

Abstract
The genesis of β-cells predominantly occurs through self-replication; therefore, understanding the regulation of cell proliferation is essential. We previously showed that the lack of nonhomologous end joining (NHEJ) DNA repair factor ligase IV leads to an accumulation of DNA damage that permanently halts β-cell proliferation and dramatically decreases insulin production, causing overt diabetes in a hypomorphic p53R172P background. In the present study, to further delineate the function of NHEJ, we analyzed mice deficient for another key NHEJ factor, Ku70, to discover the effect of cellular responses to DNA damage in pancreatic β-cells on cellular proliferation and glucose homeostasis. Analysis of Ku70−/− pancreatic β-cells revealed an accumulation of DNA damage and activation of p53-dependent cellular senescence similar to the results found in our earlier ligase IV deficiency study. To our surprise, Ku70−/− mice had significantly increased β-cell proliferation and islet expansion, heightened insulin levels, and decreased glycemia. This augmented β-cell proliferation was accompanied by an increased β-catenin level, which we propose to be responsible for this phenotype. This study highlights Ku70 as an important player not only in maintaining genomic stability through NHEJ-dependent functions, but also in regulating pancreatic β-cell proliferation, a novel NHEJ-independent function.
The inability to maintain genomic stability and control proliferation, the hallmarks of many cancers, are exacerbated in the presence of unrepaired DNA damage. One of the major pathways that repair DNA double-strand breaks (DSBs) is nonhomologous end joining (NHEJ) (1). Classically, the NHEJ pathway mends DSBs in two steps. Initially, broken DNA ends are recognized and processed, a mechanism initiated by the Ku70/80 heterodimer, which recruits DNA-dependent protein kinase and repair factor Artemis for end modification. Next, the broken DNA is ligated through a complex consisting of DNA ligase IV (Lig4), XRCC4, and Cernunnos/XLF (1). Lig4 and XRCC4 deficiencies in mice result in late embryonic lethality (2). Other NHEJ-deficient mice are viable, but they exhibit severe combined immunodeficiency because of their inability to repair the programmed DSBs created during early lymphocyte development (3). This accumulation of unrepaired DNA breaks activates p53-dependent apoptosis in developing lymphoid precursors. In the absence of p53, these double mutants lose both apoptotic and cell cycle checkpoint functions in the face of unrepaired DNA damage, succumbing to early and aggressive pro-B lymphomas with massive genomic instability (4,5).
In a previous study, we combined a hypomorphic, separation-of-function p53 mutant (p53R172P), which prevents p53-mediated apoptosis but retains a partial cell cycle arrest function (6), with NHEJ deficiency (Lig4−/−p53p/p) and showed that the mutant p53 not only rescues embryonic lethality, but also entirely eliminates lymphomagenesis in the Lig4-deficient mice (7). Further analysis of the developing lymphocytes revealed that the broken DNA ends activate a permanent cell cycle arrest, termed cellular senescence, that acts in parallel with apoptosis in suppressing tumorigenesis (7).
Although completely free of tumors, Lig4−/−p53p/p mice succumb to progressive diabetes (8). Mechanistic analysis revealed that spontaneous DNA damage accumulates in the insulin-producing β-cells of the pancreas, activating the p53/p21 axis to trigger cellular senescence. This cascade halts the proliferation of β-cells, decreases islet mass, and compromises glucose homeostasis (8). Our earlier study highlighted a crucial role for the NHEJ pathway in the prevention of broken DNA and the subsequent activation of cell cycle control in pancreatic β-cells. Recently, others attempted to coax human β-cell proliferation through adenoviral expression; of note, accumulated DNA damage activated cellular responses to halt cell cycle reentry (9,10). These studies underscore the importance of understanding DNA damage and the respective responses for the development of future therapeutics.
The genesis of adult insulin-producing β-cells predominantly occurs through self-duplication of mature cells rather than through differentiation from their progenitors (11–13). Without compensatory β-cell replication, disrupting the β-cell cycle decreases islet mass and reduces insulin production, which deregulates glucose homeostasis and ultimately leads to diabetes. Conversely, augmenting the β-cell cycle increases islet area and insulin production, which often rescues a diabetic phenotype but in some cases, results in hypoglycemia and islet hyperplasia (14). Cell cycle regulators responsible for proliferation are vital to maintaining adult β-cells, but less is known about their upstream activators. One such pathway responsible for both developing the pancreas and controlling the postnatal β-cell cycle is the canonical Wnt signaling pathway (15,16). Activating Wnt signaling through stabilizing β-catenin has been shown to increase β-cell proliferation and to elevate serum insulin levels (17,18). Conversely, increasing β-catenin inhibitors decreases β-cell proliferation levels (17,19,20). Collectively, these studies suggest that Wnt signaling plays a critical role in controlling β-cell proliferation.
Because our previous work highlighted the importance of Lig4 in preventing genomic instability in pancreatic β-cells (8), we sought to examine the role of a different NHEJ factor, Ku70, in both wild-type (Ku70−/−p53+/+) and mutant p53 (Ku70−/−p53p/p) mice. Although most notable for its role in DNA damage recognition and repair, Ku70 has been implicated in many different fundamental cellular networks (8), such as telomere maintenance (21–23), apoptosis (24–26), and transcriptional regulation (26–30). Through generating and analyzing Ku70−/− and Ku70−/−p53p/p mice, we discovered that Ku70 deficiency progressively decreases glycemia, augments β-cell replication, and increases islet size, which is in stark contrast to Lig4 deficiency. Of note, the study also revealed a stabilization of islet-specific β-catenin with elevated markers for cell cycle progression. Therefore, in addition to DNA damage repair, Ku70 has a previously undiscovered function independent of DNA end joining that directly affects the proliferation of pancreatic β-cells. Much therapeutic effort has been focused on coaxing pancreatic β-cells into proliferation; therefore, studying and identifying novel regulators of β-cell expansion may shed light on new targets and therapeutic avenues.
RESEARCH DESIGN AND METHODS
Animals.
Mutant mice (mixed C57BL/6 and 129SV) maintenance has been previously described (8). Ku70+/− mice (31) were crossed to p53p/p mice (6) to generate Ku70−/−p53p/p mice. The protocols were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center.
Measurement of blood glucose and insulin levels.
For the glucose tolerance test, mice were fasted for 16 h and injected with glucose at 1.0 g/kg of body weight. For the insulin tolerance test, mice were fasted for 6 h and injected intraperitoneally with insulin 0.75 units/kg (Sigma Aldrich, St. Louis, MO). Glucose levels were measured with a glucose analyzer (Contour; Bayer HealthCare, Tarrytown, NY). Blood insulin levels were measured by ELISA as previously described (8).
Pancreatic histology and immunostaining.
Pancreata were fixed, embedded, sectioned, and stained as previously described (8). Slides were incubated with antibodies against insulin, glucagon, cyclin D1 and D2, and p53 (Cell Signaling, Danvers, MA); proliferating cell nuclear antigen (PCNA) (Dako, Carpinteria, CA); CDK4 and p16 (Santa Cruz Biotechnology, Santa Cruz, CA); Ku70 and bromodeoxyuridine (BrdU) (AbD Serotec, Raleigh, NC); β-catenin (BD Biosciences, San Jose, CA); or γH2AX and Ki67 (Abcam, Cambridge, MA). BrdU incorporation, immunohistochemistry, immunofluorescence, and microscopy were performed as previously described (8). Mouse embryonic fibroblasts (MEFs) were generated and stained as previously described (32).
TUNEL assay.
A TUNEL assay kit (ApopTag; Millipore, Billerica, MA) was used to detect apoptosis.
Cellular senescence.
Senescence-associated β-galactosidase (SA β-gal) activity was detected by a staining kit (Cell Signaling). Glucagon was detected by immunohistochemical staining. Slides were counterstained with Nuclear Fast Red (Vector Laboratories, Burlingame, CA) as previously described (8).
Analysis of pancreatic morphology.
Pancreata sections were prepared as previously described (8). Each section was subjected to morphometric analysis with Image J software (National Institutes of Health, Bethesda, MD). Raw data were statistically analyzed as previously described (8).
Western blot analysis.
Islets were isolated as described in Li et al. (33). Islets from at least three mice per genotype and age group were pooled together, and 40 μg of protein were loaded as in Tavana et al. (32). Antibodies were Ku70; β-actin (Sigma Aldrich); CDK4, p53, total β-catenin, and phosphorylated β-catenin (Cell Signaling); activated β-catenin (Millipore); p21 (BD Pharmingen, San Jose, CA); and GAPDH and tubulin (Epitomics, Burlingame, CA). Experiments were repeated three or more times.
Statistical analysis.
Results are presented as the mean ± SEM. Differences were determined by a two-tailed unpaired Student t test with a 95% CI. The area under the curve (AUC) was calculated with Prism software (GraphPad Inc., La Jolla, CA). P < 0.05 was considered significant.
RESULTS
Ku70 expression in pancreatic β-cells.
Previously, we demonstrated high Lig4 expression in isolated islets (8) and attributed it to protecting against spontaneous genomic insults caused by intrinsic metabolic agents. Therefore, we hypothesized that NHEJ is very active in pancreatic β-cells. To analyze the expression of Ku70, we performed a Western blot in purified wild-type and mutant pancreatic islets. The results showed that the Ku70 protein is expressed in the pancreatic islets and that the expression level slightly increases with age (Fig. 1A). Consistently, immunohistochemical staining reveals high expression of Ku70 in the islets (Fig. 1A).
FIG. 1.

Accumulation of DNA damage elevates p53 and p21 in both Ku70−/− and Ku70−/−p53p/p islets, triggering cellular senescence. A: Western blot analysis of Ku70. Actin is shown as a loading control. Protein from purified islets was isolated and pooled from three or more mice per genotype and time point. Experiments were repeated three times. Mice ages were as follows: young (Y) (1–2 months), medium (M) (2.5–4 months), and old (O) (4.5–7 months). Representative immunohistochemical staining for Ku70 in a pancreatic section from a 3-month-old WT mouse (right panel). Original magnification ×400 (×40 objective and ×10 ocular). B: Immunofluorescent staining of pancreatic sections for insulin (red), γH2AX foci (green), and DAPI (blue) in samples from 6-month-old mice. Arrows indicate γH2AX-positive cells. Original magnification ×600. C: Western blot analysis of p53 and p21 in isolated islets from Y, M, and O mice. GAPDH is shown as a loading control. D: Representative immunohistochemical staining for p53 in 4-month-old mice to validate the Western blot results. Arrows indicate p53-positive cells. Original magnification ×600. E: Representative dual immunohistochemical staining for glucagon (brown) and SA β-gal (blue) of pancreatic sections from 6-month-old mice. Counterstaining was with Nuclear Fast Red. Original magnification ×600. F: Representative immunohistochemical staining for p16 in 4-month-old mice. Original magnification ×600. Ku, Ku70–/–; KuPP, Ku70–/–PP; Ku–/–PP, Ku–/–p53p/p; Ku+/–PP; WT, wild-type.
Persistent DNA damage in the pancreata of Ku70−/− and Ku70−/−p53p/p mice.
To determine whether γH2AX foci, a prominent marker of DNA damage, were present in the Ku70−/− and Ku70−/−p53p/p islets, we stained mutant pancreatic sections and compared them with littermate (Ku70+/−p53p/p) and age-matched wild-type controls. As shown in Fig. 1B, γH2AX foci and pan staining were abundantly present in samples from 6-month-old mutant mice but not in control sections. Additionally, pancreatic sections from young (1-month-old) mutant mice showed few γH2AX-positive β-cells, but none were observed in the control sections (data not shown). These results indicate that as in the Lig4−/−p53p/p mutant mice, the pancreatic islets from Ku70-deficient mice had persistent unrepaired DNA damage.
Increased expression of p53-p21 and cellular senescence but not apoptosis in Ku70−/− and Ku70−/−p53p/p pancreata.
DNA damage from NHEJ deficiency triggered p53-mediated apoptosis in lymphocytes. We previously did not detect apoptosis in the Lig4−/−p53p/p islets (8), ruling out p53-independent apoptosis, but because Lig4 deficiency led to embryonic lethality (2), we could not accurately assess the role of p53-mediated apoptosis in β-cells after persistent DNA damage. Therefore, with use of the Ku70 deficiency, we originally hypothesized that apoptosis would occur in the Ku70−/−p53+/+ but not in the Ku70−/−p53p/p sections. Of note, a TUNEL assay detected no apoptosis in either the Ku70−/− or Ku70−/−p53p/p islets and only very low levels in the wild-type sections compared with a Ku70−/− spleen (Supplementary Fig. 1). Furthermore, after 10 Gy of γ irradiation, neither the wild-type nor the Ku70−/− islets showed elevated levels of apoptosis after 24 h (data not shown). These data indicate that in the presence of DNA damage, no or very few pancreatic β-cells undergo apoptosis.
The accumulation of spontaneous unrepaired DNA damage in the Lig4−/−p53p/p β-cells triggered p53 to transactivate p21 (8). To quantitatively detect p53 and p21 levels in the wild-type, Ku70−/−, and Ku70−/−p53p/p islets, Western blot analysis was performed on pooled isolated islets. As shown in Fig. 1C, mutant mice expressed higher levels of p53 than did wild-type mice at all ages, and the p53 expression was predominantly localized in the nucleus (Fig. 1D). Expression of p21 was also elevated in all the age groups of mutant mice compared with wild-type mice (Fig. 1C).
Activated p53-p21 promotes cellular senescence in Lig4−/−p53p/p mice as a mechanism to suppress tumorigenesis not only in the lymphoid system (7), but also in the pancreatic β-cells, which inhibits insulin production in many islets (8). We next asked whether the DNA damage-induced p53-p21 activation also triggered cellular senescence in the Ku70−/− and Ku70−/−p53p/p islets. To better visualize islet-specific SA β-gal, pancreatic sections were stained with glucagon. As shown in Fig. 1E, mutant islets from older mice underwent cellular senescence, whereas no senescent cells were detected in the control pancreatic sections. These senescent islets were not driven by elevated p16 levels because the age-matched controls showed similar expression levels (Fig. 1F). Therefore, as in the Lig4−/−p53p/p islets, accumulation of spontaneous DNA damage triggered cellular senescence in the absence of Ku70.
Decreased glycemia, increased insulin levels, and normal glucose tolerance in Ku70−/− and Ku70−/−p53p/p mice.
Although persistent DNA damage activated p53-p21–induced cellular senescence, both Ku70−/− and Ku70−/−p53p/p mice did not have increased blood glucose levels and were not diabetic. Measuring of the random nonfasting serum glucose levels in Ku70−/− and Ku70−/−p53p/p mice revealed progressively decreased glycemia, which became significant starting with 4-month-old mutant mice; more specifically, 7-month-old Ku70−/− and Ku70−/−p53p/p mice showed a 39 and 38% decrease, respectively, in blood glucose levels compared with age-matched controls (P < 0.005) (Fig. 2A). To measure the nonfasting serum insulin levels, we performed ELISAs in 2- and 7-month-old mice and found elevated circulating insulin levels in both groups of mutant mice. The Ku70−/− and Ku70−/−p53p/p mice had significantly higher levels of insulin at 7 months than wild-type mice (P < 0.005), resulting in a 26–35% increase (Fig. 2B), which suggests an imbalance in pancreatic β-cell regulation.
FIG. 2.

Ku70−/− (Ku−/−) and Ku70−/−p53p/p (Ku−/−PP) mice exhibit decreased glycemic and nondiabetic phenotype. A: Random blood glucose concentrations from nonfasting animals at indicated ages. Each data point is an average of at least five animals. B: Random blood insulin concentrations measured by ELISA from nonfasting animals at 2 and 7 months of age. Each bar is an average of three animals in duplicate. C: Blood glucose concentrations measured by glucose tolerance test in 5-month-old wild-type (WT), littermate (LM), Ku−/−, and Ku−/−PP mice. Four mice from each genotype were tested. D: Blood insulin concentrations measured by ELISA from the respective glucose tolerance test. E: Blood glucose concentrations measured by insulin tolerance test in 3-month-old WT, LM, Ku−/−, and Ku70−/−PP mice. F: AUC analysis of D and E. Four mice from each genotype were tested. *P < 0.05–0.005 vs. WT or LM. **Both Ku70−/− and Ku70−/−PP are significantly (P < 0.05) different from wild-type controls.
Glucose tolerance tests were performed in young (1–2-month-old) (data not shown) and older (5-month-old) mice. Besides a lower fasting serum glucose level in mutant mice, no significant differences were seen between mutant mice and controls after glucose injection (Fig. 2C), indicating that mutant mice were able to clear glucose from the blood as efficiently as the controls. ELISA showed higher levels of secreted serum insulin on glucose stimulation in aged Ku70−/− and Ku70−/−p53p/p mice (Fig. 2D), and AUC analysis indicated a significant difference (P < 0.05) (Fig. 2F). Among 3-month-old mice injected with insulin 0.75 units/kg (insulin tolerance test), AUC analysis showed no significant differences among genotypes (Fig. 2E and F), indicating normal insulin sensitivity. Collectively, the elevated random serum insulin levels and decreased glycemia suggest an increase in islet size and β-cell proliferation in these mutant mice.
Ku70 deficiency results in β-cell expansion.
To determine whether the increased serum insulin concentrations resulted from increased β-cell proliferation, we examined the morphological changes in the pancreatic islets by determining the ratio of islet area (immunohistochemical staining positive for insulin) divided by the total pancreas area (8). For comparison, we separated the mice into three age groups: young (1.0–2.0 months), medium (2.5–4.0 months), and old (4.5–7.0 months). As shown through representative pictures and quantification in Fig. 3A and B, mutant mice had a larger islet-to-total pancreas ratio than did the age-matched controls in all age groups (P < 0.05–0.005). The increase in mutant islets was independent of the total pancreas because there was no significant difference in the ratio of pancreas weight to body mass (Supplementary Fig. 2A). Immunohistochemical staining for glucagon, indicative of α-cells, did not reveal any abnormalities in either mutant backgrounds at any time point compared with age-matched controls (Supplementary Fig. 2B). These results indicate a progressive augmentation of pancreatic islet size in the absence of Ku70, which correlates with the increased insulin production and decreased glycemic phenotype.
FIG. 3.

Ku70−/− and Ku70−/−p53p/p (Ku70−/−PP) mice present augmented pancreatic islet mass and increased proliferation. A: Islet morphometric quantification of wild-type (WT), Ku70−/−, and Ku70−/−PP mice. Multiple sections were analyzed for each pancreas from three to four mice per group. Data are mean ± SEM. *P < 0.05–0.005 vs. WT. B: Representative immunohistochemical staining for insulin in 3-month-old mice. Original magnification ×100. C: Percentage of Ki67-positive β-cells out of the total insulin-positive β-cells was measured in the Ku70−/− and Ku70−/−PP islets of 3-month-old mice and plotted in comparison with age-matched WT and littermate (LM) mice. Each bar is an average of three mice. Data are mean ± SEM. *P < 0.005 vs. WT and LM. D: Representative dual immunohistochemical staining for Ki67 (brown) and insulin (blue-gray) in 3-month-old mice. Arrows indicate Ki67-positive cells. Original magnification ×600. E: Percentage of PCNA-positive β-cells out of the total β-cells was measured in each age group as in A. Each bar is an average of five mice. Data are mean ± SEM. *P < 0.005 vs. WT. F: Representative immunohistochemical staining for PCNA in 4-month-old mice. Arrows indicate PCNA-positive cells. Original magnification ×400. G: Percentage of BrdU incorporation in insulin-positive cells was measured in 1-month-old mutant and LM mice. Multiple sections were scored, and two mice in each group were analyzed. Data are mean ± SEM. These two were significantly different (*P < 0.05). H: Representative triple immunofluorescent staining for BrdU (red), DAPI (blue), and insulin (green) in 1-month-old mice. Arrows indicate BrdU-positive cells. Ku−/−, Ku70−/−; Ku−/−PP, Ku70−/−PP. Original magnification ×1,000.
Increased islet mass owing to β-cell hyperproliferation and increased CDK4 levels in both Ku70−/− and Ku70−/−p53p/p mice.
The production of adult β-cells predominantly occurs through self-duplication of mature cells rather than through differentiation of their stem cell progenitors (11–13). Therefore, we hypothesized that the increased islet size observed in the mutant mice was caused by heightened β-cell proliferation.
Through dual Ki67 and insulin staining of pancreatic sections, Ki67-positive β-cells were counted and divided by the total number of insulin-positive β-cells to quantitatively assess the rate of β-cell proliferation. As shown in Fig. 3C and D, 3-month-old wild-type and littermate controls had no significant difference in β-cell proliferation; conversely, both Ku70−/− and Ku70−/−p53p/p mice showed a significant increase in β-cell proliferation (P < 0.02). Furthermore, islet staining for PCNA, another proliferative marker, showed that the young mutant mice had a 59–63% increase of the β-cell–specific, hyperproliferative phenotype compared with wild-type mice (P < 0.001) (Fig. 3E and F). No elevated Ki67 or PCNA staining was observed in Lig4−/−p53p/p mutant sections (data not shown), indicating that the Ki67- or PCNA-positive cells observed in the absence of Ku70 did not result from accumulated DNA damage. Finally, we performed BrdU labeling of 1-month-old Ku70−/−p53p/p and Ku70+/−p53p/p littermate mice. As shown in Fig. 3G and H, Ku70−/−p53p/p mice had a significantly higher percentage of BrdU-positive cells in the islets than did the Ku70+/−p53p/p mice (P < 0.05). Collectively, these results strongly correlate with the decreased glycemic phenotype, indicating a relationship between Ku70 deficiency and increased β-cell proliferation.
Because of the paramount role that CDK4 plays in regulating β-cell proliferation (34,35), an examination of the expression pattern would further confirm the proliferative potential of Ku70−/− and Ku70−/−p53p/p β-cells. Indeed, Western blot analysis of isolated pancreatic islets confirmed increased CDK4 protein expression in both mutant backgrounds at all time points compared with wild-type controls (Fig. 4B). Representative islet sections showed increased nuclear localization of CDK4 (Fig. 4A), corroborating the Western blot data (Fig. 4B). Furthermore, Western blot analysis demonstrated that cyclin D1 and D2 levels were slightly elevated in Ku70−/−p53p/p islets compared with those of littermate controls (Fig. 4C). Taken in concert with previous data, β-cells from Ku70−/− and Ku70−/−p53p/p showed elevated proliferation, increasing the islet area over time and resulting in a decreased glycemic phenotype.
FIG. 4.

Increased CDK4 expression and stabilization of β-catenin in Ku70−/− and Ku70−/−p53p/p (Ku70–/–PP) islets. A: Representative immunohistochemical staining for CDK4 in 3-month-old mice. Arrows indicate CDK4-positive cells. Original magnification ×1,000. B: Western blot analysis of CDK4 using protein from pooled purified islets from three different mice per time point and genotype. β-Actin is shown as a loading control. C: Western blot analyses of CDK4, cyclin D1, and cyclin D2 in Ku70−/−PP and littermate (LM) isolated islets from 1-month-old mice. Tubulin was used as a loading control. These experiments were repeated three to five times. Densitometry quantitation of this particular Western blot is shown on the right. Image J software was used to quantify each sample band and its corresponding loading control band. Their ratio is plotted. D: Western blot analysis of active and total β-catenin (bC) of protein from pooled purified islets from three different medium-aged mice per genotype. β-Actin is shown as a loading control. The experiment was repeated once. Quantitation of this particular gel is shown on the right. E: Representative immunofluorescent staining of pancreatic sections for bC (green), insulin (red), and DAPI (blue) in 6-month-old wild-type (WT), Ku70−/−, and Ku70−/−PP mice. The outline indicates islets. Original magnification ×600.
Ku70 deficiency and the progressive stabilization of β-catenin.
Canonical Wnt signaling regulates pancreatic β-cell proliferation. A hallmark for Wnt activation is the cytoplasmic accumulation of β-catenin, which eventually migrates into the nucleus to upregulate target genes like cyclin D1 and D2 and CDK4 (36). To determine the β-catenin levels in the islets, we performed immunofluorescent staining on pancreatic sections from Ku70−/− and Ku70−/−p53p/p mice and littermate and wild-type controls. Islet β-catenin levels were comparable between 1-month-old mutant mice and wild-type mice (data not shown). However, older mutant mice (6–7 months) showed higher levels of β-catenin than did the wild-type and littermate controls (Fig. 4E), which had very low levels of β-catenin. This finding is consistent with reports of a normal downregulation of the Wnt pathway in the pancreata of wild-type adult mice (37,38). To corroborate the immunofluorescent data, we analyzed purified islet protein extracts from medium-aged wild-type, Ku70−/−, and Ku70−/−p53p/p mice and observed an increase in both total and activated β-catenin levels in the absence of Ku70 (Fig. 4D). These results establish a potential link between Ku70 deficiency and increased β-catenin levels. To further demonstrate this link, we generated early passage MEFs and stained them for β-catenin. Although some nuclear localization was observed in wild-type MEFs, more cells showed stabilized β-catenin in the absence of Ku70−/− (Supplementary Fig. 3A). Additionally, total β-catenin was elevated in both Ku70−/− and Ku70−/−p53p/p MEFs, and activated β-catenin was elevated in Ku70−/−p53p/p samples compared with wild-type samples (Supplementary Fig. 3B and D). β-Catenin phosphorylation at sites Ser552 and Ser675 has been associated with enhanced β-catenin stabilization and signaling activity (39). As shown in Supplementary Fig. 3B, Ku70−/−p53p/p early passage MEFs had elevated phosphorylated β-catenin at these sites compared with wild-type MEFs. Conversely, β-catenin phosphorylation at Ser31, Ser37, Thr41, and Thr45 all indicated the initial steps of β-catenin degradation (39). As shown in Supplementary Fig. 3B and D, there was no difference in these phosphorylation sites, indicating that Ku70 did not affect the degradation of β-catenin. Furthermore, downstream Wnt transcriptional targets cyclin D2 and CDK4 were elevated in Ku70−/− and Ku70−/−p53p/p MEFs compared with wild-type MEFs (Supplementary Fig. 3C); a similar pattern was seen in the mutant islets (Fig. 4B and C). Collectively, the data suggest that Ku70 can negatively regulate Wnt signaling and that Ku70 deletion stabilizes β-catenin and increases its activity. Finally, we generated Lig4−/−Ku70−/−p53p/p mice to assess whether Ku70 loss could rescue the overt diabetes phenotype previously observed in Lig4−/−p53p/p mice. Triple-mutant mice were produced below the expected Mendelian ratio and killed early because of health problems. Nevertheless, by measuring the random blood glucose of 1-month-old mice, we observed a decrease (19%) in the blood glucose of Lig4−/−Ku70−/−p53p/p mice compared with Lig4−/−p53p/p mice (Supplementary Fig. 4), showing that Ku70 loss could increase the proliferative capacity of pancreatic β-cells in the absence of Lig4.
In summary, the findings show that the phenotype of Ku70−/−p53p/p pancreatic islets resembles that of Lig4−/−p53p/p islets in the presence of DNA damage and cellular senescence, indicating that Ku70 plays a role in NHEJ and in maintaining genomic integrity. A remarkable finding was that Ku70−/−p53p/p mice were not diabetic, which contrasts with the diabetic Lig4−/−p53p/p mice of our previous study and indicates an NHEJ-independent role for Ku70. The data demonstrate that Ku70 prevents cellular proliferation in the presence of DNA damage, possibly through inhibition of the Wnt signaling pathway. β-Catenin regulation is essential for controlling the proliferation in pancreatic β-cells; therefore, the data reveal an important direction for future drug intervention for the treatment of certain types of diabetes.
DISCUSSION
In this study, we show that Ku70, which is best-known for its role in DNA end recognition during NHEJ, has an important function in regulating β-cell replication. More specifically, Ku70 deficiency augmented islet mass, which increased insulin production and, thus, progressively decreased glycemia. Analysis revealed a stabilization of β-catenin, which was complemented by increased markers for β-cell proliferation, Ki67, PCNA, BrdU incorporation, cyclin D1 and D2, and CDK4. This finding was in stark contrast to our previous study of Lig4−/−p53p/p mice, where DNA damage-induced islet senescence led to β-cell attrition and decreased insulin production, deregulating glucose homeostasis and resulting in severe diabetes (8). The similarities of these phenotypes converge on the deficiency in NHEJ, where spontaneous DNA damage progressively accumulates and induces a p53-mediated response, halting the β-cell cycle. Therefore, the most obvious difference between these phenotypes is present early in the Ku70-deficient mice, where the β-cells showed increased proliferation and escaped cell cycle inhibition. These results allow us to dissociate the function of Ku70 in the β-cell from solely a role in the conventional NHEJ-dependent DNA repair process as seen in the deficiency of Lig4. Our proposed model is depicted in Fig. 5.
FIG. 5.
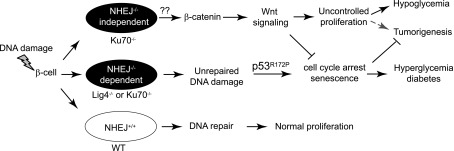
Proposed schematic depicting the different outcomes of NHEJ factors in β-cells after DNA damage. We propose that pancreatic β-cells incur DNA damage from endogenous stress. However, strong DNA repair machineries, including NHEJ, actively repair this damage. In the absence of NHEJ, DNA damage accumulates, and in p53R172P mice, the unrepaired DNA damage activates p53-p21 and drives cells into senescence. This is demonstrated in the Lig4−/−p53P/P mice. The Ku70−/−p53P/P mice, however, are different because Ku70 has a function in addition to its role in NHEJ. We propose that Ku70 inhibits cell proliferation possibly through limiting the β-catenin/Wnt signaling pathway in the presence of DNA damage. Therefore, Ku70 deficiency leads to a complicated phenotype. It resembles Lig4 deficiency in the accumulation of DNA damage, but it lacks an important cell cycle break in some cells that leads to proliferation, which rescues the diabetes phenotype.
Complementing the increased β-cell proliferation data, the results also show an early and steady elevation in CDK4 levels (Fig. 4). This is not surprising considering that CDK4 regulation is important for controlling β-cell proliferation (34,35,40). Of note, a constitutively active CDK4 mutant, CDK4R24C, presented a stark increase in β-cell proliferation and augmented islet mass, leading to islet hyperplasia (34,41). This increased islet proliferation was enough to rescue previously established diabetic models (42), collectively placing CDK4 as a central β-cell regulator. Furthermore, Rane and colleagues (43) revealed that CDK4 not only promotes proliferation in quiescent adult β-cells, but also activates early β-cell progenitors in the ductal epithelium. Further elevated CDK4 levels in the embryonic pancreas, through the CDK4R24C mutant, showed increased proliferation of early mesenchymal endocrine precursors (44). These studies underscore a pivotal role for CDK4 during embryogenesis: the regulation of pancreatic progenitors and adult β-cell regeneration by controlling the β-cell cycle. Taken together, it is tempting to speculate that the elevated CDK4 expression seen in the Ku70−/− and Ku70−/−p53p/p islets contribute to increased adult β-cell proliferation.
Many reports have linked Ku70 to transcriptional regulation either directly through binding to DNA or indirectly through interacting with other proteins (26–30,45,46). Of particular interest to the current study, researchers have speculated that Ku70 helps to regulate Wnt signaling after DNA damage occurs (30). The Wnt signaling pathway has been linked to glucose homeostasis; thus, overactive Wnt signaling could drive β-cell proliferation and increase insulin secretion (15). Islet-specific activation of β-catenin has been shown to increase β-cell proliferation and elevated insulin levels (17,18), whereas β-catenin inactivation results in islet hypoplasia (20). Disrupting β-catenin activity by deleting the Wnt coreceptor LRP5 greatly impairs β-cell proliferation (47). These studies emphasize the importance of β-catenin and Wnt signaling in controlling β-cell proliferation.
Researchers have identified Ku70 as a novel inhibitor of the β-catenin/TCF4 complex; more specifically, Ku70 physically binds to and inhibits TCF4 activation (30). Furthermore, when Ku70 was abrogated, canonical Wnt signaling increased. Conversely, after DNA was damaged, Ku70 levels increased, and TCF4 was deactivated as evidenced by a decrease in putative downstream targets. Finally, this regulation was completely independent of Ku80 levels (30). These findings may explain the present data shown in Fig. 4 and Supplementary Fig. 3. It is commonly accepted that Ku70−/− MEFs prematurely senesce because of an accumulation of DNA damage and subsequent activation of the DNA damage response (31,48). Yet in early passage MEFs, deleting Ku70 actually heightens proliferation compared with wild-type MEFs as well as rescues growth defects observed in telomere shelterin complex–deficient mice (49,50). These data combined with the present findings suggest that initial activation of Wnt signaling occurs in the absence of Ku70, which increases cell proliferation until the DNA damage accumulation becomes too excessive and prematurely activates p53-mediated cell cycle arrest, preventing tumorigenesis. Of interest, similar to many models of overactivated Wnt signaling, both Ku70−/− and Ku70−/−p53p/p mice have shown stabilized β-catenin in the epithelial cells of the colon accompanied with high-grade dysplasia and adenocarcinoma (N.P.-O. and C.Z., unpublished data). Investigation is currently ongoing to elucidate the precise mechanism of Ku70 in suppressing canonical Wnt signaling.
In summary, this study highlights an unexpected and important function of Ku70: In addition to repairing DNA damage in the NHEJ pathway, Ku70 also regulates pancreatic β-cell proliferation. The connection between the Ku70 level and stabilization of β-catenin merits further investigation, which could shed light on possible ways to enhance pancreatic β-cell proliferation.
Supplementary Material
ACKNOWLEDGMENTS
This study was financially supported in part by Robert Everett “R.E.” (Bob) Smith Educational Program Funds (to O.T.) and by The University of Texas MD Anderson Cancer Center start-up fund (to C.Z.).
No potential conflicts of interest relevant to this article were reported.
O.T. researched data and wrote the manuscript. N.P.-O. researched data, contributed to discussion, and reviewed and edited the manuscript. J.K., M.S., and S.J. researched data. C.Z. contributed to the experimental design and discussion and wrote the manuscript. C.Z. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Guillermina Lozano (The University of Texas MD Anderson Cancer Center) and Frederick Alt (Harvard Medical School) for providing the mutant mice used in this study, Sherie Mudd from the histology core facility of MD Anderson, and Jill R. Delsigne from MD Anderson for her helpful comments on the manuscript.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1218/-/DC1.
REFERENCES
- 1.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J 2009;417:639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank KM, Sekiguchi JM, Seidl KJ, et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 1998;396:173–177 [DOI] [PubMed] [Google Scholar]
- 3.Puebla-Osorio N, Zhu C. DNA damage and repair during lymphoid development: antigen receptor diversity, genomic integrity and lymphomagenesis. Immunol Res 2008;41:103–122 [DOI] [PubMed] [Google Scholar]
- 4.Zhu C, Mills KD, Ferguson DO, et al. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002;109:811–821 [DOI] [PubMed] [Google Scholar]
- 5.Difilippantonio MJ, Petersen S, Chen HT, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med 2002;196:469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu G, Parant JM, Lang G, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 2004;36:63–68 [DOI] [PubMed] [Google Scholar]
- 7.Van Nguyen T, Puebla-Osorio N, Pang H, Dujka ME, Zhu C. DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: a mouse model. J Exp Med 2007;204:1453–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavana O, Puebla-Osorio N, Sang M, Zhu C. Absence of p53-dependent apoptosis combined with nonhomologous end-joining deficiency leads to a severe diabetic phenotype in mice. Diabetes 2010;59:135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee SH, Hao E, Levine F, Itkin-Ansari P. Id3 upregulates BrdU incorporation associated with a DNA damage response, not replication, in human pancreatic β-cells. Islets 2011;3:358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieck S, Zhang J, Li Z, et al. Overexpression of hepatocyte nuclear factor-4α initiates cell cycle entry, but is not sufficient to promote β-cell expansion in human islets. Mol Endocrinol 2012;26:1590–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007;12:817–826 [DOI] [PubMed] [Google Scholar]
- 12.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004;429:41–46 [DOI] [PubMed] [Google Scholar]
- 13.Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 2004;114:963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavana O, Zhu C. Too many breaks (brakes): pancreatic β-cell senescence leads to diabetes. Cell Cycle 2011;10:2471–2484 [DOI] [PubMed] [Google Scholar]
- 15.Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta-cell biology and diabetes. Trends Endocrinol Metab 2008;19:349–355 [DOI] [PubMed] [Google Scholar]
- 16.Puri S, Hebrok M. Cellular plasticity within the pancreas—lessons learned from development. Dev Cell 2010;18:342–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rulifson IC, Karnik SK, Heiser PW, et al. Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A 2007;104:6247–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heiser PW, Lau J, Taketo MM, Herrera PL, Hebrok M. Stabilization of beta-catenin impacts pancreas growth. Development 2006;133:2023–2032 [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia 2008;51:623–631 [DOI] [PubMed] [Google Scholar]
- 20.Dessimoz J, Bonnard C, Huelsken J, Grapin-Botton A. Pancreas-specific deletion of beta-catenin reveals Wnt-dependent and Wnt-independent functions during development. Curr Biol 2005;15:1677–1683 [DOI] [PubMed]
- 21.Bertuch AA, Lundblad V. The Ku heterodimer performs separable activities at double-strand breaks and chromosome termini. Mol Cell Biol 2003;23:8202–8215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy R, Meier B, McAinsh AD, Feldmann HM, Jackson SP. Separation-of-function mutants of yeast Ku80 reveal a Yku80p-Sir4p interaction involved in telomeric silencing. J Biol Chem 2004;279:86–94 [DOI] [PubMed] [Google Scholar]
- 23.Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE. Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev 2003;17:2384–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amsel AD, Rathaus M, Kronman N, Cohen HY. Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc Natl Acad Sci U S A 2008;105:5117–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria [retraction of: Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S. In: Nat Cell Biol 2007;9:480]. Nat Cell Biol 2003;5:320–329 [DOI] [PubMed] [Google Scholar]
- 26.De Zio D, Bordi M, Tino E, et al. The DNA repair complex Ku70/86 modulates Apaf1 expression upon DNA damage. Cell Death Differ 2011;18:516–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giffin W, Torrance H, Rodda DJ, Préfontaine GG, Pope L, Hache RJ. Sequence-specific DNA binding by Ku autoantigen and its effects on transcription. Nature 1996;380:265–268 [DOI] [PubMed] [Google Scholar]
- 28.Grote J, König S, Ackermann D, et al. Identification of poly(ADP-ribose)polymerase-1 and Ku70/Ku80 as transcriptional regulators of S100A9 gene expression. BMC Mol Biol 2006;7:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebrun P, Montminy MR, Van Obberghen E. Regulation of the pancreatic duodenal homeobox-1 protein by DNA-dependent protein kinase. J Biol Chem 2005;280:38203–38210 [DOI] [PubMed] [Google Scholar]
- 30.Idogawa M, Masutani M, Shitashige M, et al. Ku70 and poly(ADP-ribose) polymerase-1 competitively regulate beta-catenin and T-cell factor-4-mediated gene transactivation: possible linkage of DNA damage recognition and Wnt signaling. Cancer Res 2007;67:911–918 [DOI] [PubMed] [Google Scholar]
- 31.Gu Y, Seidl KJ, Rathbun GA, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 1997;7:653–665 [DOI] [PubMed] [Google Scholar]
- 32.Tavana O, Benjamin CL, Puebla-Osorio N, et al. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 2010;9:3328–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nat Protoc 2009;4:1649–1652 [DOI] [PubMed] [Google Scholar]
- 34.Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 1999;22:44–52 [DOI] [PubMed] [Google Scholar]
- 35.Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol 1999;19:7011–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004;20:781–810 [DOI] [PubMed] [Google Scholar]
- 37.Murtaugh LC, Law AC, Dor Y, Melton DA. Beta-catenin is essential for pancreatic acinar but not islet development. Development 2005;132:4663–4674 [DOI] [PubMed] [Google Scholar]
- 38.Papadopoulou S, Edlund H. Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes 2005;54:2844–2851 [DOI] [PubMed] [Google Scholar]
- 39.Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J 2012;31:2714–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martín J, Hunt SL, Dubus P, et al. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 2003;22:5261–5269 [DOI] [PubMed] [Google Scholar]
- 41.Hino S, Yamaoka T, Yamashita Y, Yamada T, Hata J, Itakura M. In vivo proliferation of differentiated pancreatic islet beta cells in transgenic mice expressing mutated cyclin-dependent kinase 4. Diabetologia 2004;47:1819–1830 [DOI] [PubMed] [Google Scholar]
- 42.Miyawaki K, Inoue H, Keshavarz P, et al. Transgenic expression of a mutated cyclin-dependent kinase 4 (CDK4/R24C) in pancreatic beta-cells prevents progression of diabetes in db/db mice. Diabetes Res Clin Pract 2008;82:33–41 [DOI] [PubMed] [Google Scholar]
- 43.Lee JH, Jo J, Hardikar AA, Periwal V, Rane SG. Cdk4 regulates recruitment of quiescent beta-cells and ductal epithelial progenitors to reconstitute beta-cell mass. PLoS ONE 2010;5:e8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim SY, Rane SG. The Cdk4-E2f1 pathway regulates early pancreas development by targeting Pdx1+ progenitors and Ngn3+ endocrine precursors. Development 2011;138:1903–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenkman AB, van den Broek NJ, de Keizer PL, van Gent DC, Burgering BM. The DNA damage repair protein Ku70 interacts with FOXO4 to coordinate a conserved cellular stress response. FASEB J 2010;24:4271–4280 [DOI] [PubMed] [Google Scholar]
- 46.Nolens G, Pignon JC, Koopmansch B, et al. Ku proteins interact with activator protein-2 transcription factors and contribute to ERBB2 overexpression in breast cancer cell lines. Breast Cancer Res 2009;11:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujino T, Asaba H, Kang MJ, et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc Natl Acad Sci U S A 2003;100:229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li GC, Ouyang H, Li X, et al. Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol Cell 1998;2:1–8 [DOI] [PubMed] [Google Scholar]
- 49.Akhter S, Lam YC, Chang S, Legerski RJ. The telomeric protein SNM1B/Apollo is required for normal cell proliferation and embryonic development. Aging Cell 2010;9:1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam YC, Akhter S, Gu P, et al. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J 2010;29:2230–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.