

Abstract
The AMP-activated protein kinase (AMPK) is a highly conserved sensor of cellular energy that appears to have arisen at an early stage during eukaryotic evolution. In 2001 it was shown to be activated by metformin, currently the major drug for treatment for type 2 diabetes. Although the known metabolic effects of AMPK activation are consistent with the idea that it mediates some of the therapeutic benefits of metformin, as discussed below it now appears unlikely that AMPK is the sole target of the drug. AMPK is also activated by several natural plant products derived from traditional medicines, and the mechanisms by which they activate AMPK are discussed. One of these is salicylate, probably the oldest medicinal agent known to humankind. The salicylate prodrug salsalate has been shown to improve metabolic parameters in subjects with insulin resistance and prediabetes, and whether this might be mediated in part by AMPK is discussed. Interestingly, there is evidence that both metformin and aspirin provide some protection against development of cancer in humans, and whether AMPK might be involved in these effects is also discussed.
AMP-activated protein kinase: Evolutionary background
AMP-activated protein kinase (AMPK) was originally defined as a protein kinase from rat liver that phosphorylated and inactivated two key enzymes of mammalian fatty acid and sterol synthesis, i.e., acetyl-CoA carboxylase-1 (ACC1) and 3-hydroxy-3-methylglutaryl-CoA reductase (1). Its activation requires phosphorylation by upstream kinases at a conserved threonine residue within the kinase domain (usually termed Thr-172 because of its position in the rat sequence [2]). Phosphorylation of this site and the major site phosphorylated on ACC1 (3), detected using phosphospecific antibodies, are now almost universally used as biomarkers to monitor AMPK activation.
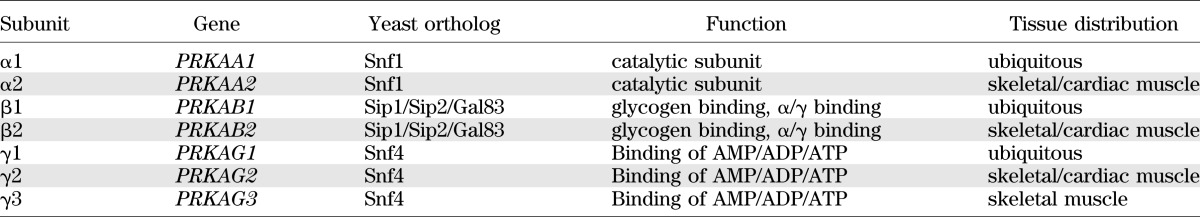
AMPK exists as heterotrimers composed of a catalytic α subunit and regulatory β and γ subunits (4,5). In humans, each of these occurs as multiple isoforms encoded by distinct genes (Table 1) such that there are at least twelve possible heterotrimeric combinations. Genes encoding the three subunits are also readily recognized in the genomes of almost all eukaryotes, from simple single-celled protozoa to humans. Based on genetic studies in lower eukaryotes, the ancestral role of this conserved pathway appears to have been in the response to glucose starvation. The AMPK-signaling pathway represents a mechanism to respond to fluctuating glucose levels that appears to have evolved much earlier than the insulin-signaling pathway, which is only found in multicellular animals. Of particular interest, in the nematode worm Caenorhabditis elegans AMPK is required for the extension of life span that is observed in response to caloric restriction or to mutations that reduce the function of the insulin-signaling pathway (6,7).
TABLE 1.
Properties of AMPK subunits. Although the β2, γ2, and γ3 subunits are most highly expressed in muscle, they are found at low levels in other tissues
Regulation of AMPK by adenine nucleotides and calcium ions
The AMPK-γ subunits contain three sites that bind adenine nucleotides and confer the ability of the kinase to act as an energy sensor (8–10). In cells not subject to energetic stress, catabolism maintains the ATP:ADP ratio at around 10:1, and this drives the adenylate kinase reaction toward ADP synthesis (ATP + AMP → 2ADP), so that AMP concentrations are very low; the typical ratios of ATP:AMP:AMP in unstressed cells are around 100:10:1. The γ subunit sites appear to bind AMP, ADP, and ATP with similar affinity, but they preferentially bind free ATP4− rather than the Mg.ATP2− complex (8). Because around 90% of ATP (but not ADP or AMP) is present as the magnesium complex, the cellular concentrations of total ADP and free ATP4− are comparable, allowing these nucleotides to compete with each other for binding to AMPK. Although the concentration of AMP is at least 10-fold lower than that of ADP or free ATP in unstressed cells, it rises markedly as the ADP:ATP ratio rises during energy stress, due to displacement of the adenylate kinase reaction toward AMP: 2ADP → ATP + AMP. Under these conditions, AMP should therefore be able to compete with ATP or ADP at the AMPK-γ subunit binding sites.
Binding of AMP or ADP (but not ATP) to the AMPK-γ subunit causes a conformational change that promotes phosphorylation of Thr-172 by upstream kinases while inhibiting dephosphorylation by upstream phosphatases (8,11,12). Stoichiometric phosphorylation of Thr-172 can cause >100-fold activation, although Thr-172 may only be partially phosphorylated in vivo even in cells experiencing metabolic stress. The effect of increased phosphorylation is amplified up to 10-fold further by allosteric activation, which is caused only by binding of AMP. This tripartite mechanism (Fig. 1) means that there can be large increases in AMPK activity in response to small increases in the AMP:ATP or ADP:ATP ratios.
FIG. 1.

Tripartite mechanism by which AMPK is activated by changes in cellular energy status. Displacement of ATP by ADP and/or AMP at one or more of the sites on the AMPK-γ subunit causes a conformational change in the heterotrimeric complex that 1) promotes phosphorylation, and 2) inhibits dephosphorylation of Thr-172, causing a large (up to 100-fold) increase in kinase activity. Binding of AMP, but not ADP, causes 3) further activation of the phosphorylated kinase of up to 10-fold. The upstream kinase LKB1 appears to be constitutively active, and increased Thr-172 phosphorylation in response to energy stress does not normally occur in tumor cells lacking LKB1. However, AMPK can also be activated by Thr-172 phosphorylation catalyzed by CaMKKβ via a mechanism that requires an increase in intracellular Ca2+ but can be independent of changes in AMP and/or ADP.
The major upstream kinase that phosphorylates Thr-172 in mammalian cells was identified to be a complex containing the tumor suppressor kinase, liver kinase B1 (LKB1) (13–15), introducing a tantalizing link between AMPK and cancer, which is considered further below. LKB1 appears to be constitutively active, but its high basal activity is required for the effect of binding of ADP or AMP to the γ subunit on net Thr-172 phosphorylation to become evident (13). Thr-172 can also be phosphorylated by the Ca2+/calmodulin-dependent kinase kinases (CaMKKs), especially CaMKKβ (16–18). This mechanism, which can act independently of changes in adenine nucleotides, is responsible for activation of AMPK in response to agonists that increase intracellular Ca2+ including thrombin in endothelial cells (19), antigen binding to the T-cell receptor (20), the hunger hormone ghrelin acting on hypothalamic neurons (21), and cannabinoids acting at CB2 receptors (22).
Metabolic consequences of AMPK activation
By sensing changes in adenine nucleotide ratios, AMPK is activated by stresses that depress cellular energy status. These include stresses that inhibit ATP production, such as hypoxia, hypoglycemia, or addition of mitochondrial poisons, as well as stresses that accelerate ATP consumption, such as muscle contraction (23). Some of the major metabolic changes elicited by AMPK activation are summarized in Table 2; the reader is referred to other reviews for details (4,5). In many cases, the effects occur both acutely via direct phosphorylation of metabolic enzymes and chronically via phosphorylation of transcription factors or coactivators that modulate gene expression.
TABLE 2.
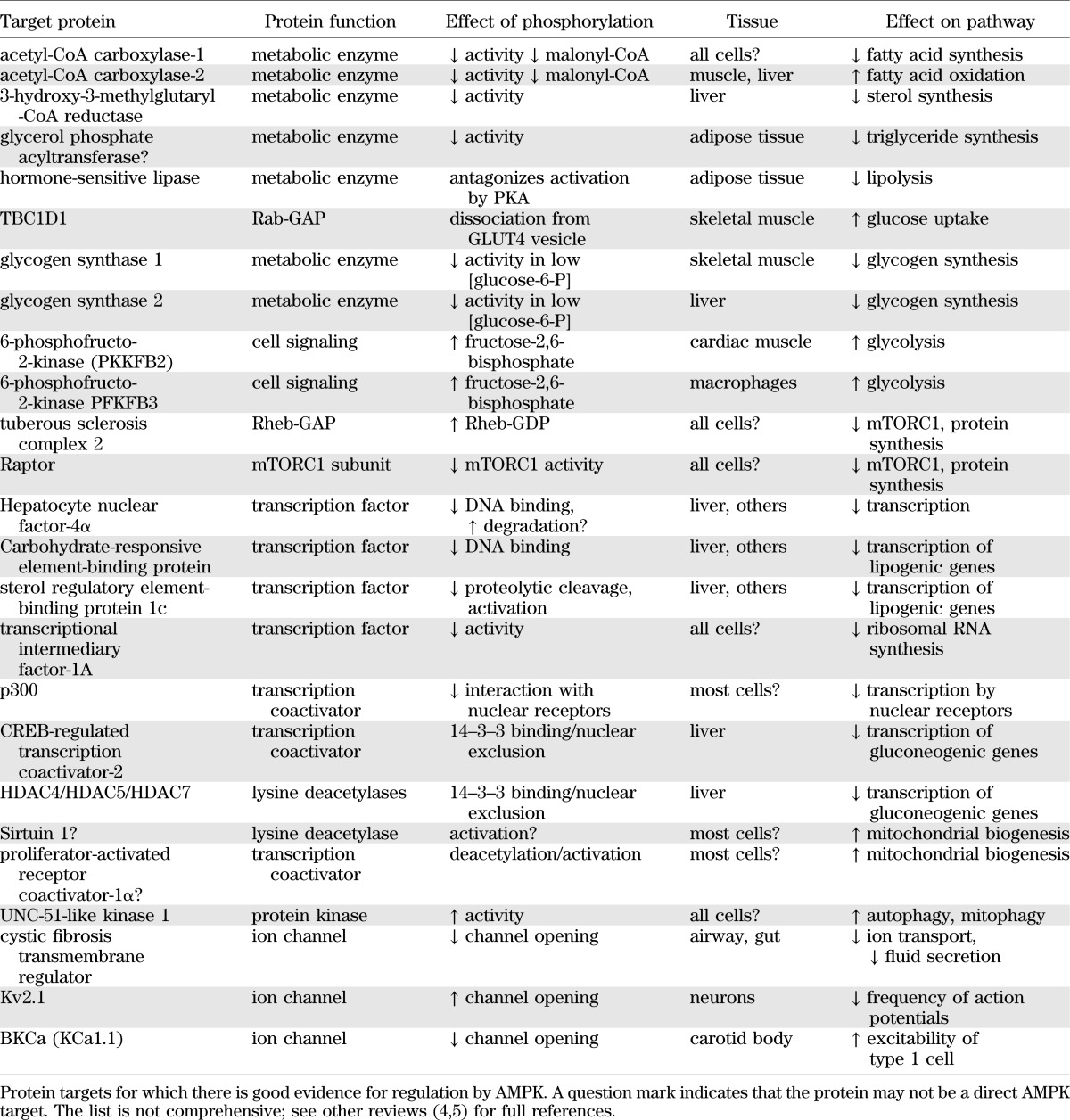
Some of the metabolic effects that ensue following AMPK activation are particularly relevant to treatment of type 2 diabetes. By inhibiting fat synthesis and promoting fat oxidation, and by enhancing mitochondrial biogenesis and disposal of damaged mitochondria by autophagy (24), AMPK activation would reverse elevated storage of triglycerides as well as deficits in mitochondrial function, both of which are associated with insulin resistance (25,26). Another important effect of AMPK is its ability to promote glucose uptake in skeletal muscle. This occurs both acutely via translocation of GLUT4 from intracellular storage vesicles to the plasma membrane, and in the longer term by upregulation of GLUT4 expression (27). The mechanism for the acute effect is similar to that by which the insulin-signaling pathway promotes glucose uptake, involving phosphorylation of the Rab-GAP protein TBC1D1 by AMPK (28,29). There has been controversy as to whether AMPK entirely accounts for the increase in glucose uptake induced by exercise or muscle contraction. Whereas experiments with mouse knockouts of single catalytic subunits (AMPK-α1 or -α2) did not support this view (30), a study involving a muscle-specific double knockout of both AMPK-β subunits, which totally ablates muscle AMPK activity, did support it (23). The latter study also revealed that mice lacking AMPK in muscle have reduced mitochondrial content and a dramatically reduced capacity for treadmill running. Human studies suggest that activation of muscle AMPK during exercise is normal in patients with type 2 diabetes (31), helping to explain why regular exercise is beneficial for them but also reinforcing the idea that pharmacological activation of AMPK would be an effective way to treat the disorder.
Activation of AMPK by metformin and other guanidine-based drugs
Guanidine and its isoprenyl derivative, galegine, are natural products from the plant Galega officinalis, which was used as a medicinal herb in medieval Europe (32). Metformin and phenformin are biguanides, synthetic derivatives of guanidine (Fig. 2). All of these compounds were tested on animals in the 1920s, but the success of insulin therapy at that time perhaps caused further studies of guanidine-based drugs to be put on hold. The biguanides were finally introduced for treatment of type 2 diabetes in the 1950s; phenformin was subsequently withdrawn in the 1970s because of a rare but life-threatening side effect of lactic acidosis, but metformin is now generally the first-choice drug for treatment of type 2 diabetes.
FIG. 2.

Structures of AMPK activators based on guanidine, including the biguanides metformin and phenformin.
Although biguanides were already known to reduce hepatic glucose production and enhance peripheral insulin sensitivity (32), the first clues to their molecular mechanism came in 2000, when it was shown that they inhibited Complex I of the mitochondrial respiratory chain (33,34). Interestingly, both drugs are cations (Fig. 2), so they accumulate in mitochondria due to the electrical gradient across the inner membrane (33). Metformin has poor plasma membrane permeability, but uptake into many cells (including hepatocytes) is promoted by the organic cation transporter OCT1, whereas phenformin uptake is less dependent on this transporter (35). Since both drugs inhibit mitochondrial ATP synthesis, they would be expected to cause increases in the ADP:ATP and AMP:ATP ratios and thus activate AMPK indirectly. AMPK activation by metformin was indeed demonstrated in 2001 (36), and this was subsequently also shown for phenformin (13) and galegine (37). The activation mechanism requires increases in AMP and/or ADP, because all three drugs fail to activate AMPK in cells expressing an AMPK-γ subunit with a mutation (R531G) that renders the complex AMP/ADP insensitive (35).
Are the therapeutic effects of metformin mediated by AMPK activation?
Whether AMPK is the metformin target that entirely accounts for its therapeutic effects now appears doubtful, at least when considering its effects on hepatic glucose production. A role for AMPK had seemed attractive because the pharmacological AMPK activator, 5-aminoimidazole-4-carboxamide riboside (AICAR) (38) downregulated expression of the gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) (39), while in mice with a liver-specific knockout of LKB1 (in which metformin no longer activated AMPK) the hypoglycemic effects of metformin were lost (40). However, in a more recent study in which both AMPK catalytic subunit isoforms were knocked out in liver, the mice had normal blood glucose and insulin, while metformin still reduced the expression of G6Pase and glucose output, and AICAR reduced the expression of both PEPCK and G6Pase, in isolated hepatocytes derived from the mice (41). This study revealed that there must be AMPK-independent pathways by which not only metformin, but also AICAR, inhibit expression of gluconeogenic enzymes and glucose output in the liver.
Evidence in favor of one such AMPK-independent mechanism (Fig. 3) was reported recently (42). The classical activator of hepatic glucose production is the starvation hormone glucagon, which increases cyclic AMP and activates cyclic AMP-dependent protein kinase A (PKA). PKA phosphorylates the liver (PFKFB1) isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, leading to a rapid drop in fructose-2,6-bisphosphate. Since the latter is an activator of the glycolytic enzyme 6-phosphofructo-1-kinase and an inhibitor of the gluconeogenic enzyme fructose-1,6-bisphosphatase, glucagon triggers an acute metabolic switch from glycolysis to gluconeogenesis. In addition, PKA enhances expression of gluconeogenic enzymes by phosphorylation of the transcription factor cyclic AMP response element-binding protein and other mechanisms. The increases in cyclic AMP are brought about by activation of adenylate cyclase by the glucagon receptor, and adenylate cyclase is directly inhibited by the metformin-induced increase in AMP, leading to a reduction in cyclic AMP formation in response to glucagon (42). Interestingly, a similar effect was observed with AICAR, a nucleoside that is taken up into cells and converted to the equivalent monophosphorylated nucleotide, ZMP (38). It seems possible that ZMP, like AMP, might inhibit adenylate cyclase, and that this might explain why AICAR was originally found to inhibit expression of PEPCK and G6Pase (39).
FIG. 3.

Schematic diagram of the proposed new mechanism by which metformin inhibits hepatic gluconeogenesis (42). Up or down arrows next to a metabolite or enzyme show the direction the concentration or activity changes in response to metformin. Metformin (whose uptake into hepatocytes is promoted by the organic cation transporter [OCT1]) accumulates in mitochondria where it inhibits Complex 1 of the respiratory chain, lowering cytoplasmic ATP and increasing ADP and AMP. AMP activates AMPK but also inhibits adenylate cyclase, reducing effects of glucagon on cAMP and PKA and thus reducing the ability of PKA to promote gluconeogenesis by phosphorylation of PFK2 and other targets regulating transcription of gluconeogenic genes. F16BP, fructose-1,6-bisphosphate; F16BPase, fructose-1,6-bisphosphatase; F26BP, fructose-2,6-bisphosphate; G6Pase, glucose-6-phosphatase; PFK1, 6-phosphofructo-1-kinase; PFK2, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase.
Interestingly, AMP is also an allosteric inhibitor of fructose-1,6-bisphosphatase (this too is mimicked by ZMP [43]) and an activator of PFK1. Because metformin increases AMP in hepatocytes (42), whereas AICAR increases ZMP (38), this represents yet another AMPK-independent mechanism by which metformin and AICAR might inhibit hepatic glucose production in vivo.
As well as inhibition of complex I of the respiratory chain (33,34), it has been proposed that metformin might act by inhibiting AMP deaminase, an enzyme that degrades cellular AMP (44). One caveat is that the reported inhibition of AMP deaminase in cell-free assays was only partial even at very high metformin concentrations (10 mmol/L). In any case, the two alternate mechanisms, involving inhibition of the respiratory chain or of AMP deaminase, would both be compatible with the results suggesting that AMPK activation (35) and adenylate cyclase inhibition (42) are mediated by increases in AMP.
To conclude this section, it now appears likely that the effects of metformin to inhibit hepatic glucose production are not mediated by AMPK but by other mechanisms such as the inhibition of adenylate cyclase and/or fructose-1,6-bisphosphatase by AMP. This does not rule out the possibility that activation of AMPK might account for other effects of metformin action, such as its ability to enhance insulin sensitivity. It might, for example, do this by stimulating fat oxidation and inhibiting fat synthesis, thus reducing triglyceride storage in liver and/or muscle.
Activation of AMPK by other natural products
In addition to galegine, a bewildering variety of other natural products—many derived from plants used as herbal medicines in Asian countries—have been reported to activate AMPK. These include resveratrol from red grapes, quercetin present in many fruits and vegetables, ginsenoside from Panax ginseng, curcumin from Curcuma longa, berberine from Coptis chinensis (used in the Chinese herbal medicine Huanglian), epigallocatechin gallate from green tea, theaflavin from black tea (45), and hispidulin from snow lotus, another plant used in Chinese herbal medicine (46). Many of these compounds are claimed to have favorable effects in type 2 diabetes and the metabolic syndrome. Although many of these compounds can be classed as polyphenols, their structures are quite variable (Fig. 4), and a puzzling feature was how such disparate structures would all be able to activate AMPK. Based on findings that berberine inhibited the respiratory chain (47), whereas resveratrol inhibited the F1 ATP synthase (48), it seemed possible that many of them activated AMPK indirectly, by inhibiting mitochondrial ATP production and thus increasing cellular AMP:ATP and/or ADP:ATP ratios, in a similar manner to the biguanides. Supporting this, AMPK activation by resveratrol, berberine, and quercetin was reduced or eliminated in cells expressing the AMP/ADP-insensitive AMPK mutant (35). Many of these natural products are secondary metabolites of plants, and some of them appear to be produced to defend plants against infection by pathogens or grazing by herbivorous animals. Production of mitochondrial poisons may be an effective deterrent, but (observing the aphorism of Paracelsus that “the dose makes the poison”) at lower doses these compounds may only cause mild inhibition of mitochondria yet still have the useful effect of activating AMPK.
FIG. 4.
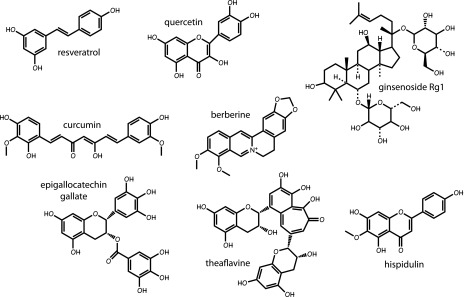
Structures of plant polyphenols that have been reported to be AMPK activators.
Small-scale pilot trials of some of these compounds have been conducted in diabetic or obese humans. Two trials of berberine in subjects newly diagnosed with type 2 diabetes (49,50) revealed favorable effects on plasma glucose, lipids, and HbA1c, although neither plasma berberine levels nor AMPK activation were assessed. Two trials of resveratrol in obese, nondiabetic males produced conflicting results. The first used a randomized, crossover design on 11 subjects given resveratrol (150 mg/day) or placebo for 30 days, and suggested that resveratrol lowered plasma glucose, insulin, and homeostasis model assessment index, as well as systolic blood pressure (51). The second used 24 male volunteers randomly assigned to resveratrol (1.5 g/day) or placebo for 4 weeks and failed to observe any significant effects on plasma glucose, insulin or homeostasis model assessment index, blood pressure, or insulin sensitivity by hyperinsulinemic-euglycemic clamp (52). Despite the 10-fold higher dose used in the second, negative study, the levels of plasma resveratrol detected were similar in both trials (1–2 μmol/L). These are around 50-fold lower than the concentrations usually used to activate AMPK in cultured cells (35), yet activation of AMPK in muscle biopsies was reported in the first study, although not in the second. Thus, it remains unclear whether clinically effective doses of these compounds are sufficiently high to activate AMPK in vivo.
Activation of AMPK by salicylate
Salicylates are natural products produced by many plants (Fig. 5), and it is now known that they are mediators released by infected plant tissues, which signal to uninfected regions to induce defense responses (53). The use of willow bark (a rich source of salicylates) was described in the 3rd millennium B.C., making it one of the oldest medicines known to humankind (54). The active component of willow bark is salicin, a β-glucosyl derivative of salicyl alcohol, the efficacy of which in rheumatic fever (an inflammatory disorder now known to be caused by streptococcal infection) was first tested by Thomas MacLagan in the author’s home town of Dundee in 1876 (55). However, the widespread use of salicylate-based drugs occurred after the development of acetyl salicylic acid, a synthetic derivative first marketed by Bayer in 1899 under the trade name aspirin (54). Another form in which salicylate can be taken orally is as the diester salsalate (Fig. 5), which has been used to treat rheumatoid arthritis. All salicylates, including salicin, aspirin, and salsalate, are rapidly converted to salicylate in vivo, with the plasma half-life of aspirin in humans being only 10–15 min, while that of the salicylate derived from it is 3–5 h (56). Salicin, aspirin, and salsalate can therefore all be regarded as prodrugs for salicylate. The one clear exception to this concerns the effect of aspirin to inhibit platelet aggregation, which it achieves by irreversible acetylation and inhibition of the cyclooxygenase COX1, thus reducing synthesis of the prothrombotic prostanoid, thromboxane A2, for the lifetime of the platelet. Most other effects of aspirin, including its anti-inflammatory effects, are most likely mediated by salicylate.
FIG. 5.

Structures of medicinal salicylates and the AMPK activator A-769662.
Salicylate, but not aspirin, was recently found to activate AMPK in cultured human cells (57). At concentrations corresponding to plasma concentrations in humans treated with high doses of aspirin or salsalate, salicylate activated wild-type AMPK and the AMP/ADP insensitive mutant equally well, showing that it worked via an AMP/ADP-independent mechanism. This suggested that it might bind directly to AMPK, which was subsequently confirmed (57). Two lines of evidence suggested that salicylate was binding at the same site as A-769662, a synthetic activator (Fig. 5) derived from a high-throughput screen (58). Thus, activation by both agents was 1) specific for AMPK complexes containing the β1 isoform and 2) abolished by a point mutation in β1. The first finding also allowed a test, by using β1-knockout mice, of whether any effects of salicylate in vivo were mediated by AMPK. AMPK is known to stimulate fat oxidation, which can be monitored in vivo by measuring the respiratory exchange ratio (RER, ratio of CO2 exhaled to O2 inhaled). When fasted mice were fed a carbohydrate meal their RER increased and then declined (as expected) when the food was withdrawn and they switched back to fat oxidation. However, the drop in RER was much more precipitous when wild-type mice were injected with salicylate or A-769662 at the time of food withdrawal, and this effect was abolished in the β1-knockout mice (57). These results confirm that the increases in whole-body fat oxidation induced by either salicylate or A-769662 were mediated by AMPK activation.
There have been numerous observational studies suggesting that metabolic parameters improved in diabetic patients who were taking salicylate-based drugs. To confirm this, Shoelson and colleagues (59) conducted two randomized controlled trials of salsalate, one for 4 weeks on obese, nondiabetic subjects and one for 12 weeks on subjects with impaired fasting glucose or impaired glucose tolerance (60). These trials showed that oral salsalate decreased fasting glucose and insulin C-peptide and increased plasma adiponectin, although in the second study the treatment did not appear to affect peripheral insulin sensitivity. It would be tempting to speculate that some of these effects of salsalate were mediated by activation of AMPK. However, salicylate treatment for 2 weeks in mice rendered insulin resistant by feeding of a high-fat diet decreased fasting glucose and insulin and improved glucose tolerance in both the wild-type and β1-knockout mice (57). This suggests that the effects of salicylate on glucose homeostasis may be AMPK independent.
Role of AMPK in cancer
The findings that the tumor suppressor LKB1 was the major upstream kinase required for AMPK activation (13–15) introduced a link between AMPK and cancer. Although it was subsequently shown that LKB1 also acted upstream of twelve other AMPK-related kinases (61,62), several indications suggest that AMPK is likely to exert most, if not all, of the tumor suppressor functions of LKB1.
AMPK activation causes a cell cycle arrest associated with stabilization of p53 and the cyclin-dependent kinase inhibitors p21WAF1 and p27CIP1 (63–65).
AMPK inhibits the synthesis of most macromolecules required for cell growth, including lipids, ribosomal RNA and proteins, the latter in part via inhibition of the mechanistic target of rapamycin complex-1 (mTORC1) (4,5).
By promoting oxidative metabolism (4,5), and inhibiting glycolysis by inhibition of mTORC1 and consequent downregulation of expression of hypoxia-inducible factor-1α (66), AMPK promotes a switch away from the rapid glycolysis observed in most tumor cells (the Warburg effect) and toward the oxidative metabolism used by most quiescent cells (67).
Consistent with the idea that AMPK exerts most of the tumor suppressor effects of LKB1, it has recently been shown that in a mouse model in which lymphomas are induced by B-cell–specific overexpression of Myc, tumor-free survival was shorter if AMPK-α1 (the only catalytic subunit expressed in B cells) was knocked out throughout the body (67).
Effect of metformin and salicylate on cancer in humans and in animal models
The original finding of a link between AMPK, LKB1, and cancer (13) led to a study involving retrospective analysis of type 2 diabetic patients, which showed that use of metformin was associated with a significantly lower incidence of cancer compared with other medications (68). This has been reproduced in several subsequent studies, with a meta-analysis indicating an overall risk reduction of 30%, with specific risk reductions being found for colon and liver cancers (69). It should be noted that these retrospective analyses merely report associations and do not prove a causal link. Some of them have also been criticized on the basis that they may be subject to time-related biases (70). However, studies in animal models support the idea that biguanides and other AMPK activators can be used to protect against tumor development and even to treat tumors once they have arisen. For example, metformin treatment delayed the onset of tumors in mice that were tumor prone as the result of heterozygous mutations in PTEN and hypomorphic mutations in LKB1 (71); interestingly, phenformin had an even more pronounced effect, as did A-769662. Since A-769662 activates AMPK by a different mechanism from the biguanides (72), this makes it likely that the delay in tumorigenesis was caused by AMPK activation rather than AMPK-independent, “off-target” effects. However, there is also evidence from another animal model that phenformin might be useful for treating cancer via AMPK-independent effects (73). This study used mice in which expression of mutant, oncogenic K-Ras could be combined with loss of either LKB1 or p53 in lung epithelial cells by inhalation of viral vectors expressing Cre recombinase. In these mice, expression of mutant K-Ras alone causes some tumors, but combination with loss of p53 or LKB1 increases the multiplicity of tumors and metastasis (74). In the new study, treatment of the mice with oral phenformin from 3 weeks after tumor initiation prolonged survival of the mice and delayed tumor progression as well as increasing expression of markers of necrosis and apoptosis in the tumors—but only in the mice in which LKB1 had been deleted (73). It was also shown by immunohistochemistry that metformin and phenformin activated AMPK in lung tumors but, as expected, only in those that still expressed LKB1. AMPK activation was greater with phenformin than metformin (possibly due its greater membrane permeability, as discussed earlier), which is why phenformin was used in the analysis of survival. In this study, phenformin is essentially acting as a cytotoxic agent that causes necrosis or apoptosis of tumor cells because, in the absence of a functional LKB1-AMPK pathway, they are more sensitive to the ATP-depleting effects of the drug. Although one of these mouse studies (71) suggests that treatment with biguanides delays onset of tumors, most likely by AMPK-dependent effects, the other (73) suggests that phenformin can be used to treat preexisting tumors that lack a functional AMPK pathway. Thus, while AMPK appears to be a tumor suppressor that protects against initial tumor development, once tumors have arisen it may, paradoxically, be easier to treat the cancer if the AMPK pathway has been lost.
Intriguingly, retrospective analysis of randomized controlled trials to study prevention of vascular events has provided evidence that aspirin also provides protection against cancer (75,76). Even though there is currently no evidence that this is caused by AMPK activation, the fact that aspirin can be regarded as a prodrug for another AMPK activator, salicylate, suggests that this possibility is worth considering.
Conclusions
The metabolic effects of AMPK activation, especially its ability to cause a metabolic switch from fat synthesis to fat oxidation and to promote muscle glucose uptake, would be expected to be beneficial in individuals with insulin resistance and/or type 2 diabetes. AMPK is activated by biguanide drugs (metformin and phenformin) and by salicylate, the major breakdown product of aspirin and salsalate. Metformin activates AMPK indirectly by inhibiting mitochondrial function, whereas salicylate binds directly to AMPK. Metformin is already the drug of first choice for treatment of type 2 diabetes, while salsalate has shown promise in randomized, controlled trials in subjects with obesity or prediabetes. However, due to their small size these drugs will only form low-affinity interactions with proteins and are likely to bind to multiple targets in vivo. An increasing number of AMPK-independent effects of metformin and salicylates are being documented, including the effects of metformin on hepatic glucose production and of salsalate on glucose homeostasis. None of this dilutes the attractiveness of AMPK as a target for novel therapies, particularly because of its ability to cause a metabolic switch from fat synthesis to fat oxidation. The Abbott compound A-769662, which appears to bind at the same site as salicylate (57) and produces favorable changes in metabolic parameters in ob/ob mice (58), represents a model for compounds that are direct activators of AMPK. At the time of writing, nearly 60 patents have been filed for small-molecule activators of AMPK, and it is hoped that some of these may enter human clinical trials soon. It seems likely that by the end of this decade we will have a much clearer picture of whether drugs that are more selective activators of AMPK than metformin or salicylate will have a place in the treatment of type 2 diabetes or cancer.
ACKNOWLEDGMENTS
Studies in the author’s laboratory are funded by a Senior Investigator Award (097726) from the Wellcome Trust and by a Programme Grant (C37030/A15101) from Cancer Research U.K.
No potential conflicts of interest relevant to this article were reported.
D.G.H. researched data and wrote the manuscript.
REFERENCES
- 1.Hardie DG, Carling D, Sim ATR. The AMP-activated protein kinase—a multisubstrate regulator of lipid metabolism. Trends Biochem Sci 1989;14:20–23 [Google Scholar]
- 2.Hawley SA, Davison M, Woods A, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 1996;271:27879–27887 [DOI] [PubMed] [Google Scholar]
- 3.Davies SP, Sim ATR, Hardie DG. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem 1990;187:183–190 [DOI] [PubMed] [Google Scholar]
- 4.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007;8:774–785 [DOI] [PubMed] [Google Scholar]
- 5.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13:251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 2004;18:3004–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009;8:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao B, Sanders MJ, Underwood E, et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011;472:230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao B, Heath R, Saiu P, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007;449:496–500 [DOI] [PubMed] [Google Scholar]
- 10.Scott JW, Hawley SA, Green KA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 2004;113:274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies SP, Helps NR, Cohen PTW, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C α and native bovine protein phosphatase-2AC. FEBS Lett 1995;377:421–425 [DOI] [PubMed] [Google Scholar]
- 12.Oakhill JS, Steel R, Chen ZP, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011;332:1433–1435 [DOI] [PubMed] [Google Scholar]
- 13.Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2003;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003;13:2004–2008 [DOI] [PubMed] [Google Scholar]
- 15.Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 2004;101:3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2005;2:9–19 [DOI] [PubMed] [Google Scholar]
- 17.Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2005;2:21–33 [DOI] [PubMed] [Google Scholar]
- 18.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 2005;280:29060–29066 [DOI] [PubMed] [Google Scholar]
- 19.Stahmann N, Woods A, Carling D, Heller R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol Cell Biol 2006;26:5933–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamás P, Hawley SA, Clarke RG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med 2006;203:1665–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011;146:992–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vara D, Salazar M, Olea-Herrero N, Guzmán M, Velasco G, Díaz-Laviada I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death Differ 2011;18:1099–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Neill HM, Maarbjerg SJ, Crane JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 2011;108:16092–16097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley DE, Goodpaster BH, Storlien L. Muscle triglyceride and insulin resistance. Annu Rev Nutr 2002;22:325–346 [DOI] [PubMed] [Google Scholar]
- 26.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 27.McGee SL, van Denderen BJ, Howlett KF, et al. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008;57:860–867 [DOI] [PubMed] [Google Scholar]
- 28.Pehmøller C, Treebak JT, Birk JB, et al. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab 2009;297:E665–E675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, Mackintosh C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J 2008;409:449–459 [DOI] [PubMed] [Google Scholar]
- 30.Jørgensen SB, Viollet B, Andreelli F, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 2004;279:1070–1079 [DOI] [PubMed] [Google Scholar]
- 31.Musi N, Fujii N, Hirshman MF, et al. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 2001;50:921–927 [DOI] [PubMed] [Google Scholar]
- 32.Bailey CJ, Campbell IW, Chan JCN, Davidson JA, Howlett HCS, Ritz P, Eds. Metformin: The Gold Standard. Chichester, U.K., John Wiley & Sons, 2007 [Google Scholar]
- 33.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000;348:607–614 [PMC free article] [PubMed] [Google Scholar]
- 34.El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000;275:223–228 [DOI] [PubMed] [Google Scholar]
- 35.Hawley SA, Ross FA, Chevtzoff C, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 2010;11:554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;108:1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mooney MH, Fogarty S, Stevenson C, et al. Mechanisms underlying the metabolic actions of galegine that contribute to weight loss in mice. Br J Pharmacol 2008;153:1669–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 1995;229:558–565 [DOI] [PubMed] [Google Scholar]
- 39.Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 2000;49:896–903 [DOI] [PubMed] [Google Scholar]
- 40.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005;310:1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010;120:2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013;494:256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vincent MF, Marangos PJ, Gruber HE, Van den Berghe G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 1991;40:1259–1266 [DOI] [PubMed] [Google Scholar]
- 44.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 2011;286:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang JT, Kwon DY, Yoon SH. AMP-activated protein kinase: a potential target for the diseases prevention by natural occurring polyphenols. New Biotechnol 2009;26:17–22 [DOI] [PubMed] [Google Scholar]
- 46.Lin YC, Hung CM, Tsai JC, et al. Hispidulin potently inhibits human glioblastoma multiforme cells through activation of AMP-activated protein kinase (AMPK). J Agric Food Chem 2010;58:9511–9517 [DOI] [PubMed] [Google Scholar]
- 47.Turner N, Li JY, Gosby A, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008;57:1414–1418 [DOI] [PubMed] [Google Scholar]
- 48.Gledhill JR, Montgomery MG, Leslie AG, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci USA 2007;104:13632–13637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Li X, Zou D, et al. Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. J Clin Endocrinol Metab 2008;93:2559–2565 [DOI] [PubMed] [Google Scholar]
- 50.Yin J, Xing H, Ye J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism 2008;57:712–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 2011;14:612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poulsen MM, Vestergaard PF, Clasen BF, et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013;62:1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thaler JS, Humphrey PT, Whiteman NK. Evolution of jasmonate and salicylate signal crosstalk. Trends Plant Sci 2012;17:260–270 [DOI] [PubMed] [Google Scholar]
- 54.Jeffreys D. Aspirin: The Remarkable Story of a Wonder Drug. London, Bloomsbury Publishing, 2004 [Google Scholar]
- 55.Maclagan TJ. The treatment of acute rheumatism by salicin. Lancet 1876;1:342–383 [PubMed] [Google Scholar]
- 56.Miners JO, Grgurinovich N, Whitehead AG, Robson RA, Birkett DJ. Influence of gender and oral contraceptive steroids on the metabolism of salicylic acid and acetylsalicylic acid. Br J Clin Pharmacol 1986;22:135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawley SA, Fullerton MD, Ross FA, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012;336:918–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006;3:403–416 [DOI] [PubMed] [Google Scholar]
- 59.Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 2008;31:289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldfine AB, Conlin PR, Halperin F, et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia 2013;56:714–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lizcano JM, Göransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 2004;23:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jaleel M, McBride A, Lizcano JM, et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett 2005;579:1417–1423 [DOI] [PubMed] [Google Scholar]
- 63.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun 2001;287:562–567 [DOI] [PubMed] [Google Scholar]
- 64.Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005;18:283–293 [DOI] [PubMed] [Google Scholar]
- 65.Liang J, Shao SH, Xu ZX, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007;9:218–224 [DOI] [PubMed] [Google Scholar]
- 66.Shackelford DB, Vasquez DS, Corbeil J, et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci USA 2009;106:11137–11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faubert B, Boily G, Izreig S, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 2013;17:113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005;330:1304–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Decensi A, Puntoni M, Goodwin P, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461 [DOI] [PubMed] [Google Scholar]
- 70.Suissa S, Azoulay L. Metformin and the risk of cancer: time-related biases in observational studies. Diabetes Care 2012;35:2665–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang X, Wullschleger S, Shpiro N, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J 2008;412:211–221 [DOI] [PubMed] [Google Scholar]
- 72.Göransson O, McBride A, Hawley SA, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem 2007;282:32549–32560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shackelford DB, Abt E, Gerken L, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013;23:143–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007;448:807–810 [DOI] [PubMed] [Google Scholar]
- 75.Rothwell PM, Price JF, Fowkes FG, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012;379:1602–1612 [DOI] [PubMed] [Google Scholar]
- 76.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012;379:1591–1601 [DOI] [PubMed] [Google Scholar]