

Abstract
Expression of the antioxidant enzyme EcSOD in normal human mammary epithelial cells was not recognized until recently. Although expression of EcSOD was not detectable in non-malignant human mammary epithelial cells (HMEC) cultured in conventional two-dimensional (2D) culture conditions, EcSOD protein expression was observed in normal human breast tissues, suggesting that the 2D-cultured condition induces a repressive status of EcSOD gene expression in HMEC. With the use of laminin-enriched extracellular matrix (lrECM), we were able to detect expression of EcSOD when HMEC formed polarized acinar structures in a 3D-culture condition. Repression of the EcSOD-gene expression was again seen when the HMEC acini were sub-cultured as a monolayer, implying that lrECM-induced acinar morphogenesis is essential in EcSOD-gene activation. We have further shown the involvement of DNA methylation in regulating EcSOD expression in HMEC under these cell culture conditions. EcSOD mRNA expression was strongly induced in the 2D-cultured HMEC after treatment with a DNA methyltransferase inhibitor. In addition, epigenetic analyses showed a decrease in the degree of CpG methylation in the EcSOD promoter in the 3D versus 2D-cultured HMEC. More importantly, >80% of clinical mammary adenocarcinoma samples showed significantly decreased EcSOD mRNA and protein expression levels compared with normal mammary tissues and there is an inverse correlation between the expression levels of EcSOD and the clinical stages of breast cancer. Combined bisulfite restriction analysis analysis of some of the tumors also revealed an association of DNA methylation with the loss of EcSOD expression in vivo. Furthermore, overexpression of EcSOD inhibited breast cancer metastasis in both the experimental lung metastasis model and the syngeneic mouse model. This study suggests that epigenetic silencing of EcSOD may contribute to mammary tumorigenesis and that restoring the extracellular superoxide scavenging activity could be an effective strategy for breast cancer treatment.
Keywords: three-dimensional culture, DNA methylation, experimental lung metastasis, spontaneous metastasis, acinar morphogenesis, laminin-enriched extracellular matrix
INTRODUCTION
Altered expression of cellular antioxidant enzymes that metabolize reactive oxygen species (ROS) are often dysregulated in human cancers.1–4 Such antioxidant enzymes include SODs (superoxide dismutases) that remove superoxide ( ) and catalase and glutathione peroxidases that neutralize hydrogen peroxide (H2O2). The impaired removal of ROS leads to a pro-oxidative environment that favors increased proliferation, survival and oncogenic progression in cancer cells.1,5 We recently demonstrated that expression of extracellular superoxide dismutase (EcSOD) is significantly repressed via aberrant DNA methylation in ~80% of lung cancers compared with normal lung tissues.6 DNA methylation is one of the epigenetic mechanisms that has been linked to aberrant gene expression of tumor suppressor genes.7 As EcSOD is the sole scavenger in lungs, the resulting increased in oxidative stress in the tumor microenvironment due to this inactivation of EcSOD expression is expected to contribute to the pathological and malignant progression of lung cancer.
In comparison with the other mammalian SODs that are ubiquitously expressed, EcSOD-gene expression is tightly controlled at a cell type and tissue-specific manner.8 Considering the cell surface and extracellular localization of EcSOD versus the cytosolic and mitochondrial distribution of CuZnSOD and MnSOD, respectively, the specific expression pattern of EcSOD highlights the differential biological roles of these SODs in tissues. In our previous breast cancer study, overexpression of EcSOD in EcSOD-null breast cancer cell lines resulted in an attenuated malignant phenotypes in vitro including decreased growth rate, clonogenic survival and invasion,9 suggesting a mammary tumor suppressive function of EcSOD. To better understand the role of EcSOD in maintaining the redox homeostasis of the normal mammary epithelial cells and in preventing the oxidative-mediated mammary tumorigenesis, it is essential to assess the expression levels of this antioxidant enzyme in the normal mammary epithelial cells versus the malignant cells. However, our failure in the detection of EcSOD-gene expression in conventionally cultured non-malignant mammary epithelial cells6 raised the fundamental question of whether EcSOD is expressed in normal mammary epithelial cells.
To further address this expression issue of EcSOD in normal mammary epithelial cells, we performed immunohistochemistry (IHC) staining and have now confirmed the expression of EcSOD in mammary epithelial cells in normal human breast tissue in contrast to our previous in vitro study using cultured cells. This discrepancy suggests that EcSOD expression could be perturbed when cells were cultured as a monolayer in the conventional two-dimensional (2D) culture system. The use of three-dimensional (3D) culture system has recently gained momentum in breast cancer studies as it provides a physiologically relevant tissue culture condition to recapitulate numerous histological features of breast epithelium in vivo. These include the formation of acinus-like spheroids with a hollow lumen, apico-basal polarization of cells, and the basal deposition of basement membrane components as well as displaying expression of genes specifically associated with mammary function.10,11
The objectives of this study were therefore, to determine the expression of EcSOD in non-malignant mammary epithelial cells using a physiologically relevant 3D-culture system, and to determine the mechanisms underlying differences in EcSOD-gene expression in monolayer culture versus acinar culture as well as in clinically derived human breast cancers. We found that tissue organization was essential for EcSOD-gene expression in non-malignant mammary epithelial cells and that reactivation of EcSOD-gene expression in the 3D-cultured cells was associated with epigenetic reprogramming. Moreover, >80% of breast tumors examined showed significantly decreased expression levels of EcSOD compared with the normal breast tissues. We have further shown that decreased expression of EcSOD in these breast tumors was associated with DNA hypermethylation in the regulatory region of the EcSOD promoter. In our in vivo metastasis models, animals treated with an adenovirus vector overexpressing the human EcSOD, AdEcSOD, showed a marked reduction of experimental lung metastasis and spontaneous metastasis compared with the empty vector treated mice.
To our knowledge, this is the first study showing the ‘plasticity’ of EcSOD-gene expression in non-malignant human mammary epithelial cells (HMEC) with a finite life-span under 3D versus 2D-cell culture conditions. The association of EcSOD-gene silencing with the loss of mammary tissue organization in vivo implicates an important role of EcSOD as a mammary tumor suppressor. Therefore, further defining the importance and mechanistic role of EcSOD in suppressing breast cancer progression will be important in developing new therapeutic interventions for this disease.
RESULTS
EcSOD expression is absent in 2D-cultured HMEC
To determine the role of EcSOD in breast cancer development, we assessed expression levels of EcSOD in cultured non-malignant mammary epithelial cells. As shown in Figure 1a, EcSOD mRNA expression was not detectable either in the HMEC or the MCF-10A cells in comparison with MRC5 human lung fibroblasts and the primary cultures of human airway epithelial cells. However, IHC staining revealed definitively positive staining for EcSOD in normal breast tissues, specifically in mammary epithelium of the lobules (Figure 1b). These observations suggest that although normal mammary epithelial cells demonstrated robust expression of EcSOD in vivo, culturing the cells in vitro somehow resulted in the loss of EcSOD-gene expression.
Figure 1.

EcSOD expression is detectable in normal breast tissue but not in non-malignant mammary epithelial cells in monolayer cultures. (a) Total RNAs were extracted and analyzed for EcSOD mRNA transcription from post-stasis HMEC, which were derived from reduction mammoplasties and a spontaneously transformed non-malignant mammary epithelial cell line, MCF-10A. In comparison with normal human lung fibroblasts (MRC5) and normal human airway epithelial cells, HMEC and MCF-10A cells showed undetectable EcSOD mRNA expression as determined by a real-time RT–PCR analysis. 18S was used as a loading control. Error bars represent s.e.m., n = three separate experiments. (b) IHC analysis demonstrates a positive staining for EcSOD in normal mammary tissue (right panel). Left panel shows H and E staining of the same normal mammary tissue.
EcSOD-gene expression is reactivated in 3D-cultured HMEC
To test the effect of culture conditions on EcSOD-gene induction, we propagated HMEC in a 3D condition and EcSOD mRNA expression was indeed activated as early as 5 days of culture, and its mRNA expression continued to accumulate until 10 days of culture (Figures 2a and b). Intriguingly, the expression of EcSOD significantly and dramatically decreased in a time-dependent manner after sub-culturing the 10-day 3D acini back into the 2D cultures (Figures 2c and d). We have further shown using immunofluorescence confocal microscopy that the 3D-cultured HMEC formed organized, differentiated and polarized acinar structures as indicated by the integrin-alpha 6 (ITG-α6) staining on the basolateral surfaces and the beta-catenin (β-catenin) labeling in the apical cell–cell junction (Figure 2e). In contrast to the 2D-cultured HMEC, EcSOD protein expression was detected in the 3D HMEC acini after 10 days of culture (Figure 2e). These data clearly demonstrate a stimulating effect of the laminin-enriched extracellular matrix (lrECM) on EcSOD-gene expression. Furthermore, this EcSOD-gene re-activation by lrECM is specific to the non-malignant mammary epithelial cells as culturing the breast cancer cell line MDA-MB231 in the 3D culture did not induce expression of EcSOD (Figure 2f). An EcSOD-expressing stable cell line of MDA-MB231, Ec.20, was generated and a strong immunofluorescence staining for EcSOD was detected in this cell line (Figure 2f).
Figure 2.

Activation of EcSOD gene expression in 3D-cultured HMEC. Total RNAs were extracted from 2D and 3D-cultured HMEC for EcSOD mRNA expression analysis. (a) Quantitative real-time RT–PCR spectrum plots showing the absence of EcSOD mRNA expression in 2D-cultured HMEC and activated EcSOD mRNA expression after 5 and 10 days of 3D culture. (b) Agarose gel electrophoresis of the qPCR products further confirms the positive mRNA transcription of EcSOD in the 3D-cultured HMEC in comparison with the 2D-cultured cells. PCR products were visualized on 2% agarose gels with ethidium bromide staining. As HMEC are derived from reduction mammoplasty from individual patients, we have also obtained two additional lots of HMECs and we were able to replicate the plasticity of EcSOD mRNA expression in 3D versus 2D culture conditions. (c) After 10 days of culture in 3D conditions, HMEC acini were extracted, suspended in a single-cell suspension and plated in 2D-culture conditions. Cells cultured in 2D conditions for 3 days were subsequently trypsinized and re-plated in 2D condition for a total of three passages (p1–p3). Total RNAs were harvested at each 2D passage. The activation of EcSOD gene expression in the 3D-cultured HMEC was dramatically decreased when cells were subsequently cultured in 2D condition as indicated by the real-time qPCR analysis. 18S was used as a control for all qPCR analyses. Three separate experiments of triplicates were performed and only the representative results from one experiment were plotted in (a) and (c). (d) The bar chart shows diminished level of EcSOD mRNA expression after just one passage (p1) in 2D culture and undetectable level in the subsequent 2D passages. (e) Confocal immunofluorescence microscopy of HMEC cells cultured in 2D and 3D conditions. After 10 days of 3D culture, HMEC cells formed organized and polarized acini as indicated by the basolateral marker, integrin α-6 and the apical cell–cell junction marker, β-catenin. EcSOD immunoreactivity was not detected in 2D culture but is present after 10 days of 3D culture. Blue = nuclear staining with DAPI. (f) EcSOD expression is absent in both 2D and 3D-cultured breast cancer cell line, MDA-MB231. An EcSOD-expressing stable cell line, Ec.20 showed a strong EcSOD immunofluorescence staining throughout the cells when cultured in both 2D and 3D conditions. Representative confocal images from three separate experiments are shown here. Green = EcSOD immunofluorescence staining; Blue = nuclear staining with DAPI. Bar scale = 20 μm.
3D-cultured HMEC display lower levels of DCFH oxidation compared with cells in monolayer culture
To determine whether levels of oxidative stress vary when HMEC are cultured under 2D versus 3D-culture conditions, we labeled cells with an oxidative-sensitive fluorescent probe, DCFH. Increased DCFH oxidation was observed in 2D-cultured HMEC in comparison with the 3D-cultured cells as shown by flow cytometry analysis in Figure 3a (mean fluorescence intensity of 2102 in 2D HMEC versus 189 in 3D HMEC). An oxidation-insensitive version of this probe that controls for dye uptake, esterase cleavage and efflux showed no difference in signal intensity, indicating that the differences in signal observed between 2D and 3D cultures was specifically due to oxidation of the probe (data not shown). This indicates that propagating the non-malignant mammary epithelial cells as a monolayer culture resulted in a heightened oxidative stress and this could in part be attributed to the loss of EcSOD expression.
Figure 3.

Monolayer HMEC showed increased ROS levels compared with 3D-cultured cells and re-activation of EcSOD gene expression in 3D-cultured HMEC was associated with a reduced promoter DNA methylation status. (a) Representative histograms of DCFH mean fluorescent intensity (MFI). HMEC cultured in both 2D and 3D conditions were labeled with oxidative-sensitive fluorescent probe, carboxy-H2DCFDA (DCFH) at 1 μg/ml for 15 min, 37 °C. DCFH fluorescence was measured by flow cytometry and MFI was analyzed using FlowJo software. (b) HMEC cultured in 2D conditions were treated for 5 days with a DNA methyltransferase inhibitor (5-Aza-dC, 4 μM) or 1 day with a histone deacetylase inhibitor (TSA, 50 ng/ml), or a combination of both treatments. Total RNAs were extracted to determine EcSOD mRNA expression by RT–PCR analysis followed by agarose gel electrophoresis. 18S was used as a loading control. (c) COBRA showing the differential sensitivity of the EcSOD promoter region to Taq1 restriction in 2D versus 3D-cultured HMEC. Following the TaqI1 digestion, the PCR products were subjected to agarose gel electrophoresis. Sensitivity to Taq1 digestion indicates methylated EcSOD promoter in the 2D HMEC while uncut product in the 3D HMEC indicates hypomethylation of the EcSOD promoter. Data shown here are representatives of three separate experiments. (d) Methylation status of the 18 CpG sites in the EcSOD promoter was determined by bisulfite genomic sequencing from 2D- and 3D-cultured HMEC. Each row represents the cytosine methylation pattern obtained from an individual clone of the bisulfite treated, PCR amplified and sequenced EcSOD promoter. Each row represents 1 of 5 clones selected for sequencing. The region of the EcSOD promoter examined within this study spanned from − 550 to + 100 of the putative transcription start site (+1) and the position of each CpG is labeled 1–18 as described previously.6 Methylation profile of the EcSOD promoter shows the largely hypermethylated status (gray squares) in 2D-cultured HMEC in comparison with that of the more hypomethylated region (clear squares) in the EcSOD-expressing 3D-cultured HMEC.
EcSOD-gene silencing in 2D-cultured HMEC is associated with promoter DNA hypermethylation
To determine whether epigenetic regulation participates in transcriptional regulation of the EcSOD gene in the 3D versus 2D-cultured HMEC, we treated 2D-cultured HMEC with a DNA methyltransferase inhibitor, 5-Aza-dC and a histone deacetylase inhibitor, trichostatin A (TSA). As shown in Figure 3b, in comparison with the untreated cells, there was a significant induction of EcSOD mRNA expression in 5-Aza-dC-treated HMEC, while there was only a slight increase in EcSOD mRNA expression in TSA-treated cells. To further confirm the association of cytosine methylation with transcriptional silencing of EcSOD in 2D-cultured HMEC, we performed a combined bisulfite restriction analysis (COBRA). Figure 3c illustrates that there was a higher degree of unmethylated EcSOD promoter in the 3D-cultured HMEC in comparison with a greater methylated status in the 2D-cultured cells. In addition, we also conducted sodium bisulfite sequencing to further analyze the 18 CpG sites at the 5′-end of the EcSOD gene as previously described.6 The methylation profile of the EcSOD promoter presented in Figure 3d shows a predominantly hypermethylated status in 2D-cultured HMEC, whereas the 3D-culture HMEC possess a hypomethylated EcSOD promoter. These results clearly demonstrated that transcriptional silencing of EcSOD gene in 2D-cultured HMEC is associated with cytosine methylation and this process is reversible when we cultured the cells in a 3D condition.
EcSOD expression is significantly downregulated in clinical breast cancer tissues and its mRNA expression levels correlated inversely with clinical stage
We next sought to determine the expression levels of EcSOD in clinical breast cancer samples. As shown in the representative pictures of EcSOD IHC staining in Figure 4a, we detected a strong and definitive EcSOD protein expression in mammary epithelial cells in the benign lobule. In contrast, the immunoreactivity of EcSOD is greatly reduced in the high-grade ductal carcinoma in situ and invasive breast cancer. The intensity of the IHC staining for EcSOD in normal versus neoplastic breast tissues was semi-quantified and shown in Table 1. To further investigate whether impaired EcSOD expression is a common event in breast cancer, we performed a TissueScan Breast Cancer cDNA Array (Origene, Inc., Rockville, MD, USA). As shown in Figure 4b, there was a decrease in EcSOD mRNA expression levels in stage I and a significant reduction in Stage II through IV when compared with the normal tissues (P<0.05). These data clearly indicate that EcSOD is a target for gene silencing in 35/43 human breast adenocarcinomas. Furthermore, COBRA analysis revealed that 4 out of 5 of the tumors examined were more sensitivity to Taq1 restriction compared with the normal breast tissue, indicating a greater degree of CpG methylation in the promoter region of EcSOD in the breast tumors (Figure 4c).
Figure 4.

EcSOD protein and mRNA expression levels are significantly reduced in a high percentage of breast cancer tissues compared with normal breast tissues. (a) Representative micrographs of EcSOD immunohistochemical staining in benign breast, ductal carcinoma in situ (DCIS), and invasive breast cancer. The intensity of the EcSOD staining was blindly scored and shown in Table 1. (b) Quantitative real-time PCR analysis of EcSOD mRNA expression levels in a panel of breast cancer tissues at various pathological stages (TissueScan Breast Cancer qPCR Array, plate BCRT102, from Origene, Inc.). This array contains 5 normal breast tissues and 43 breast tumors. The percent mRNA expression of EcSOD compared with normal tissues was plotted by pathological stage. A significant reduction of P<0.005 was observed between the normal tissues and Stage II to Stage IV of breast cancer. Beta-actin was used as a loading control. (c) Six breast tissue DNAs were purchased from Origene, Inc. (from plate BRCT102 as analyzed in Figure 4B) and COBRA analysis was performed followed by amplification using primers spanning the Taq1 site in the EcSOD promoter. The PCR products were then restricted with Taq1 and analyzed by agarose gel electrophoresis. Samples were numbered 1–6, which correspond to the labeled specimens shown in Figure 4B. The pathological state of the samples are as follows: Sample 1—normal tissue, Sample 2—stage IIA, Sample 3—stage IIA, Sample 4—stage IIB, Sample 5—stage IIB and Sample 6—stage IIIC. UC represents uncut PCR product as a negative control. Taq1-induced cut indicates methylated EcSOD promoter, whereas uncut product indicates unmethylated EcSOD promoter.
Table 1.
Semi-quantitative analysis of immune-labeling intensity of EcSOD in human breast tissues
| Tissue | N | − | + | + + | P-value vs normal breast |
|---|---|---|---|---|---|
| Normal breast | 9 | 0 | 2 | 7 | |
| DCIS | 10 | 2 | 8 | 0 | 0.001 |
| Low-grade invasive breast cancer | 7 | 6 | 1 | 0 | 0.0003 |
| High-grade invasive breast cancer | 6 | 5 | 0 | 1 | 0.003 |
Abbreviation: DCIS, ductal carcinoma in situ. Immunohistochemical staining of EcSOD was semi-quantitatively graded as negative (−), weak (+) or strong (+ +) staining in at least 50% of cells examined. P-values were calculated by the exact contingency test (3 × 2 contingency table) based on Fisher’s exact test. N represents the number of tissues examined.
Re-expression of EcSOD inhibited the stellate colony formation of breast cancer cells in 3D culture
We then determined the effect of EcSOD re-expression on the 3D morphology of the breast cancer cells. Figure 5 shows that the wild-type MDA-MB231 cells formed hallmark invasive colonies with protruding stellate structures as illustrated12 after 5 days of 3D culture. AdEmpty vector control infection did not alter the morphology of the cells, whereas overexpression of EcSOD with AdEcSOD infection significantly reduced the colony sizes and severely impaired the stellate formation in MDA-MB231 cells. As adenoviral-mediated gene expression is a transient overexpression, we generated a stable cell line of MDA-MB231 that overexpresses EcSOD, designated Ec.20. Similar abrogation of stellate formation was observed in the 3D-cultured Ec.20 cells compared with the vector control cell line, Vector.1. A biologically relevant shorter form of EcSOD that carries a deletion in the heparin-binding domain (HBD) was previously shown to have similar inhibition of in vitro growth, clonogenic survival and invasion of breast cancer cells.9 Here, we have demonstrated that re-expression of EcSODΔHBD, both transiently (with AdΔHBD infection) and stably (in ΔHBD.3 cell line) also inhibited the 3D stellate formation of MDA-MB231 cells.
Figure 5.
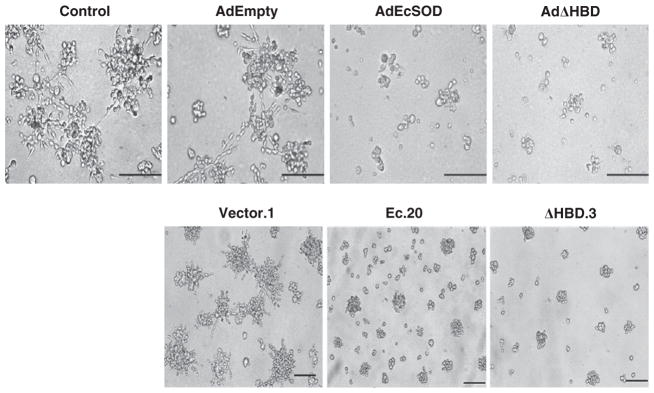
Overexpression of EcSOD inhibited stellate colony formation of MDA-MB231 cells in 3D culture. Two-dimensional cultured MDA-MB231 were infected with AdEmpty, AdEcSOD or AdΔHBD, at moi = 50 PFU/cell for 24 h, followed by reseeding the cells in 3D culture. After 5 days of 3D culture, MDA-MB231 cells formed large colonies with extensive stellate structures, which is a hallmark feature of the ‘invasive’ breast cancer 3D phenotype. While AdEmpty did not affect the 3D morphology of this cell line, overexpression of EcSOD, both the full-length and the truncated forms (with AdEcSOD and AdΔHBD adenovirus infection, moi = 50 PFU/cell) resulted in much smaller colonies which lack stellate formation. A similar 3D morphological alteration was observed when MDA-MB231 cells were generated to stably over express EcSOD, in Ec.20 (full length) and ΔHBD.3 (deletion in the HBD) where the cells formed small colonies with an absence of stellate extension. Representative micrographs of N = 3. Bar scale = 100 μm.
Overexpression of EcSOD suppressed experimental lung metastasis of MDA-MB231 cells and prolonged animal survival
As EcSOD inhibited the in vitro Matrigel invasion of MDA-MB231 cells partly by suppressing pro-oncogenic heparanase9 and heparanase expression correlates with aggressive and metastatic behavior of breast cancer cells,13 we determined whether overexpression of EcSOD will affect metastasis of MDA-MB231 in an experimental lung metastasis model. Figure 6a shows that mice given AdEmpty exhibited metastasis of MB231.luc cells in the lungs of the animals from week 2 to week 8 of the experiment, whereas mice given AdEcSOD and AdΔHBD showed much reduced lung metastasis. Photon flux of the lung metastasis was quantified and presented in Figure 6b, which indicates that overexpression of EcSOD, both the full-length and the truncated form efficiently and significantly inhibited metastasis of breast cancer cells compared with the AdEmpty group. Mean photon counts (photons/s) on week 8 = 9.39±2.1 × 107 in AdEmpty group vs 1.88±0.75 × 107 in AdEcSOD group (P = 0.0016) and 1.21±0.63 × 107 in AdΔHBD group (P = 0.0014). No significant difference was observed between the Control and AdEmpty-treated animals (P = 0.554). In addition, EcSOD-treated mice showed prolonged median survival rates compared with the AdEmpty-treated group (57 days in AdEmpty group vs 83 days in AdEcSOD group, P<0.0005 and 94 days in AdΔHBD group, P<0.0002). To ensure that EcSOD is overexpressed in mice that received the AdEcSOD and AdΔHBD inoculation, we collected plasma samples from the animals for EcSOD protein and activity analyses. As shown in Figure 6d, AdEcSOD infection induced a 2.61-fold increase in plasma EcSOD activity in the animals compared with the AdEmpty-infected group (395±122 unit/ml in AdEmpty group vs 1030±453 unit/ml in AdEcSOD group, P<0.005) while animals given the AdΔHBD increased the EcSOD activity by 10.9 fold (4290±1459 unit/ml, P<0.0005). Removal of the HBD has been demonstrated to not affect the catalytic activity of EcSOD.14 As the full-length EcSOD can be sequestered by ECM components and cell surface proteoglycan, although EcSODΔHBD lacks these affinities and hence remains in the circulation, only a moderate increase of plasma EcSOD activity was detected in AdEcSOD-treated animals. To better assess the levels of full-length EcSOD induced in the animals, we treated mice with heparin before blood collection to release most of the bound-full-length EcSOD. Comparable levels of EcSOD protein expression were induced in both the AdEcSOD and AdΔHBD-treated groups as shown by LMWH treatment in Figure 6e (top panel). Expression of the native murine form of EcSOD remained relatively constant regardless of the treatments (Figure 6e, bottom panel).
Figure 6.

Suppression of lung metastasis of MDA-MB231.luc cells by EcSOD. To induce lung metastasis, athymic nude mice were injected with 2 × 106 of MDA-MB231.luc cells in the lateral tail vein. Overexpression of EcSOD was accomplished with AdEcSOD or AdΔHBD (1 × 1010 PFU) give intramuscularly 3 days before the breast cancer cell injection. AdEmpty was used as a vector control. (a, Top panel) Representative bioluminescent images of mice taken immediately post MDA-MB231.luc cell injection. (a, Bottom panel) Mice were imaged both dorsally and ventrally every week to monitor the metastasis of breast cancer cells. EcSOD-treated groups showed reduced development of lung metastasis compared with the AdEmpty-infected group. Time course images taken from the same animal in each group are shown here. (b) Luminescent signal (photon flux) of the metastasized MDA-MB231.luc cells was plotted over time showing a significant inhibition of lung metastasis by AdEcSOD and AdΔHBD versus AdEmpty (P<0.005). Data shown are mean values and s.d. from two independent experiments with ten mice per group. P-values were calculated using two-sided Student’s t-test. (c) Survival rate of mock (Control), AdEmpty, AdEcSOD and AdΔHBD-transduced athymic nude mice were plotted using Prism stats program (Kaplan–Meier survival) with the days elapsed following tail vein injection of MDA-MB231 luc cells. Survival curve for AdEcSOD and AdΔHBD were significantly different (P<0.0005) from both Control and AdEmpty-infected groups. (d) Plasma EcSOD activity levels in mice were evaluate using the WST SOD activity kit. Mice infected with AdEcSOD and AdΔHBD showed significantly higher levels of plasma EcSOD activities compared with AdEmpty-treated animals (P<0.0005). P-values were calculated using two-sided Student’s t-test. (e) Western blot analysis showing overexpression of both the full-length and truncated EcSOD in AdEcSOD- and AdΔHBD-treated animals. Plasma samples were analyzed using either an anti-human EcSOD antibody (top) or an anti-mouse EcSOD antibody (bottom). Low molecular weight heparin (LMWH) was administered intravenously at 1000 IU/kg of body weight to release full-length (FL) EcSOD into circulation and blood was collected from the animals after 30–60 min of heparin treatment. Protein concentration loaded per lane: 2 μg for the detection of human EcSOD (huEcSOD) and 8 μg for the detection of mouse EcSOD (moEcSOD).
EcSOD reduced the rate of spontaneous metastasis of mouse mammary adenocarcinoma cells in a syngeneic mouse model
We next determined whether EcSOD will inhibit spontaneous metastasis of breast cancer cells from established primary tumors. A mouse mammary tumor cell line constructed to express luciferase, 4T1.luc was inoculated in the mammary fat pad of BALB/c mice. Figure 7a shows a representative mouse with a primary tumor (panel 1), 1 week after tumor excision (panel 2), and lung metastasis at 2 weeks post primary tumor removal (panel 3). Occurrence of metastasis to secondary sites (mainly the lungs and lymph nodes in this study) was scored and presented as percent metastasis in Figure 7b. There was an initial delay in metastasis rate in AdEcSOD and AdΔHBD-treated groups compared with the AdEmpty-treated group. When all of the animals in the AdEmpty group showed metastasized diseases at 10 weeks post primary tumor removal, only 70% of the animals in the AdEcSOD group have metastasized tumors, while 30% of the animals remained free of metastasis. Overexpression of EcSOD also prolonged survival of the animals as shown in the Kaplan–Meier plot in Figure 7c from a mean survival of 47 days in AdEmpty group to 77 days in AdEcSOD group (P<0.05) and 73 days in AdΔHBD group (P<0.05).
Figure 7.

Inhibition of spontaneous metastasis of a mouse mammary carcinoma cell line by EcSOD. (a) Representative bioluminescence images showing the progression of mouse mammary carcinoma cells, 4T1.luc in BALB/c mice at day 10 post tumor implantation (1), 7 days post tumor removal (2), and lung and lymph node metastasis at 4 weeks post tumor removal (3). (b) Occurrence of metastasis of 4T1.luc cells to secondary sites (lungs and lymph nodes) were scored and shown as percent metastasis. (c) Kaplan–Meier survival rate of the AdEmpty, AdEcSOD and AdΔHBD-transduced animals were plotted using Prism stats program with the days elapsed following surgical removal of 4T1.luc primary tumors. Survival curve for AdEcSOD and AdΔHBD were significantly different (P<0.05) from AdEmpty-infected groups. N = 8 per group.
DISCUSSION
Rapid loss of tissue-specific functions occurs when epithelial cells are dissociated and cultured as monolayers in the 2D-culture system. Our study herein clearly supports the importance of utilizing the physiologically more relevant 3D-culture substrata in studying the regulation of EcSOD-gene expression in mammary epithelial cells. We initially speculated that perhaps the non-malignant mammary epithelial cells are devoid of EcSOD as we were unable to detect any EcSOD mRNA or protein expression in HMEC and MCF-10A cells. However, upon further investigation, we detected EcSOD protein expression in normal breast tissues (Figures 1b and 4). Further supporting the positive expression of EcSOD in normal mammary epithelium in vivo, transcripts of EcSOD were detected in normal mammary gland as reported in the Expressed Sequence Tag Profile of the NCBI database (http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.2420). This discrepancy in the expression status of EcSOD in non-malignant mammary epithelial cells prompted us to reconsider the choice of our cell culture system and explore the use of 3D culture.
The observation that EcSOD is produced in normal mammary tissues is not entirely surprising as a secreted form of SOD has long been identified in bovine milk by Hicks et al.,15 who later suggested that the secreted SOD may have an important role in maintaining the stability of milk proteins by preventing oxidative-mediated protein damage.16 Here, we have convincingly demonstrated the expression of EcSOD in the normal human mammary gland and that luminal epithelial cells are likely the source that secretes this enzyme into milk.
In addition, we have shown that HMEC lose expression of EcSOD when cultured as a monolayer and this alteration is regulated via an epigenetic mechanism where DNA methylation of the promoter region of EcSOD occurs in 2D-cultured HMEC (Figure 3d). Hypermethylation of CpG islands within the promoter region of a gene is recognized as an important epigenetic mechanism of transcriptional silencing of tumor regulatory genes, in many cases tumor suppressor genes, during cancer development.17 The fact that EcSOD-gene expression is tightly controlled in a cell type-specific manner8 and its promoter shows a CpG structure similar to other genes silenced by aberrant cytosine methylation18 suggests a potential association of epigenetic regulation via DNA methylation with EcSOD-gene expression in mammary epithelial cells. The EcSOD-gene silencing via DNA methylation is a reversible process as lrECM-mediated acinar morphogenesis of the HMEC evoked reactivation of EcSOD-gene expression in the 3D-cultured HMEC.
It is important to note that the HMEC model used in this study was obtained from a commercial source that has undergone stasis or stress-induced senescence.19 Although these post-stasis HMEC still possess a finite life-span and are non-malignant, they have been shown to have acquired hundreds of transcriptional and epigenetic changes during their selection process.20,21 This suggest that loss of EcSOD expression seen in our 2D-cultured HMEC could have initiated during the progression of cells through this stasis challenge and that pre-stasis cells might still retain the expression of EcSOD.
The specific activation of EcSOD-gene expression in the 3D-cultured HMEC shown in this study is intriguing but how alteration of this epigenetic program occurs in response to the changes in the cellular microenvironment is not clear at this point. EcSOD is not the first gene reported to be reactivated in non-malignant mammary epithelial cells upon lrECM stimulation as the expression of several mammary-specific genes has previously been described as responsive to ECM signaling such as the whey acidic protein (WAP) gene22 and milk casein genes.23,24 That said, this is the first report showing the involvement of DNA demethylation process in the lrECM-induced reactivation of a gene in non-malignant mammary epithelial cells. Although enzymes responsible for active DNA demethylation remain to be identified, TET1 has recently been suggested to regulate hydroxylation of methylated cytosine, which may interfere with DNA methyltransferase 1 activity, leading to a subsequent passive loss of methylation following DNA replication.25,26 Although we have not determined the association of 5-hydroxy-methyl cytosine in the promoter region of EcSOD in the 3D-cultured HMEC, EcSOD gene, which is also known as sod3, has been identified as one of the TET1 target genes in murine embryonic stem cells.27 This suggests that TET1-induced hmC could be involved in the de-repression of EcSOD-gene expression in HMEC acini.
The ability of ECM components to promote the functional differentiation of HMEC into polar, acinar structures that leads to the expression of EcSOD likely requires several levels of control. Mechanisms in addition to the epigenetic-mediated chromatin remodeling, such as transcription regulation, are expected to be responsible for EcSOD-gene reactivation. In the case of β-casein expression in non-malignant mammary epithelial cells, lrECM and prolactin, the two key components in the 3D-culture media, regulate transcription of this gene through a cooperative action of chromatin remodeling and recruitment of SWI/SNF to associate with transcription factors such as STAT5 (signal transducers and activators of transcription protein 5), C/EBP β (CCAAT/enhancer-binding protein β), and GR (glucocorticoid receptor).24 Interestingly, the human EcSOD promoter contains seven STAT5-binding sites, four C/EBP β-binding sites and four-glucocorticoid response elements in the 2500 bp region upstream of the transcriptional start site (Supplementary Figure 1). In addition, EcSOD promoter also contains seven Rush family (SWI/SNF-related nucleophosphoproteins with a RING finger DNA-binding motif) consensus binding sites (Supplementary Figure 1). These transcription factors have been shown to be a member of the SWI/SNF complex that facilitates transcription by chromatin remodeling and has also been shown to mediate the ability of prolactin to induce gene expression.28 This suggests that expression of EcSOD in mammary acini could be activated in a similar pattern as the milk protein and that EcSOD is regulated in a mammary tissue-specific fashion, which was formerly unrecognized.
The observation that the loss of mammary tissue organization (as in monolayer culture of HMEC) promoted epigenetic silencing of EcSOD is also strongly supported and closely reflected in our clinical data where a majority of breast tumors showed significantly reduced levels of EcSOD expression when compared with the normal breast tissues. The region analyzed by the COBRA analysis (Figure 4c) corresponds to the regulatory site for the transcription factors, SP1/SP3, which have been shown to drive EcSOD expression and that we have recently identified to be hypermethylated in our lung cancer studies.6 Methylation of the CpG sites in this region interferes with the binding of these transcription factors and creates a more closed chromatin structure, hence leading to a repressive state of EcSOD expression.
Furthermore, tissue array analysis as shown in Figure 4b indicates an inverse correlation of the expression levels of EcSOD with the clinical stages of breast cancer. This implies that loss of extracellular superoxide scavenging capacity as seen in breast tumors that express significantly lowered levels of EcSOD compared with the normal breast tissue is likely be one of the underlying factors that contributes to breast tumorigenesis and metastasis. A critical event in the process of cancer invasion and metastasis is the structural alterations of the basement membrane and ECM components. The primary matrix components of the lrECM or Matrigel are laminin, collagen and heparin sulfate proteoglycan29 and these structural molecules have been reported to be sensitive to oxidative-mediated cleavage.30–32 Therefore, loss of the predominant extracellular antioxidant enzyme in breast cancer cells is expected to promote oxidative stress that compromises the structural integrity of these matrix components and assists breast cancer cells in invading through the physical barrier in the 3D-culture substrata. Here, we have shown that re-expression of EcSOD in an EcSOD-null breast cancer cell line, MDA-MB231, attenuated the formation of stellate structures in the 3D culture (Figure 5). This 3D-induced stellate formation is a hallmark feature of the invasive and aggressive phenotype of MDA-MB231.33 The reversion of the 3D morphology of the breast cancer cells to a more normal-like rounded structure suggests that EcSOD may have an important role in structural organization during acinar morphogenesis by maintaining redox homeostasis in non-malignant mammary epithelial cells and that gene silencing of EcSOD as seen in neoplastic mammary epithelial cells may promote a loss of proliferative control and induce an invasive and metastatic phenotype. Our in vivo studies further support a role of ROS in promoting malignant progression of breast cancer cells. Overexpression of EcSOD suppressed both the experimental and spontaneous metastasis of breast cancer cells (Figures 6 and 7). These findings parallel the observation that EcSOD-gene silencing is most prevalent in the more advanced stages of breast cancer (Table 1 and Figure 4b).
Taken together, this study clearly demonstrates the expression of EcSOD in normal mammary epithelial cells in vivo and in 3D-cultured HMEC. However, when lobular architecture is disrupted, as in breast tumors and in monolayer cultured HMEC, EcSOD gene is silenced through aberrant cytosine methylation. These exciting observations also suggest that increasing the ROS scavenging activity, such as with SOD mimetics, could be explored as a therapeutic strategy for metastatic breast cancer. Further validation of the causal role of EcSOD gene silencing in promoting breast cancer progression and metastasis will therefore be critical in identifying the loss of EcSOD expression as a predictive biomarker for early diagnosis and a novel avenue towards therapeutic interventions.
MATERIALS AND METHODS
Cell culture
Human mammary epithelial cell lines MCF-10A and MDA-MB231 were obtained from the American Type Cell Culture Collection (Manassas, VA, USA). Non-malignant, post-stasis human mammary epithelial cells extracted from reduction mammoplasty, HMEC, were purchased from Cell Applications Inc (San Diego, CA, USA) and used within 10 passages in this study. MCF-10A cells were maintained as recommended by the supplier. MDA-MB231 and a murine mammary carcinoma cell line, 4T1 were maintained in RPMI1640 containing 10% FBS and 1X Pen/Strep. HMEC were cultured in Human Mammary Epithelial Cell Growth medium (Cell Applications Inc.). Normal human lung fibroblasts (MRC5) and normal human airway epithelial cells were cultured as previously described.6 All cells are maintained at 37 °C, 5% O2. For the epigenetic drug treatment experiments, HMEC were seeded at 1 × 105 cells/well of six-well plate in 2D culture. After 24 h of seeding, cells were treated with 4 μM of 5-Aza-2dC for 5 days or 50 ng/ml of TSA for 1 day.
Plasmid constructions and establishment of stable cell lines
A firefly luciferase expressing stable cell line, MDA-MB231.luc, was previously generated as described.34 This stable cell line was used to simultaneously express EcSOD using the following plasmid constructs. The full-length cDNA of human EcSOD was PCR amplified with the following primers: 5′-CTGAAGCTTATGCTGGCGCTACTGTGT-3′ (forward) and 5′-TTCTCGAGTCAGGCGGCCTTGCACTC-3′ (reverse). A truncated form of EcSOD with a deletion in the HBD at the C terminus was also amplified with the same forward primer and the following reverse primer: 5′-TT CTCGAGATCACTCTGAGTGCTCCCGCGC-3′ (reverse). The PCR products were cloned into pcDNA3.1 (+) vector (Invitrogen, Life Technologies, Grand Island, NY, USA). After verification of the sequences, the plasmids containing the full-length and truncated EcSOD were transfected into MDA-MB231.luc cells using Lipofectamine 2000 transfection reagent (Life Technologies, Grand Island, NY, USA). Selection of stable cell clones was carried out in the presence of 400 μg/ml hygromycin in culture media for 14 days. Several clones were selected for expansion and EcSOD expression was verified.
3D culture
Mammary epithelial cell were propagated in 3D culture using the ‘on-top assay’ as described.33 Briefly, six-well plates were coated with lrECM or Matrigel (BD Biosciences, San Jose, CA, USA) and 2D-cultured cells were trypsinized. Single cells were seeded at 0.25 × 105 cells/cm2 for HMEC and at 0.175 × 105 cells/cm2 for MDA-MB231 in assay medium containing 5% Matrigel for the non-malignant cells and 2% Matrigel for the breast cancer cells. The 3D assay medium is DMEM/F12 containing supplements as described.35 The 3D cultures were maintained at 37 °C, 5% O2 and assay medium was replaced every 2 days.
Confocal immunofluorescence microscopy
Immunofluorescence assays were performed as described.33 In brief, cells cultured on four-chamber slides were fixed with 4% paraformaldehyde. Cells were then blocked, permeabilized and immune-stained against α6 integrin (clone GoH31 at 1/100 dilution, Chemicon, Millipore, Billerica, MA, USA), β catenin (clone 14, 1/50 dilution, BD Biosciences), or EcSOD (1/400 dilution). Cells were then incubated with 1/200 dilution of secondary antibodies conjugated to Alexa Fluor 488 (Life Technologies). Cells were mounted with VECTASHIELD Hardset Mounting medium (Vector Laboratories, Burlingame, CA, USA) containing DAPI for nuclei counter staining. Confocal analysis was performed using a Zeiss LSM 710 laser-scanning confocal system (Zeiss, Thornwood, NY, USA). Images were analyzed using NIH ImageJ (NIH, Bethesda, MD, USA) and Adobe photoshop.
Dichlorofluorescein diacetate (DCFH) staining
DCFH staining was performed as previously described.36 Briefly, 2D- and 3D-cultured HMEC were extracted and cellular oxidative status was determined by labeling cells with 1 μg/ml of oxidative-sensitive fluorescent probe (carboxy-H2DCFDA, catalog number C400, Molecular Probes, Eugene, OR, USA) for 15 min in HBSS at 37 °C. Cells were then resuspended in HBSS and analyzed using an LSR flow cytometer with the excitation of 488 nm and emission of 530/30 band-pass filter. The mean fluorescence intensity of 10 000 cells was analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA). Cell permeable and oxidation-insensitive fluorescent probe (carboxy-DCFDA, catalog number C369, Molecular Probes, Invitrogen, Life Technologies) was also used to normalize for dye retention in cells.
Cell lysate preparation and western blot analysis
Cells were isolated from 3D culture and lyzed as described.33 For 2D-cultured cells, cell lysates were prepared by scrap-harvesting the cells in the same lysis buffer. Western blot analysis was performed as previously described.37 Antibody dilutions were as follows: rabbit polyclonal anti-human EcSOD, 1/5000; rabbit polyclonal anti-mouse EcSOD, 1/2000; rabbit polyclonal anti-human β-actin (Sigma-Aldrich, St Louis, MO, USA), 1/2000; and goat anti-rabbit IgG-HRP, 1/50 000.
EcSOD activity assay
EcSOD activities in the plasma samples collected from mice were determined by measuring the inhibition of reduction of the water-soluble tetrazolium salt, WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt), which produces a water-soluble formazan dye upon reduction with a superoxide anion (Dojindo Molecular Technologies, Gaithersburg, MD, USA).
Immunohistochemistry
Archived formalin-fixed and paraffin-embedded tissue blocks of human breast cancers from 23 patients were obtained from the Department of Pathology at the University of Iowa. Analysis of anonymous tissues with no patient identifiers was approved by and in compliance with Institutional Review Board guidelines. IHC staining of EcSOD in 5 μm sections of tissues on glass slides was performed as previously described.38 Polyclonal anti-EcSOD antibody was used at 1:400 dilution (Enzo Life Sciences, Farmingdale, NY, USA).
RT–PCR
EcSOD mRNA expression was measured using ABI TaqMan primer/probe set as previously described.6 Real-time RT–PCR reactions were performed according to Applied Biosystems (Carlsbad, CA, USA) conditions on a ABI 7500 Fast machine, and differences in expression were determined as described.39
Tissuescan breast cancer qPCR array
EcSOD mRNA expression in breast tumor samples ranging from normal to stage IV was measured in a 96-well plate using the TissueScan Breast Cancer qPCR Array, plate BCRT102, from Origene, Inc. Real-Time PCR was conducted on an ABI 7000 according to manufacturer’s instruction.
Sodium bisulfite sequencing
Sodium bisulfite sequencing for EcSOD was performed as previously described.39 The 18 CpG sites of the EcSOD promoter examined herein spanned 659 bp from −544 bp upstream from the transcriptional start site downstream to + 115 bp. This correlates with nucleotide positions 24796689–24797348 of the February 2009 assembly of the UCSC genome browser. Primers used as previously described.6
COBRA analysis
Combined bisulfite restriction analyses employed the same primers as sodium bisulfite sequencing experiment to amplify EcSOD products as described.6 The amplified products were then restricted with the enzymes TaqI and run on 2% agarose gel. The breast tumor DNAs were purchased from Origene, Inc. (from plate BRCT102) and their corresponding ID numbers on the Origene TissueScan Breast Cancer qPCR Array Plate (BCRT102) are as follows: Case ID numbers: Sample 1-CI0000012704 (normal), Sample 2-CI0000011284 (stage IIA), Sample 3-CI0000010295 (stage IIA), Sample 4-CI0000006020 (stage IIB), Sample 5-CI0000011684 (stage IIB), Sample 6-CI0000017650 (stage IIIC).
Experimental lung metastasis
Eight-week-old female athymic nude mice were obtained from Harlan Laboratories Inc. (Indianapolis, IN, USA). The nude mice protocol was reviewed and approved by the Animal Care and Use Committee of the University of Iowa. As the MDA-MB231 does not produce spontaneous metastasis at an efficient rate when injected at the mammary fat pad of the nude mice, we directly introduced the MDA-MB231.luc cells (2 × 106 cells) i.v. into the lateral tail vein of the animals. Overexpression of either the full-length or HBD-deleted EcSOD was accomplished by intramuscular injection of AdEcSOD or AdΔHBD (1 × 109 PFU/mouse) 3 days before breast cancer cell injection. AdEmpty was used as an adenovirus vector control.
Spontaneous metastasis
A firefly luciferase overexpressing stable cell line of the murine mammary carcinoma cells, 4T1.luc, previously generated using retrovirus vector pQC-luc-IN40,41 was kindly provided by Dr Michael Henry. Eight-week-old female BALB/c mice (Taconic Farms, Inc., Hudson, NY, USA) were injected with 1 × 104 4T1.luc cells at the 4th right inguinal mammary fat pad. When palpable tumors were detectable~10 days post cell injection, mice were randomly grouped and given AdEmpty, AdEcSOD or AdΔHBD (1 × 109 PFU/mouse) via intramuscular injections. After 3 days of adenovirus injection, primary tumors were surgically removed and animals were monitored for metastatic diseases by bioluminescence imaging.
Bioluminescence imaging
The substrate luciferin was injected into the intraperitoneal cavity of mice at a dose of 150 mg/kg body weight (30 mg/ml luciferin), ~10 min before imaging. Mice were anesthetized with isoflurane/oxygen and placed on the imaging stage. Ventral and dorsal images were collected for 5–60 s using the IVIS200 imager (Xenogen Corporation, Alameda, CA, USA). Bioluminescence from ROI was defined manually, and data were expressed and quantified using Living Image software as photon flux (photons/s/cm2/steradian).
Statistical analysis
To determine the mean survival time, survival curves were plotted using Prism stats program (Kaplan–Meier survival) with the days of death following either the tail vein injection of MDA-MB231 or primary 4T1.luc tumor removal. P-values were calculated for the comparison of the survival curves. For some experiments, a single-factor ANOVA followed by post-hoc Tukey-test was used to determine statistical differences between means. Statistical analyses were assessed using a two-tailed Student’s t-test. P-values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
The authors thank Dr James Crapo (National Jewish Medical and Research Center, Denver, CO, USA) for providing antibodies to EcSOD and Dr Michael Henry (Department of Molecular Physiology and Biophysics, University of Iowa, IA, USA) for providing the 4T1.luc cells. We also thank Justin Fishbaugh from the Flow Cytometry Core Facility for assisting with the DCFH assays as well as the Central Microscopy Research Facility. This study was supported by NIH grant R01 CA073612 and R01 CA115438 (FE Domann), Susan G Komen for the Cure grant KG080437 (M Teoh-Fitzgerald), SFRBM Research Mini-Fellowship (M Teoh-Fitzgerald), Oberley Seed Grant (M Teoh-Fitzgerald), and Carver Research Program of Excellence in Redox Biology and Medicine, and by the Holden Comprehensive Cancer Center Breast Cancer Research Fund.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 2.Oberley LW, Oberley TD, Buettner GR. Cell division in normal and transformed cells: the possible role of superoxide and hydrogen peroxide. Med Hypotheses. 1981;7:21–42. doi: 10.1016/0306-9877(81)90018-9. [DOI] [PubMed] [Google Scholar]
- 3.Gius D, Spitz DR. Redox signaling in cancer biology. Antioxid Redox Signal. 2006;8:1249–1252. doi: 10.1089/ars.2006.8.1249. [DOI] [PubMed] [Google Scholar]
- 4.Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31(Pt 6):1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 5.Oberley LW. Mechanism of the tumor suppressive effect of MnSOD over-expression. Biomed Pharmacother. 2005;59:143–148. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Teoh-Fitzgerald ML, Fitzgerald MP, Jensen TJ, Futscher BW, Domann FE. Genetic and Epigenetic Inactivation of Extracellular Superoxide Dismutase Promotes an Invasive Phenotype in Human Lung Cancer by Disrupting ECM Homeostasis. Mol Cancer Res. 2012;10:40–51. doi: 10.1158/1541-7786.MCR-11-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teodoridis JM, Strathdee G, Brown R. Epigenetic silencing mediated by CpG island methylation: potential as a therapeutic target and as a biomarker. Drug Resist Updat. 2004;7:267–278. doi: 10.1016/j.drup.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest. 1984;74:1398–1403. doi: 10.1172/JCI111550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teoh ML, Fitzgerald MP, Oberley LW, Domann FE. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res. 2009;69:6355–6363. doi: 10.1158/0008-5472.CAN-09-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, et al. Beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I. Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J Natl Cancer Inst. 2004;96:1219–1230. doi: 10.1093/jnci/djh230. [DOI] [PubMed] [Google Scholar]
- 14.Adachi T, Kodera T, Ohta H, Hayashi K, Hirano K. The heparin binding site of human extracellular-superoxide dismutase. Arch Biochem Biophys. 1992;297:155–161. doi: 10.1016/0003-9861(92)90654-f. [DOI] [PubMed] [Google Scholar]
- 15.Hicks CL, Bucy J, Stofer W. Heat inactivation of superoxide dismutase in bovine milk. J Dairy Sci. 1979;62:529–532. doi: 10.3168/jds.S0022-0302(79)83285-3. [DOI] [PubMed] [Google Scholar]
- 16.Hicks CL. Occurrence and consequence of superoxide dismutase in milk products: a review. J Dairy Sci. 1980;63:1199–1204. doi: 10.3168/jds.S0022-0302(80)83065-7. [DOI] [PubMed] [Google Scholar]
- 17.Jones PA. DNA methylation and cancer. Oncogene. 2002;21:5358–5360. doi: 10.1038/sj.onc.1205597. [DOI] [PubMed] [Google Scholar]
- 18.Laukkanen MO, Mannermaa S, Hiltunen MO, Aittomaki S, Airenne K, Janne J, et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol. 1999;19:2171–2178. doi: 10.1161/01.atv.19.9.2171. [DOI] [PubMed] [Google Scholar]
- 19.Garbe JC, Holst CR, Bassett E, Tlsty T, Stampfer MR. Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle. 2007;6:1927–1936. doi: 10.4161/cc.6.15.4519. [DOI] [PubMed] [Google Scholar]
- 20.Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res. 2009;69:5251–5258. doi: 10.1158/0008-5472.CAN-08-4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Pan J, Li JL, Lee JH, Tunkey C, Saraf K, et al. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol Cancer. 2007;6:7. doi: 10.1186/1476-4598-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen LH, Bissell MJ. A novel regulatory mechanism for whey acidic protein gene expression. Cell Regul. 1989;1:45–54. doi: 10.1091/mbc.1.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jolivet G, Pantano T, Houdebine LM. Regulation by the extracellular matrix (ECM) of prolactin-induced alpha s1-casein gene expression in rabbit primary mammary cells: role of STAT5, C/EBP, and chromatin structure. J Cell Biochem. 2005;95:313–327. doi: 10.1002/jcb.20397. [DOI] [PubMed] [Google Scholar]
- 24.Xu R, Spencer VA, Bissell MJ. Extracellular matrix-regulated gene expression requires cooperation of SWI/SNF and transcription factors. J Biol Chem. 2007;282:14992–14999. doi: 10.1074/jbc.M610316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hewetson A, Hendrix EC, Mansharamani M, Lee VH, Chilton BS. Identification of the RUSH consensus-binding site by cyclic amplification and selection of targets: demonstration that RUSH mediates the ability of prolactin to augment progesterone-dependent gene expression. Mol Endocrinol. 2002;16:2101–2112. doi: 10.1210/me.2002-0064. [DOI] [PubMed] [Google Scholar]
- 29.Vukicevic S, Kleinman HK, Luyten FP, Roberts AB, Roche NS, Reddi AH. Identification of multiple active growth factors in basement membrane Matrigel suggests caution in interpretation of cellular activity related to extracellular matrix components. Exp Cell Res. 1992;202:1–8. doi: 10.1016/0014-4827(92)90397-q. [DOI] [PubMed] [Google Scholar]
- 30.Henrotin Y, Deberg M, Mathy-Hartert M, Deby-Dupont G. Biochemical biomarkers of oxidative collagen damage. Adv Clin Chem. 2009;49:31–55. doi: 10.1016/s0065-2423(09)49002-4. [DOI] [PubMed] [Google Scholar]
- 31.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 32.Raats CJ, Bakker MA, van den Born J, Berden JH. Hydroxyl radicals depolymerize glomerular heparan sulfate in vitro and in experimental nephrotic syndrome. J Biol Chem. 1997;272:26734–26741. doi: 10.1074/jbc.272.42.26734. [DOI] [PubMed] [Google Scholar]
- 33.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang X, Sun Z, Runne C, Madsen J, Domann F, Henry M, et al. A critical role of Gbetagamma in tumorigenesis and metastasis of breast cancer. J Biol Chem. 2011;286:13244–13254. doi: 10.1074/jbc.M110.206615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaschke RJ, Howlett AR, Desprez PY, Petersen OW, Bissell MJ. Cell differentiation by extracellular matrix components. Methods Enzymol. 1994;245:535–556. doi: 10.1016/0076-6879(94)45027-7. [DOI] [PubMed] [Google Scholar]
- 36.Case AJ, McGill JL, Tygrett LT, Shirasawa T, Spitz DR, Waldschmidt TJ, et al. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free Radic Biol Med. 2011;50:448–458. doi: 10.1016/j.freeradbiomed.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teoh ML, Sun W, Smith BJ, Oberley LW, Cullen JJ. Modulation of reactive oxygen species in pancreatic cancer. Clin Cancer Res. 2007;13:7441–7450. doi: 10.1158/1078-0432.CCR-07-0851. [DOI] [PubMed] [Google Scholar]
- 38.Shan W, Zhong W, Zhao R, Oberley TD. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic Biol Med. 2010;49:2078–2087. doi: 10.1016/j.freeradbiomed.2010.10.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose SL, Fitzgerald MP, White NO, Hitchler MJ, Futscher BW, De Geest K, et al. Epigenetic regulation of maspin expression in human ovarian carcinoma cells. Gynecol Oncol. 2006;102:319–324. doi: 10.1016/j.ygyno.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 40.Svensson RU, Barnes JM, Rokhlin OW, Cohen MB, Henry MD. Chemotherapeutic agents up-regulate the cytomegalovirus promoter: implications for bioluminescence imaging of tumor response to therapy. Cancer Res. 2007;67:10445–10454. doi: 10.1158/0008-5472.CAN-07-1955. [DOI] [PubMed] [Google Scholar]
- 41.Drake JM, Gabriel CL, Henry MD. Assessing tumor growth and distribution in a model of prostate cancer metastasis using bioluminescence imaging. Clin Exp Metastasis. 2005;22:674–684. doi: 10.1007/s10585-006-9011-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.