

Abstract
Leptin is a key regulator of glucose metabolism in mammals, but the mechanisms of its action have remained elusive. We now show that signaling by extracellular signal–regulated kinase (ERK) and its upstream kinase MEK in the ventromedial hypothalamus (VMH) mediates the leptin-induced increase in glucose utilization as well as its insulin sensitivity in the whole body and in red-type skeletal muscle of mice through activation of the melanocortin receptor (MCR) in the VMH. In contrast, activation of signal transducer and activator of transcription 3 (STAT3), but not the MEK-ERK pathway, in the VMH by leptin enhances the insulin-induced suppression of endogenous glucose production in an MCR-independent manner, with this effect of leptin occurring only in the presence of an increased plasma concentration of insulin. Given that leptin requires 6 h to increase muscle glucose uptake, the transient activation of the MEK-ERK pathway in the VMH by leptin may play a role in the induction of synaptic plasticity in the VMH, resulting in the enhancement of MCR signaling in the nucleus and leading to an increase in insulin sensitivity in red-type muscle.
Leptin is an adipocyte-derived hormone that plays an important role in glucose metabolism in peripheral tissues as well as in overall energy metabolism in mammals (1,2). Treatment with leptin ameliorates diabetes in lipodystrophic mice and humans (3–5) as well as type 1 (6,7) and obesity-unrelated type 2 diabetes (8) in rodents. Although the antidiabetic effects of leptin are known to be mediated by the central nervous system (9–11), the mechanism by which leptin stimulates glucose utilization in muscle has remained unclear.
Neurons in the arcuate hypothalamic nucleus (ARC) and ventromedial hypothalamus (VMH) contribute to the effects of leptin on glucose metabolism. Restoration of expression of the Ob-Rb receptor for leptin in proopiomelanocortin (POMC) neurons of db/db mice (which lack Ob-Rb) normalizes blood glucose concentration (12,13). The hyperinsulinemia and insulin resistance characteristic of these animals remain unaffected, however, suggesting that other brain regions may also regulate glucose metabolism. We previously showed that injection of leptin into the VMH increases glucose uptake by skeletal muscle (mainly the red type), brown adipose tissue (BAT), and the heart, but not by white adipose tissue, through activation of the melanocortin receptor (MCR) in the VMH (14). These effects of leptin were manifest at 6 h after injection (14) and were abolished by attenuation of sympathetic nerve signaling through surgical denervation or through administration of either a blocker of sympathetic nerve activity (guanethidine) or the β-adrenergic antagonist propranolol (11,15). Furthermore, whereas leptin injection into the VMH increased glucose uptake in muscle, BAT, and the heart, injection into the ARC increased glucose uptake in BAT alone, and injection into the dorsomedial hypothalamus (DMH) or paraventricular hypothalamus (PVH) had no effect (14). The effect of leptin on muscle glucose uptake is thus dependent on Ob-Rb activation in the VMH, as well as on Ob-Rb activation in the ARC.
Activation of Ob-Rb stimulates intracellular signaling pathways, including those mediated by signal transducer and activator of transcription 3 (STAT3), phosphoinositide 3-kinase (PI3K), and extracellular signal–regulated kinase 1 or 2 (ERK1/2) (1,2,16). Leptin also downregulates the activity of AMP-activated protein kinase in the ARC and PVH, an effect that contributes to the anorexic action of leptin (17). With the use of a hyperinsulinemic-euglycemic clamp and measurement of 2-deoxyglucose (2DG) uptake, we have now examined the role of leptin signaling in the VMH in the acute effects of leptin injected into the periphery or the VMH on glucose metabolism in skeletal muscle of lean mice. Our results reveal that signaling by ERK and its upstream kinase MEK in the VMH mediates the leptin-induced increase in glucose utilization and its insulin sensitivity both in the whole body and in red-type skeletal muscle through activation of MCR in the VMH. In contrast, leptin in the VMH was found to enhance the insulin-induced suppression of endogenous glucose production (EGP), which largely reflects hepatic glucose production, through a STAT3-dependent, MCR-independent pathway in this nucleus.
RESEARCH DESIGN AND METHODS
Animals.
Male FVB mice (CLEA Japan, Tokyo, Japan) were studied at 12–16 weeks of age. The animals were housed individually in plastic cages at 24 ± 1°C with lights on from 0600 to 1800 h, and they were maintained with free access to a laboratory diet (Oriental Yeast, Tokyo, Japan) and water. All animal experiments were approved by the ethics committee for animal experiments of the National Institute for Physiological Sciences.
Surgical procedures.
A chronic double-walled stainless steel cannula was implanted stereotaxically and either unilaterally into the right side of the VMH or bilaterally into the VMH as described previously (14,18). Bilateral cannula placement was performed for examination of the effects of bilateral injection of the MEK inhibitor U0126 or a STAT3 inhibitor into the VMH in those on systemic injection of leptin. Unilateral cannula implantation was performed for all other studies. For the hyperinsulinemic-euglycemic clamp, polyethylene catheters were inserted into the right carotid artery and jugular vein of mice. Animals were handled repeatedly during the recovery period (2 weeks) after cannula implantation. Correct placement of the cannula tips was verified microscopically in brain sections, with >95% of animals manifesting correct placement; the few animals with incorrect cannula placement were excluded from analysis. Food was removed immediately before the administration of inhibitors, leptin, or the MCR agonist melanotan-II (MT-II).
Administration of leptin, MT-II, and inhibitors.
Leptin (5 ng) (PeproTech, Rocky Hill, NJ), the MCR agonist MT-II (10 pmol) (Phoenix Pharmaceuticals, Burlingame, CA), or the MCR antagonist SHU9119 (10 pmol) (Phoenix Pharmaceuticals) in 0.1 µL of physiological saline was injected with a Hamilton microsyringe into the right side of the VMH of freely moving mice through the unilateral cannula. The MEK inhibitor U0126 (10 μmol/L) (Cell Signaling Technology, Beverly, MA) or the PI3K inhibitor LY294002 (10 μmol/L) (Merck, Darmstadt, Germany) in 0.1 µL of 0.01% DMSO was injected into the same side of the VMH at 1 h before injection of leptin or MT-II. Cell-permeable SH2 domain–binding phosphopeptide (STAT3 inhibitor, Merck) (0.1 µL of a 250 μmol/L solution in saline) was injected into the VMH twice, at 1 h and 5 min before leptin injection. In some experiments, 0.1 µL of U0126 (10 μmol/L in 0.01% DMSO) or the STAT3 inhibitor (250 μmol/L in saline) was injected into the VMH bilaterally at 1 h or at both 1 h and 5 min, respectively, before intraperitoneal injection of leptin (5 mg/kg). Control mice were injected with the same volume of saline or 0.01% DMSO into the VMH or with intraperitoneal saline as appropriate.
Hyperinsulinemic-euglycemic clamp and measurement of associated 2-[14C]DG uptake.
Four hours after leptin or MT-II injection, the hyperinsulinemic-euglycemic clamp protocol was initiated in conscious and unrestrained mice. The protocol was modified slightly from that described on the website of the Mouse Metabolic Phenotyping Center at Vanderbilt University (http://www.mc.vanderbilt.edu/root/vumc.php?site=mmpc&doc=32773). The 120-min basal period (t = –120 to 0 min) was initiated at 1300 h and was followed by a 105-min clamp period (t = 0–105 min) beginning at 1500 h (Fig. 1A). A priming dose of [3-3H]glucose (5 μCi) (American Radiolabeled Chemicals, St. Louis, MO) was administered via the jugular vein catheter at t = –120 min and was followed by infusion of the tracer at a rate of 0.05 µCi/min for 2 h. The clamp period was initiated at t = 0 min by primed and continuous infusion of bovine insulin (bolus of 16 mU/kg followed by a rate of 5 mU ⋅ kg–1 ⋅ min–1) (Sigma-Aldrich Japan, Tokyo, Japan) through the jugular vein catheter. The rate of [3-3H]glucose infusion was increased to 0.1 µCi/min for the remainder of the experiment in order to minimize changes in specific activity relative to the equilibration period. Blood was collected every 5–10 min from the carotid artery catheter, and blood glucose was monitored (One Touch Ultra; Lifescan, Johnson & Johnson). Glucose (30%) was infused at a variable rate via the jugular vein catheter in order to maintain blood glucose levels at 130–150 mg/dL. Withdrawn erythrocytes were suspended in sterile 0.9% saline and returned to each animal.
FIG. 1.

Effects of leptin injection into the VMH on whole-body and muscle glucose metabolism in mice during a hyperinsulinemic-euglycemic clamp. A: Experimental protocol for the hyperinsulinemic-euglycemic clamp. B: Phosphorylation of STAT3 (on Tyr705), ERK (Thr202/Tyr204), and Akt (Ser473) in the VMH at 30 min after the unilateral injection of leptin (5 ng in 0.1 µL) or saline (0.1 µL) into the VMH. The data were evaluated with the ratio of phosphorylated form to the total protein and expressed as percent increase of the ratio to that of the saline-injected group. Representative immunoblots with antibodies to the phosphorylated (p) or total (t) forms of each protein are shown above densitometric quantitation of the relative phosphorylated/total protein ratio. *P < 0.05 vs. corresponding value for saline-injected group. C: Blood glucose levels during the basal and clamp periods. The clamp period begins at time 0. D: GIR required to maintain euglycemia during the clamp period. *P < 0.05 vs. corresponding value for saline-injected group. E: Rate of glucose disappearance (Rd) during the basal and clamp periods in mice injected with leptin or saline into the VMH unilaterally. F: Rate of glucose appearance (Ra) during the basal and clamp periods as well as the percentage suppression of Ra induced by insulin infusion. Ra reflects EGP. G: Rates of whole-body glycolysis and glycogen synthesis during the clamp period. H: 2DG uptake in muscle during the basal and clamp periods. I: Rates of glycolysis and glycogen synthesis in muscle during the clamp period. *P < 0.05 vs. basal, saline in VMH. †P < 0.05 vs. basal, leptin in VMH. #P < 0.05 vs. clamp, saline in VMH (D–I). All quantitative data are means ± SEM (n = 6 or 7 mice).
Tissue 2DG uptake was measured as described previously (14). For assessment of 2DG uptake during the basal period, mice were infused with 2-[14C]DG (5 µCi) (American Radiolabeled Chemicals) at t = –45 min through the jugular vein catheter. At t = –40, –30, –20, –10, and 0 min, an arterial blood sample (50 µL) was collected for assessment both of the rate of blood glucose appearance (Ra), which reflects EGP, and of 2DG uptake. For measurement of 2DG uptake during the clamp period, another group of mice was infused with 2-[14C]DG (5 µCi) at t = 60 min, and blood samples (50 µL) were collected at t = 65, 75, 85, 95, and 105 min. Immediately after collection of the final blood sample (t = 0 or 105 min), mice were killed with an overdose of pentobarbital sodium, and the soleus, red (Gastro-R) and white (Gastro-W) portions of the gastrocnemius, epididymal white adipose tissue (epiWAT), and liver were rapidly dissected. Gastro-R was dissected from the inner surface of the gastrocnemius attached to the soleus, whereas Gastro-W was dissected from the outer surface of the muscle. The rate of disappearance of blood glucose (Rd), which reflects whole-body glucose utilization, as well as Ra, the rates of whole-body glycolysis and glycogen synthesis, and the rates of glycolysis and glycogen synthesis in muscle were determined as described previously (19,20). Rd is equal to Ra plus the glucose infusion rate (GIR) during the clamp period, whereas Rd is equal to Ra during the basal period. Plasma concentrations of insulin (Insulin ELISA; Shibayagi, Gunma, Japan) and glucagon (Glucagon EIA kit; Yanaihara Institute, Shizuoka, Japan) were measured with the use of kits. Plasma epinephrine and norepinephrine concentrations were measured by high-performance liquid chromatography as described previously (18). Glycogen phosphorylase a activity in liver was measured as described previously (21) and was expressed as the ratio of activity in the absence of AMP to that in the presence of 3 mmol/L AMP.
Immunoblot analysis.
The right side of the ARC, VMH, or DMH sampled at 30 min after leptin injection into the VMH or at 1 h after intraperitoneal injection of leptin was dissected from a 1-mm-thick sagittal section prepared from the midline of the fresh brain and was subjected to immunoblot analysis as described previously (14) (Supplementary Fig. 1). The primary antibodies included those to Tyr705-phosphorylated STAT3 (pSTAT3), Thr202- and Tyr204-phosphorylated p44/42 MAPK (pERK1/2), and Ser473-phosphorylated Akt (pAkt) (all from Cell Signaling Technology); those to Ser549-phosphorylated synapsin (Thermo Scientific Pierce, Rockford, IL); and those to total forms of these various proteins (Cell Signaling Technology). All antibodies were used at a dilution of 1:1,000.
Immunofluorescence analysis of phosphorylated STAT3 and ERK.
At 1 h after intraperitoneal leptin injection, mice were anesthetized and perfused transcardially with 4% paraformaldehyde in 0.1 mol/L phosphate buffer. Brain tissue was removed, fixed again, and embedded in OCT compound (Sakura Finetechnical, Tokyo, Japan). Phosphorylated forms of STAT3 and ERK in the same cryosections (thickness of 7 μm) were detected by consecutive incubations with rabbit polyclonal antibodies to Tyr705-phosphorylated STAT3 (1:100 dilution) (Cell Signaling Technology) and Alexa Fluor 568–labeled goat antibodies to rabbit immunoglobulin G (1:400 dilution) (Life Technologies, Carlsbad, CA), and then with rabbit polyclonal antibodies to Thr202- and Tyr204-phosphorylated ERK1/2 (1:500 dilution) (Cell Signaling Technology) and Alexa Fluor 488–labeled goat antibodies to rabbit immunoglobulin G (1:400 dilution) (Life Technologies). Sections were examined with a fluorescence microscope (Olympus AX-70) and a laser confocal microscope (Digital Eclipse C1; Nikon). Staining was absent in control sections processed without primary antibodies.
Extraction of RNA and reverse transcription PCR analysis.
Reverse transcription and real-time PCR analysis with Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) were performed as described previously (14,18). The sequences of PCR primers (forward and reverse, respectively) were 5′-CATGGGCGCAGCAGGTGTATACT-3′ and 5′-CAAGGTAGATCCGGGACAGACAG-3′ for glucose-6-phosphatase (G6Pase), 5′-GGTGTTTACTGGGAAGGCATC-3′ and 5′-CAATAATGGGGCACTGGCTG-3′ for PEPCK, and 5′-AACTTTGGCATTGTGGAAGG-3′ and 5′-ACACATTGGGGGTAGGAACA-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data were normalized by the corresponding abundance of GAPDH mRNA.
Statistical analysis.
Data are presented as means ± SEM. Statistical comparisons between two groups and among multiple groups were performed with Student t test and with ANOVA followed by Tukey HSD post hoc test, respectively. A P value of <0.05 was considered statistically significant.
RESULTS
Leptin injection into the VMH stimulates whole-body glucose metabolism.
We examined the effects of leptin injection into the VMH on glucose metabolism with the use of a hyperinsulinemic-euglycemic clamp (Fig. 1A). The dose of leptin was selected on the basis of the results of our previous study (14). We also selected the dose of insulin as 5 mU ⋅ kg–1 ⋅ min–1 for the clamp on the basis of the results of preliminary experiments showing that this dose increased the rate of glucose disappearance (Rd, reflecting whole-body glucose utilization) about twofold and suppressed the rate of glucose appearance (Ra, reflecting EGP) by about one-half. Immunoblot and immunohistochemical analyses revealed that leptin injection into the VMH increased the phosphorylation of STAT3, ERK1/2, and Akt (which functions downstream of PI3K) in this brain region but not in the ARC or DMH (Fig. 1B and Supplementary Fig. 2). Preliminary data revealed that the phosphorylation of ERK1/2 peaked at 30 min and returned to the control level at 6 h after leptin injection into the VMH (data not shown).
During the clamp period, the plasma insulin concentration increased 1.78- and 1.63-fold in mice injected with saline or leptin, respectively, with these values not differing significantly (Supplementary Table 1). The blood glucose concentration was maintained constant by glucose infusion (Fig. 1C). Leptin injection into the VMH necessitated an increase in GIR (Fig. 1D). The increase in GIR was associated with an increase in Rd (Fig. 1E) and a decrease in Ra (Fig. 1F). Leptin injection also significantly increased whole-body glycolysis and tended to increase glycogen synthesis (Fig. 1G). Leptin enhanced the insulin-induced increase in 2DG uptake in muscle, with this effect being more pronounced in red-type muscle (soleus and Gastro-R) than in white-type muscle (Gastro-W) (Fig. 1H). The increase in 2DG uptake in red-type muscle was accompanied by an increase in glycolysis and glycogen synthesis in the muscle tissue (Fig. 1I).
In the basal condition, leptin injection into the VMH increased Rd (Fig. 1E) without affecting the blood glucose level (Fig. 1C). This effect was accompanied by an increase in Ra (Fig. 1F), given that Ra is equal to Rd in the steady-state condition. Leptin increased 2DG uptake in soleus and to a lesser extent in Gastro-R, but not in Gastro-W, similar to its effects during the clamp period (Fig. 1H). Plasma insulin levels did not differ between control and leptin-injected groups during the basal period (Supplementary Table 1). Leptin injection into the VMH thus increased glucose turnover at the whole-body level during the basal period. These results suggested that leptin injection into the VMH increases whole-body and muscle glucose utilization under both basal and clamp conditions, whereas it suppresses hepatic EGP in a manner dependent on plasma insulin concentration.
Leptin in the VMH induces glucose utilization and enhances insulin-induced suppression of EGP differentially via MEK-ERK and STAT3 pathways.
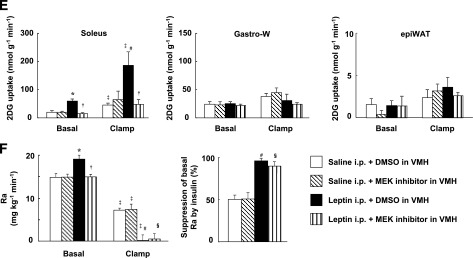
We examined the effects of injection of a MEK inhibitor (U0126), a STAT3 inhibitor (cell-permeable SH2 domain–binding phosphopeptide), and a PI3K inhibitor (LY294002) into the VMH. Whereas determination of the acute effects of specific inhibitors minimizes the influence of adaptive responses of neuronal circuits in the brain, the specificity of such agents depends on their concentration. We therefore first determined the doses of inhibitors that preferentially attenuated the activation of their target molecules (Fig. 2A). Plasma insulin and blood glucose levels were not affected by injection of these inhibitors into the VMH, or by similar injection of leptin, the MCR agonist MT-II, or the MCR antagonist SHU9119 (Supplementary Fig. 3 and Supplementary Table 1). The MEK inhibitor U0126 and the STAT3 inhibitor, but not the PI3K inhibitor LY294002, suppressed the leptin-induced increase in GIR during the clamp period, with the effect of U0126 being greater than that of the STAT3 inhibitor (Fig. 2B). None of these three inhibitors affected GIR in response to insulin infusion alone. U0126 inhibited the effect of leptin on Rd but not on Ra during the clamp period, whereas the STAT3 inhibitor attenuated the effect of leptin on Ra but not on Rd (Fig. 2C and D). LY294002 had no effect on either Rd or Ra. The leptin-induced increase in whole-body glycolysis was suppressed by U0126 but not by the other inhibitors (Fig. 2E). Furthermore, the MEK inhibitor, but not the STAT3 inhibitor or PI3K inhibitor, attenuated leptin-induced 2DG uptake, glycolysis, and glycogen synthesis in soleus and Gastro-R, but not in Gastro-W, during the clamp period (Fig. 3A and C–E). Uptake of 2DG by epiWAT was not affected by leptin or by the inhibitors (Fig. 3B and F). U0126 also suppressed the increase in Rd (equal to that in Ra) induced by leptin during the basal period (data not shown).
FIG. 2.

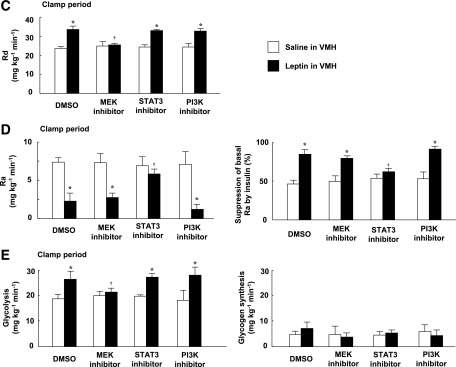
Leptin injection into the VMH increases whole-body insulin sensitivity via MEK-ERK and STAT3 signaling in the VMH. A: Phosphorylation of ERK (Thr202/Tyr204), STAT3 (Tyr705), and Akt (Ser473) in the VMH at 30 min after unilateral injection of leptin or saline into the VMH. The MEK inhibitor U0126 or PI3K inhibitor LY294002 was injected unilaterally into the VMH at 1 h before leptin injection, whereas the STAT3 inhibitor was injected into the VMH at both 1 h and 5 min before leptin injection. DMSO at 0.1% was injected as a control for the inhibitors. The data were evaluated with the ratio of phosphorylated form to the total protein and expressed as percent increase of the ratio to that of the saline-injected group. Representative immunoblots with antibodies to the phosphorylated (p) or total (t) forms of the proteins are shown above the quantitative data, which are means ± SEM (n = 6 or 7 mice). *P < 0.05 vs. corresponding saline-injected group. †P < 0.05 vs. corresponding value for leptin + DMSO in VMH. B–E: Effects of leptin and leptin signaling inhibitors on the time course of the increase in GIR (B), on Rd during the clamp period (C), on Ra during the clamp period and on the percentage suppression of Ra induced by insulin infusion (D), and on the rate of whole-body glycolysis or glycogen synthesis during the clamp period (E) for mice subjected to the hyperinsulinemic-euglycemic clamp protocol. The mean of the GIR values from 60 to 105 min is shown in the bar graphs (B). All data are means ± SEM (n = 6 mice). *P < 0.05 vs. corresponding value for saline-injected group. †P < 0.05 vs. corresponding value for leptin + DMSO in VMH. ‡P < 0.05 for leptin + MEK inhibitor in VMH vs. leptin + STAT3 inhibitor in VMH.
FIG. 3.

Leptin injection into the VMH increases glucose utilization in red-type muscle via MEK-ERK signaling in the VMH, whereas it enhances insulin-induced suppression of glycogen phosphorylase a activity in the liver via STAT3 in the VMH. Effects of leptin and the MEK inhibitor U0126 on 2DG uptake (A and B) as well as on glycolysis and glycogen synthesis (C) in soleus, Gastro-R, Gastro-W, or epiWAT during the clamp period of the hyperinsulinemic-euglycemic clamp protocol. Effects of leptin, the STAT3 inhibitor, and the PI3K inhibitor LY294002 on 2DG uptake in soleus (D), Gastro-W (E), or epiWAT (F) during the clamp period. G: Effects of leptin and leptin signaling inhibitors on glycogen phosphorylase a activity in liver during the clamp period. All tissue samples were obtained from the mice studied in Fig. 2B–E. All data are means ± SEM (n = 6 mice). *P < 0.05 vs. corresponding value for saline-injected group. †P < 0.05 vs. corresponding value for leptin + DMSO in VMH.
Insulin downregulated the amounts of PEPCK and glucose-6-phosphatase (G6Pase) mRNAs in the liver during the clamp period, whereas leptin and the leptin signaling inhibitors did not affect the hepatic abundance of these mRNAs (Supplementary Fig. 4A). In contrast, leptin attenuated the activity of glycogen phosphorylase a in the liver during the clamp period, and this effect was blocked by the STAT3 inhibitor but not by the MEK inhibitor or the PI3K inhibitor (Fig. 3G). These results suggested that leptin in the VMH enhances the insulin-induced suppression of hepatic glucose production via STAT3 signaling in the VMH.
The plasma glucagon concentration did not differ between control and leptin-injected mice either with or without insulin infusion (Supplementary Fig. 4B). Injection of inhibitors of leptin signaling pathways into the VMH also had no effect on the plasma glucagon level. Furthermore, plasma norepinephrine and epinephrine concentrations had decreased to undetectable levels at the end of the clamp period in both control and leptin-injected mice (data not shown), probably as a result of adequate training and handling of the animals as well as the high plasma insulin concentration. Whereas humoral factors may contribute to the leptin-induced suppression of EGP and hepatic glycogen phosphorylase a activity, the effect of leptin on EGP may be mediated by the autonomic nervous system, including the vagus nerve, as described previously (8,22,23).
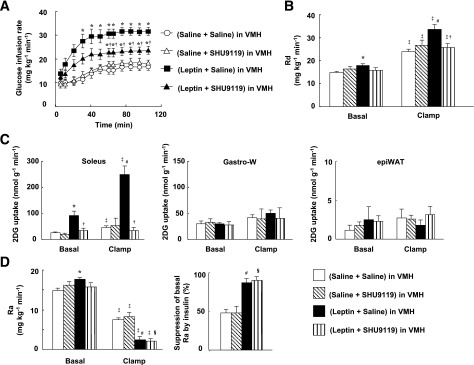
Systemic leptin increases glucose utilization via MEK-ERK signaling in the VMH.
The intraperitoneal injection of leptin (5 mg/kg) increased the amounts of pERK1/2 and pSTAT3, but not pAkt, in the VMH (Fig. 4A). Leptin also increased the amounts of pERK1/2 and pSTAT3 in the ARC as well as pSTAT3 in the DMH (Supplementary Fig. 5A). The intraperitoneal injection of leptin increased the number of cells positive for both pSTAT3 and pERK in the VMH (Fig. 4B) as well as in the ARC (Supplementary Fig. 5B). Systemic leptin thus activates ERK and STAT3 in the same neurons in the VMH as well as in the ARC, probably in a manner dependent on Ob-Rb. Prior bilateral injection of the MEK inhibitor U0126 into the VMH suppressed the increase in ERK phosphorylation in the VMH induced by intraperitoneal injection of leptin without affecting the phosphorylation of STAT3 or Akt in the VMH (Fig. 4A) or of any of the three signaling molecules in the ARC or DMH (Supplementary Fig. 5A).
FIG. 4.

Systemic injection of leptin increases whole-body glucose utilization and glucose uptake in soleus muscle via MEK-ERK signaling in the VMH. A: Phosphorylation of STAT3 (on Tyr705), ERK (Thr202/Tyr204), and Akt (Ser473) in the VMH at 1 h after intraperitoneal injection of leptin (5 mg/kg) or saline (leptin i.p. or saline i.p.). The MEK inhibitor U0126 or DMSO (0.01%) was injected into the VMH bilaterally 1 h before injection of leptin or saline. The data were evaluated with the ratio of phosphorylated form to the total protein and expressed as percent increase of the ratio to that of saline i.p. + DMSO group. Representative immunoblots with antibodies to the phosphorylated (p) or total (t) forms of the proteins are shown above the quantitative data (n = 6 or 7 mice). *P < 0.05 vs. corresponding value for saline i.p. + DMSO in VMH. §P < 0.05 vs. corresponding value for saline i.p. + MEK inhibitor in VMH. †P < 0.05 vs. leptin i.p. + DMSO in VMH. B: Immunohistofluorescence analysis of phosphorylated forms of STAT3 (Tyr705) and ERK (Thr202/Tyr204) in the VMH at 1 h after intraperitoneal injection of saline (c, e, and g) or leptin (a, d, f, and h). Panels d, f, and h are digital zoom images corresponding to the boxed area in panel a, whereas panels c, e, and g represent the equivalent area in the VMH. Immunofluorescence of pSTAT3 (c and d), pERK (e and f), and both phosphorylated proteins (a, g, and h) is shown. The dashed trace in panels a and b represents the VMH. Panel b shows a Nissl-stained section of the medial hypothalamus from a C57BL/6 mouse and is modified with permission from Hof et al. (40). 3V, third ventricle. Scale bars, 20 µm. C–F: Effects of injection of the MEK inhibitor U0126 into the VMH on changes in glucose metabolism induced by intraperitoneal injection of leptin in mice subjected to the hyperinsulinemic-euglycemic clamp protocol. U0126 or DMSO was injected into the VMH bilaterally 1 h prior to leptin or saline injection (n = 5 or 6 mice). C: Glucose infusion rate during the clamp period. *P < 0.05 vs. corresponding value for saline i.p. + DMSO in VMH. †P < 0.05 vs. corresponding value for leptin i.p. + DMSO in VMH. D: Rd in basal and clamp periods. E: 2DG uptake in soleus, Gastro-W, and epiWAT during the basal and clamp periods. F: Ra during the basal and clamp periods as well as the percentage suppression of Ra induced by insulin infusion. The Ra in the basal period is equal to Rd in the basal period. *P < 0.05 vs. corresponding value for saline i.p. + DMSO in VMH in the basal period. #P < 0.05 vs. corresponding value for saline i.p. + DMSO in VMH in the clamp period. §P < 0.05 vs. corresponding value for saline i.p. + MEK inhibitor in VMH in the clamp period. ‡P < 0.05 vs. corresponding value for the basal period. †P < 0.05 vs. corresponding value for leptin i.p. + DMSO in VMH (D–F). All quantitative data are means ± SEM.
Injection of U0126 into the VMH partially inhibited the increase in GIR induced by intraperitoneal injection of leptin (Fig. 4C). This effect of U0126 was associated with suppression of the leptin-induced increases in Rd (Fig. 4D) and 2DG uptake in the soleus (Fig. 4E). The MEK inhibitor did not affect the leptin-induced enhancement of the suppression of Ra by insulin (Fig. 4F). In the basal period, U0126 injection into the VMH inhibited the leptin-induced increases in Rd (Fig. 4D), 2DG uptake in the soleus (Fig. 4E), and Ra (Fig. 4F), the latter being equal to the increase in Rd (Fig. 4D). Blood glucose and plasma insulin levels during the basal or clamp periods were not affected by intraperitoneal injection of leptin with or without prior injection of U0126 into the VMH (Supplementary Fig. 3 and Supplementary Table 1). The enhancement by leptin of the insulin-induced suppression of hepatic EGP was only partially inhibited by injection of the STAT3 inhibitor into the VMH (data not shown). Hepatic EGP is thus regulated by leptin via other brain sites or peripheral tissues as well as via the VMH.
MCR in the VMH mediates leptin-induced glucose utilization (but not suppression of EGP) in a manner independent of MEK-ERK signaling.
Injection of the MCR antagonist SHU9119 into the VMH resulted in partial inhibition of the increase in GIR induced by leptin injection into the VMH (Fig. 5A). This inhibition of GIR by SHU9119 was associated with suppression of the leptin-induced increases in Rd (Fig. 5B) and 2DG uptake in soleus muscle (Fig. 5C). SHU9119 did not affect the enhancement by leptin of the insulin-induced suppression of Ra (Fig. 5D). During the basal period, SHU9119 injection into the VMH inhibited the leptin-induced increases in Rd (Fig. 5B), 2DG uptake in soleus (Fig. 5C), and Ra (Fig. 5D), with the latter being equal to the increase in Rd (Fig. 5B).
FIG. 5.
MCR in the VMH mediates leptin-induced whole-body glucose utilization and glucose uptake in soleus muscle, but not leptin-induced enhancement of the suppressive effect of insulin on EGP. The effects of the MCR antagonist SHU9119 injected into the VMH on changes in glucose metabolism induced by unilateral injection of leptin into the VMH were evaluated with the hyperinsulinemic-euglycemic clamp. SHU9119 or saline was injected into the VMH 1 h prior to leptin or saline injection. A: GIR during the clamp period. *P < 0.05 vs. corresponding value for saline + saline in VMH. †P < 0.05 vs. corresponding value for leptin + saline in VMH. B: Rd during basal and clamp periods. C: 2DG uptake in soleus, Gastro-W, and epiWAT during basal and clamp periods. D: Ra during basal and clamp periods as well as the percentage suppression of Ra by insulin infusion. Ra in the basal period is equal to Rd in the basal period. *P < 0.05 vs. corresponding value for saline + saline in VMH in the basal period. #P < 0.05 vs. corresponding value for saline + saline in VMH in the clamp period. §P < 0.05 vs. corresponding value for saline + SHU9119 in VMH in the clamp period. ‡P < 0.05 vs. corresponding value for the basal period. †P < 0.05 vs. corresponding value for leptin + saline in VMH (B–D). All data are means ± SEM (n = 6 or 7 mice).
Injection of the MCR agonist MT-II into the VMH increased both GIR (Fig. 6A) and Rd (Fig. 6B) during the clamp period. Injection of MT-II into the VMH also enhanced insulin-induced 2DG uptake in soleus but not in Gastro-W or epiWAT (Fig. 6C). However, the MT-II–induced increases in GIR, Rd, and 2DG uptake in soleus were not inhibited by injection of the MEK inhibitor U0126 into the VMH (Fig. 6A–C). Moreover, MT-II did not enhance the suppression of Ra induced by insulin during the clamp period (Fig. 6D). Consistent with our previous results showing that MT-II increased 2DG uptake in red-type skeletal muscle (14), MT-II increased both Rd (Fig. 6B) and Ra (Fig. 6D) in the basal period, and these effects were not inhibited by U0126. Whereas MT-II did not increase glucose uptake in Gastro-W during the clamp period, we previously found that MT-II induced a small (1.3-fold) increase in 2DG uptake in white muscle in the absence of insulin infusion (14). The effect of MT-II as well as leptin on glucose uptake and its insulin sensitivity in white skeletal muscle is thus markedly smaller than in red muscle. These data thus suggested that the increases in whole-body glucose utilization as well as in glucose utilization by red-type muscle induced by leptin in the VMH, but not the enhancement by leptin of the suppression of EGP by insulin, are mediated by MCR in the VMH.
FIG. 6.

An MCR agonist in the VMH increases whole-body glucose utilization and glucose uptake in soleus muscle but does not enhance the suppressive effect of insulin on EGP. The effects of injection of the MCR agonist MT-II into the VMH on glucose metabolism were evaluated with the hyperinsulinemic-euglycemic clamp. The MEK inhibitor U0126 or DMSO (0.01%) was injected into the VMH unilaterally at 1 h before injection of MT-II or saline. A: GIR during the clamp period. *P < 0.05 vs. corresponding value for saline + DMSO in VMH. B: Rd during the basal and clamp periods. C: 2DG uptake in soleus, Gastro-W, and epiWAT during the clamp period. D: Ra during the basal and clamp periods as well as the percentage suppression of Ra induced by insulin infusion. Ra in the basal period is equal to Rd in the basal period. *P < 0.05 vs. corresponding value for saline + DMSO in VMH in the basal period. #P < 0.05 vs. corresponding value for saline + DMSO in VMH in the clamp period. §P < 0.05 vs. corresponding value for saline + MEK inhibitor in VMH in the clamp period. ‡P < 0.05 vs. corresponding value for the basal period (B and D). E: Phosphorylation of synapsin in the VMH after injection of leptin and the MEK inhibitor U0126. The MEK inhibitor U0126 or DMSO (0.01%) was injected into the VMH unilaterally at 1 h before injection of leptin or saline without the hyperinsulinemic-euglycemic clamp. The VMH was collected at 30 min after leptin injection. The data were evaluated with the ratio of phosphorylated form (p) to the total (t) protein and expressed as percent increase of the ratio to that of saline + DMSO in the VMH group. *P < 0.05 vs. corresponding value for saline + DMSO in VMH. †P < 0.05 vs. corresponding value for leptin + DMSO in VMH. ns, not significant. All data are means ± SEM (n = 6 or 7 mice).
Leptin has previously been shown to increase the density of hippocampal synapses and of N-methyl-d-aspartate–sensitive glutamate receptors at these synapses in a manner dependent on ERK signaling (24). Finally, we examined the effects of leptin on the phosphorylation of synapsin, which contributes to synapse formation, in the VMH. Leptin injection into the VMH increased the phosphorylation of synapsin in the VMH in a manner dependent on MEK-ERK signaling (Fig. 6E).
DISCUSSION
Central or peripheral administration of leptin has beneficial effects on diabetes in lipodystrophic mice and humans (3–5) as well as on type 1 (6,7) and obesity-unrelated type 2 diabetes in rodents (8). We have now shown that MEK-ERK signaling in the VMH mediates the leptin-induced acute increase in glucose uptake in red-type muscle as well as the insulin sensitivity of this process, and that it thereby contributes to the leptin-induced increase in whole-body glucose utilization (Fig. 7). In contrast, leptin in the VMH enhances insulin-induced suppression of hepatic EGP through the action of STAT3 in the VMH and the inhibition of glycogen phosphorylase a activity in the liver (Fig. 7). Given that leptin stimulates hepatic EGP under basal conditions, it reciprocally regulates this process in a manner dependent on the plasma insulin concentration. MCR activation in the VMH contributes to the leptin-induced increase in whole-body and muscle glucose utilization but not to its suppression of EGP. Activation of the MEK-ERK pathway in the VMH appears to occur upstream of and to be necessary for the activation of MCR signaling in the VMH and is required for the increase in muscle glucose uptake.
FIG. 7.
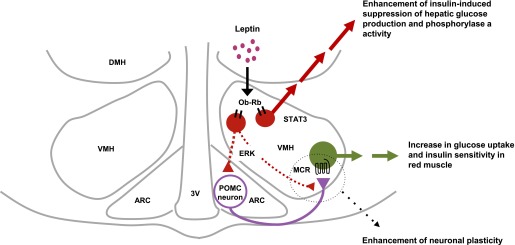
Model for the mechanism of regulation of glucose metabolism in muscle and liver by leptin in the VMH. Ob-Rb in the VMH plays a key role in the regulation of glucose metabolism and insulin sensitivity in muscle and liver by leptin. Leptin-activated MEK-ERK signaling in the VMH increases insulin sensitivity and glucose utilization in red muscle through activation of MCR in the VMH. Ob-Rb–expressing VMH neurons likely activate POMC neurons either in the ARC itself (14) or at their synaptic connections with VMH neurons through the MEK-ERK pathway. The MEK-ERK pathway then stimulates synaptic plasticity for POMC neurons and MCR-expressing neurons in the VMH. Whereas other brain sites may contribute to the leptin-induced enhancement of the suppressive effect of insulin on hepatic glucose production, leptin-activated STAT3 signaling in the VMH mediates this enhancement by inhibiting glycogen phosphorylase a activity in liver. 3V, third ventricle.
We previously showed that injection of leptin into the VMH increased muscle glucose uptake in mice at 6 h but not at 3 h after the injection, whereas intracerebroventricular injection of the MCR agonist MT-II increased muscle glucose uptake within 3 h (14). Similarly, intravenous or intracerebroventricular injection of leptin was found to induce a slow but progressive increase in sympathetic nerve activity in peripheral tissues (25,26), whereas intracerebroventricular injection of α-melanocyte–stimulating hormone (α-MSH) resulted in immediate activation of sympathetic nerves (26). We previously showed that the increase in muscle glucose uptake induced by leptin injection into the VMH is mediated by the sympathetic nervous system and β-adrenergic receptors (β-ARs) (15). We also found that injection of the hypothalamic neuropeptide orexin into the VMH stimulates glucose uptake in the muscle of mice, similar to the effect of leptin, and that this action of orexin is mediated via sympathetic nerves and β2-ARs (18). The orexin-induced increase in glucose uptake was blunted in β-AR–deficient mice (β-less mice), whereas restoration of β2-AR expression in the muscle of these mice resulted in recovery of the orexin effect. Furthermore, preliminary data revealed that the phosphorylation of ERK1/2 peaked at 30 min and returned to the control level at 6 h after leptin injection into the VMH (data not shown). These observations suggest that leptin exerts its effects on glucose metabolism by altering neuronal plasticity in the VMH through the activation of MEK-ERK signaling in this region. Consistent with this notion, we found that leptin injection into the VMH increased the phosphorylation of synapsin, which contributes to synapse formation, in the VMH in a manner dependent on MEK-ERK signaling.
We previously showed that injection of leptin into the VMH induced expression of the transcription factor c-FOS in the ARC as well as in the VMH at 6 h after the injection (14). Intense stimulation of VMH neurons by a photoactivatable caged form of glutamate increased the electrical activity of POMC neurons in the ARC (27). Moreover, the ventrolateral region of the VMH and the VMH shell contain a high number of dendrites that harbor a substantial number of axons and boutons immunoreactive for α-MSH, and MT-II and SHU9119 reciprocally regulate the activity of VMH neurons (28). Brain-derived neurotrophic factor is expressed in the ventrolateral region of the VMH and acts downstream of MCR in the VMH (29). Brain-derived neurotrophic factor enhances neuronal plasticity through retrograde action. Early activation of the MEK-ERK signaling pathway in the VMH by leptin may thus induce synaptic plasticity in the VMH, resulting in the enhancement of MCR signaling in the VMH via POMC neurons in the ARC and leading to an increase in insulin sensitivity in red-type muscle (Fig. 7).
STAT3 and the leptin receptor Ob-Rb in VMH neurons are implicated in the homeostatic regulation of glucose metabolism by leptin. Ablation of leptin receptors in steroidogenic factor 1 (SF1)–expressing VMH neurons of mice induced insulin resistance before the onset of obesity (30). Conversely, SF1-specific ablation of suppressor of cytokine signaling-3 (SOCS-3), a feedback inhibitor of the leptin-induced JAK-STAT3 signaling pathway, improved glucose homeostasis in mice (31). Furthermore, intracerebroventricular injection of a STAT3 inhibitor suppressed leptin-induced enhancement of insulin sensitivity in the liver, and abolishment of Ob-Rb–dependent STAT3 signaling (in s/s mice) results in pronounced hepatic insulin resistance (32). Our data suggest that STAT3 in the VMH regulates liver insulin sensitivity. However, other brain regions, such as the ARC and brain stem, may also contribute to such regulation, given that the enhancement of insulin-induced suppression of hepatic EGP elicited by intraperitoneal injection of leptin was only partially attenuated by injection of the STAT3 inhibitor in the VMH.
We found that leptin suppressed EGP during the clamp period of the hyperinsulinemic-euglycemic clamp protocol, whereas it increased EGP during the basal period. The insulin-dependent reciprocal regulation of EGP by leptin may explain why leptin does not induce hypoglycemia in the postprandial state. Insulin inhibits hepatic EGP via the central nervous system (33) as well as through the inhibition of agouti-related peptide–containing neurons in the ARC (34). Insulin in the brain modulates leptin signaling in the hypothalamus (35). The increased plasma insulin levels during the prandial state may thus influence the effect of leptin on hepatic EGP in both the brain and liver.
PI3K signaling in the hypothalamus also regulates glucose and lipid, as well as energy, metabolism (36–38). Ablation of the p110α catalytic subunit of PI3K in POMC neurons was found to impair glucose metabolism (36). In contrast, ablation of this subunit in SF1-expressing neurons did not affect glucose homeostasis (37). These observations as well as our present data suggest that the PI3K pathway in Ob-Rb–expressing neurons in other brain regions, such as the ARC, rather than in the VMH, might contribute to the effects of leptin on glucose metabolism. Furthermore, the PI3K pathway in Ob-Rb–expressing neurons in the VMH might contribute to the chronic rather than the acute effects of leptin on glucose metabolism.
We previously showed that whereas leptin injection into the VMH increased glucose uptake in muscle, BAT, and the heart, injection into the ARC increased glucose uptake in BAT but not in muscle or heart, and injection into the DMH or PVH had no effect (14). Injection of MT-II either into the VMH or intracerebroventricularly increased glucose uptake in muscle, BAT, and the heart, whereas injection into the PVH increased glucose uptake in BAT alone, and injection into the DMH or ARC had no effect (14). Thus, whereas leptin-induced glucose uptake in BAT and muscle is mediated by Ob-Rb and MCR in the VMH, glucose uptake in BAT may also be mediated by Ob-Rb in POMC neurons in the ARC and then MCR in the PVH. PVH neurons project to the solitary nucleus and to the raphe nucleus, and thereby stimulate thermogenesis in BAT (39). Further investigation is necessary to explore the neuronal pathway responsible for leptin-induced glucose uptake in muscle from the VMH to the hindbrain and muscle tissue.
In summary, we have found that the MEK-ERK pathway in the VMH plays an important role in leptin-induced glucose uptake in red-type muscle as well as whole-body glucose utilization. MCR in the VMH also mediates these effects of leptin. In contrast, activation of STAT3 in the VMH by leptin enhances the insulin-induced suppression of hepatic EGP by inhibiting glycogen phosphorylase a activity. These results suggest that VMH neurons mediate the antidiabetic effects of leptin in the control of muscle as well as liver glucose metabolism. They thus provide important insight into the mechanism of the antidiabetic effects of leptin in humans as well as rodents.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Grants-in-Aid for Scientific Research (B) (21390067 and 24390058 to Y.M.), Grants-in-Aid for Young Scientists (B) (20790656 and 22790875 to S.O. and 23790282 to C.T.), and a Grant-in-Aid for Scientific Research on Innovative Areas (Research in a Proposed Research Area, “Molecular Basis and Disorders of Control of Appetite and Fat Accumulation;” 22126005 to Y.M.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, as well as by the Specific Research Fund of the National Institutes for Natural Sciences (to Y.M.).
No potential conflicts of interest relevant to this article were reported.
C.T. researched data, contributed to discussion, and wrote and edited the manuscript. T.S., H.K., and S.S. researched data, contributed to discussion, and reviewed and edited the manuscript. S.O. contributed to discussion. E.A.C., T.S., Y.O.-O., S.Y., K.T., L.T., and K.S. researched data. Y.M. designed the study, contributed to discussion, and wrote and edited the manuscript. Y.M. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank N. Kawai and K. Nagatani for laboratory management, the Center for Analytical Instruments at the National Institutes for Basic Biology (Okazaki, Japan) for biochemical analysis, and K.W. Brocklehurst, an independent scientific editorial consultant (Washington, DC), for editorial assistance.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1629/-/DC1.
REFERENCES
- 1.Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 2008;70:537–556 [DOI] [PubMed] [Google Scholar]
- 2.Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev 2011;91:389–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999;401:73–76 [DOI] [PubMed] [Google Scholar]
- 4.Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002;346:570–578 [DOI] [PubMed] [Google Scholar]
- 5.Ebihara K, Masuzaki H, Nakao K. Long-term leptin-replacement therapy for lipoatrophic diabetes. N Engl J Med 2004;351:615–616 [DOI] [PubMed] [Google Scholar]
- 6.Chinookoswong N, Wang JL, Shi ZQ. Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes 1999;48:1487–1492 [DOI] [PubMed] [Google Scholar]
- 7.Fujikawa T, Chuang J-C, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA 2010;107:17391–17396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Wu X, Camacho R, Schwartz GJ, LeRoith D. Intracerebroventricular leptin infusion improves glucose homeostasis in lean type 2 diabetic MKR mice via hepatic vagal and non-vagal mechanisms. PLoS ONE 2011;6:e17058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 1997;389:374–377 [DOI] [PubMed] [Google Scholar]
- 10.Minokoshi Y, Kim Y-B, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002;415:339–343 [DOI] [PubMed] [Google Scholar]
- 11.Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 1999;48:287–291 [DOI] [PubMed] [Google Scholar]
- 12.Huo L, Gamber K, Greeley S, et al. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab 2009;9:537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berglund ED, Vianna CR, Donato J, Jr, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest 2012;122:1000–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toda C, Shiuchi T, Lee S, et al. Distinct effects of leptin and a melanocortin receptor agonist injected into medial hypothalamic nuclei on glucose uptake in peripheral tissues. Diabetes 2009;58:2757–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes 1999;48:1706–1712 [DOI] [PubMed] [Google Scholar]
- 16.Rahmouni K, Sigmund CD, Haynes WG, Mark AL. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes 2009;58:536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004;428:569–574 [DOI] [PubMed] [Google Scholar]
- 18.Shiuchi T, Haque MS, Okamoto S, et al. Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab 2009;10:466–480 [DOI] [PubMed] [Google Scholar]
- 19.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 2006;55:390–397 [DOI] [PubMed] [Google Scholar]
- 20.Park S-Y, Cho Y-R, Kim H-J, et al. Mechanism of glucose intolerance in mice with dominant negative mutation of CEACAM1. Am J Physiol Endocrinol Metab 2006;291:E517–E524 [DOI] [PubMed] [Google Scholar]
- 21.Shimazu T, Fukuda A. Increased activities of glycogenolytic enzymes in liver after splanchnic-nerve stimulation. Science 1965;150:1607–1608 [DOI] [PubMed] [Google Scholar]
- 22.Shimazu T. Glycogen synthetase activity in liver: regulation by the autonomic nerves. Science 1967;156:1256–1257 [DOI] [PubMed] [Google Scholar]
- 23.German J, Kim F, Schwartz GJ, et al. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology 2009;150:4502–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Malley D, MacDonald N, Mizielinska S, Connolly CN, Irving AJ, Harvey J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol Cell Neurosci 2007;35:559–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest 1997;100:270–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunbar JC, Lu H. Leptin-induced increase in sympathetic nervous and cardiovascular tone is mediated by proopiomelanocortin (POMC) products. Brain Res Bull 1999;50:215–221 [DOI] [PubMed] [Google Scholar]
- 27.Sternson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH —> arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci 2005;8:1356–1363 [DOI] [PubMed] [Google Scholar]
- 28.Fu L-Y, van den Pol AN. Agouti-related peptide and MC3/4 receptor agonists both inhibit excitatory hypothalamic ventromedial nucleus neurons. J Neurosci 2008;28:5433–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu B, Goulding EH, Zang K, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci 2003;6:736–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology 2008;149:2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang R, Dhillon H, Yin H, et al. Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis without affecting body weight. Endocrinology 2008;149:5654–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buettner C, Pocai A, Muse ED, Etgen AM, Myers MG, Jr, Rossetti L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab 2006;4:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue H, Ogawa W, Asakawa A, et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab 2006;3:267–275 [DOI] [PubMed] [Google Scholar]
- 34.Könner AC, Janoschek R, Plum L, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 2007;5:438–449 [DOI] [PubMed] [Google Scholar]
- 35.Plum L, Belgardt BF, Brüning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest 2006;116:1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill JW, Xu Y, Preitner F, et al. Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology 2009;150:4874–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Hill JW, Fukuda M, et al. PI3K signaling in the ventromedial hypothalamic nucleus is required for normal energy homeostasis. Cell Metab 2010;12:88–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warne JP, Alemi F, Reed AS, et al. Impairment of central leptin-mediated PI3K signaling manifested as hepatic steatosis independent of hyperphagia and obesity. Cell Metab 2011;14:791–803 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Kong D, Tong Q, Ye C, et al. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell 2012;151:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hof PR, Young WG, Bloom FE, Belichenko PV, Celio MR. Comparative Cytoarchitectonic Atlas of the C57BL/6 and 129/Sv Mouse Brains. Amsterdam, the Netherlands, Elsevier, 2000 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.