

Abstract
Expansion of pancreatic β-cells is a key goal of diabetes research, yet induction of adult human β-cell replication has proven frustratingly difficult. In part, this reflects a lack of understanding of cell cycle control in the human β-cell. Here, we provide a comprehensive immunocytochemical “atlas” of G1/S control molecules in the human β-cell. This atlas reveals that the majority of these molecules, previously known to be present in islets, are actually present in the β-cell. More importantly, and in contrast to anticipated results, the human β-cell G1/S atlas reveals that almost all of the critical G1/S cell cycle control molecules are located in the cytoplasm of the quiescent human β-cell. Indeed, the only nuclear G1/S molecules are the cell cycle inhibitors, pRb, p57, and variably, p21: none of the cyclins or cdks necessary to drive human β-cell proliferation are present in the nuclear compartment. This observation may provide an explanation for the refractoriness of human β-cells to proliferation. Thus, in addition to known obstacles to human β-cell proliferation, restriction of G1/S molecules to the cytoplasm of the human β-cell represents an unanticipated obstacle to therapeutic human β-cell expansion.
Both type 1 and type 2 diabetes ultimately result from β-cell deficiency. Although β-cell replacement in humans can reverse diabetes, the paucity of β-cells available from adult or juvenile human cadaveric islets, or from hES cell or iPS cell sources, makes this approach untenable for β-cell replacement therapy on a public health scale. Accordingly, a major goal of diabetes research is to develop means to induce human β-cell proliferation and expansion, targeting either endogenous human β-cells or β-cells grown ex vivo. This desire to expand human β-cells is complicated by the fact that while there are many models of β-cell replication in juvenile rodents, adult cadaveric human β-cells—the major source of β-cells available for research and therapeutic manipulation—are notoriously refractory to induction of replication: indeed, no growth factors, mitogens, or (patho)physiologic maneuvers (such as pregnancy, partial pancreatectomy, or obesity) are known that are able to induce high rates of adult human β-cell proliferation (1–12). Equally perplexingly, we have little understanding as to why this is. This is particularly surprising because in contrast to the intractable quiescence of adult human β-cells, fetal and neonatal human β-cells can and do replicate transiently from ~5 months antepartum to ~6 months postpartum (13–15). Yet, even here replication is very low: in the 3% range (13–15). Further, we are only beginning to understand the physiological signals or mechanisms that activate and then inactivate this perinatal β-cell proliferation. As one example, we have only recently learned that loss of the platelet-derived growth factor (PDGF) receptor-α in adult human β-cells, with the resultant loss of ability to activate mitogen-activated protein kinase and methylation (Ezh2) and downstream cell cycle (p16) machinery, may underlie the refractoriness of human β-cells to proliferation (16).
With the goal of understanding how best to encourage human β-cells to replicate, we and others previously delineated the repertoire of G1/S regulatory proteins present in the adult human islet and have used this information to develop a working model of the “human islet G1/S proteome” (12,14–29), hoping that it might be useful in developing therapeutic approaches to manipulating human β-cell proliferation. Since many, and perhaps most, G1/S molecules are regulated at the level of protein stability, rather than or in addition to transcription (24,26,29), we have focused in this G1/S model on immunoblots of whole human islets rather than exploring mRNA expression of these molecules. The G1/S model has proven useful in predicting approaches to driving human β-cell proliferation in in vitro and in vivo systems. For example, the model accurately predicted that it should be possible to induce pRb phosphorylation (and thus its inactivation) and thereby markedly activate adult human β-cell replication (10–15% as assessed using BrdU incorporation or Ki67 immunohistochemistry) by overexpression of combinations of G1/S cyclins and cdks such as the d-cyclins, cyclin E, or cdks 2, 4, or 6 both in cultured adult human β-cells and in transplanted adult human β-cells in vivo (21–23,26). Further, it is also possible to use cyclin/cdk combinations to induce human β-cell proliferation not only constitutively or continuously but also using doxycycline-inducible delivery systems to transiently induce human β-cell proliferation in a regulated, reversible fashion that mimics the transitory replication that occurs in embryonic and neonatal life (28).
However, the human islet G1/S proteome model is not perfect. One major limitation is that it was derived from immunoblots of whole human islets. This is problematic because it is well-known that human islets are composed of many cells types in addition to β-cells. Indeed, β-cells frequently comprise <50% of all islet cells (30–32). Thus, while it may serve as a human islet G1/S proteome, it is not necessarily an accurate human β-cell G1/S proteome. Accordingly, we next wanted to rigorously define which of the 25 or more G1/S molecules are actually present in the human β-cell. More specifically, we wanted to use immunohistochemical and subcellular fractionation approaches to develop a “human adult β-cell G1/S proteome atlas” that accurately defines and displays which G1/S molecules are present in the human β-cell, that might suggest hypotheses as to which molecules maintain human β-cell quiescence or drive proliferation, and that might effectively reveal targets for therapeutic induction of human β-cell proliferation.
Here, we describe this human β-cell G1/S proteome atlas that we believe is a valuable resource for the β-cell research community. One major finding is that each of the G1/S proteins, previously reported to be present in human islets, is actually present in the human β-cell, permitting the deployment of an accurate human β-cell G1/S proteome model. A second major and wholly unanticipated finding is that the majority of the G1/S molecules, which we had presumed to be nuclear proteins, are in fact cytoplasmic in location in human β-cells, presumably unavailable to drive cell cycle progression. This may represent an unanticipated obstacle to induction of proliferation of human β-cell.
RESEARCH DESIGN AND METHODS
Human cadaveric islets.
Human islets were purchased from the National Institutes of Health– and JDRF-supported Integrated Islet Distribution Program (iidp.coh.org) and also generously provided by Dr. Tatsuya Kin at the University of Alberta (33). One hundred and sixty-four different cadaveric islet preparations were used for these studies and those in the accompanying manuscript (11). The mean ± SEM age of the donors was 43.0 ± 1.0 years, BMI 29.8 ± 0.4 kg/m2, and purity 82.8 ± 1.1%. Five of the 164 were under the age of 20 years, at 9, 12, 16, 17, and 19 years old. Upon arrival, islets were incubated in RPMI medium (Gibco, Grand Isle, NY) containing 5.5 mmol/L glucose, 1% penicillin, and streptomycin with 10% FBS until they were used for experiments. The use of human cadaver islets was approved in advance by the University of Pittsburgh School of Medicine Institutional Review Board.
Intact human pancreas.
Intact human normal adult pancreas was obtained from two sources. One was the Network for Pancreatic Organ Donors developed by the JDRF (www.jdrfnpod.org). These pancreas specimens were fixed in 10% formalin. The second was Dr. Rita Bottino in the Transplant Institute at the University of Pittsburgh School of Medicine. These samples were fixed in 4% paraformaldehyde overnight at 4°C prior to embedding and sectioning.
Adenovirus production and transduction.
Adenoviruses, all under control of the CMV promoter, were prepared using human cDNAs encoding human cdk4, human cdk6, human cdk1, human cdk2, human cyclins D1–D3, and human cyclin E1 as previously described (21–23,26,28,32). Ad.human p27 was provided by Dr. B. Law (University of Florida). Ad.human cyclin A2 was purchased from Vector Biolabs (Philadelphia, PA). Dispersed islets on coverslips were transduced with either Ad.LacZ or relevant control adenoviruses (100 multiplicity of infection) for 2 h; cultured for 24, 48, and 72 h as described in the figures; and immunolabeled as described below.
Human islet cell dispersion.
Human islets were aliquoted into 400 islet equivalents (IEQ) (1 IEQ = 125 µm) and washed twice in PBS. As previously described (21–23,26,28,32), islets were resuspended with 400 µL solution of 1 mg/mL trypsin and then incubated for 20 min at 37°C. During the digestion, the islets were dispersed by pipetting up and down every 5 min for 10 s. Complete medium (RPMI medium [Gibco, Grand Isle, NY] containing 5.5 mmol/L glucose, 1% penicillin, and streptomycin with 10% FBS) was then added to stop the digestion. The cells were then centrifuged for 3 min at 600g. The pellet was then resuspended with 400 µL complete medium, and cells were plated on coverslip with 50 µL cell suspension per coverslip. Cells were then allowed to attach for 2 h at 37°C or were transduced with adenovirus for 2 h as detailed in results. After 2 h, 500 µL complete medium was added in each well to terminate the adenoviral transduction. Cells were cultured for 24–72 h as described below.
Immunocytochemistry
Dispersed islets.
Islets were dispersed to single cells as described above, plated on coverslips, cultured for 48–72 h, and then fixed in fresh 4% paraformaldehyde for 15 min at 25°C. As a positive control, when possible dispersed islets were transduced with the adenoviruses above (100 multiplicity of infection) for 2 h, cultured on coverslips, and fixed at 48–72 h. Cells were washed with PBS and incubated in blocking buffer (1.0% BSA, 0.5% Triton, and 5% normal goat serum in PBS) for 1 h at 25°C. Primary antisera were exposed to the fixed cells on coverslips overnight at 4°C in blocking buffer, and secondary antisera were exposed for 2 h at 25°C in secondary buffer (1% BSA, 0.5% Triton in PBS). Primary antisera and secondary antisera are described in Supplementary Table 1. When reliable and specific human antisera were available, results were confirmed with a second primary antiserum; this was done for cdk6, cyclins D1 and D3, p16, p27, and p57. No antigen retrieval was performed. Labeled cells were then visualized using laser confocal microscopy. Results shown are representative of three to six separate human islet preparations.
Intact pancreas.
Sections were deparaffinized (xylene) and hydrated (ethanol to water), incubated for 30 min in boiling citrate buffer (10 mmol/L Na citrate, 0.5% Tween 20, pH 6.0) for antigen retrieval, and then cooled at 25°C. Blocking, primary, and secondary steps were performed as described above. Results shown are representative of three human cadaver pancreata.
Subcellular fractionation.
For preparation of cytoplasmic and nuclear fractions of whole human cadaveric islets, 4,000 IEQ were pelleted and washed twice with ice-cold PBS. The islet pellet was then resuspended with 250 µL cytoplasm extraction buffer (50 mmol/L HEPES, pH 7.5; 150 mmol/L NaCl; 1% Triton; 1.5 mmol/L MgCl2; 10 mmol/L EGTA; 10% glycerol; 0.1 mmol/L Na3VO4; 1% phenylmethylsulfonyl fluoride; and 20 µg/mL aprotinin), kept on ice for 5 min, and then centrifuged at 16,000g for 10 min at 4°C, yielding a supernatant (cytoplasmic fraction) and a nuclear pellet. The cytoplasmic fraction was transferred into a fresh tube and centrifuged again at 16,000g for 10 min at 4°C. The nuclear pellet was washed three times with PBS and then resuspended in 0.25 mol/L sucrose buffer (0.25 mol/L sucrose; 10 mmol/L MgCl2; 10 mmol/L HEPES, pH 7.5; and 1% Triton), layered over 0.35 mol/L sucrose buffer (0.35 mol/L sucrose; 10 mmol/L MgCl2; 10 mmol/L HEPES, pH7.5; and 1% Triton), and centrifuged at 4°C for 5 min at 1,430g. The nuclear pellet was then resuspended in 50 μL nuclear extract buffer (20 mmol/L HEPES, pH 7.5; 0.1 mmol/L EDTA; 5 mmol/L MgCl2; 0.5 mol/L NaCl; 20% glycerol; 1% NP-40; 0.1 mmol/L Na3VO4; 1% phenylmethylsulfonyl fluoride; and 20 µg/mL aprotinin) and placed on ice on a rotating shaker for 4 h, sonicated, and centrifuged at 16,000g for 10 min at 4°C. The supernatant (nuclear fraction) was collected and transferred into a fresh tube. The nuclear and cytoplasmic extracts were compared with extracts of whole islets prepared as previously described (21,22). Results shown are representative of three to six separate human islet preparations.
Immunoblotting.
Islet extracts were resolved using 10 or 12% SDS-PAGE and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Antibodies used to detect the G1/S molecules are described in detail in Supplementary Table 1. Each experiment shown is representative of three to six human islet preparations.
RESULTS
Localization of the pocket proteins and E2F family of transcription factors in human β-cells.
Our initial objective was to comprehensively document which of the G1/S family of proteins are present in the human β-cell, using immunohistochemical colabeling for insulin and cell cycle proteins in dispersed cadaveric adult human islet cells. As can be seen in Fig. 1, the retinoblastoma protein pRb is present in the human β-cell (as well as in non-β-cells) and, as anticipated, is principally in the nuclear compartment, being observed in the nuclei of 65 ± 7% (mean ± SEM) of human β-cells in five different islet preparations. The other two pocket proteins, p107 and p130, are also present in the β-cell but, surprisingly, appear in the cytoplasmic compartment and not the nuclear compartment. The specificity of the labeling for the pocket proteins was confirmed for pRb (using SaOS2 human osteosarcoma cells, which are null for pRb) and supported for p107 and p130 by the absence of labeling when the primary antisera for p107 and p130 were omitted.
FIG. 1.

The “pocket proteins.” Human islets were dispersed as described in research design and methods and labeled for insulin (green), for nuclei with DAPI (blue), and for each of the G1/S molecules (red). SaOS2 cells, which lack Rb, serve as a negative control for pRb. Labeling with no primary antibody (Ab) or with primary antibody plus blocking peptide is shown as negative controls for p107 and p130. Each panel is representative of 3–5 experiments on different human islet preparations. Note that pRb is present not only in β-cells (insulin+) but also in other (insulin−) islet cell types. Ins, insulin.
The E2F family of transcription factors, E2Fs1–8, was examined next (Fig. 2). E2Fs1–7 were observed in the human β-cell, whereas E2F8 was not easily detected. Surprisingly, despite being transcription factors, each was absent from the nuclear compartment but was predominantly present in the cytoplasmic compartment. The only exception to this was E2F4 (Fig. 2C), which was predominantly cytoplasmic but also nucleolar, as confirmed with nucleolin coimmunolabeling (Fig. 3). The specificity was supported, where possible, by blocking the primary antibody using the cognate antigenic peptide or, where no antigenic peptide was available, by omitting the primary antibody. E2F1–3 immunochemistry was also enhanced by their adenoviral overexpression as shown in Fig. 2B, where it can be seen that labeling for each of these E2Fs increased in intensity, and most remained cytoplasmic, even when overexpressed—an exception being E2F2, which also became nuclear in some cells.
FIG. 2.
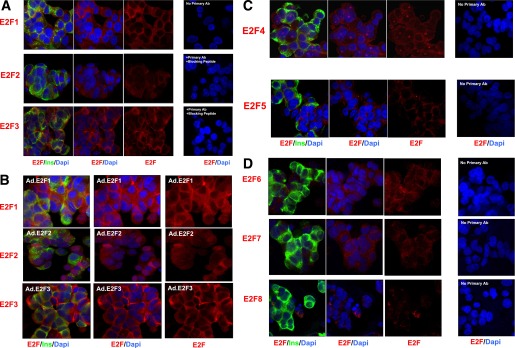
The E2F transcription factors. A: The proproliferative E2Fs 1–3. B: Adenoviral overexpression of E2Fs enhances E2F immunocytochemistry. C and D: E2Fs 4–8. Each panel is representative of 3–5 experiments on different human islet preparations. Labeling with no primary antibody (Ab) or with primary antibody plus blocking peptide is shown as negative controls. Ins, insulin.
FIG. 3.

E2F4 both is cytoplasmic and colocalizes with nucleoli. Coimmunolabeling for E2F4 and nucleolin suggests that E2F4 is a cytoplasmic as well as nucleolar molecule.
Collectively, these observations indicate that with the exception of pRb, all of the other pocket proteins and most of the E2Fs are located in the cytoplasm of the human β-cell.
Cyclins A and E and cdk1 and -2 in the human β-cell.
We next examined the “late G1/S cyclins,” cyclins E and A, and their cdk partners, cdk1 and cdk2 (Fig. 4A). Once again, each of these molecules was present in the β-cell, but each was present predominantly in the cytoplasm. Specificity was confirmed where possible with competing peptides (Fig. 4A) or supported by omission of primary antisera, and each was enhanced by overexpression (Fig. 4B). Notably, several of these, particularly cyclin E and cdk1, appeared nuclear when overexpressed.
FIG. 4.

The late G1/S cyclins/Cdks. A: Each of the cyclin A, cyclin E, and cdk1 and -2 members of late G1/S complexes is present in the cytoplasmic, but not nuclear, compartment. B: Adenoviral overexpression enhances the abundance of the late G1/S molecules and, in some cases, e.g., cyclin E and cdk2, leads to their nuclear appearance. Each panel is representative of 3–5 experiments on different human islet preparations. Labeling with no primary antibody or with primary antibody plus blocking peptide is shown as negative controls. Ins, insulin.
d-Cyclins and cdks 4 and 6 in the human β-cell.
We and others had previously reported that the d-cyclins and their cognate cdks, cdk4 and cdk6, are present in the human islet, with the exception of cyclin D2, which was present at very low abundance if at all (2,12,22,24). Figure 5A confirms that cyclins D1 and D3 are easily detectable in human β-cells, whereas cyclin D2 is not observed. As reported previously (22), cyclins D1 and D3 appear exclusively in the cytoplasmic compartment. Similar results were observed for cdk4 and cdk6 (Fig. 5B), both of which are clearly present in the human β-cell, as anticipated, but each of which is present exclusively in the cytoplasmic compartment, confirming previous reports (22). For determination of whether the techniques used could observe the d-cyclins in the nucleus if they were present, immunohistochemistry for each of the three d-cyclins was performed after adenoviral overexpression of each. In each case, nuclear presence was readily observed (Fig. 5C). The intensity of labeling increased with adenoviral overexpression of cdk4 and cdk6 as well (Fig. 5C).
FIG. 5.

The early G1/S cyclins/Cdks. A: The d-cyclins. Note that cyclins D1 and D3 are principally cytoplasmic and that cyclin D2 is not apparent. B: Cdks 4 and 6. Again, these two molecules appear in the cytoplasm and not the nuclear compartment. Comparable results have previously been described (4) and are included here for completeness. C: Adenoviral overexpression of d-cyclins and cdk4 and -6 enhances their abundance and, for the d-cyclins, their nuclear presence. Each panel is representative of 3–5 experiments on different human islet preparations. Labeling with no primary antibody (Ab) or with primary antibody plus blocking peptide is shown as negative controls. Ins, insulin.
INK4 and CIP/KIP families in the human β-cell.
Attention was then turned to the cyclin-dependent kinase inhibitors: the four INK4 family members (p15, p16, p18, and p19) and the three CIP/KIP members (p21, p27, and p57). Controls for specificity included competing peptides where available or omission of primary antiserum. As shown in Fig. 6A, each of the INK4 members proved to be present in the human β-cell but again appeared to be predominantly cytoplasmic and not nuclear. In the case of p16, it was observed primarily in the cytoplasm but also was seen in ~10% of β-cell nuclei, as quantified in the accompanying manuscript (34). Among the CIP/KIP members (Fig. 6B), p27 was present in β-cells but was cytoplasmic in location as well. Interestingly, p21 was occasionally observed to be nuclear in some human islet preparations but not in others. For the eight subjects assessed for p21 in this and the accompanying manuscript, the ages were 23, 34, 35, 39, 39, 42, 45, and 45 years old, with a mean of 37.8 years. The BMIs of the subjects were 24, 25, 25.6, 26.6, 26.9, 26.9, 29, and 32.5 kg/m2, with a mean BMI of 27.1 kg/m2. These values were not significantly different from those of the group as a whole, and there was no correlation between age, BMI, and nuclear versus cytoplasmic location of p21. The specificity of the p21 and p27 immunostaining was supported by lack of primary antiserum (Fig. 6B), and adenoviral overexpression enhanced their immunolabeling (Fig. 6C). Remarkably, p57, in contrast to all of the other G1/S molecules except pRb, was almost exclusively nuclear (Fig. 6B). Moreover, unlike other cell cycle molecules examined, it appeared to be principally located in β-cells and absent in other cell types, as has been reported by Kassem et al. (35). The nuclear-cytoplasmic distribution of the INK4s and CIP/KIPs is quantified and statistically validated in the accompanying manuscript (34). Interestingly, cdk6, p27, and p16 also display a cytoplasmic localization in α- and Δ-cells, as shown in Supplementary Fig. 1.
FIG. 6.
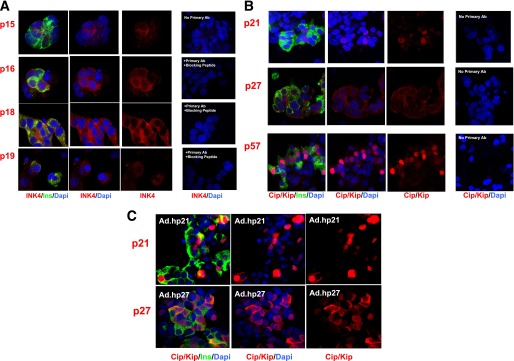
The cell cycle inhibitors. A: The INK4 family. B: The CIP-KIP family. C: Adenoviral overexpression of human p21 and p27. Each panel is representative of 3–5 experiments on different human islet preparations. Labeling with no primary antibody (Ab) or with primary antibody plus blocking peptide is shown as negative controls. Ins, insulin.
Parenthetically, the ability to observe p57, as with pRb, in the nuclear compartment indicates that the immunolabeling techniques used were able to detect endogenous G1/S molecules in the nuclear compartment if they were present in that compartment.
Subcellular fractionation independently confirms that all G1/S molecules except pRb, p57, and p21 are cytoplasmic and not nuclear in human β-cells.
The predominant nuclear localization of G1/S molecules was unanticipated. For independent confirmation of this intracellular distribution, whole human islets were subjected to subcellular fractionation into cytoplasmic and nuclear components. Figure 7 reveals the results for each G1/S molecule in each fraction: nuclear and cytoplasmic compared with whole unfractionated human islets. The cytoplasmic marker, tubulin, confirmed the cytoplasmic fraction of each prep, and a nuclear marker (histone 3 or RNA polymerase) confirmed the nuclear fraction of each prep; there was little contamination between nuclear and cytoplasmic fractions.
FIG. 7.
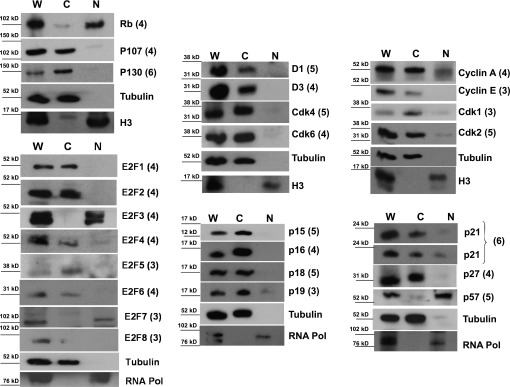
Subcellular fractionation of human islets. Human islets were fractionated into cytoplasmic fractions (C), as confirmed with heat-shock protein 90 (Hsp90) or tubulin, and nuclear fractions (N) confirmed with histone 3 (H3) or RNA polymerase (RNA Pol) and compared with the initial prep of whole (W) human islets. Two representative examples for p21 are shown from two different cadaver islet preparations: one in which p21 is primarily cytoplasmic and one in which a nuclear component is also visible. Note that all of the proteins that appeared cytoplasmic in Figs. 1–6 are independently confirmed to be cytoplasmic and those that appeared nuclear are confirmed to be nuclear, with only two exceptions: E2F3 and E2F7. Note also that the only molecules that are nuclear are the cell cycle inhibitors pRb and p57 and, in some preparations, p21. Each immunoblot was performed using 3–6 different human islet preparations, as indicated by the adjacent numbers in parentheses.
Using this independent technique, once again pRb was clearly nuclear, as was p57. p21 was either occasionally partly nuclear or entirely cytoplasmic, depending on the human islet preparation studied. The remainder of the G1/S molecules—even the E2F transcription factors—was principally or entirely cytoplasmic. Thus, there was near-perfect concordance between results obtained using whole islets and dispersed islets and between results obtained using immunohistochemistry or immunoblots of fractionated islet extracts: of the 27 molecules examined, two were principally nuclear, 24 were exclusively cytoplasmic, and one, p21, varied among human islet preparations. The only exceptions to this near-perfect correlation were E2F3 and E2F7, which appeared to be nuclear by immunoblot but cytoplasmic by immunohistochemistry. It is uncertain which of these observations is most accurate. Importantly, the only G1/S molecules in the human β-cell nucleus are the cell cycle inhibitors, pRb, p57, and p21. In contrast, all of the cell cycle activators appear in the cytoplasmic compartment.
Examination of intact human pancreas confirms the subcellular location of G1/S molecules.
It is conceivable that the enzymatic and mechanical rigors of human islet isolation from cadaver pancreases, culturing human islets in 10% FBS, or islet cell dispersal of these into single cells on coverslips might lead to nonphysiological or artifactual localization or shifting of G1/S molecules into the cytoplasm. For determination of whether the results described herein were also observed in intact normal human β-cells, pancreas sections were obtained from the Network for Pancreas Organ Donors (nPOD), and immunohistochemistry was performed on these intact human pancreatic sections. These nPOD sections were fixed in 10% formalin, rendering them difficult to immunolabel. Therefore, only two antisera, those for pRb and p18, were able to lead to clear visualization of G1/S molecules. As can be seen in Fig. 8, for these two antisera the results in intact pancreas faithfully reproduced the results observed in β-cells from isolated islets: pRb was principally nuclear, and p18 was principally cytoplasmic. In an effort to extend these observations to additional G1/S molecules, additional fresh human pancreas samples were generously provided by Dr. R. Bottino at the University of Pittsburgh and fixed overnight in 4% paraformaldehyde, as had been used for the isolated dispersed β-cells in the preceding figures. As shown in Fig. 8, p107 appeared in the cytoplasm and not the nucleus of β-cells, as in dispersed β-cells. Conversely, p57 was nuclear and not cytoplasmic in β-cells—exactly as had been observed in dispersed β-cells and by immunoblot and as described by Kassem et al. (35). In addition, although p16 was less well visualized, it appeared to be cytoplasmic in most β-cells (Fig. 8), although occasional β-cells contained p16 in the nucleus, as shown in the inset. The remaining G1/S molecules were not able to be visualized using these longer-term formalin- or paraformaldehyde-fixed intact pancreas sections. Thus, all five results available from intact pancreas corresponded to those in isolated islets (subcellular fractionation) as well as in dispersed human β-cells on coverslips.
FIG. 8.

Confirmation that subcellular localization observed in isolated and dispersed human β-cells is also observed in intact human islets in situ. Human pancreatic sections obtained from nPOD (pRb and p18, fixed in 10% formalin) or fresh human pancreas blocks fixed in 4% paraformaldehyde (p57, p107, and p16) were labeled as described in research design and methods and confirm the observation in dispersed human islets and subcellular fractionation studies. Each panel is representative of 3–5 experiments on different human pancreas preparations. The inset in the p16 panel shows that it is generally cytoplasmic but occasionally nuclear, particularly in smaller islets. See the accompanying article (34) for quantification. Ins, insulin.
DISCUSSION
Although all agree that induction of adult human β-cell proliferation is important, achieving this goal in a therapeutically meaningful manner has been frustratingly difficult. In part, this is the result of an incomplete understanding of the mechanistic details of cell cycle progression in the human β-cell, i.e., the lack of a “wiring diagram” or accurate model of the relevant regulatory molecules. Prior models of the control of the G1/S transition were based on extracts of whole islets (12,18–24,26), which are comprised of multiple different cell types (30–32). Here, we endeavored to develop a comprehensive β-cell–specific G1/S model of cell cycle control. The two broad results of this endeavor, based on 164 separate human cadaver islet preparations, are that we now have an atlas of G1/S molecules that are present in the human β-cell. Equally importantly, we now have a complete and very substantially revised working model of G1/S control in the human β-cell. More specifically, the atlas, the model, and the data presented herein 1) reveal that most G1/S molecules are located in the cytoplasm in human β-cells and are not nuclear as anticipated; 2) reveal that the only nuclear G1/S molecules—pRb, p57, and sometimes p21—are all cell cycle inhibitors; 3) may provide unanticipated insight into why human β-cells are quiescent and resistant to proliferation; and 4) may have broad applicability beyond β-cells, extending to quiescent mammalian cells in general.
The prior model of the human β-cell G1/S control was in part correct in assuming that most of the G1/S molecules are present in the β-cell. It was incorrect, however, in assuming that they are nuclear. Thus, the new “atlas” presented in Figs. 1–8 provides a more accurate cell biological description of the quiescent human β-cell, showing that all of the G1/S molecules, except cyclin D2, as reported previously (12,21,22,24), are found in β-cells but are found in the cytoplasm, with three notable exceptions: pRb, p57, and in some human islet preparations, p21. Thus, we now have a reasonably complete and concrete model for understanding human β-cell G1/S control and for hypothesis generation as to approaches to therapeutic induction of proliferation.
The observation that the majority of G1/S molecules are cytoplasmic was unanticipated. Yet, it is strongly supported by three independent methods: first, immunohistochemistry with appropriate negative (no primary antibody, human cell lines that lack specific G1/S molecules, and competition with excess antigenic peptide) controls; second, subcellular fractionation of human islets; and, third, confirmation that this is not an artifact of islet isolation and dispersal, since the same pattern is observed, when technically possible, in intact islets in intact pancreas. The observation that pRb is nuclear was expected, since it is widely viewed as a nuclear protein (36–38). Similarly, the observation that p57 is nuclear was also anticipated, since this has been reported previously by Kassem et al. (35). Interestingly, here as well as in the study by Kassem et al. (35), p57 is not only abundant in the nuclei of β-cells (it is present in 30–40% of β-cell nuclei in both reports) but is also apparently restricted to β-cells compared with other endocrine and exocrine cell types. Again, the observation that the only G1/S molecules that are consistently nuclear are these two cell cycle inhibitors may provide insight into why human β-cells are quiescent.
Most cell cycle biologists, as illustrated in current reviews of G1/S control (36–42), have considered the G1/S molecules to be nuclear molecules, perhaps best exemplified by the E2Fs that are transcription factors. But it is also true that most who study G1/S biology are focused on replication in rapidly proliferating cells such as cancer cells, embryonic cells, or cells in lower organisms that are replicating rapidly or are prokaryotes. Thus, it was a surprise to observe these molecules in the cytoplasm. On the other hand, scattered reports indicate that many G1/S molecules may be present in the cytoplasm of β-cells or other quiescent cells. For example, we had previously noted that p21 is principally cytoplasmic in the quiescent human β-cell but could be translocated to the nucleus by overexpression of cdk4 and cyclin D1 (32). Others have reported that d-cyclins are present in the cytoplasm of the mouse β-cell and that they can translocate into the nucleus with glucose-induced β-cell proliferation (43,44). E2Fs have also been reported to be present in the cytoplasm of quiescent cells, such as differentiated skeletal muscle cells and keratinocytes (45,46). In other mammalian cell types, p107 and p130 have been observed under some conditions in the cytoplasm of prostate cells (47), and p53 has also been observed in the cytoplasm of many cell types and to undergo cytoplasm-to-nuclear translocation (48). Cdk6 has also been reported to exist in the cytoplasmic compartment of astrocytes (49) and lymphocytes (41), as have cyclins A and E and cdks 1 and 2 in HeLa cells (50). Thus, there is ample precedent for G1/S molecules being cytoplasmic proteins. Interestingly, the possibility exists that the association between cytoplasmic localization of G1/S molecules and the enforcement of quiescence of the cell types involved may represent a broad biologic principle among quiescent mammalian cells—not solely restricted to β-cell biology.
Construction of this atlas and the model described in the accompanying article (34) is a work in progress and has limitations, as discussed in the discussion section of the accompanying article.
In conclusion, these studies provide both a G1/S cell cycle molecule atlas for the human β-cell and a working model for hypothesis generation. The atlas suggests that restraint or tethering of cell cycle molecules in the cytoplasm may be a regulatory step that restrains or prevents replication in adult human β-cells. It further suggests that if one were able to develop mechanisms to direct cyclins and cdks to traffic into the nucleus, one might be able to effectively drive human β-cell proliferation. This hypothesis is demonstrated to be true in the accompanying article (34).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Beta Cell Biology Consortium through National Institutes of Health Grant U-01 DK 089538. It was also supported by R-01 DK55023 and JDRF grants 1-2008-39 and 34-2008-630, American Diabetes Association Research Grant 7-12-BS-046, and a University of Pittsburgh Junior Faculty Award to N.M.F.-T. Human islets were generously supplied by the National Institute of Diabetes and Digestive and Kidney Diseases, the JDRF-supported Integrated Islet Distribution Program, and Dr. Tatsuya Kin at the University of Alberta. The authors also thank nPOD and Dr. Rita Bottino at the University of Pittsburgh for supplying intact human pancreas samples.
No potential conflicts of interest relevant to this article were reported.
N.M.F.-T. researched data, contributed to the discussion, and wrote the manuscript. J.W.K., F.S., R.T., R.W., M.T., G.C., A.E.C., and K.K.T. researched data. D.K.S. contributed to the discussion. A.F.S. contributed to the discussion and wrote the manuscript. N.M.F.-T. and A.F.S. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Adolfo Garcia-Ocaña, PhD; Rupangi C. Vasavada, PhD; and Laura C. Alonso, MD, at the University of Pittsburgh School of Medicine for many helpful discussions during the preparation of these studies.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0777/-/DC1.
D.K.S. and A.F.S. are currently affiliated with the Diabetes Obesity and Metabolism Institute, Mount Sinai School of Medicine, New York, New York.
See accompanying original article, p. 2460.
REFERENCES
- 1.Perl S, Kushner JA, Buchholz BA, et al. Significant human β-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010;95:E234–E239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–110 [DOI] [PubMed] [Google Scholar]
- 3.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008;10(Suppl. 4):32–42 [DOI] [PubMed] [Google Scholar]
- 4.Meier JJ, Menge BA, Breuer TGK, et al. Functional assessment of pancreatic beta-cell area in humans. Diabetes 2009;58:1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-Cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 2013;36:111–117 [DOI] [PMC free article] [PubMed]
- 6.Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010;53:2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menge BA, Tannapfel A, Belyaev O, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes 2008;57:142–149 [DOI] [PubMed] [Google Scholar]
- 8.Parnaud G, Bosco D, Berney T, et al. Proliferation of sorted human and rat beta cells. Diabetologia 2008;51:91–100 [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Remedi MS, Pappan KL, et al. Glycogen synthase kinase-3 and mammalian target of rapamycin pathways contribute to DNA synthesis, cell cycle progression, and proliferation in human islets. Diabetes 2009;58:663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maedler K, Schumann DM, Schulthess F, et al. Aging correlates with decreased beta-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for Fas and pancreatic duodenal homeobox-1. Diabetes 2006;55:2455–2462 [DOI] [PubMed] [Google Scholar]
- 11.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004;429:41–46 [DOI] [PubMed] [Google Scholar]
- 12.Davis DB, Lavine JA, Suhonen JI, et al. FoxM1 is up-regulated by obesity and stimulates beta-cell proliferation. Mol Endocrinol 2010;24:1822–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008;57:1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Köhler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of β-cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab 2011;300:E221–E230 [DOI] [PubMed] [Google Scholar]
- 15.Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 2000;49:1325–1333 [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Gu X, Liu Y, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 2011;478:349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic beta-cell proliferation. Annu Rev Cell Dev Biol 2006;22:311–338 [DOI] [PubMed] [Google Scholar]
- 18.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev 2006;27:356–370 [DOI] [PubMed] [Google Scholar]
- 19.Halvorsen TL, Beattie GM, Lopez AD, Hayek A, Levine F. Accelerated telomere shortening and senescence in human pancreatic islet cells stimulated to divide in vitro. J Endocrinol 2000;166:103–109 [DOI] [PubMed] [Google Scholar]
- 20.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006;443:453–457 [DOI] [PubMed] [Google Scholar]
- 21.Fiaschi-Taesch NM, Bigatel TA, Sicari BM, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes 2009;58:882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiaschi-Taesch NM, Salim F, Kleinberger J, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 2010;59:1926–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guthalu Kondegowda N, Joshi-Gokhale S, Harb G, et al. Parathyroid hormone-related protein enhances human ß-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes 2010;59:3131–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavine JA, Raess PW, Davis DB, et al. Overexpression of pre-pro-cholecystokinin stimulates beta-cell proliferation in mouse and human islets with retention of islet function. Mol Endocrinol 2008;22:2716–2728 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 2011;54:572–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karslioglu E, Kleinberger JW, Salim FG, et al. cMyc is a principal upstream driver of beta-cell proliferation in rat insulinoma cell lines and is an effective mediator of human beta-cell replication. Mol Endocrinol 2011;25:1760–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong L, Georgia S, Tschen SI, Nakayama K, Nakayama K, Bhushan A. Essential role of Skp2-mediated p27 degradation in growth and adaptive expansion of pancreatic beta cells. J Clin Invest 2007;117:2869–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takane KK, Kleinberger JW, Salim FG, Fiaschi-Taesch NM, Stewart AF, Stewart AF. Regulated and reversible induction of adult human β-cell replication. Diabetes 2012;61:418–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fatrai S, Elghazi L, Balcazar N, et al. Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 2006;55:318–325 [DOI] [PubMed] [Google Scholar]
- 30.Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren P-O, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA 2006;103:2334–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brissova M, Fowler MJ, Nicholson WE, et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005;53:1087–1097 [DOI] [PubMed] [Google Scholar]
- 32.Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF. Induction of beta-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 2004;53:149–159 [DOI] [PubMed] [Google Scholar]
- 33.Kin T, O’Gorman D, Schroeder A, et al. Human islet distribution program for basic research at a single center. Transplant Proc 2011;43:3195–3197 [DOI] [PubMed] [Google Scholar]
- 34.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human β-cell replication: a revised model of human β-cell G1/S control. Diabetes 2013;62:2460–2470 [DOI] [PMC free article] [PubMed]
- 35.Kassem SA, Ariel I, Thornton PS, et al. p57(KIP2) expression in normal islet cells and in hyperinsulinism of infancy. Diabetes 2001;50:2763–2769 [DOI] [PubMed] [Google Scholar]
- 36.Cobrinik D. Pocket proteins and cell cycle control. Oncogene 2005;24:2796–2809 [DOI] [PubMed] [Google Scholar]
- 37.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004;18:2699–2711 [DOI] [PubMed] [Google Scholar]
- 38.Berns A. Tumour suppressors: timing will tell. Nature 2003;424:140–141 [DOI] [PubMed] [Google Scholar]
- 39.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 2005;30:630–641 [DOI] [PubMed] [Google Scholar]
- 40.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009;28:2925–2939 [DOI] [PubMed] [Google Scholar]
- 41.Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol 2004;5:31–37 [DOI] [PubMed] [Google Scholar]
- 42.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999;13:1501–1512 [DOI] [PubMed] [Google Scholar]
- 43.He LM, Sartori DJ, Teta M, et al. Cyclin D2 protein stability is regulated in pancreatic beta-cells. Mol Endocrinol 2009;23:1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alonso LC, Yokoe T, Zhang P, et al. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes 2007;56:1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivanova IA, Vespa A, Dagnino L. A novel mechanism of E2F1 regulation via nucleocytoplasmic shuttling: determinants of nuclear import and export. Cell Cycle 2007;6:2186–2195 [DOI] [PubMed] [Google Scholar]
- 46.Gill RM, Hamel PA. Subcellular compartmentalization of E2F family members is required for maintenance of the postmitotic state in terminally differentiated muscle. J Cell Biol 2000;148:1187–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claudio PP, Zamparelli A, Garcia FU, et al. Expression of cell-cycle-regulated proteins pRb2/p130, p107, p27(kip1), p53, mdm-2, and Ki-67 (MIB-1) in prostatic gland adenocarcinoma. Clin Cancer Res 2002;8:1808–1815 [PubMed] [Google Scholar]
- 48.Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004;303:1010–1014 [DOI] [PubMed] [Google Scholar]
- 49.Kohrt DM, Crary JI, Gocheva V, Hinds PW, Grossel MJ. Distinct subcellular distribution of cyclin dependent kinase 6. Cell Cycle 2009;8:2837–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackman M, Kubota Y, den Elzen N, Hagting A, Pines J. Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol Biol Cell 2002;13:1030–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.