Nakagawa SH, Jia W, Xu B, Hua QX, Chu YC, Wang RY, Katsoyannis PG. Protein structure and the spandrels of San Marco: insulin’s receptor-binding surface is buttressed by an invariant leucine essential for protein stability. Biochemistry. 2002;41:809–819. doi: 10.1021/bi011839+. [DOI] [PubMed] [Google Scholar]
- 75.Hua QX, Chu YC, Jia W, Phillips NB, Wang RY, Katsoyannis PG, Weiss MA. Mechanism of insulin chain combination. Asymmetric roles of A-chain α-helices in disulfide pairing. J Biol Chem. 2002;277:43443–43453. doi: 10.1074/jbc.M206107200. [DOI] [PubMed] [Google Scholar]
- 76.Nakagawa SH, Zhao M, Hua QX, Hu SQ, Wan ZL, Jia W, Weiss MA. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry. 2005;44:4984–4999. doi: 10.1021/bi048025o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakagawa SH, Hua QX, Hu SQ, Jia W, Wang S, Katsoyannis PG, Weiss MA. Chiral mutagenesis of insulin. Contribution of the B20-B23 β-turn to activity and stability. J Biol Chem. 2006;281:22386–22396. doi: 10.1074/jbc.M603547200. [DOI] [PubMed] [Google Scholar]
- 78.Hua QX, Nakagawa SH, Hu SQ, Jia W, Wang S, Weiss MA. Toward the active conformation of insulin. Stereospecific modulation of a structural switch in the B chain. J Biol Chem. 2006;281:24900–24909. doi: 10.1074/jbc.M602691200. [DOI] [PubMed] [Google Scholar]
- 79.Wan Z, Huang K, Whittaker J, Weiss MA. The structure of a mutant insulin uncouples receptor binding from protein allostery. An electrostatic block to the TR transition. J Biol Chem. 2008;283:21198–21210. doi: 10.1074/jbc.M800235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baldwin TO, Ziegler MM, Chaffotte AF, Goldberg ME. Contribution of folding steps involving the individual subunits of bacterial luciferase to the assembly of the active heterodimeric enzyme. J Biol Chem. 1993;268:10766–10772. [PubMed] [Google Scholar]
- 81.Huang Y, Liang Z, Feng Y. The relationship between the connecting peptide of recombined single chain insulin and its biological function. Sci China. 2001;44:593–600. doi: 10.1007/BF02879353. [DOI] [PubMed] [Google Scholar]
- 82.Hober S, Forsberg G, Palm G, Hartmanis M, Nilsson B. Disulfide exchange folding of insulin-like growth factor I. Biochemistry. 1992;31:1749–1756. doi: 10.1021/bi00121a024. [DOI] [PubMed] [Google Scholar]
- 83.Creighton TE. Protein folding coupled to disulphide bond formation. Biol Chem. 1997;378:731–744. doi: 10.1515/bchm.1997.378.8.731. [DOI] [PubMed] [Google Scholar]
- 84.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 85.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 86.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 87.Qiao ZS, Min CY, Hua QX, Weiss MA, Feng YM. In vitro refolding of human proinsulin. Kinetic intermediates, putative disulfide-forming pathway, folding initiation site, and protential role of C-peptide in folding process. J Biol Chem. 2003;278:17800–17809. doi: 10.1074/jbc.M300906200. [DOI] [PubMed] [Google Scholar]
- 88.Hober S, Hansson A, Uhlen M, Nilsson B. Folding of insulin-like growth factor I is thermodynamically controlled by insulin-like growth factor binding protein. Biochemistry. 1994;33:6758–6761. doi: 10.1021/bi00188a003. [DOI] [PubMed] [Google Scholar]
- 89.Milner SJ, Carver JA, Ballard FJ, Francis GL. Probing the disulfide folding pathway of insulin-like growth factor-I. Biotechnol Bioeng. 1999;62:693–703. [PubMed] [Google Scholar]
- 90.Hua QX, Narhi L, Jia W, Arakawa T, Rosenfeld R, Hawkins N, Miller JA, Weiss MA. Native and non-native structure in a protein-folding intermediate: spectroscopic studies of partially reduced IGF-I and an engineered alanine model. J Mol Biol. 1996;259:297–313. doi: 10.1006/jmbi.1996.0320. [DOI] [PubMed] [Google Scholar]
- 91.Hua QX, Nakagawa SH, Jia W, Hu SQ, Chu YC, Katsoyannis PG, Weiss MA. Hierarchical protein folding: asymmetric unfolding of an insulin analogue lacking the A7-B7 interchain disulfide bridge. Biochemistry. 2001;40:12299–12311. doi: 10.1021/bi011021o. [DOI] [PubMed] [Google Scholar]
- 92.Chu YC, Burke GT, Chanley JD, Katsoyannis PG. Possible involvement of the A20-A21 peptide bond in the expression of the biological activity of insulin. 2 [21-Asparagine diethylamide-A]insulin. Biochemistry. 1987;26:6972–6975. doi: 10.1021/bi00396a017. [DOI] [PubMed] [Google Scholar]
- 93.Kristensen C, Kjeldsen T, Wiberg FC, Schaffer L, Hach M, Havelund S, Bass J, Steiner DF, Andersen AS. Alanine scanning mutagenesis of insulin. J Biol Chem. 1997;272:12978–12983. doi: 10.1074/jbc.272.20.12978. [DOI] [PubMed] [Google Scholar]
- 94.Gill R, Verma C, Wallach B, Urso B, Pitts J, Wollmer A, De Meyts P, Wood S. Modelling of the disulphide-swapped isomer of human insulin-like growth factor-1: implications for receptor binding. Protein Eng. 1999;12:297–303. doi: 10.1093/protein/12.4.297. [DOI] [PubMed] [Google Scholar]
- 95.Chen Y, You Y, Jin R, Guo ZY, Feng YM. Sequences of B-chain/domain 1-10/1-9 of insulin and insulin-like growth factor 1 determine their different folding behavior. Biochemistry. 2004;43:9225–9233. doi: 10.1021/bi049710y. [DOI] [PubMed] [Google Scholar]
- 96.Huang QL, Zhao J, Tang YH, Shao SQ, Xu GJ, Feng YM. The sequence determinant causing different folding behaviors of insulin and insulin-like growth factor-1. Biochemistry. 2007;46:218–224. doi: 10.1021/bi0616798. [DOI] [PubMed] [Google Scholar]
- 97.Liu M, Ramos-Castaneda J, Arvan P. Role of the connecting peptide in insulin biosynthesis. J Biol Chem. 2003;278:14798–14805. doi: 10.1074/jbc.M212070200. [DOI] [PubMed] [Google Scholar]
- 98.Zhang BY, Liu M, Arvan P. Behavior in the eukaryotic secretory pathway of insulin-containing fusion proteins and single-chain insulins bearing various B-chain mutations. J Biol Chem. 2003;278:3687–3693. doi: 10.1074/jbc.M209474200. [DOI] [PubMed] [Google Scholar]
- 99.Liu M, Li Y, Cavener D, Arvan P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem. 2005;280:13209–13212. doi: 10.1074/jbc.C400475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu M, Hua QX, Hu SQ, Jia W, Yang Y, Saith SE, Whittaker J, Arvan P, Weiss MA. Deciphering the hidden informational content of protein sequences: foldability of proinsulin hinges on a flexible arm that is dispensable in the mature hormone. J Biol Chem. 2010;285:30989–31001. doi: 10.1074/jbc.M110.152645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Noiva R. Protein disulfide isomerase: the multifunctional redox chaperone of the endoplasmic reticulum. Semin Cell Dev Biol. 1999;10:481–493. doi: 10.1006/scdb.1999.0319. [DOI] [PubMed] [Google Scholar]
- 102.Zhu YL, Abdo A, Gesmonde JF, Zawalich KC, Zawalich W, Dannies PS. Aggregation and lack of secretion of most newly synthesized proinsulin in non-β-cell lines. Endocrinology. 2004;145:3840–3849. doi: 10.1210/en.2003-1512. [DOI] [PubMed] [Google Scholar]
- 103.Markussen J. Comparative reduction/oxidation studies with single chain des-(B30) insulin and porcine proinsulin. Int J Pept Protein Res. 1985;25:431–434. doi: 10.1111/j.1399-3011.1985.tb02197.x. [DOI] [PubMed] [Google Scholar]
- 104.Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:200–213. doi: 10.1038/ncpendmet0778. [DOI] [PubMed] [Google Scholar]
- 105.Slingerland AS, Hattersley AT. Mutations in the Kir6.2 subunit of the KATP channel and permanent neonatal diabetes: new insights and new treatment. Ann Med. 2005;37:186–195. doi: 10.1080/07853890510007287. [DOI] [PubMed] [Google Scholar]
- 106.Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–466. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 107.Yoshinaga T, Nakatome K, Nozaki J, Naitoh M, Hoseki J, Kubota H, Nagata K, Koizumi A. Proinsulin lacking the A7-B7 disulfide bond, Ins2Akita, tends to aggregate due to the exposed hydrophobic surface. Biol Chem. 2005;386:1077–1085. doi: 10.1515/BC.2005.124. [DOI] [PubMed] [Google Scholar]
- 108.Herbach N, Rahtkolb B, Kemter E, Pichl L, Klaften M, De Angelis MH, Halban P, Wolf E, Aigner B, Wanker R. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe β-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56:1268–1276. doi: 10.2337/db06-0658. [DOI] [PubMed] [Google Scholar]
- 109.Shoelson S, Haneda M, Blix P, Nanjo A, Sanke T, Inouye K, Steiner D, Rubenstein A, Tager H. Three mutant insulins in man. Nature. 1983;302:540–543. doi: 10.1038/302540a0. [DOI] [PubMed] [Google Scholar]
- 110.Shoelson SE, Polonsky KS, Zeidler A, Rubenstein AH, Tager HS. Human insullin B24 (Phe→Ser), secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J Clin Invest. 1984;73:1351–1358. doi: 10.1172/JCI111338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol Metab. 2011;22:266–274. doi: 10.1016/j.tem.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thomas SE, Dalton L, Malzer E, Marciniak SJ. Unravelling the story of protein misfolding in diabetes mellitus. World J Diabetes. 2011;2:114–118. doi: 10.4239/wjd.v2.i7.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yuan Q, Tang W, Zhang X, Hinson JA, Liu C, Osei K, Wang J. Proinsulin atypical maturation and disposal induces extensive defects in mouse Ins2+/Akita β-cells. PLoS One. 2012;7:e35098. doi: 10.1371/journal.pone.0035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Despa F. Endoplasmic reticulum overcrowding as a mechanism of β-cell dysfunction in diabetes. Biophys J. 2010;98:1641–1648. doi: 10.1016/j.bpj.2009.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care. 1990;13:600–609. doi: 10.2337/diacare.13.6.600. [DOI] [PubMed] [Google Scholar]
- 116.Schwartz GP, Burke GT, Katsoyannis PG. A superactive insulin: [B10-aspartic acid]insulin(human) Proc Natl Acad Sci U S A. 1987;84:6408–6411. doi: 10.1073/pnas.84.18.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carroll RJ, Hammer RE, Chan SJ, Swift HH, Rubenstein AH, Steiner DF. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc Natl Acad Sci U S A. 1988;85:8943–8947. doi: 10.1073/pnas.85.23.8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gross DJ, Halban PA, Kahn CR, Weir GC, Villa-Komaroff L. Partial diversion of a mutant proinsulin (B10 aspartic acid) from the regulated to the constitutive secretory pathway in transfected AtT-20 cells. Proc Natl Acad Sci U S A. 1989;86:4107–4111. doi: 10.1073/pnas.86.11.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chan SJ, Seino S, Gruppuso PA, Schwartz R, Steiner DF. A mutation in the B chain coding region is associated with impaired proinsulin conversion in a family with hyperproinsulinemia. Proc Natl Acad Sci U S A. 1987;84:2194–2197. doi: 10.1073/pnas.84.8.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hua QX, Shoelson SE, Kochoyan M, Weiss MA. Receptor binding redefined by a structural switch in a mutant human insulin. Nature. 1991;354:238–241. doi: 10.1038/354238a0. [DOI] [PubMed] [Google Scholar]
- 121.Olsen HB, Ludvigsen S, Kaarsholm NC. Solution structure of an engineered insulin monomer at neutral pH. Biochemistry. 1996;35:8836–8845. doi: 10.1021/bi960292+. [DOI] [PubMed] [Google Scholar]
- 122.Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DC, Mercola DA, Vijayan M. Atomic positions in rhombohedral 2-zinc insulin crystals. Nature. 1971;231:506–511. doi: 10.1038/231506a0. [DOI] [PubMed] [Google Scholar]
- 123.Baker EN, Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DM, Hubbard RE, Isaacs NW, Reynolds CD. The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos Trans R Soc Lond B Biol Sci. 1988;319:369–456. doi: 10.1098/rstb.1988.0058. [DOI] [PubMed] [Google Scholar]
- 124.Betts S, King J. There’s a right way and a wrong way: in vivo and in vitro folding, misfolding and subunit assembly of the P22 tailspike. Structure. 1999;7:R131–R139. doi: 10.1016/s0969-2126(99)80078-1. [DOI] [PubMed] [Google Scholar]
- 125.Weigele PR, Haase-Pettingell C, Campbell PG, Gossard DC, King J. Stalled folding mutants in the triple β-helix domain of the phage P22 tailspike adhesin. J Mol Biol. 2005;354:1103–1117. doi: 10.1016/j.jmb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 126.Liu M, Wan ZL, Chu YC, Aladdin H, Klaproth B, Choquette MH, Hua QX, Mackin RB, Rao JS, De Meyts P, et al. Crystal structure of a “non-foldable” insulin: Impaired folding efficiency and ER stress despite native activity. J Biol Chem. 2009;284:35259–35272. doi: 10.1074/jbc.M109.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brader ML, Dunn MF. Insulin hexamers: new conformations and applications. Trends Biochem Sci. 1991;16:341–345. doi: 10.1016/0968-0004(91)90140-q. [DOI] [PubMed] [Google Scholar]
- 128.Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GKW, Smith BJ, Watson CJ, Žáková L, Kletvikova E, Jiráček J, et al. How insulin engages its primary binding site on the insulin receptor surface. Nature. 2013;493:241–245. doi: 10.1038/nature11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mirmira RG, Nakagawa SH, Tager HS. Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J Biol Chem. 1991;266:1428–1436. [PubMed] [Google Scholar]
- 130.Hua QX, Xu B, Huang K, Hu SQ, Nakagawa S, Jia W, Wang S, Whittaker J, Katsoyannis PG, Weiss MA. Enhancing the activity of insulin by stereospecific unfolding. Conformational life cycle of insulin and its evolutionary origins. J Biol Chem. 2009;284:14586–14596. doi: 10.1074/jbc.M900085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xu B, Hu SQ, Chu YC, Wang S, Wang RY, Nakagawa SH, Katsoyannis PG, Weiss MA. Diabetes-associated mutations in insulin identify invariant receptor contacts. Diabetes. 2004;53:1599–1602. doi: 10.2337/diabetes.53.6.1599. [DOI] [PubMed] [Google Scholar]
- 132.De Meyts P. The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signaling. Diabetologia. 1994;37:S135–S148. doi: 10.1007/BF00400837. [DOI] [PubMed] [Google Scholar]
- 133.Schäffer L. A model for insulin binding to the insulin receptor. Eur J Biochem. 1994;221:1127–1132. doi: 10.1111/j.1432-1033.1994.tb18833.x. [DOI] [PubMed] [Google Scholar]
- 134.De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov. 2002;1:769–783. doi: 10.1038/nrd917. [DOI] [PubMed] [Google Scholar]
- 135.Malaguarnera R, Belfiore A. The insulin receptor: a new target for cancer therapy. Front Endocrinol (Lausanne) 2011;2:93. doi: 10.3389/fendo.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ptitsyn OB. How does protein synthesis give rise to the 3D-structure. FEBS Lett. 1991;285:176–181. doi: 10.1016/0014-5793(91)80799-9. [DOI] [PubMed] [Google Scholar]
- 137.Matagne A, Radford SE, Dobson CM. Fast and slow tracks in lysozyme folding: insight into the role of domains in the folding process. J Mol Biol. 1997;267:1068–1074. doi: 10.1006/jmbi.1997.0963. [DOI] [PubMed] [Google Scholar]
- 138.Yokota A, Izutani K, Takai M, Kubo Y, Noda Y, Koumoto Y, Tachibana H, Segawa S. The transition state in the folding-unfolding reaction of four species of three-disulfide variant of hen lysozyme: the role of each disulfide bridge. J Mol Biol. 2000;295:1275–1288. doi: 10.1006/jmbi.1999.3442. [DOI] [PubMed] [Google Scholar]
- 139.Lazaridis T, Karplus M. “New view” of protein folding reconciled with the old through multiple unfolding simulations. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 140.Mirny LA, Abkevich VI, Shakhnovich EI. How evolution makes proteins fold quickly. Proc Natl Acad Sci U S A. 1998;95:4976–4981. doi: 10.1073/pnas.95.9.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]