

Abstract
Excess fructose intake causes hypertriglyceridemia and hepatic insulin resistance in sedentary humans. Since exercise improves insulin sensitivity in insulin-resistant patients, we hypothesized that it would also prevent fructose-induced hypertriglyceridemia. This study was therefore designed to evaluate the effects of exercise on circulating lipids in healthy subjects fed a weight-maintenance, high-fructose diet. Eight healthy males were studied on three occasions after 4 days of 1) a diet low in fructose and no exercise (C), 2) a diet with 30% fructose and no exercise (HFr), or 3) a diet with 30% fructose and moderate aerobic exercise (HFrEx). On all three occasions, a 9-h oral [13C]-labeled fructose loading test was performed on the fifth day to measure [13C]palmitate in triglyceride-rich lipoprotein (TRL)-triglycerides (TG). Compared with C, HFr significantly increased fasting glucose, total TG, TRL-TG concentrations, and apolipoprotein (apo)B48 concentrations as well as postfructose glucose, total TG, TRL-TG, and [13C]palmitate in TRL-TG. HFrEx completely normalized fasting and postfructose TG, TRL-TG, and [13C]palmitate concentration in TRL-TG and apoB48 concentrations. In addition, it increased lipid oxidation and plasma nonesterified fatty acid concentrations compared with HFr. These data indicate that exercise prevents the dyslipidemia induced by high fructose intake independently of energy balance.
It is currently suspected that overconsumption of fructose, in the form of either sugar or high-fructose corn syrup, may promote obesity and favor the development of metabolic diseases such as type 2 diabetes and dyslipidemia (1,2). This is supported by a large number of studies in rodents, which demonstrate that a high-sucrose diet causes obesity, diabetes, dyslipidemia, and hepatic steatosis (3) and that this effect is mainly due to the fructose component of sucrose (4,5). Consistent with this hypothesis, epidemiological studies have shown that high intakes of sugar, fructose, or sweetened beverages are associated with the development of obesity (6,7), diabetes (8), hypertriglyceridemia (9), an increase in small dense atherogenic LDL particles (10), high blood pressure (11), albuminuria (12), and nonalcoholic fatty liver diseases (13). Several short-term studies have further documented that hypercaloric, high-fructose diets can cause increases in a number of cardiometabolic risk factors in humans, such as fasting and postprandial hypertriglyceridemia (14–18), ectopic lipid deposition in liver cells (19,20), impaired postprandial glucose homeostasis (18), and hepatic insulin resistance (21,22). Some of these effects may be related, at least in part, to the fact that fructose can be converted into fatty acids, which has been demonstrated after both acute (23) and chronic (18) fructose feeding. Exercise is very efficient at reducing the metabolic dysfunctions associated with obesity (24,25), and although many of these effects appear to be related to enhanced energy expenditure and improved energy balance (26,27), there is growing evidence that such improvements are independent of the changes in energy balance or body composition (28,29). Exercise has also been shown to prevent the accumulation of triglyceride-rich lipoprotein (TRL)-triglycerides (TG) and improve the plasma atherogenic lipid profile in healthy subjects fed a high-carbohydrate diet (30). The purpose of this study was to investigate whether exercise would similarly prevent fructose-induced metabolic effects.
RESEARCH DESIGN AND METHODS
Eight healthy, nonobese, male volunteers aged 21.5 ± 2.7 years, with mean ± SD body weight 68.5 ± 7.0 kg, height 1.76 ± 0.03 m, and BMI 22.1 ± 1.9 kg/m2, were studied. The subjects were sedentary, defined as undergoing <60 min exercise per week, and nonsmokers, who were not taking medication and had no history of diabetes. The experimental protocol was approved by the ethics committee of Lausanne University School of Medicine. All participants provided informed, written consent. The experimental, randomized crossover design is illustrated in Fig. 1. Each of the eight volunteers was studied on three different occasions after having followed three different diet and physical activity programs during 4 days as follows:
FIG. 1.

Experimental design of the study: Ex, exercise; OF, oral fructose test.
• Control (C): Subjects received a low-fructose, weight-maintenance diet containing 1.5 times their basal energy requirements calculated using the Harris-Benedict equation (31), which was composed of 50% complex carbohydrates, 5% sugars (mainly lactose), 30% fat, and 15% protein, and performed minimal physical activity (<30 min walking/day and no other exercise).
• High-fructose diet (HFr): Subjects received a weight-maintenance, high-fructose diet containing 1.5 times their basal energy requirements composed of 20% complex carbohydrates, 5% nonfructose sugars, 30% fructose provided as lemon-flavored drinks, 30% fat, and 15% protein and performed minimal physical activity.
• High-fructose diet plus exercise (HFrEx): Subjects received a high-fructose diet containing 1.7 times their basal energy requirements and completed two 30-min cycling exercise sessions at a power output of 125 W—one at noon and one at 5:00 p.m. on each day during 4 days. The physical activity factor of 1.7 was calculated at the time of protocol submission for a 22-year-old male weighing 75 kg and measuring 175 cm, with a predicted basal metabolic rate of 1.2845 kcal/min calculated using the Harris-Benedict equation (31). The predicted 24-h sedentary energy requirement was calculated assuming a nighttime energy expenditure of 8 h at a physical activity level of 1.0 and a daytime energy expenditure of 16 h at a physical activity level of 1.75, totaling 1.5 times basal energy requirements over 24 h or 2,775 kcal/day. The predicted 24-h energy requirements for HFrEx was estimated to be 3,205 kcal/day by assuming an exercise energy efficiency of 25% and equivalent to an extra 430 kcal/day, which corresponds to 1.7 times basal energy requirements.
Diets were calculated individually for each subject by a research nutritionist and were distributed each day as prepacked food items and drinks to be consumed at home. Subjects were instructed to consume all the food and beverages that were provided according to detailed instructions and not to consume anything else except unsweetened tea. All exercise sessions were performed at the Department of Physiology under supervision of one of the investigators.
Oral [13C]fructose tests.
On the fifth day of each condition, subjects reported to the Clinical Research Centre at 7:00 a.m. after an 11-h fast. Upon their arrival, subjects were weighed and their body composition was measured using bioelectrical impedance plethysmography (Imp DF50; ImpediMed, Eight Mile Plains, Australia). After subjects were lying in a bed, a catheter was inserted into a vein of the right arm for blood sampling and was maintained patent by a slow infusion of normosaline. Another catheter was inserted into a vein of the left arm and was used for the administration of a primed continuous infusion of [6,6-2H2]glucose (bolus, 2 mg/kg body wt, and continuous infusion, 0.02 mg/kg/min; Cambridge Isotope Laboratories, Andover, MA) throughout the test. Oral loads of fructose (0.2 g/kg fat-free mass, enriched with 0.1% [U-13C6]fructose; Cambridge Isotope Laboratories) were given every hour for 9 h. No other food was provided, and fructose intake corresponded to 78 ± 5% total energy expenditure during the experimental period. A bolus of 100 μmol/kg [1,1,2,3,3-2H5]glycerol (Cambridge Isotope Laboratories) was administered, and TRL-TG kinetics was calculated from the modeling of the decrease in [2H5]glycerol enrichment in TRL-TG over time. This method provides a single value over several hours. For this reason, the bolus of [2H5]glycerol was administered after the administration of the third dose of fructose (time 120 min) to allow 2 h for stimulation of hepatic fructose uptake and metabolism. Blood samples were collected at baseline (t = 0) and after 60, 120, 140, 160, 180, 240, 300, 360, 420, 480, and 540 min. Blood pressure was measured at baseline using an automatic blood pressure device (Omron 907; Omron, Hoofddorp, the Netherlands). Energy expenditure and net substrate oxidation rates were monitored over the last 3 h of the test by open-circuit indirect calorimetry (Deltatrac II; Datex Instrument, Helsinki, Finland). However, owing to technical problems with the indirect calorimeter, results could be obtained for only six of the eight participants. Urine was collected throughout the day to determine urea nitrogen excretion rate.
Analytic procedures.
Plasma was immediately separated from blood cells by centrifugation at 3,600g for 10 min at 4°C, and aliquots were stored at −20°C until assayed. Plasma metabolites (glucose, TG, nonesterified fatty acids [NEFAs], cholesterol, and lactate) (Randox Laboratories, Crumlin, U.K.), β-hydroxybutyric acid (BHB) (Roche Diagnostics Hitachi, Rotkreuz, Switzerland), and urinary urea (Randox Laboratories, Crumlin, U.K.) were measured by enzymatic methods; insulin and glucagon (Millipore, Billerica, MA) were assessed by radioimmunoassay. Apolipoprotein (apo)B48 was measured by ELISA using a kit from Shibayagi, Shibukawa, Japan.
The TRL fraction (Svedberg flotation unit [Sf] >20) was separated by ultracentrifugation (17 h at 45,000 rpm at 4°C) in an Optima L-90 K ultracentrifuge (Beckman Coulter, Brea, CA) in a fixed-angle rotor (50.3 Ti; Beckman Coulter). After plasma deproteinization and partial purification over anion- and cation-exchange resins, plasma [2H5]glycerol and [6,6-2H2]glucose were acetylated in the presence of acetic anhydride and pyridine and their enrichments were measured by GC-MS (Agilent Technologies, Santa Clara, CA) in chemical ionization mode, with selective monitoring of m/z 331 and 333 for glucose and 159 and 164 for glycerol. For [13C]palmitate enrichment and concentration, total lipids were extracted from plasma and fatty acid methyl esters (FAMEs) were prepared from TG fractions. The ratio of 13C to 12C in the FAME derivatives was ascertained by using Δ Plus XP GC-combustion isotope ratio MS (Thermo Electron, Bremen, Germany). Tricosanoic acid methyl ester was used as an isotopic enrichment standard, and a quality-control sample (certified standard of eicosanoic acid FAME; Department of Geological Sciences, Indiana University, Bloomington, IN) was run with each set of samples.
LDL size and subclasses were determined in frozen samples from baseline measurements. For analysis of LDL size and subclasses, nondenaturing PAGE of plasma was performed and analyzed and LDL subclass distribution (Class I-IVb) was calculated as previously described (10).
Calculations.
TRL-TG production was calculated as follows: the fractional turnover rate (FTR) of TRL-TG was determined by using compartmental modeling analysis as previously described (32). The rate of TRL-TG secretion (in millimoles per hour), which represents the amount of TRL-TG entering the bloodstream, was calculated by multiplying the FTR of TRL-TG (in pools/hour) by the pool of TRL-TG in plasma (in millimoles). The clearance rate of TRL-TG (in millimeters plasma per minute), which is an index of the removal efficiency of TRL-TG from the systemic circulation, was calculated by dividing the TRL-TG secretion rate (in millimoles per minute) by the TRL-TG concentration (in millimoles per milliliter).
Plasma [13C]TRL-palmitate (nmol/L) was calculated as follows: [13C]TRL-TG-palmitate isotopic enrichment [atom percent excess] × weight % palmitate × [TRL-TG (nmol/L)]. [13C]TRL-palmitate production (nanomoles per hour) was calculated by multiplying the FTR of TRL-TG (pools/hour) by the pool of [13C]TRL-palmitate (nanomoles).
The contribution of gluconeogenesis from fructose to endogenous glucose production [EGP(F) in grams per kilogram per hour] was calculated as follows:
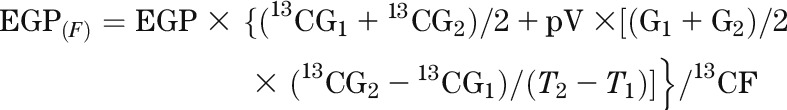 |
where EGP is the endogenous glucose production (grams per kilograms per hour) calculated with [6,6-2H2]glucose (20), 13CG is the isotopic enrichment of plasma glucose (atom percent excess), G is the glucose concentration (grams/liter), 13CF is the isotopic enrichment of oral fructose, p is the pool fraction (set at 0.75), V is the glucose distribution space (set at 0.2 times body weight), and T is the time (hours).
Glycogen synthesis (GS) was estimated as:
where net CHOox (oxidized carbohydrates) was calculated using standard indirect calorimetry equations (20).
Statistical analysis.
All values are expressed as means ± SEM. Log or box-cox transformation was applied to skewed data before statistical analysis. Values obtained at time 0 were used as fasting values. Changes in body weight, body fat content, blood pressure, and fasting parameters were assessed by using ANOVA for repeated measures, followed by Tukey post hoc tests for multiple comparisons.
Metabolic effects of fructose loading in the C condition were expressed as the percent change from T = 0 to T = 540 min. The effects of fructose loading over time, dietary condition with or without exercise, and their interaction were evaluated by a two-way ANOVA for repeated measures with interaction, followed by Tukey post hoc tests for multiple comparisons.
TRL-TG production and clearance, [13C]TRL-palmitate production, and averaged postfructose gluconeogenesis from fructose and GS were compared using ANOVA for repeated measures, followed by Tukey post hoc tests.
The prespecified primary outcome was postfructose [13C]TRL-palmitate concentrations. Power analysis was based on our previous study comparing the effects of oral fructose in male and females (33) and indicated that a sample size of six was required to detect an effect size of 0.5 with a power of 80%.
All statistical calculations were performed with Stata 10 (Stata, College Station, TX). P < 0.05 was considered statistically significant.
RESULTS
Anthropometric variables and fasting metabolic parameters.
There was no difference in body weight (C 68.7 ± 2.6 kg, HFr 68.8 ± 2.7 kg, and HFrEx 68.9 ± 2.7 kg), body fat content (C 17.9 ± 2.3%, HFr 17.8 ± 2.3%, and HFrEx 17.7 ± 2.2%), or blood pressure (C 118/63 ± 3/2 mmHg, HFr 115/65 ± 2/2 mmHg, and HFrEx 119/61 ± 3/2 mmHg) after the three different dietary conditions.
HFr significantly increased fasting plasma TG, TRL-TG, apoB48, and glucose concentrations (all P < 0.05 [Fig. 2, Fig. 3, and Table 1]). However, it did not significantly change fasting cholesterol, HDL cholesterol, insulin, glucagon, NEFA, glycerol, BHB, or lactate concentrations (NS [Fig. 2, Fig. 3, and Table 1]). LDL size and LDL subclass distribution were not different after the HFr diet compared with the C diet, except for a small increase in LDL IIa (P < 0.05) (Table 1).
FIG. 2.

Mean ± SEM total TG (A), TRL-TG (B), NEFA (C), glycerol (D), BHB (E), and [13C]TRL-palmitate (F) concentrations over time after oral loads of fructose taken hourly (n = 8). *HFr significantly different from C at baseline, ‡HFrEx significantly different from HFr at baseline, aHFr significantly different from C (Tukey post hoc test, P < 0.05), bHFrEx significantly different from C (Tukey post hoc test, P < 0.05), cHFrEx significantly different from HFr (Tukey post hoc test, P < 0.05).
FIG. 3.

Mean ± SEM glucose (A), insulin (B), and lactate (C) concentrations over time after fructose oral loads taken hourly (n = 8). *HFr significantly different from C at baseline, aHFr significantly different from C (Tukey post hoc test, P < 0.05), bHFrEx significantly different from C (Tukey post hoc test, P < 0.05), cHFrEx significantly different from HFr (Tukey post hoc test, P < 0.05).
TABLE 1.
Fasting plasma hormone and substrate concentrations at t = 0 (n = 8)
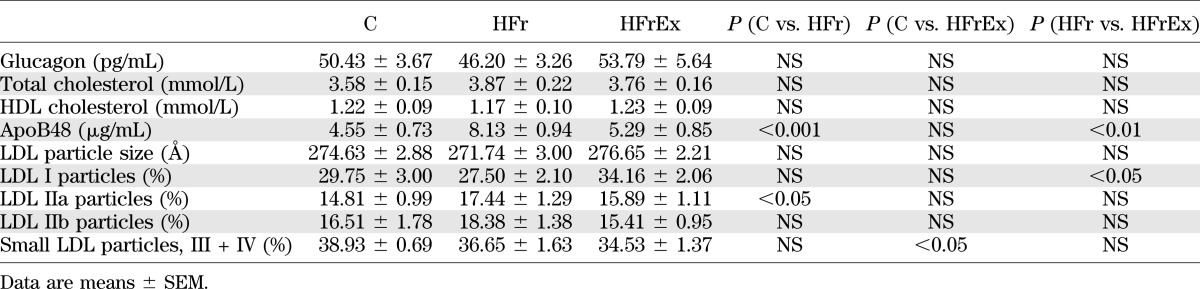
HFrEx significantly blunted the effects of HFr on plasma TG and normalized fasting total TG, TRL-TG, and apoB48 concentrations. It also significantly increased LDL-I particles (P < 0.05 vs. HFr) (Table 1) and decreased small LDL particles (III + IV) compared with C (P < 0.05 vs. C) (Table 1). Fasting plasma glucose concentrations, however, remained unchanged compared with HFr. HFrEx also increased fasting BHB concentrations (P < 0.05 vs. HFr). All other parameters were unchanged (Fig. 2, Fig. 3, and Table 1).
Metabolic effects of fructose loading.
To further focus on the postprandial metabolic effects of fructose, we measured several metabolic parameters relevant to lipid and glucose metabolism during a 9-h fructose loading test. In C, fructose ingestion caused a modest increase in total TG and TRL-TG (P < 0.05) (Fig. 2A and B) and a marked suppression of NEFA (P < 0.005) (Fig. 2C), glycerol (P < 0.01, Fig. 2D), and BHB concentrations (P < 0.0005) (Fig. 2E). [13C]TRL-palmitate concentrations increased (P < 0.05), indicating conversion of fructose into lipids (Fig. 2F). Although a substantial amount of ingested fructose was converted into glucose and released in the systemic circulation, plasma glucose (Fig. 3A) and insulin concentrations were not increased (Fig. 3B) compared with baseline. There was also a significant increase in lactate concentration, most likely from splanchnic fructose metabolism (P < 0.0005) (Fig. 3C).
Compared with C, HFr enhanced postfructose total TG and TRL-TG concentrations (P < 0.0001) (Fig. 2A and B) and [13C]TRL-palmitate concentration (P < 0.0001) (Fig. 2F) and decreased postfructose NEFA concentrations (P < 0.05) (Fig. 2C). TRL-TG secretion was not significantly increased, but TRL-TG clearance tended to decrease (Table 2). [13C]TRL-palmitate production was increased 2.9-fold (Table 2) compared with C, but the difference did not reach statistical significance owing to unexpected, very high interindividual variations. Gluconeogenesis from fructose was similar to that with C (Table 3), but glucose concentrations were significantly increased (P < 0.0001) (Fig. 3A).
TABLE 2.
TRL-TG kinetics (n = 8)
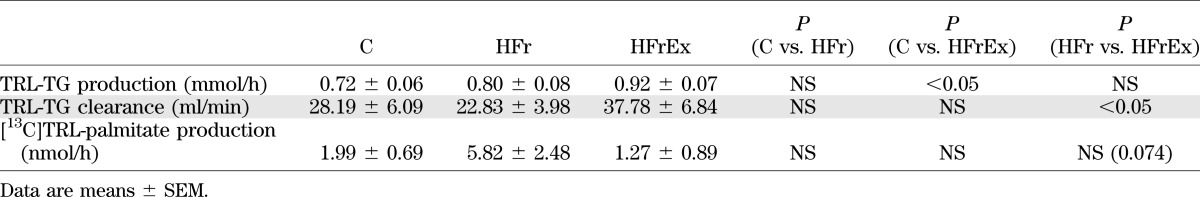
TABLE 3.
Energy expenditure, net substrate oxidation, and calculated net glycogen synthesis (n = 6)
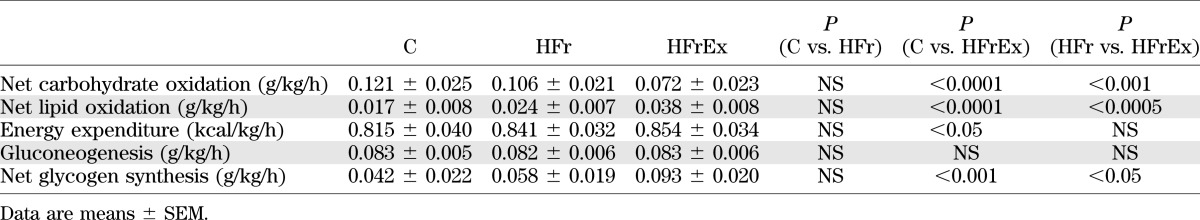
In the HFrEx condition, postfructose total TG, TRL-TG, and [13C]TRL-palmitate concentrations were completely normalized (all NS compared with C) (Fig. 2A, B, and F). TRL-TG clearance was increased by 66% compared with HFr (P < 0.05 vs. HFr) (Table 2). TRL-TG secretion was not significantly changed, but [13C]TRL-palmitate production was markedly decreased compared with HFr (P = 0.074 vs. HFr) (Table 2). Furthermore, NEFA and glycerol concentrations were increased compared with both HFr and C (both P < 0.0001 vs. HFr) (Fig. 2C and D). Gluconeogenesis from fructose and plasma glucose concentrations remained increased to the same extent as HFr (NS) (Table 3 and Fig. 3A), while insulin concentrations were slightly decreased (P < 0.05 vs. HFr) (Fig. 3B). Lactate concentrations were significantly decreased compared with those observed with C (P < 0.005) (Fig. 3C). Net lipid oxidation was increased (P < 0.0005 vs. HFr) (Table 3) while net carbohydrate oxidation was decreased (P < 0.001 vs. HFr) (Table 3), and net GS was nearly doubled compared with HFr and C (P < 0.05 vs. HFr) (Table 3). Energy expenditure was increased with HFrEx but only compared with C (P < 0.05) (Table 3). Mean LDL size was not significantly different from HFr or C, but the amount of large, less atherogenic LDL subclass I particles was significantly increased after HFrEx compared with HFr (P < 0.05) (Table 1), and small dense LDL particles (III + IV) were significantly decreased compared with C (P < 0.05) (Table 1).
DISCUSSION
The current study was specifically designed to evaluate whether fructose, when administered together with a weight-maintenance diet, causes significant alterations of blood lipids and whether these effects can be modulated by exercise. For this purpose, we selected a design in which fructose was administered to the same subjects on three occasions, i.e., after having consumed a low-fructose diet without exercise and after having received a high-fructose diet with and without exercise. Total energy intake was adapted to meet energy expenditure on all three occasions. Fructose intake corresponded to 30% total energy intake on both occasions but was higher in absolute values when subjects exercised.
With low physical activity, an HFr diet over 4 days led to an increase in total plasma TG and TRL-TG concentrations both after an overnight fast and after fructose loading. There was no significant change in TRL-TG kinetics after fructose loading, most likely due to the small number of subjects included and to a relatively large interindividual variability. It appears likely, however, that decreased TRL-TG clearance contributed to the rise in TRL-TG, since this parameter was previously reported to decrease after fructose ingestion (34) and showed a strong trend toward a 22% reduction. Increased TRL-TG production has also been reported after high-carbohydrate diets (35) and may have contributed as well.
During the fructose-loading experiments, oral fructose was labeled with 13C, and incorporation of 13C into TRL-TG palmitate could be documented. Furthermore, the increase in [13C]TRL-palmitate concentration was markedly enhanced when fructose was ingested after consumption of a high-fructose diet versus a low-fructose diet without exercise. This suggests that chronic fructose intake upregulated hepatic de novo lipogenesis. However, it is also possible that an increase in gut fructose absorption, induced by chronic fructose intake, contributed to increase the total fructose load delivered to the liver (36).
In humans, carbohydrate-induced de novo lipogenesis occurs essentially in the liver, although massive carbohydrate overfeeding may also stimulate this pathway in adipose tissue (37,38). Hepatic de novo lipogenesis has further been suggested to make a significant contribution to fructose- or sucrose-induced hypertriglyceridemia through enhanced secretion of hepatic VLDL (21,39). Interestingly, fasting apoB48 concentrations nearly doubled with HFr. Since this apolipoprotein is exclusively synthesized in enterocytes, this observation indicates that a high-fructose diet stimulates the secretion of intestinal, chylomicron-like particles even after an overnight fast. Activation of this pathway has indeed been documented in high fructose–fed hamsters (40). However, the contribution of intestinal lipogenesis to the fructose-induced increase in TRL-TG cannot be estimated from the mere increase in apoB48 concentration.
When the high-fructose diet was associated with exercise, all the effects of fructose on lipoprotein metabolism were totally prevented. There was indeed complete normalization of fasting and postprandial TRL-TG concentration, which was essentially due to a 65% increase in TRL-TG clearance and suggests that exercise-enhanced lipoprotein lipase activity facilitates the disposal of lipids in adipose cells or skeletal muscle fibers. In addition to this accelerated TRL-TG removal from the circulation, exercise also very dramatically decreased [13C]TRL-palmitate concentrations and secretion. This indicates that exercise inhibited de novo lipogenesis and that this may also have contributed to the TG-lowering effects of exercise. Consumption of high-fructose or high-sucrose diets has also been shown to decrease the concentration of LDL subclass I particles, which have a lower atherogenic potential than other LDL subclasses (10,41). In the current study, HFr failed to significantly alter LDL particle size and the proportion of LDL subclass I, possibly due to the short duration of diet administration. However, LDL subclass I was significantly increased with HFrEx. This is consistent with previous reports showing that exercise has beneficial effects on lipoprotein profiles in obese dyslipidemic patients (42) and in healthy subjects fed a high-carbohydrate diet (30) and adds to the evidence that it can efficiently prevent the adverse consequences of a high-fructose diet.
Unexpectedly, exercise restored normal fasting apoB48 concentrations, indicating that exercise regulated not only hepatic but also intestinal fructose metabolism. However, the functional significance of gut fructose metabolism remains to be more fully evaluated.
All of these effects of exercise were observed even though the additional energy expended during physical activity (∼430 kcal/day) was compensated for by increased total energy and fructose intakes indicating that exercise potently impacts on fructose metabolic pathways independently of changes in overall energy balance.
We can only speculate on the mechanisms leading to these effects. Exercise may indeed cause multiple metabolic alterations, which converge to reduce plasma TG concentrations. First, acute exercise enhances fructose conversion into glucose and lactate and their use as energy providing substrates by the working muscle (43), which may have decreased the availability of fructose carbons for de novo lipogenesis during the days preceding the oral fructose test. Second, exercise-induced hepatic and muscle glycogen depletion can be expected to result in an enhanced conversion of fructose into hepatic glycogen when fructose is subsequently ingested, thus diverting fructose away from hepatic de novo lipogenesis. Our observation that exercise increased net GS after fructose loading is entirely consistent with this hypothesis. Third, acute exercise increases LPL activity in skeletal muscle (44,45), resulting in enhanced TRL-TG clearance. Finally, exercise stimulates lipolysis, and the NEFA, released into the systemic circulation, may activate the nuclear receptors peroxisome proliferator–activated receptor-α in the liver and possibly in enterocytes as well to stimulate fat oxidation and reduce hepatic and intestinal de novo lipogenesis (46).
Besides its effect on plasma TG, HFr also increased slightly, but significantly, fasting and postfructose plasma glucose concentrations. Other studies have also documented that short-term fructose overfeeding increases basal hepatic glucose output and impairs the ability of low-dose insulin to suppress hepatic glucose output (21,22). These alterations of glucose homeostasis can be observed within a few days of fructose overfeeding and may be due to fructose increasing hepatic glycogen stores (47). It may therefore correspond to a physiological adaptation to important changes in sugar intake rather than to pathological adverse effects of fructose.
Our study has several limitations that need be addressed. First, energy intake was calculated to match energy requirements based on calculated energy expenditure. This was done using a well-accepted, reliable equation to predict basal energy expenditure (31) and a physical activity level of 1.5. However, since the 24-h energy expenditure was not actually measured, it is possible that energy balance was not reached for every subject. This may have affected our results to some extent, since the effect of exercise to lower plasma TG is known to be significantly blunted when energy expended during exercise is fully replaced compared with negative energy balance (48). Second, incorporation of 13C administered as [13C6]fructose into TRL-TG-palmitate was used as an indirect estimate of de novo lipogenesis. This is a relatively crude method that does not allow a quantitative estimate of fatty acid synthesis owing to the fact that the isotopic enrichment of intrahepatic acetyl-CoA, the actual precursor for fatty acid synthesis, was not measured. There may also be an underestimation of hepatic fatty acid synthesis due to the short period of tracer administration, which may have been insufficient for equilibration of newly synthesized fatty acids and delayed secretion of TG from the intrahepatic pool into VLDL (49). Third, this method provides an estimate of the contribution of de novo lipogenesis to TRL-palmitate secretion but not to intrahepatic TG storage, and we cannot directly assess the effect of fructose or exercise on the latter pathway. Fourth, we cannot discard the hypothesis that exercise may alter plasma lipoprotein metabolism irrespective of dietary fructose content. Finally, this study evaluated the effects of a very high fructose intake (30% total energy, which far exceeds the average U.S. per capita daily fructose consumption [~10% total energy] [50]) over a 4-day period and, hence, does not provide information on the effects of fructose in the general population or over longer periods of time. It does, however, demonstrate that exercise can efficiently reduce some potentially adverse effects of fructose.
In summary, we observed that substitution of fructose for starch in a weight-maintenance diet increased plasma total and TRL-TG. These alterations may increase cardiometabolic risk but were observed at a very high fructose intake (∼200 g/day), largely exceeding the average fructose intake observed in the population (50). Even with such a high intake, exercise completely prevented fructose-induced alterations of lipid metabolism.
ACKNOWLEDGMENTS
This study was supported by grant 320030_138428 from the Swiss National Foundation for Science.
L.T. has received research support from Nestle SA and Ajinomoto Co., Inc., for research unrelated to this article. No other potential conflicts of interest relevant to this article were reported.
L.E. designed the study, recruited participants, performed tests, analyzed data, performed statistical analysis, drafted the manuscript, and revised the manuscript. V.L. analyzed data, performed statistical analysis, and revised the manuscript. F.T. and V.C. recruited participants, performed tests, and revised the manuscript. L.H. analyzed the isotopic enrichment of plasma lipids and revised the manuscript. P.S. designed the study and revised the manuscript. B.M. and B.W.P. calculated VLDL-TG kinetics and revised the manuscript. B.A.F. analyzed the isotopic enrichment of plasma lipids and revised the manuscript. P.A.G. analyzed LDL subfractions and revised the manuscript. V.G. revised the manuscript. K.B. analyzed LDL subfractions and revised the manuscript. L.T. designed the study, analyzed LDL subfractions, drafted the manuscript, and revised the manuscript. L.T. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the 76th and 77th Scientific Sessions and Annual Meetings of the American Society of Nutrition in conjunction with Experimental Biology, San Diego, California, 21–25 April 2012, and Boston, Massachusetts, 19–24 April 2013.
The authors thank the staff of the Department of Physiology of Lausanne and of the Clinical Research Centre for their outstanding assistance and all the volunteers for their participation.
Footnotes
Clinical trial reg. no. NCT01121003, clinicaltrials.gov.
REFERENCES
- 1.Tappy L, Lê KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev 2010;90:23–46 [DOI] [PubMed] [Google Scholar]
- 2.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr 2004;79:537–543 [DOI] [PubMed] [Google Scholar]
- 3.Bizeau ME, Pagliassotti MJ. Hepatic adaptations to sucrose and fructose. Metabolism 2005;54:1189–1201 [DOI] [PubMed] [Google Scholar]
- 4.Wei Y, Wang D, Pagliassotti MJ. Fructose selectively modulates c-jun N-terminal kinase activity and insulin signaling in rat primary hepatocytes. J Nutr 2005;135:1642–1646 [DOI] [PubMed] [Google Scholar]
- 5.Wei Y, Pagliassotti MJ. Hepatospecific effects of fructose on c-jun NH2-terminal kinase: implications for hepatic insulin resistance. Am J Physiol Endocrinol Metab 2004;287:E926–E933 [DOI] [PubMed] [Google Scholar]
- 6.Wharton CM, Hampl JS. Beverage consumption and risk of obesity among Native Americans in Arizona. Nutr Rev 2004;62:153–159 [DOI] [PubMed] [Google Scholar]
- 7.Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB. Changes in diet and lifestyle and long-term weight gain in women and men. N Engl J Med 2011;364:2392–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulze MB, Manson JE, Ludwig DS, et al. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 2004;292:927–934 [DOI] [PubMed] [Google Scholar]
- 9.Welsh JA, Sharma A, Abramson JL, Vaccarino V, Gillespie C, Vos MB. Caloric sweetener consumption and dyslipidemia among US adults. JAMA 2010;303:1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aeberli I, Zimmermann MB, Molinari L, et al. Fructose intake is a predictor of LDL particle size in overweight schoolchildren. Am J Clin Nutr 2007;86:1174–1178 [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Caballero B, Mitchell DC, et al. Reducing consumption of sugar-sweetened beverages is associated with reduced blood pressure: a prospective study among United States adults. Circulation 2010;121:2398–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoham DA, Durazo-Arvizu R, Kramer H, et al. Sugary soda consumption and albuminuria: results from the National Health and Nutrition Examination Survey, 1999-2004. PLoS ONE 2008;3:e3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Assy N, Nasser G, Kamayse I, et al. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol 2008;22:811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crapo PA, Kolterman OG, Henry RR. Metabolic consequence of two-week fructose feeding in diabetic subjects. Diabetes Care 1986;9:111–119 [DOI] [PubMed] [Google Scholar]
- 15.Sievenpiper JL, Carleton AJ, Chatha S, et al. Heterogeneous effects of fructose on blood lipids in individuals with type 2 diabetes: systematic review and meta-analysis of experimental trials in humans. Diabetes Care 2009;32:1930–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bantle JP. Clinical aspects of sucrose and fructose metabolism. Diabetes Care 1989;12:56–61 [DOI] [PubMed] [Google Scholar]
- 17.Stanhope KL, Bremer AA, Medici V, et al. Consumption of fructose and high fructose corn syrup increase postprandial triglycerides, LDL-cholesterol, and apolipoprotein-B in young men and women. J Clin Endocrinol Metab 2011;96:E1596–E1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lê KA, Ith M, Kreis R, et al. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr 2009;89:1760–1765 [DOI] [PubMed] [Google Scholar]
- 20.Theytaz F, Noguchi Y, Egli L, et al. Effects of supplementation with essential amino acids on intrahepatic lipid concentrations during fructose overfeeding in humans. Am J Clin Nutr 2012;96:1008–1016 [DOI] [PubMed] [Google Scholar]
- 21.Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005;54:1907–1913 [DOI] [PubMed] [Google Scholar]
- 22.Aeberli I, Hochuli M, Gerber PA, et al. Moderate amounts of fructose consumption impair insulin sensitivity in healthy young men: a randomized controlled trial. Diabetes Care 2013;36:150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parks EJ, Skokan LE, Timlin MT, Dingfelder CS. Dietary sugars stimulate fatty acid synthesis in adults. J Nutr 2008;138:1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katzmarzyk PT, Leon AS, Wilmore JH, et al. Targeting the metabolic syndrome with exercise: evidence from the HERITAGE Family Study. Med Sci Sports Exerc 2003;35:1703–1709 [DOI] [PubMed] [Google Scholar]
- 25.Albright A, Franz M, Hornsby G, et al. American College of Sports Medicine position stand. Exercise and type 2 diabetes. Med Sci Sports Exerc 2000;32:1345–1360 [DOI] [PubMed] [Google Scholar]
- 26.Okura T, Nakata Y, Ohkawara K, et al. Effects of aerobic exercise on metabolic syndrome improvement in response to weight reduction. Obesity (Silver Spring) 2007;15:2478–2484 [DOI] [PubMed] [Google Scholar]
- 27.Anderssen SA, Carroll S, Urdal P, Holme I. Combined diet and exercise intervention reverses the metabolic syndrome in middle-aged males: results from the Oslo Diet and Exercise Study. Scand J Med Sci Sports 2007;17:687–695 [DOI] [PubMed] [Google Scholar]
- 28.Zotou E, Magkos F, Koutsari C, et al. Acute resistance exercise attenuates fasting and postprandial triglyceridemia in women by reducing triglyceride concentrations in triglyceride-rich lipoproteins. Eur J Appl Physiol 2010;110:869–874 [DOI] [PubMed] [Google Scholar]
- 29.Tsekouras YE, Magkos F, Kellas Y, Basioukas KN, Kavouras SA, Sidossis LS. High-intensity interval aerobic training reduces hepatic very low-density lipoprotein-triglyceride secretion rate in men. Am J Physiol Endocrinol Metab 2008;295:E851–E858 [DOI] [PubMed] [Google Scholar]
- 30.Koutsari C, Karpe F, Humphreys SM, Frayn KN, Hardman AE. Exercise prevents the accumulation of triglyceride-rich lipoproteins and their remnants seen when changing to a high-carbohydrate diet. Arterioscler Thromb Vasc Biol 2001;21:1520–1525 [DOI] [PubMed] [Google Scholar]
- 31.Harris JA, Benedict FG. A Biometric Study of Human Basal Metabolism. Proc Natl Acad Sci USA 1918;4:370–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patterson BW, Mittendorfer B, Elias N, Satyanarayana R, Klein S. Use of stable isotopically labeled tracers to measure very low density lipoprotein-triglyceride turnover. J Lipid Res 2002;43:223–233 [PubMed] [Google Scholar]
- 33.Tran C, Jacot-Descombes D, Lecoultre V, et al. Sex differences in lipid and glucose kinetics after ingestion of an acute oral fructose load. Br J Nutr 2010;104:1139–1147 [DOI] [PubMed] [Google Scholar]
- 34.Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr 2007;85:1511–1520 [DOI] [PubMed] [Google Scholar]
- 35.Parks EJ, Hellerstein MK. Carbohydrate-induced hypertriacylglycerolemia: historical perspective and review of biological mechanisms. Am J Clin Nutr 2000;71:412–433 [DOI] [PubMed] [Google Scholar]
- 36.Bode C, Eisenhardt JM, Haberich FJ, Bode JC. Influence of feeding fructose on fructose and glucose absorption in rat jejunum and ileum. Res Exp Med (Berl) 1981;179:163–168 [DOI] [PubMed] [Google Scholar]
- 37.Aarsland A, Chinkes D, Wolfe RR. Hepatic and whole-body fat synthesis in humans during carbohydrate overfeeding. Am J Clin Nutr 1997;65:1774–1782 [DOI] [PubMed] [Google Scholar]
- 38.Minehira K, Vega N, Vidal H, Acheson K, Tappy L. Effect of carbohydrate overfeeding on whole body macronutrient metabolism and expression of lipogenic enzymes in adipose tissue of lean and overweight humans. Int J Obes Relat Metab Disord 2004;28:1291–1298 [DOI] [PubMed] [Google Scholar]
- 39.Hudgins LC, Hellerstein MK, Seidman CE, Neese RA, Tremaroli JD, Hirsch J. Relationship between carbohydrate-induced hypertriglyceridemia and fatty acid synthesis in lean and obese subjects. J Lipid Res 2000;41:595–604 [PubMed] [Google Scholar]
- 40.Haidari M, Leung N, Mahbub F, et al. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J Biol Chem 2002;277:31646–31655 [DOI] [PubMed] [Google Scholar]
- 41.Aeberli I, Gerber PA, Hochuli M, et al. Low to moderate sugar-sweetened beverage consumption impairs glucose and lipid metabolism and promotes inflammation in healthy young men: a randomized controlled trial. Am J Clin Nutr 2011;94:479–485 [DOI] [PubMed] [Google Scholar]
- 42.Kraus WE, Houmard JA, Duscha BD, et al. Effects of the amount and intensity of exercise on plasma lipoproteins. N Engl J Med 2002;347:1483–1492 [DOI] [PubMed] [Google Scholar]
- 43.Lecoultre V, Benoit R, Carrel G, et al. Fructose and glucose co-ingestion during prolonged exercise increases lactate and glucose fluxes and oxidation compared with an equimolar intake of glucose. Am J Clin Nutr 2010;92:1071–1079 [DOI] [PubMed] [Google Scholar]
- 44.Gill JM, Hardman AE. Exercise and postprandial lipid metabolism: an update on potential mechanisms and interactions with high-carbohydrate diets (review). J Nutr Biochem 2003;14:122–132 [DOI] [PubMed] [Google Scholar]
- 45.Seip RL, Angelopoulos TJ, Semenkovich CF. Exercise induces human lipoprotein lipase gene expression in skeletal muscle but not adipose tissue. Am J Physiol 1995;268:E229–E236 [DOI] [PubMed] [Google Scholar]
- 46.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 1999;103:1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsson LH, Hultman E. Liver and muscle glycogen in man after glucose and fructose infusion. Scand J Clin Lab Invest 1974;33:5–10 [DOI] [PubMed] [Google Scholar]
- 48.Harrison M, O’Gorman DJ, McCaffrey N, et al. Influence of acute exercise with and without carbohydrate replacement on postprandial lipid metabolism. J Appl Physiol 2009;106:943–949 [DOI] [PubMed] [Google Scholar]
- 49.Vedala A, Wang W, Neese RA, Christiansen MP, Hellerstein MK. Delayed secretory pathway contributions to VLDL-triglycerides from plasma NEFA, diet, and de novo lipogenesis in humans. J Lipid Res 2006;47:2562–2574 [DOI] [PubMed] [Google Scholar]
- 50.Vos MB, Kimmons JE, Gillespie C, Welsh J, Blanck HM. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med 2008;10:160. [PMC free article] [PubMed] [Google Scholar]