

Abstract
We studied the protective effect of stromal cell-derived factor-1β (SDF-1β) on cardiac cells from lipotoxicity in vitro and diabetes in vivo. Exposure of cardiac cells to palmitate increased apoptosis by activating NADPH oxidase (NOX)–associated nitrosative stress and endoplasmic reticulum (ER) stress, which was abolished by pretreatment with SDF-1β via upregulation of AMP-activated protein kinase (AMPK)–mediated p38 mitogen-activated protein kinase (MAPK) phosphorylation and interleukin-6 (IL-6) production. The SDF-1β cardiac protection could be abolished by inhibition of AMPK, p38 MAPK, or IL-6. Activation of AMPK or addition of recombinant IL-6 recaptured a similar cardiac protection. SDF-1β receptor C-X-C chemokine receptor type 4 (CXCR4) antagonist AMD3100 or CXCR4 small interfering RNA could not, but CXCR7 small interfering RNA completely abolished SDF-1β’s protection from palmitate-induced apoptosis and activation of AMPK and p38 MAPK. Administration of SDF-1β to diabetic rats, induced by feeding a high-fat diet, followed by a small dose of streptozotocin, could significantly reduce cardiac apoptosis and increase AMPK phosphorylation along with prevention of diabetes-induced cardiac oxidative damage, inflammation, hypertrophy, and remodeling. These results showed that SDF-1β protects against palmitate-induced cardiac apoptosis, which is mediated by NOX-activated nitrosative damage and ER stress, via CXCR7, to activate AMPK/p38 MAPK–mediated IL-6 generation. The cardiac protection by SDF-1β from diabetes-induced oxidative damage, cell death, and remodeling was also associated with AMPK activation.
Intracellular accumulation of long-chain fatty acids in nonadipose tissues is associated with cellular dysfunction and cell death and may ultimately contribute to the pathogenesis of disease. For example, lipotoxic accumulation of long-chain fatty acids in the heart of the Zucker diabetic fatty rat leads to the development of pathogenic changes (1). Similarly, the pathogenic changes in the heart of diabetic patients are also associated with the increased cardiac triglyceride content and contributes to arrhythmia occurrence and reduced contractile function or sudden death (2). In cultured cardiac cells, palmitate induced cardiac cell death (3,4). Because palmitate and stearate, but not unsaturated fatty acids, are precursors for de novo ceramide synthesis, fatty acid–induced apoptosis was assumed to probably occur through ceramide; however, some studies did not support this notion (5,6). Chinese hamster ovary cells did not require de novo ceramide synthesis for palmitate-induced apoptosis, and palmitate supplementation rather overgenerated reactive oxygen species or reactive nitrogen species that initiate apoptosis (5). Other later studies also reported the importance of palmitate-induced oxidative and nitrosative damage in the induction of apoptotic cell death (3,7,8).
Reportedly, palmitate induced endoplasmic reticulum (ER) stress and apoptosis in multiple tissues (9), and AMP-activated protein kinase (AMPK) activation inhibited palmitate-induced ER stress and apoptotic effects (9,10). Terai et al. (11) demonstrated the preventive effect of AMPK activation on hypoxia-induced ER stress and apoptosis in cardiac cells: hypoxia-induced C/EBP homologous protein (CHOP) expression and caspase 12 cleavage were significantly inhibited by pretreatment with 5-aminoimidazole-4-carboxyamide-1-β-d-ribofuranoside (AICAR), a pharmacological activator of AMPK. In parallel, adenovirus expressing dominant-negative AMPK significantly attenuated AICAR’s cardioprotection (11). Another study showed the antiapoptotic effect of AMPK activation on tumor necrotic factor-α (TNF-α) (12). Furthermore, the AMPK antiapoptotic effect seemed associated with p38 mitogen-activated protein kinase (MAPK) and interleukin-6 (IL-6) (13,14). Therefore, AMPK activation is an attractive approach in the prevention and/or treatment of cardiac diseases. However, concerns have recently been raised about AICAR-mediated AMPK upregulation (15): 1) chemical AICAR unselectively stimulates AMPK phosphorylation in all cells when it is chronically administered in vivo, and 2) AICAR as an AMPK-specific activator will persistently upregulate AMPK, which is undesirable because it usually induces apoptosis (15–19).
Chemokine stromal cell-derived factor (SDF-1), also known as chemokine (C-X-C motif) ligand 12 (CXCL12), regulates many essential biological processes, including cardiac and neuronal development, stem cell motility, neovascularization, angiogenesis, and tumorigenesis (20,21). Generally, SDF-1 mediates these disparate processes predominantly through CXC receptor 4 (CXCR4) and/or CXCR7 (22). Six variants of SDF-1, including SDF-1α, β, γ, δ, ε, and θ have been identified to date (23,24). SDF-1α and β are mostly involved in cardiovascular diseases (25–27). Previous studies predominantly showed the role of SDF-1α or β in directing stem cells into the damaged heart to repair tissue damage (21,28–30). Whether SDF-1 directly protects cardiac cells from lipotoxic effects has not yet been studied.
Therefore the objectives of the current study are to determine:
-
1)
whether SDF-1β protects cardiac cells from palmitate-induced nitrosative damage, ER stress, and apoptotic cell death;
-
2)
whether any protective effect of SDF-1β against palmitate-induced cell death is mediated by activation of the AMPK-related protective pathway;
-
3)
whether p38 MAPK and IL-6 are involved in the protective effect of SDF-1β from palmitate-induced cell death;
-
4)
which subtype of SDF-1 receptors is required for SDF-1β’s protection from these palmitate-induced pathogenic effects; and
-
5)
whether the antiapoptotic effect of SDF-1β on cardiac cells in vitro can be seen in the diabetic heart and whether the protective effect on cardiac cell death can lead to a prevention of cardiac pathogenic changes.
An understanding of these issues is very important for developing SDF-1β as a potential activator of AMPK to be used chronically in vivo. This peptide is an attractive candidate for clinical development because if it can activate AMPK, it will transiently activate AMPK only in cells expressing its receptor, unlike AICAR, which persistently and unselectively activates AMPK in all the cells in the body. Therefore, in vitro cultured cardiac H9C2 cells and also the primary cultures of neonatal cardiomyocytes, combined with pharmacological inhibitors and small interfering RNA (siRNA) approaches, were used. Diabetic rats were induced by being fed a high-fat diet (HFD) for 8 weeks, followed by a small dose of streptozotocin (STZ), as reported by others (31,32).
RESEARCH DESIGN AND METHODS
Cell culture.
Embryonic rat heart–derived H9C2 cells (CRL-1446), purchased from ATCC (Manassas, VA), were maintained in the conditions as instructed by ATCC. H9C2 cells were exposed to palmitate, with and without SDF-1β, and some of these cultures were pretreated with scavengers, inhibitors, or siRNA according to experimental needs, which will be described for each study. Palmitate (Sigma-Aldrich, St. Louis, MO) was dissolved in 50% ethanol, heated at 70°C for 2 min, and added to 2% fatty acid–free BSA (Sigma-Aldrich) in medium as stock solution (2.5 mmol/L). Before use, the stock palmitate solution was gently rotated for 1 h at 37°C and further diluted to the required concentrations for treatment. SDF-1β, prepared as reported previously (21,22), was dissolved in PBS to the required concentrations. For some experiments, primary cultures of neonatal cardiomyocytes were prepared and used as described previously (33).
Animal models.
Type 2 diabetes was induced in male Wistar rats (220–240 g), purchased from the Jilin University Animal Center, Changchun, China, by feeding an HFD containing 60% kcal from fat for 8 weeks, followed with an intraperitoneal injection of a small dose of STZ (Sigma-Aldrich) at 25 mg/kg body weight, based on published protocol (31,32). Control rats were fed a control diet containing 10% kcal from fat. Blood glucose levels >250 mg/dL were considered as diabetes. After diabetes onset, rats were divided into four groups: control (n = 6), SDF-1β control (SDF, n = 6), diabetes (DM, n = 9), and diabetes plus SDF-1β (DM/SDF, n = 7). SDF-1β was given by tail vein at 5 mg/kg body weight twice a week for 6 weeks. All animal protocols were approved by the Jilin University Animal Ethics Committee.
ELISA and other quantification assays.
Cell Death Detection ELISA kit was used to measure histone-bound DNA fragments for cultured cells, following the provided instruction. IL-6 ELISA kit (Thermo Scientific, Barrington, IL) was used to detect the concentration of IL-6 in culture supernatants, following the kit’s instruction.
Glycated hemoglobin (HbA1c %) was determined by the quantification kit (Roche Diagnostics, Mannheim, Germany). Quantification kits were used to assay plasma triglyceride and total cholesterol (Jiancheng, Nanjing, China), and contents of malondialdehyde (MDA) and superoxide dismutase (SOD) in cardiac tissues (Jiancheng).
Western blotting.
Western blotting was performed according to our previous studies (21,22). The first antibodies used at 1:1000 dilution included anti-cleaved caspase 3, anti-Bax, anti–Bcl-2, anti–apoptosis-induced factor, anti–phospho-p38(Thr180/Tyr182), anti-p38, anti–phospho-Akt(Ser473), anti-Akt, anti–phospho-AMPKα(Thr172), and anti-AMPKα (all from Cell Signaling, Beverly, MA), anti-CHOP, anti–phospho-extracellular signal–related kinase (p-ERK), anti-ERK, anti–transforming growth factor-β1 (TGF-β1), anti-vascular cell adhesion molecule (VCAM), anti-intracellular adhesion molecule 1 (ICAM-1), anti-plasminogen activator inhibitor type 1 (PAI-1), anti–TNF-α, and anti-collagen I, III, and IV (all from Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti–atrial natriuretic peptide (ANP; EMD Millipore, Billerica, MA), anti–3-nitrotyrosine (Chemicon, Billerica, MA), anti- 78 kDa glucose-regulated protein (GRP78; Abcam, Cambridge, MA), and anti-caspase 12 (Exalpha Biologicals, Shirley, MA).
Real-time PCR analysis of gene expression.
Total RNA was extracted from H9C2 cells using Trizol reagent. Random-primed cDNA was prepared using a commercial cDNA kit following the manufacturer’s protocol. Real-time quantitative PCR (qPCR) was performed with appropriate dilution of cDNA using the Applied Biosystems PRISM 7700 sequence detector and TaqMan gene expression assay kit (Applied Biosystems, Carlsbad, CA). Primers (CXCR4: Rn00573522_s1; CXCR7: Rn00584358_m1; ANP: Rn00561661_m1; TNF-α: Rn99999017_m1; β-actin: Rn01768120_m1) for qPCR were purchased from Applied Biosystems. The housekeeping gene β-actin was used as an internal reference.
siRNA transfection.
siRNAs specific for rat p38β MAPK (target sequence: GCA AUG UAG CAG UGA AUGA), IL-6 (target sequence: UAU GAG GUC UAC UCG GCAA), and CXCR7 (target sequence: ACA GCA UAU GGA ACG GAAA), along with nontargeting siRNA as a negative control (Dharmacon, Lafayette, CO) and Silencer Specific siRNA for rat CXCR4 (target sequence: UUC GUU UUC AUC CCG GAA GCA), along with the negative control (Ambion Life Technologies, Carlsbad, CA), were transfected into H9C2 cells for 48 h by transfection reagent before palmitate treatment. Transfection efficiency was assessed by ELISA kit and Western blot analysis for the target genes or proteins.
Cardiac fibrosis and staining by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
Cardiac tissues were fixed with 10% formalin and embedded in paraffins for preparing 5-μm tissue sections. Sections were stained by Sirius red for collagen, for which 0.1% Sirius red F3BA and 0.25% Fast Green FCF were used. Cardiac sections in paraffin were processed for a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay using an ApopTag in situ detection kit (Boster, Wuhan, China). Apoptotic cell death was quantitatively analyzed under a microscope (original magnification ×20) by counting TUNEL-positive cardiac cells relative to the total number of cardiac cells in each of five randomly selected fields of view from each of three slides for each rat, as described previously (34,35).
Statistical analysis.
Data were collected from the repeated experiments at least three times for in vitro studies and from at least six animals for in vivo studies and are presented as mean ± SD. One-way ANOVA was used to determine whether differences existed, and if so, a post hoc Tukey test was used for analysis of the differences among groups, with Origin 7.5 laboratory data analysis and graphing software. Statistical significance was considered as P < 0.05.
RESULTS
Palmitate-induced nitrosative stress-mediated ER stress and cell death.
Although palmitate induction of cardiac cell death has been reported (36,37), there remains the need for a detailed study on the dose-effect and time-course of palmitate-induced cell death. The effect of palmitate at 31.25–250 μmol/L on cell death was measured by DNA fragmentation (Fig. 1A) and cleaved caspase 3 (Fig. 1B), showing significant dose-dependent increases. Considering that Wang et al. (38) also demonstrated the induction of apoptotic cell death by palmitate at concentrations as low as 50 μmol/L in H9C2 cells, we selected a palmitate dose of 62.5 μmol/L to perform the following studies. Exposure of H9C2 cells to palmitate at 62.5 μmol/L for 2.5 to 15 h induced a time-dependent increase of cell death from 5 h, examined by DNA fragmentation (Fig. 1C) or 10 h, examined by cleaved caspase 3 (Fig. 1D), to 15 h as the longest time point in the current study.
FIG. 1.
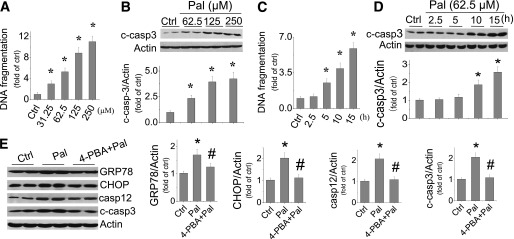
Palmitate-induced ER stress is associated cardiac cell death. H9C2 cells were exposed to palmitate at different doses (A and B) or at 62.5 μmol/L for different times (C and D). Cell death was evaluated by DNA fragmentation with cell death ELISA assay (A and D) and cleaved caspase 3 with Western blotting assay (B, C, and E). E: ER stress was examined by detecting its makers (GRP78, CHOP, and caspase 12) with Western blotting assay 12 h after cells were exposed to palmitate at 62.5 μmol/L, with and without ER stress inhibitor 4-PBA at 100 μmol/L for 12 h. Data are presented as mean ± SD from at least three separate experiments. c-casp3, cleaved caspase 3; Ctrl, control; Pal, palmitate. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
In the next study, H9C2 cells were exposed to palmitate at 62.5 μmol/L for 15 h, and the expression of apoptosis-induced factor, Bax, and Bcl-2 was examined; no difference among groups with and without palmitate treatment was observed (Supplementary Fig. 1). Recent studies implicated a possible role of ER stress in palmitate-induced cell death (36,37). Here, exposure of cells to palmitate at 62.5 μmol/L for 12 h also significantly increased the expression of GRP78, CHOP, and caspase 12, suggesting the induction of ER stress and ER stress-related cell death (Fig. 1E). Treatment of cells with ER stress inhibitor 4-phenylbutyrate (4-PBA) at 100 μmol/L for 12 h completely prevented palmitate-induced ER stress (GRP78, CHOP, and caspase 12) and caspase 3 activation (Fig. 1E), suggesting the dependence of ER stress in palmitate-induced cell death.
Next, whether palmitate-induced ER stress-related cell death is associated with nitrosative damage was measured by protein nitration, 3-NT. Palmitate significantly increased the 3-NT content in multiple groups of proteins from 22 to 148 kDa (Fig. 2A), which could be attenuated by pretreatment with NADPH oxidase (NOX) inhibitor (apocynin), SOD mimic (manganese[III] tetrakis[1-methyl-4-pyridyl]porphyrin [MnTMPyP]), NOS inhibitor (l-NG-nitro-l-arginine methyl ester [l-NAME]) or peroxynitrite scavenger (urate), suggesting the involvement of NOX-involved generation of superoxide and associated peroxynitrite (Fig. 2B).
FIG. 2.

Palmitate-induced nitrosative stress and damage is the cause of cell death. A: H9C2 cells were exposed to palmitate at 62.5 μmol/L at the indicated times, and nitrosative stress and damage was assayed by 3-NT accumulation with Western blot. Because 3-NT showed multiple groups of nitrated proteins within the mass range of 22–148 kDa, all bands within the range were quantitatively analyzed with densitometry. Inhibitive effects of different inhibitors or scavengers on palmitate-induced 3-NT accumulation (B) and ER stress (C) were detected in palmitate-treated cells. H9C2 cells were pretreated with NOX inhibitor (apocynin, 100 μmol/L), superoxide mimic (MnTMPyP, 75 μmol/L), NOS inhibitor (l-NAME, 100 μmol/L), or peroxynitrite scavenger (urate 100 μmol/L) separately for 1 h and then exposed to 62.5 μmol/L palmitate with the presence of the inhibitor or scavenger for another 6 h to examine 3-NT (B) or for another 12 h to examine ER stress (C). D: Effect of inhibition of ER stress with its inhibitor 4-PBA at 100 μmol/L on palmitate-induced 3-NT was examined at 6 h after exposed to palmitate, with and without 4-PBA. E: Palmitate-induced NOX-mediated nitrosative stress, ER stress, and cell death are illustrated based on the results from Figs. 1 and 2. Data are presented as mean ± SD from at least three separate experiments. casp12, caspase 12; Ctrl, control; Pal, palmitate. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
Inhibition of NOX, superoxide, NO, or peroxynitrite with their inhibitors or scavengers also completely abolished palmitate-induced ER stress (CHOP and caspase 12 expression, Fig. 2C). However, inhibition of ER stress with its inhibitor 4-PBA did not affect palmitate-induced nitrosative damage (3-NT accumulation, Fig. 2D), suggesting that the nitrosative stress is the cause of ER stress and related cell death, as illustrated in Fig. 2E.
Protective effect of SDF-1β on palmitate-induced cardiac cell death, which is independent of Akt and ERK1/2 cell survival signaling.
First, the effect of palmitate at 62.5 μmol/L on SDF-1 expression in H9C2 cells (Fig. 3A) and primary culture of neonatal cardiomyocytes (Fig. 3B) was examined. Exposure of these cells to palmitate induced a significant decrease in SDF-1 expression. Next the effect of exogenous SDF-1β at different dose levels (25, 50, and 100 nmol/L) on palmitate-induced cell death was examined by detecting caspase 3 cleavage (Fig. 3C) and DNA fragmentation (Fig. 3D). SDF-1β protected palmitate-induced cell death in a dose-dependent manner. The antiapoptotic effect of SDF-1β observed in H9C2 cells was also confirmed in the primary cultures of neonatal cardiomyocytes that were exposed to 62.5 μmol/L palmitate and 100 nmol/L SDF-1β (Fig. 3E). Furthermore, SDF-1β at 100 nmol/L protected H9C2 cells from spontaneous and palmitate-induced apoptosis (Supplementary Fig. 2) and ER stress and related cell death (Fig. 3F).
FIG. 3.

Palmitate downregulates SDF-1β expression, and SDF-1β protects from palmitate-induced ER stress and cell death. SDF-1β expression was measured with Western blotting in H9C2 cells (A) and in primary culture of neonatal cardiomyocytes (B) exposed to palmitate at 62.5 μmol/L for 15 h. Effects of SDF-1β at 25, 50, or 100 nmol/L on palmitate-induced cell death were determined by cleaved caspase 3 (C) and DNA fragmentation (D) in H9C2 cells that were pretreated by SDF-1β for 1 h and then treated by SDF-1β and palmitate together for 15 h. E: Apoptotic cell death was also measured by caspase 3 cleavage in the primary culture of neonatal cardiomyocytes that were exposed to palmitate at 62.5 μmol/L for 15 h, with and without SDF-1β at 100 nmol/L. F: Effects of SDF-1β on palmitate-induced ER stress and its related cell death at 12 h were determined for the expression of GRP78, CHOP, and caspase 12. Data are presented as mean ± SD from at least three separate experiments. c-casp3, cleaved caspase 3; Ctrl, control; Pal, palmitate. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
Here, we also found that palmitate stimulated the expression of several inflammatory cytokines, including VCAM, PAI-1, and TNF-α, which were completely prevented by SDF-1β (Supplementary Fig. 3A–C). Neither palmitate nor SDF-1β affected ICAM-1 expression (Supplementary Fig. 3D). In addition, palmitate stimulated the expression of TGF-β1 and ANP, both of which were completely prevented by SDF-1β treatment (Supplementary Fig. 3E and F).
Because several cellular functions of SDF-1β depend on Akt and ERK1/2 signaling pathways (21), whether these two pathways were required for the protection by SDF-1β from palmitate-induced cardiac cell death was examined. Exposure to SDF-1β rapidly increased the phosphorylation of Akt and ERK1/2 at 15 min, and then they gradually decreased to normal levels at 2 h (Supplementary Fig. 4A and B). However, inhibition of Akt with phosphatidyl inositol 3-kinase (PI3K) inhibitor (LY294002 at 50 μmol/L, Supplementary Fig. 4C) or ERK1/2 with its inhibitor (U0126 at 10 μmol/L, Supplementary Fig. 4D) did not affect palmitate-induced cell death with or without SDF-1β.
Protective effect of SDF-1β on palmitate-induced cardiac cell death is mediated by AMPK-dependent upregulation of p38β MAPK.
Because SDF-1β plays an important role in protecting the heart under various conditions (13,14,39), its effect on p38 MAPK expression was examined in the cells exposed to palmitate or palmitate/SDF-1β for 3 to 15 h. The p38 MAPK phosphorylation was significantly upregulated in the cells exposed to palmitate/SDF-1β (Fig. 4A) or SDF-1β alone for 6 h (Fig. 4B) but not in the cells exposed to palmitate only (Fig. 4A). A p38 MAPK inhibitor (SB203580) was given before cells were treated with palmitate or palmitate/SDF-1β for 15 h (Fig. 4C). Inhibition p38 MAPK did not affect palmitate-induced cell death but completely abolished the protective effect of SDF-1β on palmitate-induced cell death (Fig. 4C).
FIG. 4.

Protective effect of SDF-1β on palmitate-induced cardiac cell death is mediated by p38β MAPK activation. H9C2 cells were pretreated by SDF-1β for 1 h and then treated by SDF-1β and palmitate together for different times. Phosphorylated p38 and total p38 were detected from the cells treated with palmitate at 62.5 μmol/L with SDF-1β for 3 to 15 h or palmitate alone for 6 h (A) and also treated with SDF-1β, with and without palmitate, for 6 h (B). C: Inhibitory effects of p38 with its inhibitor (SB203580, 10 μmol/L) on palmitate-induced cell death were examined by measuring cleaved caspase 3. Inhibitory effects of p38-specific siRNA on SDF-1β’s protection from palmitate-induced cell death and ER stress (D) and 3-NT accumulation (E) were examined by measuring the activation of caspase 3 and expression of CHOP and caspase 12 (D) and 3-NT (E). For this study, cells were pretreated with specific p38β siRNA and nontarget siRNA for 48 h and then treated with palmitate and Pal/SDF-1β for 12 h for cell death and ER stress (D) or for 6 h for 3-NT (E). F: The possibility of p38 siRNA on palmitate-induced cell death was eliminated by comparing effects of p38 siRNA with and without palmitate. Data are presented as mean ± SD from at least three separate experiments. c-casp3, cleaved caspase 3; casp12, caspase 12; Ctrl, control; p-p38, phosphorylated p38; Pal, palmitate. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
The antibody used to detect p38 phosphorylation in Fig. 4A and B detects p38α and p38β MAPKs, and the p38 inhibitor used in the study of Fig. 4C also unselectively inhibits the α and β isoforms of p38 MAPK. Therefore, p38β MAPK siRNA was used to define the specific role of p38β MAPK in the protection of SDF-1β from palmitate-induced cell death. Figure 4D shows that p38β MAPK siRNA completely abolished the protective effect of SDF-1β on palmitate-induced ER stress and cell death but control siRNA did not (Fig. 4D). There was no influence of p38 MAPK siRNA on palmitate-induced and spontaneous apoptosis (Fig. 4F). Specific inhibition of p38β MAPK also abolished the protective effect of SDF-1β on palmitate-induced nitrosative damage at 6 h after exposure to palmitate/SDF-1β (Fig. 4E).
We next examined the effect of palmitate on AMPK expression and function in H9C2 cells by Western blotting (Fig. 5A). Palmitate at 62.5 μmol/L did not affect AMPK expression but significantly decreased its phosphorylation at 3 and 6 h postexposure (Fig. 5A). In contrast to palmitate, SDF-1β significantly increased AMPK phosphorylation at 3 h postexposure (Fig. 5B). Inhibition of AMPK phosphorylation by exposure to palmitate at 62.5 μmol/L for 3 h could be abolished by cotreatment with SDF-1β (Fig. 5C). In the neonatal cardiomyocyte primary cultures, the inhibition of AMPK phosphorylation by exposure to palmitate at 62.5 μmol/L for 3 h was also abolished by cotreatment with SDF-1β (Supplementary Fig. 5).
FIG. 5.

Protective effect of SDF-1β on palmitate-induced cardiac cell death is mediated by AMPK activation. H9C2 cells were treated with palmitate at 62.5 μmol/L (A) or SDF-1β at 100 nmol/L (B) for the indicated times, and phosphorylated AMPK (p-AMPK) and total AMPK were detected by Western blotting assays. C: Effects of SDF-1β on AMPK expression and phosphorylation were examined in the cells with and without exposure to palmitate at 62.5 μmol/L for 3 h in presence or absence of SDF-1β at 100 nmol/L. H9C2 cells were pretreated by AMPK inhibitor (Com. C, 10 μmol/L) (D) or AMPK activator (AICAR, 250 μmol/L) (E) for 1 h, and then treated with SDF-1β/palmitate or palmitate with the presence of compound C or AICAR for 15 h to examine the expression and activation of AMPK and p38 as well as caspase 3 activation by Western blot assay. Data are presented as mean ± SD from at least three separate experiments. c-casp 3, cleaved caspase 3; Com. C, compound C; Ctrl, control; Pal, palmitate; p-AMPK, phosphorylated AMPK; p-p38, phosphorylated p38. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
To address whether the preservation of AMPK function is the mechanism responsible for the protection by SDF-1β from palmitate-induced cell death, cells were pretreated by AMPK inhibitor compound C at 10 μmol/L (Fig. 5D) or AMPK activator AICAR at 250 μmol/L (Fig. 5E) for 1 h and then cotreated with SDF-1β/palmitate or palmitate for 15 h. By Western blotting assay, we showed that inhibition of AMPK function significantly abolished the preservation of p38 MAPK phosphorylation by SDF-1β with palmitate and also abolished the protective effect of SDF-1β on palmitate-induced cell death (Fig. 5D). In contrast to compound C, activation of AMPK could significantly stimulate p38 AMPK activation and reduced palmitate-induced cell death (Fig. 5E), which recaptures the protective effect of SDF-1β on palmitate-induced cell death. Activation of AMPK with AICAR in normal cells also reduced spontaneous apoptosis (Fig. 5E).
IL-6 is required for the protective effect of SDF-1β–activated AMPK/p38β signaling on palmitate-induced cardiac cell death.
Previous studies have indicated that p38β MAPK protected the heart by increasing IL-6 generation (13,39). We therefore evaluated the role of IL-6 in the protection by SDF-1β from palmitate-induced cell death, first by measuring IL-6 levels in the medium of cultured cells exposed to palmitate at 62.5 μmol/L, with or without SDF-1β at 100 nmol/L for 5 to 15 h. Medium IL-6 levels were increased only in the cells exposed to SDF-1β with palmitate for 10 or 15 h but not in the cells exposed to palmitate alone (Fig. 6A). Exposure of the cells to SDF-1β alone, AMPK activator or inhibitor alone, or p38 MAPK inhibitor alone did not affect IL-6 levels (Fig. 6B). However, as with SDF-1β, the AMPK activator AICAR with palmitate significantly increased the IL-6 level (Fig. 6B), whereas the increased IL-6 content in the cells exposed to palmitate and SDF-1β was abolished by an AMPK inhibitor or a p38 MAPK inhibitor (Fig. 6B). This study suggests that the increased IL-6 level by SDF-1β in palmitate-treated cells is mediated by activation of AMPK and p38 MAPK.
FIG. 6.

Protective effect of SDF-1β on palmitate-induced cardiac cell death is mediated by IL-6. A and B: IL-6 concentrations in the medium of cultured cells by different treatments were determined by ELISA assay. A: H9C2 cells were treated with palmitate at 62.5 μmol/L alone for 15 h or with palmitate and SDF-1β at 100 nmol/L for indicated times. B: H9C2 cells were treated with palmitate (62.5 μmol/L), SDF-1β (100 nmol/L), compound C (10 μmol/L), AICAR (250 μmol/L), or p38 inhibitor (SB203580, at 10 μmol/L) alone or combined as indicated, for 15 h. C: Cells were pretreated with specific IL-6 siRNA and nontarget siRNA for 48 h before treatment with palmitate and SDF-1β for examining the expression of CHOP, caspase 12, and cleaved caspase 3 at 12 h. D: H9C2 cells were treated by palmitate at 62.5 μmol/L, with or without rIL-6, at indicated doses for 15 h and then cleaved caspase 3 was examined. E: H9C2 cells were pretreated with rIL-6 for 1 h and then continually treated with rIL-6 in the absence or presence of palmitate at 62.5 μmol/L for another 12 h to measure ER stress by Western blotting of CHOP and caspase 12. Data are presented as mean ± SD from at least three separate experiments. c-casp3, cleaved caspase 3; Com.c, compound C; Ctrl, control; p-AMPK, phosphorylated AMPK; p-p38, phosphorylated p38; Pal, palmitate. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
The requirement of IL-6 for the protective effect of SDF-1β on palmitate-induced ER stress and related cell death was defined with its specific siRNA because the protective effect of SDF-1β on palmitate-induced increases in CHOP, caspase 12, and cleaved caspase 3 was abolished by IL-6 siRNA but not by control siRNA (Fig. 6C). Furthermore, the direct role of IL-6 in the protective effect of SDF-1β on palmitate-induced ER stress and cell death was also addressed by direct addition of human recombinant IL-6 (rIL-6) into the cultures. Addition of rIL-6 at 1 or 2 ng/mL, but not at 4 ng/mL, significantly reduced palmitate-induced cardiac cell death measured at 15 h (Fig. 6D). The addition of 2 ng/mL rIL-6 completely protected from palmitate-induced CHOP and caspase 12 expression at 12 h postexposure (Fig. 6E). Therefore, the protective effect of SDF-1β on palmitate-induced ER stress and cell death is mediated by its activation of AMPK/p38β MAPK–-mediated IL-6 generation.
Protective effect of SDF-1β on palmitate-induced cell death is CXCR7 receptor dependent.
The protective effect of SDF-1β on palmitate-induced ER stress and cell death was not mediated by its receptor CXCR4 because when cells were exposed to SDF-1β/palmitate with the presence of CXCR4 blocker AMD3100 at 0.5–5 μmol/L, the protective effect of SDF-1β on palmitate-induced cell death and AMPK phosphorylation was not affected and was even slightly enhanced (Fig. 7A, Supplementary Fig. 6A). To rule out nonspecific effects of ADM3100 on CXCR4 receptors (40), we further confirmed that siRNA-induced disruption of CXCR4 expression did not affect the protection of cardiac tissue by SDF-1β against palmitate-induced cell death and AMPK inactivation (Fig. 7B).
FIG. 7.

Protective effect of SDF-1β on palmitate-induced cell death is mediated through CXCR7 receptor. A: H9C2 cells were pretreated with AMD3100 at 0.5–5.0 μmol/L for 1 h before treatment with SDF-1β, palmitate, and AMD3100 together for 3 h to measure AMPK inactivation by the ratio of phosphorylation or for 15 h to measure cell death by caspase 3 activation. B: Cells were pretreated with specific CXCR4 siRNA (50 nmol/L) and negative control for 48 h before treatments with palmitate and SDF-1β for 3 h to detect inactivation of AMPK or for 15 h for measuring cell death with caspase 3 activation. C: Cells were pretreated with specific CXCR7 siRNA at indicated doses and nontarget siRNA for 48 h before treatments with palmitate and SDF-1β for another 15 h for cell death measurement with DNA fragmentation assay. D–F: siRNA at 12.5 nmol/L was used for the other studies. Cells were pretreated with specific CXCR7 siRNA and nontarget siRNA for 48 h before treatments with palmitate and SDF-1β for another 3 h to detect expression and activation of AMPK (D), for another 6 h to detect the expression and activation of p38 MAPK (E), and for another 15 h to detect cell death by measurement of cleaved caspase 3 (F). Data are presented as mean ± SD from at least three separate experiments. c-casp3, cleaved caspase 3; Ctrl, control; p-AMPK, phosphorylated AMPK; p-p38, phosphorylated p38. *P < 0.05 vs. control group; #P < 0.05 vs. palmitate group.
CXCR7 is reported to exist in a few organs, including the heart (20,41). We also confirmed the expression of CXCR7 in cardiac H9C2 cells by real-time PCR at baseline, although CXCR4 is about 100-fold higher than CXCR7 expression (Supplementary Fig. 6B). CXCR4 and CXCR7 mRNA expression were not response to palmitate (Supplementary Fig. 6C and D). CXCR7-specific siRNA was preapplied to the cells treated with palmitate and/or SDF-1β (Fig. 7C), which abolished SDF-1β protection from palmitate-induced cell death in a dose-dependent manner, measured by DNA fragmentation measurement. Because CXCR7 siRNA at 25 nmol/L also has an apoptotic effect in normal cells (Fig. 7C), CXCR7 siRNA at 12.5 nmol/L was used in the following studies. Pretreatment of the cells with CXCR7 siRNA completely abolished the activation of AMPK (Fig. 7D) and p38 MAPK (Fig. 7E) by SDF-1β with the presence of palmitate. Pretreatment of the cells with CXCR7 siRNA also completely abolished the protection by SDF-1β from palmitate-induced caspase 3 activation (Fig. 7F). These results suggest that SDF-1β protection from palmitate-induced cell death via activation of AMPK and p38 MAPK is mediated by CXCR7, not CXCR4, receptor.
Protective effect of SDF-1β on diabetes-induced cardiac cell death and other pathological changes, which were associated with upregulation of AMPK phosphorylation.
The above in vitro studies clearly indicate the protective effect of SDF-1β on palmitate-induced cardiac cell death. We also found a reduction of cardiac expression of SDF-1 in HFD-induced type 2 diabetic mice (Supplementary Fig. 7). To ensure whether SDF-1β can afford a protective effect on the heart in vivo, a type 2 diabetic rat model was induced with 8-week HFD feeding, followed by a small dose of STZ, as used by others (31,32). Three days after STZ treatment, hyperglycemic rats and age-matched control rats were given with SDF-1β at 5 mg/kg body weight or the same value of PBS twice a week for 6 weeks. At the end of 6 weeks SDF-1β treatment, rat body weights, blood glucose levels, HbA1c %, and plasma cholesterol and triglyceride levels, cardiac triglyceride, and heart weight were significantly increased in the DM group, which were not different from those in the DM/SDF group (Fig. 8A–F, Supplementary Fig. 8A). However, the heart weight-to-body weight ratio was significantly decreased in the DM group but not in the DM/SDF group (Fig. 8G). The cardiac incidence of TUNEL-positive cells was significantly increased in the hearts of the DM group, which was significantly, although not completely, attenuated by 6 weeks of SDF-1β treatment (Fig. 8H). There was also a significant increase in caspase 3 activation, with a significant decrease of AMPK phosphorylation in the hearts of the DM group but not of the DM/SDF group (Fig. 8I).
FIG. 8.

Protective effect of SDF-1β on diabetes-induced cardiac cell death and remodeling. Diabetic rats were induced by being fed an HFD for 8 weeks, followed by a small dose of STZ at 25 mg/kg. At hyperglycemia onset, diabetic and age-matched control rats were treated with SDF-1β at 5 mg/kg body weight twice weekly for 6 weeks. At the end of treatment, body weight (A), blood glucose (B), HbA1c (C), plasma total cholesterol (D) and triglyceride (E), cardiac triglyceride (F), and heart weight (HW)-to-body weight (BW) ratio (G) were determined. H: Cardiac apoptotic cell death was examined by TUNEL staining, from which the TUNEL-positive cells (arrows) were quantitatively analyzed. I: The expression of cleaved caspase 3 (c-casp3), phosphorylated AMPK (p-AMPK), and total AMPK were examined by Western blotting assay. J: Cardiac fibrosis was reflected by Western blotting of collagen I, III, and IV expression. Cardiac mRNA expression of hypertrophy marker ANP (K) and inflammation cytokine TNF-α (L) was examined by real-time PCR. Cardiac oxidative stress was measured by biochemical assays for the increased MDA (M) and decreased SOD contents (N). Data are presented as mean ± SD (n = 6 at least in each group). Ctrl, control. *P < 0.05 vs. control group; #P < 0.05 vs. DM. (A high-quality color representation of this figure is available in the online issue.)
Apoptotic cell death in the diabetic heart is a well-known cause for several pathological changes (42,43), which, therefore, were examined here. Western blotting for collagen I, III, and IV (Fig. 8J), as well as the collagen I-to-III ratio (Supplementary Fig. 8B) and Sirius red staining for total collagen accumulation (Supplementary Fig. 8C) showed a significant increase in cardiac fibrosis in the hearts of the DM group but not significantly in DM/SDF group. Furthermore, the existence of cardiac hypertrophy in the DM group, but not in the DM/SDF group, was also reflected in increased mRNA expression levels of the hypertrophic molecular marker ANP (Fig. 8K) and increased sizes of myofibers examined by hematoxylin and eosin staining (Supplementary Fig. 8D). In addition, a significant increase in cardiac inflammation was detected by increased mRNA expression of TNF-α in the DM group that was partially attenuated in the DM/SDF group (Fig. 8L). Cardiac oxidative stress, shown by an increased level of MDA (Fig. 8M) and decreased SOD content (Fig. 8N), was significantly elevated in the DM group but not in the DM/SDF group.
DISCUSSION
Our findings indicate that palmitate-induced apoptotic death in the cardiac cell is mediated by ER stress that was caused by NOX activation associated nitrosative stress, based on the following evidence:
-
1)
Palmitate induced significant increases in 3-NT at 3–9 h (Fig. 2A) and ER stress (increased GRP78, CHOP, and caspase 12 expression) at 12 h (Fig. 1E), and induction of apoptotic cell death, peaking at 15 h (Fig. 1A and B).
-
2)
Inhibition of NOX activation-mediated superoxide generation and peroxynitrite formation significantly prevented palmitate-induced 3-NT accumulation, ER stress, and apoptosis (Fig. 2).
-
3)
Inhibition of ER stress with 4-PBA completely inhibited ER stress and caspase 3 activation (Fig. 1E) but did not affect palmitate-induced 3-NT accumulation (Fig. 2D).
In addition, we have provided the following innovative findings.
Protective effect of SDF-1β on palmitate-induced cardiac apoptotic cell death and the underlying signaling mechanism.
Early studies showed an induction of cell death by SDF-1 in lymphocytes (44). Recent studies demonstrated that SDF-1 improved some hematological cell survival (45). The antiapoptotic effect of SDF-1 was predominantly dependent on Akt and/or ERK1/2 activation in pancreatic β-cells, bone marrow stem cells, or cardiac cells (46–48). However, whether SDF-1 protects cardiac cells from fatty acids, such as palmitate, has not been reported.
We found here that SDF-1β significantly protects against palmitate-induced cardiac nitrosative damage, ER stress, and cell death through activation of AMPK/p38 MAPK–mediated generation of the IL-6 pathway. SDF-1β significantly increased Akt and ERK1/2 phosphorylation, but neither one is required for the protection by SDF-1β from palmitate-induced cell death (Supplementary Fig. 4). In contrast, SDF-1β activated p38 MAPK phosphorylation at 6 h after treatment (Fig. 4A and B). Inhibition of p38 MAPK with SB203580, which inhibits α and β isoforms of p38 MAPK (Fig. 4C), or specific siRNA for p38β MAPK (Fig. 4D) abolished the protective effect of SDF-1β from palmitate-induced ER stress and cell death, suggesting the direct requirement of p38β MAPK for the cardiac protection by SDF-1β (Supplementary Fig. 9).
The MAPK signaling pathway allows cells to process a wide range of external signals and respond appropriately, generating a plethora of different biological effects. The diversity and specificity of cellular outcomes is achieved by functionally distinct p38 MAPK isoforms (49). In the heart, several investigations have demonstrated the proapoptotic effect p38α MAPK activation during ischemia (50). Huang et al. (51) reported that cardiac apoptosis and lactate dehydrogenase activity in cell culture supernatants decreased markedly and cardiac viability increased significantly in the group treated with p38α MAPK anti-sense. A recent study also demonstrated that during hypoxia, followed by reoxygenation stress, cardiomyocytes undergo p53-dependent apoptosis after phosphorylation of p53 by p38α MAPK, leading to p38β suppression. Estrogen could protect cardiomyocytes from apoptosis by inhibiting p38α MAPK–mediated p53 apoptotic signaling (52). This study supports the early notion that p38α MAPK plays a proapoptotic role and p38β MAPK plays an antiapoptotic roles, as reported by Venkatakrishnan et al. (53). Here we also demonstrated that p38β MAPK activation was required for SDF-1β’s cardiac protection from palmitate-induced ER stress and cell death (Fig. 4), which is consistent with the studies cited above.
IL-6 is a proinflammatory cytokine and also activates signaling pathways that mediate cardiac protection (54). Using cardiac myocytes, Craig et al. (39) found the involvements of p38 MAPK and IL-6 in the protection of cardiac cells from apoptosis. Here, we provide evidence that IL-6 is required for the protection by SDF-1β from palmitate-induced apoptotic cell death:
-
1)
SDF-1β induced a significant increase in IL-6 at 10–15 h (Fig. 6A).
-
2)
Pretreatment of SDF-1β/palmitate-treated cells with IL-6 specific siRNA completely abolished the protective effect of SDF-1β on palmitate-induced ER stress and cell death (Fig. 6C).
-
3)
Addition of human rIL-6 into palmitate-treated cells significantly prevented palmitate-induced ER stress and cell death (Fig. 6D and E).
A few previous studies have shown the requirement of AMPK for the stimulation of p38β MAPK (13,55). We extend these findings to provide the first evidence that SDF-1β–induced activation of AMPK stimulates p38 MAPK–dependent IL-6 generation to protect cardiac cells against palmitate-induced ER stress and apoptosis (Supplementary Fig. 9). We found that palmitate significantly decreased AMPK phosphorylation at 3 or 6 h after treatment and that SDF-1β significantly increased AMPK phosphorylation in normal cells and preserved the AMPK phosphorylation level in palmitate-treated cells (Fig. 5A–C). Whether AMPK activation is pro- or antiapoptotic depends on several factors, including the type of cell, the type of challenge, and the duration of AMPK activation (11–19). For instance, transitory AMPK activation is often antiapoptotic (11–14), whereas persistent AMPK activation is typically proapoptotic (15–19).
SDF-1 regulates many essential biological processes, which has been considered predominantly through CXCR4 (21,22). However, recent studies from us and others have implicated that CXCR7 has different functions compared with CXCR4 and also plays an important role in mediating cell survival or the antiapoptotic effect of SDF-1 (22). Bakondi et al. (56) and Hattermann et al. (57) have separately reported the important role of CXCR7 in mediating the antiapoptotic effect of SDF-1. They found that in astroglia, stimulation of CXCR7 prevented camptothecin- and temozolomide-induced apoptosis and that the selective CXCR7 antagonist CCX733 reduced the antiapoptotic effects of CXCL12 (57).
We show here for the first time that in addition to CXCR4, CXCR7 also expresses in cardiac H9C2 cells. Blocking the interaction of SDF-1 with CXCR4 by AMD3100 at dose range of 0.5–5.0 μmol/L did not affect and even slightly at high-dose levels enhanced the protective effect of SDF-1β on palmitate-induced cell death (Fig. 7A), which may be due to its binding to CXCR7 with allosteric effect (40). To rule out the nonspecific banding of AMD3100 to its receptors, CXCR4 expression was downregulated using its specific siRNA, with no discernible effect (Fig. 7B), but downregulation of CXCR7 expression with its specific siRNA (Fig. 7C–F) completely abolished the protective effect of SDF-1β on palmitate-induced cell death and also completely eliminated the stimulation by SDF-1β of AMPK and p38 MAPK activation in the cells treated by palmitate. These findings suggest that the protective effect of SDF-1β on palmitate-induced ER stress and cell death by activation of AMPK/p38β MAPK–mediated generation of IL-6 was mediated by CXCR7 receptor.
Protective effect of SDF-1β on diabetes-induced cardiac cell death and other pathogenic changes.
Several studies have demonstrated the critical role of apoptotic cell death in the development of pathogenic changes in the heart of diabetic or nondiabetic subjects (42,43). We speculated that the significant antiapoptotic effect of SDF-1β might be applicable to the prevention of diabetes-induced cardiac cell death and even other pathogenic changes in vivo, which was confirmed in the hearts of diabetic rats in the current study. We showed that diabetes induced by the HFD, followed by a small dose of STZ, significantly increased the plasma glucose, total cholesterol, and triglyceride levels as well as cardiac triglyceride level. Diabetes also induced a significant increase of cardiac cell death and a decrease of AMPK phosphorylation, along with significant increases in cardiac oxidative damage, inflammation, hypertrophy, and remodeling (fibrosis). Treatment of these diabetic rats with SDF-1β for 6 weeks did not affect the systemic and cardiac hyperlipidemic profiles but significantly attenuated the diabetic increase of cardiac cell death and reduction of AMPK phosphorylation, leading to a significant prevention of diabetes-induced pathogenic changes in the heart, including oxidative damage, inflammation, hypertrophy, and remodeling (fibrosis). Although we did not measure cardiac function here, we assume that these pathogenic changes would lead to a development of cardiac dysfunction in vivo, because several other studies have demonstrated the development of diabetic dysfunction in animal models (58–60) that were similar to the model used in the current study.
The antiapoptotic and preserving-AMPK functional effects of SDF-1β on diabetes were incomplete, thus resulting in an incomplete prevention of diabetes-induced fibrosis. This partial prevention may be related. First, the dosage of SDF-1β at 5 mg/kg body weight twice weekly for 6 weeks may be not enough to efficiently prevent diabetes-induced cell death. Although previous studies (46,61) have demonstrated the protective effect of SDF-1 on cardiac damage induced by acute ischemia and reperfusion, they gave SDF-1 in perfused hearts. One recent study gave mice an intravenous injection of bifunctional protein consisting of an SDF-1 domain and a glycoprotein VI at 10 mg/kg (62), which remains higher than that used in the current study.
Second, we gave the SDF-1β to diabetic rats after the rats were fed the HFD for 8 weeks, which already significantly increased body weight and systemic lipid levels and may also increase cardiac cell death. In addition, compared with the protective effect of SDF-1β on diabetes-induced TUNEL-positive cells (Fig. 8H), the protective effect of SDF-1β on the diabetes-induced increase in caspase 3 cleavage and the decrease in AMPK phosphorylation were small (Fig. 8I). This asymmetry may be attributed to the time-point difference between these molecular events. For instance, apoptotic cell death is a late event that occurs after AMPK activation and caspase 3 cleavage. AMPK activation is an early and transient signaling event (AMPK was downregulated at 3 and 6 h, whereas apoptotic cell death was seen from 5 to 15 h [the longest time in the current study] in palmitate-treated cells, Fig. 1C; AMPK activation was also only seen at 3 h in SDF-1β–treated cells, Fig. 5A and B). Therefore, it will be difficult to see the significant upregulation of phosphorylated AMPK at the same time point when apoptotic cells can be seen significantly. Caspase-3 cleavage is also transient, whereas apoptotic cell death (TUNEL-positive cells) is the final step and lasts a relatively long time. In addition, TUNEL-positive cells may include apoptotic and necrotic cells. These potential reasons will be explored in future studies.
The relative low dose of palmitate used here for the in vitro studies may limit its relevance to in vivo conditions. A few studies have shown the apoptotic effect of palmitate at 125–250 μmol/L, measured by flow cytometry and TUNEL assay, respectively (36,37). We showed here that exposure of H9C2 cells to palmitate at dose range of 31.25–250 μmol/L induced a significant dose-dependent increase of cell death (Fig. 1A and B). Wang et al. (38) also demonstrated the induction of apoptotic cell death by palmitate at concentrations as low as 50 μmol/L in H9C2 cells . The discrepancy between these results may be due to differences in cell death detection as a function of the assays used and/or cell culture conditions (36–38). We showed the increased cell death induced by palmitate at 31.25 μmol/L with the DNA fragmentation ELISA assay (Fig. 1A) but not with the caspase 3 cleavage assay (data not shown). The apoptotic effect of 62.5 μmol/L palmitate after 5 h was estimated by examining DNA fragmentation (Fig. 1C) but was estimated after 10 h by quantifying cleaved caspase 3 (Fig. 1D). H9C2 cells were incubated in high-glucose Dulbecco's modified Eagle's medium for these studies, but in two previous studies (36,37) that showed the resistance of cells to palmitate-induced cell death, the medium was supplemented with 10% FBS for preculture and 5% BSA during the palmitate treatment period, whereas we supplemented with 10% FBS for preculture and 2% BSA for the treatment period. The lower BSA during the palmitate treatment period might make the cells more susceptible to palmitate-induced cytotoxic effects. For instance, Wang et al. (38) used 0.1% BSA during the palmitate treatment period and detected high sensitivity of these cells to palmitate-induced cell death. These results suggest that cell culture conditions and experimental methods may determine cell susceptibility to palmitate-induced cell death.
None of these in vitro cultured conditions can be directly translated to in vivo or clinically relevant conditions. Therefore, we did not try to directly translate our in vitro studies to the in vivo animal model or to explain clinical observations. Instead, these in vitro studies elucidate an important cellular event and signaling pathway. The possibly preventive effect of SDF-1β on diabetes-induced cardiac cell death and other pathogenic changes needs to be directly examined in an animal model, such as the rat model here.
In summary, the current study has investigated the protective effect of SDF-1β on palmitate-induced cardiac ER stress and cell death in vitro and diabetes-induced cardiac cell death in vivo for the first time. The cardiac protective effect of SDF-1β in vitro is mediated through its interaction with CXCR7 and activation of AMPK and p38 MAPK–mediated IL-6 production (Supplementary Fig. 9). Although whether this in vitro mechanism is true for the heart in vivo remains a systemic investigation, the antiapoptotic cell death in the heart of diabetic rats was also associated with upregulation of AMPK phosphorylation. The antiapoptotic effect and upregulation of AMPK in the heart was also associated with significant prevention of diabetic cardiac remodeling. These findings provide a novel insight into understanding the mechanism for SDF-1’s cardiac protection, which should be considered both its angiogenesis and cardiac cytotoxic protection.
Supplementary Material
ACKNOWLEDGMENTS
This study was partly supported by a key project from the Science and Technology Department of Jilin Province (11ZDGG003 to W.L.), a basic award from the Department of Defense (W81XWH-10-1-0677 to L.C. and Y.T.), a National Science and Technology Major Project of New Drug, a regular National Natural Science Foundation of China (NSFC) project and a Wenzhou Science and Technology project (2012ZX09103301-016, 30971209, Y20100001 to Y.T. and L.C.), a youth scientific project from NSFC (81200239 to X.Y.), a Basic Science Award, and Junior Faculty Awards from the American Diabetes Association (01-11-BS-17 to L.C. and 1-13-JF-53 to Y.T.). Y.Z. is a recipient of a scholarship under the State Scholarship Fund from the China Scholarship Council.
No potential conflicts of interest relevant to this article were reported.
Y.Z., S.X., Y.L., C.L., J.C., and X.Y. researched data and reviewed the manuscript. Y.T. discussed project progression, researched data, and reviewed the manuscript. X.L., G.W., and W.L. discussed project progression and reviewed the manuscript. L.C. contributed to the initial formulation of the project, discussed project progression, and wrote, reviewed, and edited the manuscript. L.C. is the guarantor of this work, and, as such, had full access to all the data in the study and takes full responsibility for the integrity of data and the accuracy of data analysis.
The authors acknowledge the editorial assistance of Dr. Nickolas Mellen, Department of Pediatrics of the University of Louisville.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1233/-/DC1.
REFERENCES
- 1.Zhou YT, Grayburn P, Karim A, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 2000;97:1784–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med 1994;330:913–919 [DOI] [PubMed] [Google Scholar]
- 3.Rabkin SW, Klassen SS. Palmitate-induced NO production has a dual action to reduce cell death through NO and accentuate cell death through peroxynitrite formation. Prostaglandins Leukot Essent Fatty Acids 2008;78:147–155 [DOI] [PubMed] [Google Scholar]
- 4.Tsang MY, Cowie SE, Rabkin SW. Palmitate increases nitric oxide synthase activity that is involved in palmitate-induced cell death in cardiomyocytes. Nitric Oxide 2004;10:11–19 [DOI] [PubMed] [Google Scholar]
- 5.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem 2001;276:14890–14895 [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 2006;291:E275–E281 [DOI] [PubMed] [Google Scholar]
- 7.Wang XL, Zhang L, Youker K, et al. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 2006;55:2301–2310 [DOI] [PubMed] [Google Scholar]
- 8.Yuan H, Zhang X, Huang X, et al. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010;5:e15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology 2010;151:576–585 [DOI] [PubMed] [Google Scholar]
- 10.Kim DS, Jeong SK, Kim HR, Kim DS, Chae SW, Chae HJ. Metformin regulates palmitate-induced apoptosis and ER stress response in HepG2 liver cells. Immunopharmacol Immunotoxicol 2010;32:251–257 [DOI] [PubMed] [Google Scholar]
- 11.Terai K, Hiramoto Y, Masaki M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 2005;25:9554–9575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kewalramani G, Puthanveetil P, Wang F, et al. AMP-activated protein kinase confers protection against TNF-alpha-induced cardiac cell death. Cardiovasc Res 2009;84:42–53 [DOI] [PubMed] [Google Scholar]
- 13.Du JH, Xu N, Song Y, et al. AICAR stimulates IL-6 production via p38 MAPK in cardiac fibroblasts in adult mice: a possible role for AMPK. Biochem Biophys Res Commun 2005;337:1139–1144 [DOI] [PubMed] [Google Scholar]
- 14.Tang CH, Chiu YC, Tan TW, Yang RS, Fu WM. Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B pathway. J Immunol 2007;179:5483–5492 [DOI] [PubMed] [Google Scholar]
- 15.Kim M, Tian R. Targeting AMPK for cardiac protection: opportunities and challenges. J Mol Cell Cardiol 2011;51:548–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005;18:283–293 [DOI] [PubMed] [Google Scholar]
- 17.Okoshi R, Ozaki T, Yamamoto H, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem 2008;283:3979–3987 [DOI] [PubMed] [Google Scholar]
- 18.Calvert JW, Gundewar S, Jha S, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008;57:696–705 [DOI] [PubMed] [Google Scholar]
- 19.Paiva MA, Gonçalves LM, Providência LA, Davidson SM, Yellon DM, Mocanu MM. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc Drugs Ther 2010;24:25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerrits H, van Ingen Schenau DS, Bakker NE, et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis 2008;46:235–245 [DOI] [PubMed] [Google Scholar]
- 21.Tan Y, Li Y, Xiao J, et al. A novel CXCR4 antagonist derived from human SDF-1beta enhances angiogenesis in ischaemic mice. Cardiovasc Res 2009;82:513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai X, Tan Y, Cai S, et al. The role of CXCR7 on the adhesion, proliferation and angiogenesis of endothelial progenitor cells. J Cell Mol Med 2011;15:1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu L, Cecil J, Peng SB, et al. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene 2006;374:174–179 [DOI] [PubMed] [Google Scholar]
- 24.Altenburg JD, Broxmeyer HE, Jin Q, Cooper S, Basu S, Alkhatib G. A naturally occurring splice variant of CXCL12/stromal cell-derived factor 1 is a potent human immunodeficiency virus type 1 inhibitor with weak chemotaxis and cell survival activities. J Virol 2007;81:8140–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De La Luz Sierra M, Yang F, Narazaki M, et al. Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood 2004;103:2452–2459 [DOI] [PubMed] [Google Scholar]
- 26.Janowski M. Functional diversity of SDF-1 splicing variants. Cell Adhes Migr 2009;3:243–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho TK, Tsui J, Xu S, Leoni P, Abraham DJ, Baker DM. Angiogenic effects of stromal cell-derived factor-1 (SDF-1/CXCL12) variants in vitro and the in vivo expressions of CXCL12 variants and CXCR4 in human critical leg ischemia. J Vasc Surg 2010;51:689–699 [DOI] [PubMed] [Google Scholar]
- 28.Tang J, Wang J, Yang J, et al. Mesenchymal stem cells over-expressing SDF-1 promote angiogenesis and improve heart function in experimental myocardial infarction in rats. Eur J Cardiothorac Surg 2009;36:644–650 [DOI] [PubMed] [Google Scholar]
- 29.Ghadge SK, Mühlstedt S, Ozcelik C, Bader M. SDF-1α as a therapeutic stem cell homing factor in myocardial infarction. Pharmacol Ther 2011;129:97–108 [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Luther K. Genetically manipulated progenitor/stem cells restore function to the infarcted heart via the SDF-1α/CXCR4 signaling pathway. Prog Mol Biol Transl Sci 2012;111:265–284 [DOI] [PubMed] [Google Scholar]
- 31.Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006;55:1695–1704 [DOI] [PubMed] [Google Scholar]
- 32.Si Y, Zhao Y, Hao H, et al. Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: identification of a novel role in improving insulin sensitivity. Diabetes 2012;61:1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai L, Wang J, Li Y, et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes 2005;54:1829–1837 [DOI] [PubMed] [Google Scholar]
- 34.Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 2002;51:1938–1948 [DOI] [PubMed] [Google Scholar]
- 35.Cai L, Wang Y, Zhou G, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol 2006;48:1688–1697 [DOI] [PubMed] [Google Scholar]
- 36.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 2006;47:2726–2737 [DOI] [PubMed] [Google Scholar]
- 37.Borradaile NM, Buhman KK, Listenberger LL, et al. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol Cell 2006;17:770–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, McLennan SV, Allen TJ, Tsoutsman T, Semsarian C, Twigg SM. Adverse effects of high glucose and free fatty acid on cardiomyocytes are mediated by connective tissue growth factor. Am J Physiol Cell Physiol 2009;297:C1490–C1500 [DOI] [PubMed] [Google Scholar]
- 39.Craig R, Larkin A, Mingo AM, et al. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem 2000;275:23814–23824 [DOI] [PubMed] [Google Scholar]
- 40.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol 2009;75:1240–1247 [DOI] [PubMed] [Google Scholar]
- 41.Sierro F, Biben C, Martínez-Muñoz L, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A 2007;104:14759–14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol 2003;3:219–228 [DOI] [PubMed] [Google Scholar]
- 43.Gürtl B, Kratky D, Guelly C, et al. Apoptosis and fibrosis are early features of heart failure in an animal model of metabolic cardiomyopathy. Int J Exp Pathol 2009;90:338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colamussi ML, Secchiero P, Gonelli A, Marchisio M, Zauli G, Capitani S. Stromal derived factor-1 alpha (SDF-1 alpha) induces CD4+ T cell apoptosis via the functional up-regulation of the Fas (CD95)/Fas ligand (CD95L) pathway. J Leukoc Biol 2001;69:263–270 [PubMed] [Google Scholar]
- 45.Kortesidis A, Zannettino A, Isenmann S, Shi S, Lapidot T, Gronthos S. Stromal-derived factor-1 promotes the growth, survival, and development of human bone marrow stromal stem cells. Blood 2005;105:3793–3801 [DOI] [PubMed] [Google Scholar]
- 46.Hu X, Dai S, Wu WJ, et al. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation 2007;116:654–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yano T, Liu Z, Donovan J, Thomas MK, Habener JF. Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes 2007;56:2946–2957 [DOI] [PubMed] [Google Scholar]
- 48.Yin Q, Jin P, Liu X, et al. SDF-1α inhibits hypoxia and serum deprivation-induced apoptosis in mesenchymal stem cells through PI3K/Akt and ERK1/2 signaling pathways. Mol Biol Rep 2011;38:9–16 [DOI] [PubMed] [Google Scholar]
- 49.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J 2010;429:403–417 [DOI] [PubMed] [Google Scholar]
- 50.Saurin AT, Martin JL, Heads RJ, et al. The role of differential activation of p38-mitogen-activated protein kinase in preconditioned ventricular myocytes. FASEB J 2000;14:2237–2246 [DOI] [PubMed] [Google Scholar]
- 51.Huang Y, Zheng J, Fan P, Zhang X. Transfection of antisense p38 alpha gene ameliorates myocardial cell injury mediated by hypoxia and burn serum. Burns 2007;33:599–605 [DOI] [PubMed] [Google Scholar]
- 52.Liu H, Pedram A, Kim JK. Oestrogen prevents cardiomyocyte apoptosis by suppressing p38α-mediated activation of p53 and by down-regulating p53 inhibition on p38β. Cardiovasc Res 2011;89:119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venkatakrishnan CD, Tewari AK, Moldovan L, et al. Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am J Physiol Heart Circ Physiol 2006;291:H2680–H2691 [DOI] [PubMed] [Google Scholar]
- 54.Smart N, Mojet MH, Latchman DS, Marber MS, Duchen MR, Heads RJ. IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc Res 2006;69:164–177 [DOI] [PubMed] [Google Scholar]
- 55.Cacicedo JM, Benjachareonwong S, Chou E, Yagihashi N, Ruderman NB, Ido Y. Activation of AMP-activated protein kinase prevents lipotoxicity in retinal pericytes. Invest Ophthalmol Vis Sci 2011;52:3630–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bakondi B, Shimada IS, Peterson BM, Spees JL. SDF-1α secreted by human CD133-derived multipotent stromal cells promotes neural progenitor cell survival through CXCR7. Stem Cells Dev 2011;20:1021–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hattermann K, Held-Feindt J, Lucius R, et al. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res 2010;70:3299–3308 [DOI] [PubMed] [Google Scholar]
- 58.Ménard SL, Croteau E, Sarrhini O, et al. Abnormal in vivo myocardial energy substrate uptake in diet-induced type 2 diabetic cardiomyopathy in rats. Am J Physiol Endocrinol Metab 2010;298:E1049–E1057 [DOI] [PubMed] [Google Scholar]
- 59.Ti Y, Xie GL, Wang ZH, et al. TRB3 gene silencing alleviates diabetic cardiomyopathy in a type 2 diabetic rat model. Diabetes 2011;60:2963–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai SZ, Sun J, Wu H, et al. Decrease in calcium-sensing receptor in the progress of diabetic cardiomyopathy. Diabetes Res Clin Pract 2012;95:378–385 [DOI] [PubMed] [Google Scholar]
- 61.Huang C, Gu H, Zhang W, Manukyan MC, Shou W, Wang M. SDF-1/CXCR4 mediates acute protection of cardiac function through myocardial STAT3 signaling following global ischemia/reperfusion injury. Am J Physiol Heart Circ Physiol 2011;301:H1496–H1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ziegler M, Elvers M, Baumer Y, et al. The bispecific SDF1-GPVI fusion protein preserves myocardial function after transient ischemia in mice. Circulation 2012;125:685–696 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.