

Abstract
Rationale
Rossdeutsch et al. describe a requirement for Thymosin β4 (Tβ4) in vascular development. Impaired mural cell migration, differentiation, partial embryonic lethality, and hemorrhaging were observed following analysis of two lines of mice, one of which was germline null for Tβ4, and another in which Tβ4 was knocked down by endothelial specific expression of Tβ4 shRNA. These data are in direct contrast to our published global and cardiac specific Tβ4 knockout lines. Thus the role of Tβ4 needs to be clarified to understand its importance in cardiovascular development.
Objective
To investigate and clarify the role of Tβ4 in vascular smooth muscle cell development and vessel stability.
Methods and Results
Examination of Tβ4 global knockouts did not demonstrate embryonic hemorrhaging, altered mural cell development or lethality. Endothelial specific knockouts also did not exhibit any embryonic lethality and were viable to adulthood.
Conclusions
Analysis of our Tβ4 global and cardiac- and endothelial-specific knockout models demonstrated that Tβ4 is dispensable for embryonic viability and vascular development.
Keywords: Thymosin Beta 4, Cardiac Development, Vascular Biology, Vascular Smooth Muscle
Introduction
Understanding signaling factors that regulate formation of the vasculature can lend significant insight into both development and disease. Thymosin β4 (Tβ4) is a 43 amino acid factor initially found to interact with G-actin and regulate F-actin formation1. Recent studies have indicated that administration of Tβ4 to either post-ischemia reperfusion or myocardial infarction models can improve cardiovascular function and abrogate scar formation, primarily through de novo vasculature formation2-4. Early studies using an shRNA knockdown approach suggested that Tβ4 acted as a key regulator of blood vessel formation (angiogenesis, vasculogenesis and arteriogenesis)5. However, our studies using both global and cardiac specific knockout approaches found Tβ4 to be dispensable for embryonic viability and vessel development6.
In a recent paper, Rossdeutsch et al. describe a requirement for Tβ4 in vascular development7. Impaired mural cell migration and differentiation was observed following analysis of two lines of mice, one of which was germline null for Tβ4, and another in which Tβ4 was knocked down by endothelial specific expression of Tβ4 shRNA. Rossdeutsch et al. report that global knockout of Tβ4 resulted in partial lethality at the start of the study that was decreased in subsequent generations later during the study, an incompletely penetrant hemorrhagic phenotype and decreased mural cell overage of the aorta7. Tβ4 knockdown in endothelium also resulted in partial lethality and decreased mural cell coverage7. These data are in direct contrast to our published Tβ4-knockout model6. Thus, to clarify the role of Tβ4 in vascular formation we have further analyzed our global knockouts of Tβ4 and an endothelial-specific Tβ4 knockout not previously reported. Consistent with our previous results, we did not observe an evident phenotype, alteration in vascular development or embryonic lethality. We conclude from these data that Tβ4 is not required for embryonic viability or vascular development.
Materials and Methods
Animal Care
All animal procedures were performed and approved by the UCSD Animal Care and Use Committee.
Generation of tβ4 Floxed, Knockout and Endothelial-Specific Knockout mice
tβ4-targeted mice were generated and used as previously described to a C57/B6J background6. Endothelial-specific knockout mice were generated by crossing tβ4f/f female mice to Tie2-Cre male mice.
Immunofluorescence Analyses
Frozen sections from E14.5, E13.5 and E12.5 embryos were isolated and stained as previously described6.
Results
Mural Cell Coverage and Development Are Not Altered in Global Null tβ4 Mutants
To clarify the role of Tβ4 in large vessel development, we examined our global tβ4- knockout mice from E12.5 to E14.5 (Figures 1-2). Initial observation in these stages revealed no gross abnormalities or hemorrhage between wild type and knockout samples. To examine large vessel defects we first examined aortas in E14.5 embryos (Figure 1). At this stage, no differences were observed between examined wild type and knockout embryos. To rule out developmental and mural cell recruitment defects, E12.5 and E13.5 aortas were stained with antibodies to αSMA and PDGFRβ, two markers of vascular smooth muscle cells (Figures 2&3) 6-8. No differences were observed between control and Tβ4 null embryos.
Figure 1. tβ4-loss does not alter developing vasculature at E14.5.
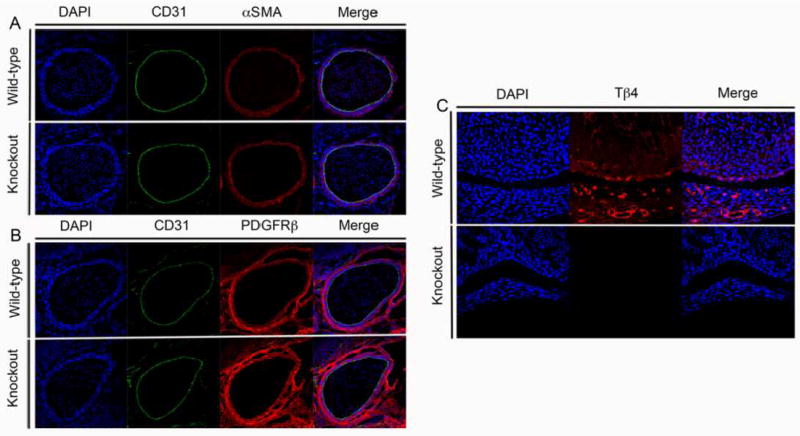
A&B∷ E14.5 Wildtype and Tβ4 knockout aorta. A:Blue Dapi, Green CD31 Red αSMA or B: Blue Dapi, Green CD31, Red PDGFRβ. 40X C: 13.5 Wildtype and Tβ4 knockout left ventricle. Blue Dapi, Red Tβ4. 20X
Figure 2. tβ4-loss does not alter developing vasculature at E12.5 or E13.5.
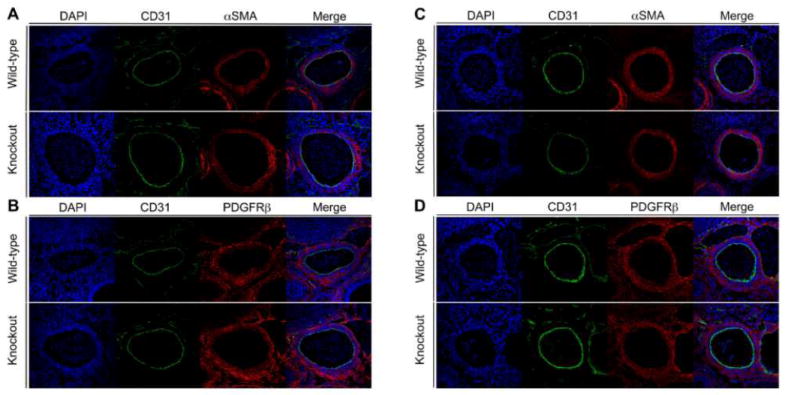
A&B: E12.5 Wildtype and Tβ4 knockout aortae stained in A:Blue Dapi, Green CD31 Red αSMA or B: Blue Dapi, Green CD31, Red PDGFRβ. C&D: E13.5 Wild type and Tβ4 knockout aorta stained in C:Blue Dapi, Green CD31 Red αSMA or D: Blue Dapi, Green CD31, Red PDGFRβ. 40X
Endothelial-Specific Deletion of tβ4 Does Not Result in Embryonic Lethality
Rossdeutsch et al. observed 12% lethality in embryos with endothelial specific shRNA mediated knockdown of Tβ4. As stated in previous publications, shRNA knockdown can have off-target effects6,9. To further investigate this possibility, we used the same Tie2-Cre employed by Rossduetsch et al.7,10. Our tβ4f/f females6 were bred to Tie2-Cre male mice11. Male Tie2-Cre mice were also utilized by Rossdeutsch et al. in their knockdown studies. Out of 34 postnatal male mice, 18 were tβ4f/y and 16 were tβ4f/y;Tie2-Cre+, demonstrating lack of embryonic lethality in endothelial specific knockouts of Tβ4. These data are similar to our global and cardiac specific knockouts of Tβ46; where we did not observe any postnatal lethality or vascular phenotype.
Discussion
Rossdeutsch and co-workers report that global tβ4-knockout mice present with hemorrhage and defective mural cell recruitment, resulting in partial lethality between E10.5 and E14.57. These data are in direct contrast to our published global tβ4-knockout model6. In the Discussion section of their manuscript, Rossdeutsch and co-workers refer to our paper7, and state that, from our data,“Tβ4 knockout aortas had an apparent reduction in α-SMA+ cell coverage within the vessel wall relative to wild-type controls at E14.5”7. We believe this to be a misinterpretation of our data. In our publication6 we did not report, nor observe, differences in mural cell coverage in any of our samples at E14.5. Data showing αSMA staining of aortic vascular smooth muscle were meant to illustrate maintenance of vascular smooth muscle within the aorta, and were not meant to be a quantitative assessment. However, given the observations by Rossdeutsch et al, we have specifically examined aortas in multiple sections from E12.5 to E14.5 Tβ4 null and control embryos and found no differences between control and Tβ4 null embryos (Figures 1 and 2). We also did not observe any hemorrhaging in any of the embryos in our studies. Thus, our data do not support an essential role for Tβ4 in vessel development.
Rossdeutsch et al. also suggest that differences in observed phenotypes between our Tβ4 null mutants and theirs can be attributed to either strain background dependent effects or distinct gene targeting strategies7. Initially, Rossdeutsch et al. observed 40% lethality of their global null Tβ4 mice in a mixed C57/B6J and 129Sv background, with reduction to 20% lethality when bred into a pure C57/B6J background. However, our global null mice were bred into a C57/B6J background and yet did not display any phenotype6. It is possible that differences in observed phenotypes between the two Tβ4 null mutant lines may result from variables such as environment, diet, and infectious state which may differ between the two mouse facilities.
An alternative explanation to strain or facility-dependent phenotype is that the targeted ES cells used to establish the Rossdeutsch mutant mouse line contained a Tβ4- independent lethal mutation(s) that may have been bred out over successive generations. Moreover, the possibility raised by Rossdeutsch et al. that differences in targeting strategies, in particular the retention of the Neo cassette in the Rossdeutsch et al. targeted allele, could also account for discrepant phenotypes is a possibility. However, if this is the case, it is unlikely to reflect differences in targeting Tβ4 function, as exon 2, encompassing 75% of the coding sequence, is deleted in both knockouts and similarly Tβ4 protein is absent, as demonstrated by protein analysis, in both knockouts. Therefore, if the different phenotypes are a result of retention of the Neo gene in the Rossdeutsch Tβ4 null allele, observed phenotypes may result from non-Tβ4 related functions.
Rossdeutsch et al. argue that there are no “off-target” effects with their shRNA gene silencing approach and state “the differences between our global knockout and knockdown model are probably attributed to the fact that RNAi targeting in vivo, when sufficiently optimal to abrogate expression of the target gene, can result in a more severe phenotype than a corresponding global-null. Genetic ablation via homologous recombination through the germline, leading to complete loss of function from the outset in development, may be partially compensated for by functional orthologues, whereas RNAi-mediated efficient knockdown, occurring rapidly and at a defined developmental stage, may not be permissive for compensation.”7 Our previously published cardiac specific (Nkx2.5-Cre, αMHC-Cre), 6 and endothelial cell specific (Tie2-Cre) (reported here) ablations of our Tβ4 floxed alleles, have demonstrated that targeted deletion during development does not result in an evident phenotype, thus calling into question this argument by Rossdeutsch et al. to address discrepancies between their global Tβ4 null and their conditional shRNA-Tβ4 knockdown phenotypes.
In summary, our data with global and conditional Tβ4 knockout mice suggest that the shRNA knockdown approach used by Rossdeutsch et al may have ‘off-target’ effects which result in observed phenotypes. The phenotype observed in their global Tβ4 knockout mice may result from perturbation of Tβ4-independent events. Together, these data do not support an essential role for Tβ4 in vessel development or embryonic viability.
Acknowledgments
Source of Funding
J.C. and S.M.E are funded by grants from the National Heart, Lung, and Blood Institute. J.C. is the American Heart Association Endowed Chair in Cardiovascular Research. T.M-M. and I.B. are supported by AHA postdoctoral fellowships (11POST7310066) and (12POST12030256). Microscopy work was performed at the UCSD Neuroscience Microscopy Shared Facility and was supported by NIH grant (P30 NS047101).
Non-standard Abbreviations and Acronyms
- αSMA
α-smooth muscle actin
- CD31
Platelet endothelial cell adhesion molecule
- ES
Embryonic Stem (Cells)
- PDGFRβ
Platelet-derived growth factor subunit b
- ShRNA
short hairpin RNA
- Tβ4
thymosin beta 4
Footnotes
Disclosures
None
References
- 1.Fan Y, Gong Y, Ghosh PK, Graham LM, Fox PL. Spatial coordination of actin polymerization and ILK-Akt2 activity during endothelial cell migration. Dev Cell. 2009;16:661–74. doi: 10.1016/j.devcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bock-Marquette I, Saxena A, White MD, Dimaio JM, Srivastava D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432:466–472. doi: 10.1038/nature03000. [DOI] [PubMed] [Google Scholar]
- 3.Hinkel R, El-Aouni C, Olson T, Horstkotte J, Mayer S, Müller S, Willhauck M, Spitzweg C, Gildehaus FJ, Münzing W, Hannappel E, Bock-Marquette I, DiMaio JM, Hatzopoulos AK, Boekstegers P, Kupatt C. Thymosin beta4 is an essential paracrine factor of embryonic endothelial progenitor cell-mediated cardioprotection. Circulation. 2008;117:2232–2240. doi: 10.1161/CIRCULATIONAHA.107.758904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock-Marquette I, Shrivastava S, Pipes GC, Thatcher JE, Blystone A, Shelton JM, Galindo CL, Melegh B, Srivastava D, Olson EN, DiMaio JM. Thymosin beta4 mediated PKC activation is essential to initiate the embryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J Mol Cell Cardiol. 2009;46:728–38. doi: 10.1016/j.yjmcc.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smart N, Risebro CA, Melville AA, Moses K, Schwartz RJ, Chien KR, Riley PR. Thymosin beta 4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445:177–82. doi: 10.1038/nature05383. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee I, Zhang J, Moore-Morris T, Lang S, Shen T, Dalton ND, Gu Y, Peterson KL, Evans SM, Chen J. Thymosin Beta 4 is dispensable for murine cardiac development and function. Circ Res. 2012;110:456–464. doi: 10.1161/CIRCRESAHA.111.258616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossdeutsch A, Smart N, Dubé KN, Turner M, Riley PR. Essential role for Thymosin β4 in regulating vascular smooth muscle cell development and vessel wall stability. Circ Res. 2012;111:e89–e102. doi: 10.1161/CIRCRESAHA.111.259846. [DOI] [PubMed] [Google Scholar]
- 8.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–621. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- 9.Mohr SE, Perrimon N. RNAi screening: new approaches, understandings, and organisms. Sci Rep. 2012;2:428. doi: 10.1002/wrna.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]