

Abstract
Although exercise reduces several cardiovascular risk factors associated with obesity/diabetes, the metabolic effects of exercise on the heart are not well-known. This study was designed to investigate whether high-intensity interval training (HIT) is superior to moderate-intensity training (MIT) in counteracting obesity-induced impairment of left ventricular (LV) mechanoenergetics and function. C57BL/6J mice with diet-induced obesity (DIO mice) displaying a cardiac phenotype with altered substrate utilization and impaired mechanoenergetics were subjected to a sedentary lifestyle or 8–10 weeks of isocaloric HIT or MIT. Although both modes of exercise equally improved aerobic capacity and reduced obesity, only HIT improved glucose tolerance. Hearts from sedentary DIO mice developed concentric LV remodeling with diastolic and systolic dysfunction, which was prevented by both HIT and MIT. Both modes of exercise also normalized LV mechanical efficiency and mechanoenergetics. These changes were associated with altered myocardial substrate utilization and improved mitochondrial capacity and efficiency, as well as reduced oxidative stress, fibrosis, and intracellular matrix metalloproteinase 2 content. As both modes of exercise equally ameliorated the development of diabetic cardiomyopathy by preventing LV remodeling and mechanoenergetic impairment, this study advocates the therapeutic potential of physical activity in obesity-related cardiac disorders.
Obesity, sedentary lifestyle, and reduced aerobic capacity are known predictors of heart failure and a major challenge to the health care system of the Western society (1,2). In addition to increasing the risk of cardiovascular disease, obesity and diabetes have been associated with the development of a distinct cardiomyopathy with ventricular remodeling and the progression to cardiac dysfunction (3). Reduced mechanical efficiency is an important hallmark of obesity/diabetic cardiomyopathy (4,5), and recent experimental studies have demonstrated diabetes-related inefficiency to be due to impaired mechanoenergetics, where myocardial oxygen consumption (MVo2) for nonmechanical processes is increased (6,7). Several obesity- or diabetes-induced changes, including increased fatty acid oxidation (4,8,9), impaired calcium handing (10,11), increased oxidative stress (12), and mitochondrial dysfunction (13), are factors that, most likely, contribute to increased MVo2. Although exercise has been reported to induce mitochondrial and cellular adaptations that could potentially influence myocardial oxygen-consuming processes (such as increased antioxidant capacity, reduced oxidative stress, increased mitochondrial efficiency, and improved myocardial Ca2+ homeostasis [10,14,15]), no studies have assessed the consequences of exercise-induced adaptations in terms of left ventricular (LV) mechanical efficiency and mechanoenergetics properties in a model of obesity and insulin resistance.
High-intensity exercise training induces a more pronounced increase in aerobic capacity and more evident cardiovascular adaptations compared with low- and moderate-intensity training (MIT) in healthy subjects (16–18). In a recent study by Tjønna et al. (19), high-intensity training (HIT) was also found to be superior to MIT in reducing cardiovascular risk factors in patients with metabolic syndrome. In addition, we found that high- but not MIT altered myocardial substrate utilization (decrease in fatty acid oxidation and increase in glucose oxidation) and increased mitochondrial respiratory capacity and LV mechanoenergetic properties in hearts from lean mice (18). Based on these findings, we hypothesized that exercise of high intensity would be superior to isocaloric moderate-intensity exercise in counteracting the unfavorable metabolic and functional changes that occur in the heart during obesity.
RESEARCH DESIGN AND METHODS
Mice with diet-induced obesity (DIO mice) were produced by initially feeding male C57BL/6J mice (5–6 weeks; Charles River Laboratories) a high-fat diet (60% kcal from fat, cat. no. 58Y1; TestDiet, London, U.K.) for 9 weeks. The diet was then changed to a palatable Western diet (35% kcal from fat, cat. no. 5AA7-1814152; TestDiet), and the mice were subjected to high-intensity interval training (HIT) (DIOHIT) (n = 10), isocaloric moderate-intensity continuous training (MIT) (DIOMIT) (n = 10), or a sedentary lifestyle (DIOSED) (n = 15). Lean control mice fed a standard control diet (10% kcal from fat, cat. no. 58Y2, TestDiets) over the entire period were also included (CON) (n = 15). The experiments were approved by the local authority of the National Animal Research Authority in Norway (identification no. 2348/2010). The mice were treated in accordance with the guidelines on accommodation and care of animals formulated by the European Convention for the Protection of Vertebrate Animals for Experimental and Other Scientific Purposes. All mice received chow ad libitum and free access to drinking water and were housed at 23°C on a reversed light/dark cycle so that the exercise occurred during the dark period.
Exercise protocol and determination of aerobic capacity.
Treadmill running (25° inclination) was performed 5 days/week for 8–10 weeks as previously described by Hafstad et al. (18). Aerobic capacity was assessed as Vo2max using a metabolic chamber equipped with a treadmill (18). HIT consisted of 10 bouts of 4-min high-intensity running, corresponding to 85–90% of Vo2max, interspersed by 2 min active rest. The interval pace was increased gradually from 12 to 23 m/min over the first 8 weeks. MIT consisted of distance-matched continuous running, corresponding to 65–70% of Vo2max, where the average running time was close to 2 h. The pace during MIT was increased gradually from 7.5 to 10 m/min over the first 8 weeks.
Glucose tolerance test and plasma and tissue samples.
For glucose tolerance test, blood was collected from the saphenous vein in fasted (4 h) animals and measured (glucometer, Ascensia Contour; Bayer Healthcare, Berlin, Germany) before (0 min) and 15, 30, 60, and 120 min after administration of a glucose solution (1.3 g/kg body wt i.p.). Plasma glucose and free fatty acids and liver triglycerides were analyzed as previously described (20).
Ventricular function and mechanoenergetics.
Myocardial glucose and fatty acid oxidation rates were measured in isolated perfused working hearts using radiolabeled isotopes (8). Total mechanical work (pressure-volume area [PVA]), stroke work (SW), and parameters of LV function were assessed by a 1.0-F micromanometer conductance catheter (21). MVo2 was measured using fiber-optic oxygen probes as previously described (21). Steady-state values of PVA, SW, and MVo2 were obtained at several workloads, and the regression between PVA and MVo2 was used to identify myocardial oxygen cost for work-independent MVo2 and work-dependent MVo2 (22). The relationship between SW and MVo2 was used to evaluate changes in mechanical efficiency. Finally, MVo2 was measured in unloaded retrograde perfused hearts before (MVo2 unloaded) and after KCl-arrest to measure oxygen cost for basal metabolism (MVo2 BM) and for calculating the oxygen consumption for processes related to excitation-contraction coupling (MVo2 ECC) (7). After the ex vivo perfusion protocol, heart tissue samples were harvested for tissue staining and histological and mRNA analysis.
Myocardial superoxide and collagen content.
LV tissue (15-μm sections) was stained with dihydroethidium (DHE) (10 mmol/L DHE, 30 min, 37°C) for evaluation of superoxide production (23). DHE florescence was detected by a Leica DFC320 camera (Mannheim, Germany) mounted on a fluorescent microscope (Leitz Aristoplan, Wetzlar, Germany) with a N2.1 filter (excitation filter BP 515–560, suppression filter LP590; Leica). Approximately 30 pictures were taken from each section and analyzed with Image J (NIH, Bethesda, MD). Formalin-fixed LV sections were paraffin embedded and sliced for Sirius Red staining of collagen fibers as previously described (24).
Immunohistochemistry.
Immunohistochemistry was performed on Zinc salt-based fixed, paraffin-embedded LV tissue using a polyclonal primary antibody against matrix metalloproteinase (MMP)-2 (Novus Biologicals, Littleton, CO). Horseradish peroxidase–labeled secondary antibody and diaminobenzidine substrate were used for visualization according to the product manual (Polylink-2 HRP Plus Rabbit DAB Detection System for Immunohistochemistry; Golden Bridge International, Mukilteo, WA). MMP-2 staining was almost exclusively seen within the cardiomyocytes and was subjectively double-blindly scored and quantified for relative amount and density of stained fibers.
Electron microscopy and stereology.
Fixated (McDowells) LV tissues were embedded in Epoxy resin according to standard methods. Ultra-thin sections (70 nm) were placed on carbon-coated Formvar films on 200 mesh copper grids, contrasted with 5% uranylacetat and, subsequently, Reynolds lead citrate. Ten to fifteen electron microscopy images (JEOL 1010 transmission electron microscope, ×6,000) were obtained from three different sections from each heart using the principle of Systematic Uniform Sampling. Stereology (using a 2.5 × 2.5 µm grid square lattice) was performed by counting points falling on the interstitium, myocyte, and mitochondria. For quantification of lipid content, the total number of droplets were counted in each picture.
Real-time quantitative PCR.
Real-time quantitative qPCR analysis was performed on LV tissue samples using an ABI PRISM 7900 HT Fast real-time thermal cycler as previously described (18,20). Details about primer/probes sequences are given in supplemented data.
Mitochondrial respiration and citrate synthase activity.
In a separate group of mice (CON, DIOSED, and DIOHIT), we investigated mitochondrial respiration in isolated cardiac mitochondria using a slight modification of the method of Palmer et al. (25). Respiration was measured in an oxygraph (Oxygraph 2k; Oroboros Instruments, Austria) using glutamate (5 mmol/L) and malate (2.5 mmol/L) as substrates. Maximal mitochondrial respiration capacity (Vmax) was obtained after addition of 300 µmol/L ADP, and oligomycin (4 ug/mL) was added to study mitochondrial proton leak (Voligo). All respiration rates were adjusted to citrate synthase activity (18). The mitochondrial respiration coupling (P-to-O ratio) was calculated from the ratio of molecules of ADP phosphorylated to each oxygen molecule consumed (26).
Statistical analysis.
Data are expressed as means ± SEM. Differences between groups were analyzed using one-way ANOVA with multiple comparisons versus DIOSED (Holm-Sidak method as post hoc test). Where normality test failed (Shapiro-Wilk test), a Mann-Whitney rank sum test was performed. A Pearson product moment correlation was performed to investigate the association between MVo2 unloaded and cardiac DHE staining (SigmaStat 13). A linear mixed model was used in the regression analysis of the relationship between MVo2 and PVA or SW (SPSS 19; IBM).
RESULTS
Diet-induced obesity.
After 9 weeks on a high-fat diet, C57BL/6J mice showed increased body weight, increased fasting glucose, and reduced glucose tolerance, while aerobic capacity (whole-body Vo2max) was similar to controls (Supplementary Table 1 and Supplementary Fig. 1). Despite marked changes in myocardial substrate utilization (i.e., increased fatty acid oxidation with a concomitant decrease in glucose oxidation [see Supplementary Fig. 2]), LV function was not impaired (Supplementary Table 1). A significant parallel upward shift of the SW-MVo2 relationships (Supplementary Fig. 3B) revealed reduced mechanical efficiency as previously reported in hearts from high fat–fed rats (5). In accordance with previous findings (7), we found that the decrease in mechanical efficiency was due to impaired LV mechanoenergetics, where MVo2 for noncontractile work (the y-intercept of the PVA-MVo2 relationships) was increased (Supplementary Fig. 3A), while LV contractile efficiency (the inverse slope of the PVA-MVo2 relationships) was unaltered. The increased work-independent MVo2 was confirmed by a 69% increase in MVo2 in retrogradely perfused mechanically unloaded hearts (MVo2 unloaded). This was due to an increase in the oxygen cost for basal metabolism (MVo2 BM, measured in electrically arrested hearts), as well as in processes related to ECC (MVo2 ECC, Supplementary Fig. 4).
DIOSED mice continued to gain weight after the next 8–10 weeks so that at the end of the protocol they showed a 43% higher body weight compared with CON mice (Supplementary Fig. 5). The increased body weight was accompanied by visceral obesity (increased perirenal fat) and an enlarged liver with steatosis (Table 1). DIOSED mice also displayed reduced aerobic capacity (Table 1 and Supplementary Fig. 6), reduced glucose tolerance (Table 1 and Supplementary Fig. 7), and increased plasma levels of glucose and free fatty acids (Table 1). Obesity was also associated with increased mRNA expression of tumor necrosis factor-α (tnfα) in perirenal fat, indicative of a low-grade inflammatory state (Table 1). The hearts from DIOSED mice also showed higher work-independent MVo2, as revealed both by analyzing the PVA-MVo2 relationships (increased y-intercept [Fig. 1A]) and by measuring MVo2 in mechanically unloaded hearts (Fig. 1C). Again, increased MVo2 was ascribed to increased oxygen cost for BM and ECC (Fig. 1D and E). The impaired mechanoenergetic properties resulted in mechanical inefficient hearts, i.e., a significant upward parallel shift of the MVo2-SW relationship (Fig. 1B). The metabolic phenotype with increased myocardial fatty acid oxidation rates and decreased glucose oxidation rates was maintained in DIOSED mice (Fig. 2).
TABLE 1.
Animal characteristics of CON, DIOSED, DIOMIT, and DIOHIT mice
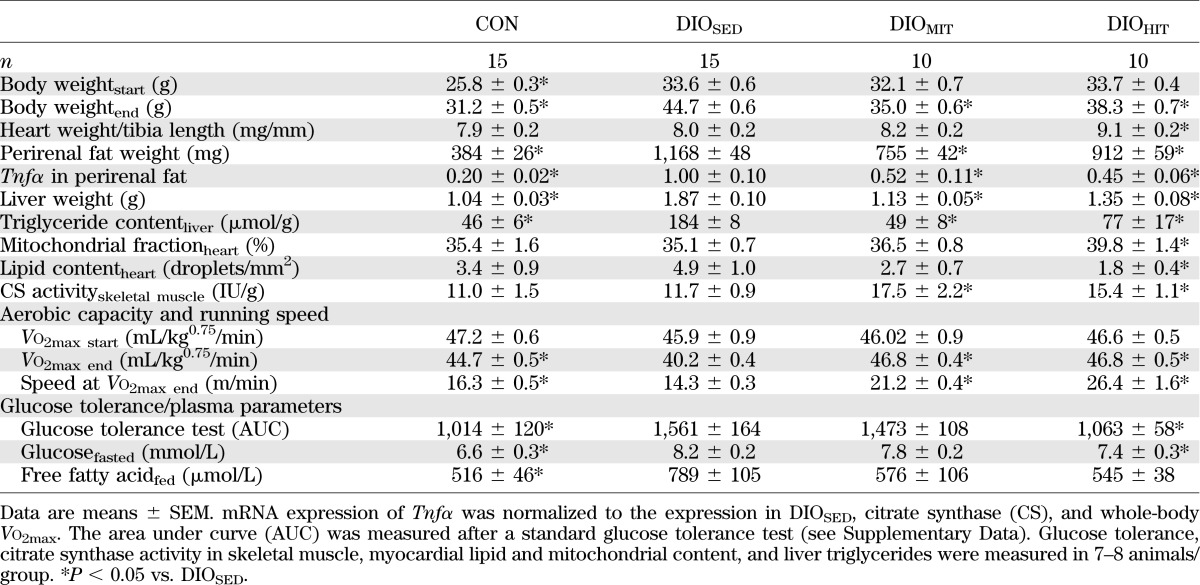
FIG. 1.

Individual values of MVo2 in relation to total cardiac work (assessed as PVA) (A) or SW (B) in isolated working hearts from CON (n = 13), DIOSED (n = 10), DIOMIT (n = 8), and DIOHIT (n = 7) mice at different workloads (preload, 4–10 mmHg; afterload, 40–50 mmHg). The table gives the mean ± SEM values of the y-intercept and the slope of the PVA-MVo2 relationships (r2 = 0.89 ± 0.01) obtained after mixed-model analysis. MVo2 was also measured in retrogradely perfused unloaded hearts (paced at 7 Hz) before (MVo2 unloaded) (C) and after (MVo2 BM) (D) electrical arrest. MVo2 ECC (E) was calculated as the difference between MVo2 unloaded and MVo2 BM. Values are means ± SEM. n = 8–14 in each group; *P < 0.05 vs. DIOSED; wwt, wet weight.
FIG. 2.

Absolute myocardial fatty acid (A) and glucose (B) oxidation rates and work-adjusted oxidation rates to cardiac output (C and D) measured in isolated perfused working hearts from CON, DIOSED, DIOMIT, and DIOHIT mice. Preload and afterload were set to 8 and 50 mmHg, respectively. Values are means ± SEM. n = 9–14 in each group; *P < 0.05 vs. DIOSED; wwt, wet weight.
In a follow-up study, assessment of mitochondrial respiration (Fig. 3) showed reduced mitochondrial respiratory capacity (Vmax) and impaired mitochondrial respiratory coupling (reduced P-to-O ratio) in hearts from DIOSED mice. Obesity was, however, not associated with altered oxygen consumption during state 4 respiration (Voligo), which may suggest an unchanged proton leak.
FIG. 3.

Respiration in isolated cardiac mitochondria measured using 5 mmol/L glutamate and 2.5 mmol/L malate as substrates from CON, DIOSED, and DIOHIT mice. Maximal respiratory capacity was obtained after addition of 300 µmol/L ADP (Vmax) (A), and mitochondrial proton leak was assessed after addition of 4 μg/mL oligomycin (Voligo) (B). The relationship between molecules of ADP phosphorylation and atom oxygen consumed (P-to-O ratio) and the respiratory coupling ration (i.e., Vmax/Voligo) are given in C and D, respectively. Values are means ± SEM. n = 8–15 in each group; *P < 0.05 vs. DIOSED; cs, citrate synthase; RCR, respiratory coupling ratio.
Finally, as shown in Table 2, isolated hearts from DIOSED mice showed impaired LV systolic function (i.e., reduced dP/dtmax and preload recruitable stroke work index [PRSWi]), as well as impaired early diastolic function (i.e., increased diastolic relaxation time constant and reduced dP/dtmin). A leftward shift of the pressure-volume (P-V) loop indicated obesity-induced LV concentric remodeling, and an elevated end-diastolic P-V relationship (EDPVR) revealed ventricular stiffening (Fig. 4). In concert with the observed changes in LV function, these hearts showed obesity-induced fibrosis (Fig. 5C and Supplementary Fig. 8) and increased myocardial intracellular staining of MMP-2 (Fig. 5A and B). Finally, DHE staining showed elevated myocardial reactive oxygen species (ROS) content (Fig. 6A and B) in the sedentary DIO mice.
TABLE 2.
Steady-state and load-independent parameters of LV function obtained in isolated perfused working hearts from CON, DIOSED, DIOMIT, and DIOHIT mice
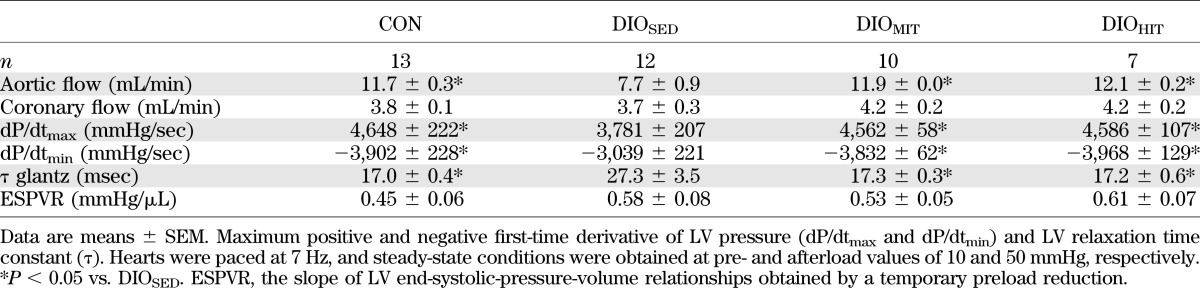
FIG. 4.

LV function measured in isolated perfused and electrically paced (7 Hz) working hearts, using a 1-Fr conductance catheter inserted into the left ventricle in CON, DIOSED, DIOMIT, and DIOHIT mice. A: Representative LV P-V loops (preload 8 mmHg, afterload 50 mmHg). Steady-state values of SW (B) and LV end-diastolic pressure (LVEDP) (C) at different LV end-diastolic volumes (LVEDV) obtained at three different preload conditions (6, 8, and 10 mmHg). PRSWi (D) and slope of the EDPVR (E) respectively, obtained by a temporary preload reduction. Values are means ± SEM (n, as given in Table 2); *P < 0.05 vs. DIOSED.
FIG. 5.

A: Immunohistochemical staining (scale bar: 50 µm) (A) and mean intensity (B) of myocardial MMP-2 from lean CON, DIOSED, DIOMIT, and DIOHIT mice. Myocardial collagen content measured with Sirius Red staining (C) and myocardial mRNA expression of collagen III (col III) type 1α (D). Values are means ± SEM. n = 6 in each group; *P < 0.05 vs. DIOSED.
FIG. 6.

DHE staining (scale bar: 100 µm) (A) and mean fluorescence intensity (B) of LV myocardium from lean CON, DIOSED, DIOMIT, and DIOHIT mice. Myocardial mRNA expression of superoxide dismutase (mn-sod) (D). Values are means ± SEM, n = 6, in each group; *P < 0.05 vs. DIOSED. C: Correlation (r2 = 0.68, P < 0.0001) between MVo2 unloaded and myocardial DHE staining.
Effects of exercise on obesity and glucose tolerance.
Exercise training reduced weight gain in DIO mice (Supplementary Fig. 6) so that the body weights of the DIOHIT and DIOMIT mice at the end of the training protocol were only 14 and 21% above CON mice, respectively (Table 1). Both training protocols also reduced visceral obesity, plasma free fatty acid, liver weight, and steatosis and showed anti-inflammatory action (reduced perirenal fat tnfα expression) (Table 1). Only HIT significantly improved glucose tolerance (Table 1 and Supplementary Fig. 7). HIT was also superior to MIT in increasing the running pace, while both training protocols increased Vo2max and citrate synthase activity in skeletal muscle (gastrocnemius) to the same extent (Table 1 and Supplementary Fig. 6).
Effects of exercise on LV mechanoenergetics.
Both HIT and MIT induced a significant parallel downward shift of the SW-MVo2 and the PVA-MVo2 relationships, showing that exercise normalized the obesity-induced mechanical inefficiency (Fig. 1B) by decreasing work-independent MVo2 (y-intercept [Fig. 1A]). This finding was further supported by measurements of MVo2 in mechanically unloaded hearts, which also showed that both training protocols reduced MVo2 BM, while the reduction in MVo2 ECC was statistically significant only after HIT (Fig. 1C–E). None of the training protocols altered contractile efficiency (inverse slope of the PVA-MVo2 relationships [Fig. 1A]). Myocardial ROS content was reduced after both training protocols (Fig. 6A and B) and accompanied by increased mRNA levels of mitochondrial superoxide dismutase (mn-sod) (Fig. 6D). Interestingly, we found a significant (P < 0.001) correlation between ROS content and MVo2 unloaded (Fig. 6C), suggesting that oxidative stress plays an important role in inducing processes leading to increased MVo2.
Effects of exercise myocardial substrate utilization and mitochondrial respiration.
Both MIT and HIT significantly increased absolute and work-adjusted rates of myocardial glucose oxidation (Fig. 2). While absolute rates of fatty acid oxidation were unaltered, work adjustment revealed a significant decrease in fatty acid oxidation rates after MIT and HIT. These findings indicate that both modes of exercise induced a mild change in substrate utilization toward an increased myocardial use of glucose. Exercise was also found to normalize the obesity-induced impairment of mitochondrial capacity (Vmax), as well as efficiency (P-to-O ratio). Interestingly, we also found exercise to induce a mild proton leak (Voligo) without any change in respiratory coupling ratio (Fig. 3).
Effects of exercise on ventricular function and cardiac histology.
Both HIT and MIT showed increased myocardial expression of myosin heavy chain α (mchα, Supplementary Table 2). Only HIT, however, induced a physiological cardiac hypertrophy, as reflected by increased heart weight (Table 1) with no increase in the gene expression of brain-like natriuretic peptide (bnp), atrial natriuretic peptide (anf), or myosin heavy chain β (mhcβ) in the heart (Supplementary Table 2). We did not find exercise to induce changes in the mRNA expression of cardiac sarcoplasmic reticulum calcium ATPase 2 (serca2) or the rhyanodine receptor (ryr2) (Supplementary Table 2). LV pressure-volume analysis revealed normalization of LV function after both MIT and HIT. Both early and late diastolic function (τ, dP/dtmin, and EDPVR) and systolic function (aortic flow, dP/dtmax, and PRSWi) were improved (Fig. 4 and Table 2). Hearts from exercised DIO mice did not display a leftward shift in the LV P-V loop, suggesting no concentric remodeling (Fig. 4A). MIT was found to significantly reduce myocardial collagen content (Fig. 5C and Supplementary Fig. 8), and there was a trend toward reduced mRNA expression of collagen type III-1 α (Fig. 5D). Immunohistochemistry also revealed that the obesity-induced increase in myocardial intracellular MMP-2 content was reduced by both modes of exercise (Fig. 5A and B). The mRNA expression of tissue inhibitor of metallopeptidase 1 was, however, unaltered (Supplementary Table 2).
DISCUSSION
Exercise has been shown to reduce cardiovascular risk factors associated with type 2 diabetes (27). The cardiac effects of exercise in obesity/diabetes are, however, not well studied, and to our knowledge, this is the first study addressing the impact of exercise intensity on functional, metabolic, and mechanoenergetic cardiac adaptations in a model of obesity/diabetes. The main findings of the study are that exercise of both high and moderate intensity improved LV mechanical efficiency, by abrogating the obesity-induced increase in MVo2, and that these changes were accompanied by prevention of obesity-induced LV remodeling and dysfunction.
Obesity/diabetes-induced cardiac phenotype.
The current study used a mouse model of cardiomyopathy induced by 20 weeks’ feeding on high-energy rich diets. As anticipated, we found that 9 weeks on the high-fat diet altered myocardial substrate utilization, reduced LV mechanical efficiency, and impaired mechanoenergetics (4–6,8,28). As LV dysfunction was not evident at this stage, this study shows for the first time that not only altered substrate utilization (8,28) and mechanical inefficiency (4,5) but also LV mechanoenergetic impairment precede development of cardiac dysfunction in obesity/diabetes.
The diabetic cardiac metabolic phenotype persisted in mice kept on a sedentary lifestyle for another 10 weeks. In addition, these hearts developed diastolic and systolic dysfunction and showed a leftward shift of the LV P-V loop indicative of development of concentric remodeling (6,29). In accordance with previous studies on diabetes-induced cardiac remodeling, the hearts exhibited increased fibrosis, impaired metalloproteinase expression, and elevated oxidative stress (12,30). Obesity was also found to reduce mitochondrial maximal respiratory capacity and efficiency (P-to-O ratio)—findings that are in accordance with a recent report by Cole et al. (5) in high-fat fed rats and suggest that decreased respiratory coupling can contribute to the impaired cardiac efficiency observed following obesity.
Systemic effects of exercise training.
Exercise training is considered a cornerstone in the treatment of insulin resistance and obesity, where the impact of exercise mode and intensity has been suggested to play a decisive role. In lean subjects, high-intensity has been shown to be superior to moderate-intensity exercise training in terms of increased aerobic capacity (16–18). Although the numbers of studies directly examining the effect of isocaloric high- and MIT on aerobic capacity in metabolic disorders are limited, a superior effect of high intensity has been suggested (19,31,32).
In contrast to this view, we found both intensities to equally increase aerobic capacity in this model of obesity and insulin resistance. The ability of subjects on high-fat diets to perform HIT has, however, been questioned (33), and it can therefore not be excluded that diet might have limited the potentiation of aerobic capacity in response to high-intensity exercise in the current study.
While MIT was as effective as isocaloric HIT in reducing obesity, only exercise of high intensity improved glucose tolerance. This finding supports a recent pilot study by Tjønna et al. (19) where high-intensity aerobic interval training in patients with metabolic syndrome improved glycemic control to a greater extent than MIT—a response most likely due to exercise-induced adaptations in skeletal muscle (19,31,34).
Cardiac effects of exercise training.
Despite the notion that exercise training has beneficial effects in patients with cardiovascular disease, its documented effect on ventricular energetics is sparse. Improved LV function and mechanical efficiency have been reported in heart failure patients enrolled in a 5-month endurance and strength-training program (35). In a recent study, we found that only exercise of high, but not moderate, intensity was able to induce metabolic and energetic adaptations in hearts from normal (nonobese) mice (18). Hence, we anticipated that exercise training of high intensity would be superior to moderate intensity in counteracting obesity-induced LV metabolic, energetic, and functional changes. Surprisingly, however, we found both modes of exercise to be equally effective in ameliorating LV mechanical and energetic dysfunction. Exercise improved both LV diastolic and systolic function, and improved LV mechanical efficiency owing to a reduction of the oxygen cost for nonmechanical purposes (i.e., BM and ECC).
The development of both ventricular dysfunction and mechanoenergetic impairments in diabetes/obesity is clearly multifactorial and complex and has been suggested to involve alterations in myocardial substrate utilization and calcium handling, as well as oxidative stress, mitochondrial dysfunction, and structural remodeling (11,12,30,36–38). Many of these processes are likely to be influenced by exercise training, and here we focus on how they may be linked to the exercise-induced improvements of LV mechanoenergetics and prevention of LV dysfunction. It should be noted, however, that owing to the pleiotropic effects of exercise in a model of obesity (both systemic and direct cardiac effects), the current study cannot pinpoint one underlying mechanism leading to the observed cardiac effects, and additional mechanistic studies will be warranted.
First, although obesity-induced increase in myocardial fatty acid supply and/or utilization is believed to contribute to the increased myocardial oxygen consumption (5,7,9,21,36), this change can only partly account for the decrease in O2 consumption (18,39). This study shows, however, for the first time, that exercise abrogated obesity-induced mitochondrial respiratory uncoupling. Surprisingly, the improved mitochondrial efficiency in response to exercise training was accompanied by a mild mitochondrial proton leak during state 4 (oligomyocin-inhibited) respiration, which was also recently reported in skinned cardiac fibers from high-fat fed UCP3KO mice (40). Although the role of this proton leak is unclear, there are substantial data suggesting that a mild proton leak will, owing to reduced mitochondrial potential, reduce mitochondrial ROS formation (15,41). Thus, both enhancement of endogenous antioxidant capacity (14,15) and decreased mitochondrial ROS production are candidates for the observed exercise-induced decrease in myocardial oxidative stress.
Diabetes is also associated with impaired myocardial Ca2+ handling, including increased ryanodine receptor (RyR2) Ca2+ leak (10,11), which most likely contributes to the increased oxygen consumption demonstrated in the present and in previous studies (4–7,9), and exercise has been reported to improve myocardial Ca2+ handling in lean and diabetic models (10,31,42,43). In cardiomyocytes from type 2 diabetic mice, exercise training enhanced synchronization of sarcoplasmic reticulum Ca2+ release and reduced diabetes-induced RyR2 Ca2+ leak (10)—adaptations that could provide oxygen-sparing effects. Although we did not find exercise to induce changes in the gene expression of RyR2 or SERCA, improvements in Ca2+ homeostasis can be achieved by posttranscriptional regulation of calcium-handling proteins, as well as by changes in the intracellular redox environment. ROS has been shown to activate RyR2 and inhibit SERCA (44), and it is therefore tempting to suggest that the positive correlation between myocardial ROS and myocardial O2 consumption is linked to ROS-mediated changes in Ca2+ handling, which is supported by the recent study showing changes in MVo2 to predict improvement of LV relaxation (45). Amelioration of the diabetes-induced myocardial Ca2+ dysregulation (10) most likely also plays a key role in the enhancement of systolic and early diastolic LV function in exercised mice, while the decreased fibrosis will reduce LV chamber stiffness and thus improve late diastolic function. Our results are in line with a previous study in the aging heart, where exercise decreased fibrosis and normalized MMP-2 regulation (46). Although the underlying mechanisms of these changes are unclear, the current study suggests that exercise may have exerted such effects through improved inflammatory status and ameliorated oxidative stress, as both ROS and TNF-α can increase the expression of MMP-2 (47,48) and induce fibrosis (49). Future studies are, however, required to address this issue.
In a recent study, substantial weight loss (1 year of dieting) was also found to improve myocardial energetics and diastolic function in obese subjects (50). In the current study, DIO mice already exhibited decreased LV mechanoenergetic features before the start of exercise. As exercise did not reduce body weight but, rather, attenuated weight gain in DIO mice, we suggest that the observed exercise-induced improvements in LV mechanoenergetics are not only a result of decreased obesity per se.
Conclusion.
The current study demonstrates that prevention of LV dysfunction by exercise training in diet-induced obesity involves restored mechanical efficiency and improved mechanoenergetic properties. These changes are most likely related to improvements in mitochondrial efficiency and capacity, reduction in oxidative stress, and reversal of ventricular remodeling. Despite previous reports of superior effects of HIT compared with MIT with respect to reducing cardiovascular risk factors associated with metabolic disorders, the current study shows that both intensities equally ameliorated the obesity-induced functional and structural changes in the heart, underlining the profound therapeutic potential of physical activity in obesity and diabetes-related cardiovascular disease.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Norwegian Research Council and the Northern Norway Regional Health Authority (Helse Nord RHF, UNIKARD), as well as by the Norwegian Health Association.
No potential conflicts of interest relevant to this article were reported.
A.D.H. designed the study, analyzed data, wrote the manuscript, and reviewed and edited the manuscript. J.L., E.H.-O., and A.C.H. analyzed data and reviewed and edited the manuscript. T.S.L. reviewed and edited the manuscript. E.A. designed the study, analyzed data, wrote the manuscript, and reviewed and edited the manuscript. E.A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the American Heart Association Scientific Sessions, Orlando, Florida, 12–16 November 2011, and at the 10th Annual Meeting of the Society of Heart and Vascular Metabolism, Oxford, U.K., 24–27 June 2012.
The authors acknowledge valuable contributions from Knut Steinnes, Ahmed Murtaz Khalid, Martin Hagve, Elisabeth Boerde, Randi Olsen, Sigurd Lindal, and Elin Mortensen (Department of Medical Biology, University of Tromsø).
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1580/-/DC1.
REFERENCES
- 1.Keteyian SJ, Brawner CA, Savage PD, et al. Peak aerobic capacity predicts prognosis in patients with coronary heart disease. Am Heart J 2008;156:292–300 [DOI] [PubMed] [Google Scholar]
- 2.Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med 2002;347:305–313 [DOI] [PubMed] [Google Scholar]
- 3.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 1974;34:29–34 [DOI] [PubMed] [Google Scholar]
- 4.Peterson LR, Herrero P, Schechtman KB, et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004;109:2191–2196 [DOI] [PubMed] [Google Scholar]
- 5.Cole MA, Murray AJ, Cochlin LE, et al. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol 2011;106:447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 2006;55:466–473 [DOI] [PubMed] [Google Scholar]
- 7.Boardman N, Hafstad AD, Larsen TS, Severson DL, Aasum E. Increased O2 cost of basal metabolism and excitation-contraction coupling in hearts from type 2 diabetic mice. Am J Physiol Heart Circ Physiol 2009;296:H1373–H1379 [DOI] [PubMed] [Google Scholar]
- 8.Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes 2003;52:434–441 [DOI] [PubMed] [Google Scholar]
- 9.Hafstad AD, Khalid AM, How OJ, Larsen TS, Aasum E. Glucose and insulin improve cardiac efficiency and postischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am J Physiol Endocrinol Metab 2007;292:E1288–E1294 [DOI] [PubMed] [Google Scholar]
- 10.Stølen TO, Høydal MA, Kemi OJ, et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res 2009;105:527–536 [DOI] [PubMed] [Google Scholar]
- 11.Belke DD, Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep 2004;6:424–429 [DOI] [PubMed] [Google Scholar]
- 12.Fukuda M, Nakamura T, Kataoka K, et al. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens 2010;28:340–352 [DOI] [PubMed] [Google Scholar]
- 13.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005;112:2686–2695 [DOI] [PubMed] [Google Scholar]
- 14.Muthusamy VR, Kannan S, Sadhaasivam K, et al. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med 2012;52:366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bo H, Jiang N, Ma G, et al. Regulation of mitochondrial uncoupling respiration during exercise in rat heart: role of reactive oxygen species (ROS) and uncoupling protein 2. Free Radic Biol Med 2008;44:1373–1381 [DOI] [PubMed] [Google Scholar]
- 16.Helgerud J, Høydal K, Wang E, et al. Aerobic high-intensity intervals improve VO2max more than moderate training. Med Sci Sports Exerc 2007;39:665–671 [DOI] [PubMed] [Google Scholar]
- 17.Kemi OJ, Loennechen JP, Wisløff U, Ellingsen O. Intensity-controlled treadmill running in mice: cardiac and skeletal muscle hypertrophy. J Appl Physiol 2002;93:1301–1309 [DOI] [PubMed] [Google Scholar]
- 18.Hafstad AD, Boardman NT, Lund J, et al. High intensity interval training alters substrate utilization and reduces oxygen consumption in the heart. J Appl Physiol 2011;111:1235–1241 [DOI] [PubMed] [Google Scholar]
- 19.Tjønna AE, Lee SJ, Rognmo O, et al. Aerobic interval training versus continuous moderate exercise as a treatment for the metabolic syndrome: a pilot study. Circulation 2008;118:346–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khalid AM, Hafstad AD, Larsen TS, et al. Cardioprotective effect of the PPAR ligand tetradecylthioacetic acid in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 2011;300:H2116–H2122 [DOI] [PubMed] [Google Scholar]
- 21.How OJ, Aasum E, Kunnathu S, Severson DL, Myhre ES, Larsen TS. Influence of substrate supply on cardiac efficiency, as measured by pressure-volume analysis in ex vivo mouse hearts. Am J Physiol Heart Circ Physiol 2005;288:H2979–H2985 [DOI] [PubMed] [Google Scholar]
- 22.Suga H. Ventricular energetics. Physiol Rev 1990;70:247–277 [DOI] [PubMed] [Google Scholar]
- 23.Sumitomo-Ueda Y, Aihara K, Ise T, et al. Heparin cofactor II protects against angiotensin II-induced cardiac remodeling via attenuation of oxidative stress in mice. Hypertension 2010;56:430–436 [DOI] [PubMed] [Google Scholar]
- 24.Aljabri MB, Songstad NT, Lund T, et al. Pregnancy protects against antiangiogenic and fibrogenic effects of angiotensin II in rat hearts. Acta Physiol (Oxf) 2011;201:445–456 [DOI] [PubMed] [Google Scholar]
- 25.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 1977;252:8731–8739 [PubMed] [Google Scholar]
- 26.Estabrook RW. Mitochondrial respiratory control and polarographic measurement of ADP/O ratios. Methods Enzymol 1967;10:41–47 [Google Scholar]
- 27.Marwick TH, Hordern MD, Miller T, et al. Council on Clinical Cardiology, American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee. Council on Cardiovascular Disease in the Young. Council on Cardiovascular Nursing. Council on Nutrition, Physical Activity, and Metabolism. Interdisciplinary Council on Quality of Care and Outcomes Research Exercise training for type 2 diabetes mellitus: impact on cardiovascular risk: a scientific statement from the American Heart Association. Circulation 2009;119:3244–3262 [DOI] [PubMed] [Google Scholar]
- 28.Chatham JC, Seymour AM. Cardiac carbohydrate metabolism in Zucker diabetic fatty rats. Cardiovasc Res 2002;55:104–112 [DOI] [PubMed] [Google Scholar]
- 29.Devereux RB, Roman MJ, Paranicas M, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation 2000;101:2271–2276 [DOI] [PubMed] [Google Scholar]
- 30.Van Linthout S, Seeland U, Riad A, et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 2008;103:319–327 [DOI] [PubMed] [Google Scholar]
- 31.Haram PM, Kemi OJ, Lee SJ, et al. Aerobic interval training vs. continuous moderate exercise in the metabolic syndrome of rats artificially selected for low aerobic capacity. Cardiovasc Res 2009;81:723–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang CL, Wu YT, Chou CH. Effect of aerobic interval training on exercise capacity and metabolic risk factors in people with cardiometabolic disorders: a meta-analysis. J Cardiopulm Rehabil Prev 2011;31:378–385 [DOI] [PubMed] [Google Scholar]
- 33.Fleming J, Sharman MJ, Avery NG, et al. Endurance capacity and high-intensity exercise performance responses to a high fat diet. Int J Sport Nutr Exerc Metab 2003;13:466–478 [DOI] [PubMed] [Google Scholar]
- 34.Hawley JA, Gibala MJ. Exercise intensity and insulin sensitivity: how low can you go? Diabetologia 2009;52:1709–1713 [DOI] [PubMed] [Google Scholar]
- 35.Stolen KQ, Kemppainen J, Ukkonen H, et al. Exercise training improves biventricular oxidative metabolism and left ventricular efficiency in patients with dilated cardiomyopathy. J Am Coll Cardiol 2003;41:460–467 [DOI] [PubMed] [Google Scholar]
- 36.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207–258 [DOI] [PubMed] [Google Scholar]
- 37.Pereira L, Matthes J, Schuster I, et al. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006;55:608–615 [DOI] [PubMed] [Google Scholar]
- 38.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation 2007;115:3213–3223 [DOI] [PubMed] [Google Scholar]
- 39.Boardman NT, Larsen TS, Severson DL, Essop MF, Aasum E. Chronic and acute exposure of mouse hearts to fatty acids increases oxygen cost of excitation-contraction coupling. Am J Physiol Heart Circ Physiol 2011;300:H1631–H1636 [DOI] [PubMed] [Google Scholar]
- 40.Boudina S, Han YH, Pei S, et al. UCP3 regulates cardiac efficiency and mitochondrial coupling in high fat-fed mice but not in leptin-deficient mice. Diabetes 2012;61:3260–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azzu V, Brand MD. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci 2010;35:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kemi OJ, Ellingsen O, Ceci M, et al. Aerobic interval training enhances cardiomyocyte contractility and Ca2+ cycling by phosphorylation of CaMKII and Thr-17 of phospholamban. J Mol Cell Cardiol 2007;43:354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kemi OJ, Haram PM, Loennechen JP, et al. Moderate vs. high exercise intensity: differential effects on aerobic fitness, cardiomyocyte contractility, and endothelial function. Cardiovasc Res 2005;67:161–172 [DOI] [PubMed] [Google Scholar]
- 44.Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci 2011;16:553–567 [DOI] [PubMed] [Google Scholar]
- 45.Lin CH, Kurup S, Herrero P, et al. Myocardial oxygen consumption change predicts left ventricular relaxation improvement in obese humans after weight loss. Obesity (Silver Spring) 2011;19:1804–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwak HB, Kim JH, Joshi K, Yeh A, Martinez DA, Lawler JM. Exercise training reduces fibrosis and matrix metalloproteinase dysregulation in the aging rat heart. FASEB J 2011;25:1106–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu J, Ben QW, Yao WY, et al. BMP2 induces PANC-1 cell invasion by MMP-2 overexpression through ROS and ERK. Front Biosci 2012;17:2541–2549 [DOI] [PubMed] [Google Scholar]
- 48.Zhang HS, Wang SQ. Salvianolic acid B from Salvia miltiorrhiza inhibits tumor necrosis factor-alpha (TNF-alpha)-induced MMP-2 upregulation in human aortic smooth muscle cells via suppression of NAD(P)H oxidase-derived reactive oxygen species. J Mol Cell Cardiol 2006;41:138–148 [DOI] [PubMed] [Google Scholar]
- 49.Dai DF, Santana LF, Vermulst M, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009;119:2789–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rider OJ, Francis JM, Tyler D, Byrne J, Clarke K, Neubauer S. Effects of weight loss on myocardial energetics and diastolic function in obesity. Int J Cardiovasc Imaging. 27 December 2012. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.