

Abstract
By most standard engineering practice principles, it is premature to credibly discuss the “engineering” of a human cornea. A professional design engineer would assert that we still do not know what a cornea is (and correctly so), therefore we cannot possibly build one. The proof resides in the fact that there are no clinically viable corneas based on classical tissue engineering methods available. This is possibly because tissue engineering in the classical sense (seeding a degradable scaffolding with a population synthetically active cells) does not produce conditions which support the generation of organized tissue. Alternative approaches to the problem are in their infancy and include the methods which attempt to recapitulate development or to produce corneal stromal analogs de novo which require minimal remodeling. Nonetheless, tissue engineering efforts, which have been focused on producing the fundamental functional component of a cornea (organized alternating arrays of collagen or “lamellae”) may have already provided valuable new insights and tools relevant to development, growth, remodeling and pathologies associated with connective tissue in general. This is because engineers ask a fundamentally different question (How can that be done?) than do biological scientists (How is that done?). The difference in inquiry has prompted us to closely examine (and to mimic) development as well as investigate collagen physicochemical behavior so that we may exert control over organization both in cell-culture (in vitro) and on the benchtop (de novo). Our initial results indicate that reproducing corneal stroma-like local and long-range organization of collagen may be simpler than we anticipated while controlling spacing and fibril morphology remains difficult, but perhaps not impossible in the (reasonably) near term.
1. Introduction
1.1 Tissue Engineering
The term “tissue engineering” is thought to have been officially coined and defined in its modern sense by Robert Nerem in1988 at the Lake Granlibakken NSF workshop on the topic of engineering tissue(Viola et al., 2003). However, the concept was brought to widespread attention and formalized in a review paper in Science in 1993 (Langer and Vacanti, 1993) which paraphrased the definition: Tissue engineering is an interdisciplinary field that applies the principles of engineering and the life sciences toward the development of biological substitutes that restore, maintain or improve tissue function.
In the subsequent years since the field of tissue engineering was formalized, there has been an enormous scientific effort to produce tissue constructs for clinical use (Vacanti, 2006). The success of the approach has thus far been modest in relation to the expenditure of time and resources (Nerem, 2006) with few tissue engineered constructs approved for clinical use in the US (the most successful of these being artificial skin). The myriad limitations of current tissue engineering methods are enumerated by Dr. Vacanti in a recent article in the journal Tissue Engineering (Vacanti, 2006). Nonetheless, tissue engineering in general does hold great promise and the potential for producing tissue engineered replacements for diseased or injured corneas in the reasonably near term does exist. This is because corneal tissue is reasonably “simple”, thin and avascular (Ko et al., 2007). However, all tissues are extremely complex at some level (in the cornea it is at the level of matrix molecule organization). Thus, it is critical that our approach to this endeavour be circumspect and tempered with the appropriate scientific skepticism.
The term “engineering” as it relates to tissue engineering
An engineer is defined as “One who contrives, designs, or invents” (first definition, OED second edition 1989). In the sense of this definition, the term “tissue engineering” seems a perfectly reasonable description for what has been attempted in the field since its inception. However, in practice, the gerund “engineering” carries the implication that a particular task can be accomplished given the currently available knowledge-base, adequate funding and adequate manpower. Indeed professional engineers usually have a large toolbox of established principles (i.e. boiler codes, steam tables, mechanical property indices etc. for mechanical engineers) from which to draw upon to produce rationally designed solutions to practical problems. Unlike practitioners of most engineering disciplines (mechanical, chemical, civil and electrical), tissue engineers have a relatively empty toolbox at the moment. It is thus no surprise that things are progressing slowly and have been largely empirical in nature (because we are busy filling the toolbox!). Unfortunately, some declared attempts to produce “engineered” constructs as solutions to complex and poorly understood physiological/pathological problems are very premature. The problem resides with the semantic connotation of the term “tissue engineering”, which carries the implication of “doability” and may create expectations that cannot be met. The consequences of this semantics “gap” between what most tissue engineers actually do and what individuals outside the field expect of them, have yet to be fully realized (and may be dire with regard to future funding of the field). Certainly, what our laboratories have been doing in the past few years is not engineering corneas or even corneal stromas. We have been relegated to “filling the toolbox” for future corneal tissue engineers.
1.2 Motivation
Corneal disease and/or injury is the second leading cause of vision loss and affects more than 10 million people worldwide (Whitcher et al., 2001). Trachoma is the leading cause of corneal blindness in the third world (4.5 million people) followed by corneal injury (1.5 million people). The vast majority of these cases might benefit from a suitable corneal replacement (provided there was adequate follow up care – which is no small concern). In the developed world, conditions which indicate the use of donor corneal grafts include Fuch’s dystrophy, keratoconus, pseudo and aphakic bullous keratopathy, corneal stromal dystrophies, corneal scarring and herpes simplex virus. There were approximately 33,000 corneal transplants per year performed in the United States as of the year 2000 (Aiken-O’neill and Mannis, 2002). Though the donor cornea potential far outstrips the actual need of grafts, there are legitimate cultural, logistical and technical difficulties associated with the procurement of quality donor graft material. Under the best conditions, donor grafts are typically variable in quality and usually fail due to immunological rejection or endothelial decompensation resulting in an 18% failure rate for initial grafts (Thompson et al., 2003). In addition, LASIK surgery constitutes a rising threat to the number of viable donor corneas (LASIK procedures disqualify donor tissue for transplantation).
Therefore, aside from the academic challenge, there is appreciable clinical interest in the development of a suitable replacement for donor graft material. The cornea is a fairly simple, avascular, sparsely-populated and multilaminar structure, which would seem to be an attractive target for tissue engineers. The fact that the cornea is thin and avascular is encouraging as it does not appear that significant diffusion limitations (a particularly vexing problem for tissue engineers in general (Vacanti, 2006)) will hamper the effort. However, the cornea is more complex than is apparent at first glance. There are three generally different cell types (epithelial, fibroblasts/keratocytes and endothelial) which require successful and compatible culturing techniques if a cornea is to be constructed by tissue engineering methods. In addition, at the heart of the cornea lies a complex, highly-organized stromal extracellular matrix which provides the principal functions of the corneal tissue. Before the design of any engineered replacement for a tissue (or for any device) can begin, it is important to fully understand the function (and structure) of what it is that is to be replaced.
1.3 Understanding corneal form and function
As part of the design process for the engineering of a tissue replacement, it is first necessary to attain a detailed understanding of the function that is performed by the native tissue in the context of its physiological environment. Our focus for this article is the corneal stroma. Nonetheless, it is instructive to review the functional role that the stroma plays within the whole cornea and the functional role of the cornea within the visual system. We will start with a brief review of corneal structure and function. There are three principal design requirements that the natural cornea must satisfy: 1) protection of the fragile intraocular contents, 2) transparency to visible light and 3) formation of a nearly perfect optical interface to refract light onto the retina. Over the millennia, Nature has evolved an elegant structure which is capable of simultaneously meeting all three of these critical design criteria.
1.3.1 Corneal Structure
Gross anatomy
The cornea comprises a highly-structured, membrane bound but relatively acellular, transparent collagenous tissue that joins the more disorganized and opaque sclera at the limbus. The diameter of the human cornea is about 12 mm and the average radius of curvature of the central anterior surface is 7.8 mm. The cornea is roughly 520 microns thick at the center and 650 microns thick in the periphery. It is bounded anteriorly by a stratified squamous epithelium and posteriorly by an actively pumping monolayer of non-proliferative cells which are referred to as the corneal endothelium. At the heart of the cornea is the stromal tissue which comprises 90% of the total thickness where the three principal design requirements of the cornea (protection, transmission and refraction) are simultaneously satisfied by the long and short-range extracellular matrix organization.
Microscopic anatomy
The three major functional layers of the cornea are the epithelium, stroma and endothelium. Figure 1 shows the orderly structure of the corneal tissue in cross section.
Figure 1.

The cornea (approximately 500 microns thick in humans) is a layered structure comprising the epithelium (E), Bowman’s layer (Bw), stroma (St), Descemet’s membrane (De), and the endothelium (En). (with permission from Klyce and Beuerman, 1997)
Epithelium
The corneal epithelium is a 50 micron thick, “tight”, stratified, squamous, multilaminar epithelium comprising three distinct cellular strata. The stratum germinatum is the posteriormost layer and the only epithelial cell layer capable of undergoing mitosis (Hanna and O’brien, 1960). The middle layer comprises daughter or wing cells which are pushed anteriorly during epithelial desquamation. The surface layer of squamous cells forms the complete tight junctions which generate the primary chemical and antigen protective barrier in the cornea. This continual desquamation of the corneal epithelium depends on a stable supply of stem cells which reside in limbal niches at the junction between the sclera and the cornea (Schermer et al., 1986). A healthy epithelium has 5 to 7 layers of cells and rests on a basement membrane comprising laminin, type IV collagen, identifiable hemidesmosomes and anchoring fibrils.
That the cornea requires a functional epithelium is demonstrated by pathological conditions which chronically disrupt the ocular surface mucosa (ocular cicatricial pemphigoid, Stevens-Johnson syndrome etc), disrupt tear production (Sjogren’s) or by injuries which destroy the stem cell niches in the limbus (severe chemical or alkali burns). Losing the epithelial barrier typically results in corneal opacity and the loss of the surface air/tear interface for refraction. Natural or tissue engineered grafts cannot succeed without a functional epithelium.
Nonetheless, we assert that the epithelium is there to protect the stroma from invasion (and to maintain the tear/air interface). It is the stromal architecture that produces the necessary aberration-free curvature to generate the refractive power. In addition, given the significant advances in corneal epithelial culturing onto various substrates (Zieske et al., 1994), and the fact that donor corneas (which are de-epithelialized) gain epithelial coverage from the host, the epithelium is not currently a critical concern for corneal tissue engineers. It is important to mention the fact that there is a considerable effort underway to innervate engineered corneal constructs (Li et al., 2003; Li et al., 2005). The rationale for this approach is derived from data which suggest that denervation of the corneal stroma leads to poorly formed epithelium without proper cellular stratification (Alper, 1975).
Endothelium
The corneal endothelium is a transporting monolayer of about 400,000 hexagonal cells, 20 microns across and 4–6 microns in height. The endothelium maintains corneal transparency by keeping the corneal stroma in a state of relative deturgescence via a complex pump-leak mechanism ((Maurice, 1972) for pump discovery and see (Bonanno, 2003) for extensive review of endothelial ion transport). Without the endothelium the cornea has a natural tendency to imbibe fluid which can cause swelling, opacity and blindness. This dehydrating mechanism is part of a sophisticated corneal transport system which includes both limiting layers and is described in detail in Ruberti and Klyce (Ruberti and Klyce, 2002). As with the epithelium, an engineered construct that is perfectly mimetic of the natural stroma could not function without a patent, active endothelial layer. However, it may be quite possible to design a collagen-based cornea that does not swell by incorporating an analog to the sutural fibers in the dogfish (Smelser, 1962). Nonetheless, an endothelium is a far greater concern for tissue engineers than the epithelium, as it has been refractory to expansion in culture and the host endothelium will not effectively repopulate and deturgesce a bare donor stromal graft. Recently, researchers have been able to cultivate untransformed endothelial cells and expand them multiple times in a specialized culture medium (Engelmann et al., 1999; Engelmann et al., 2004; Joyce and Zhu, 2004; Mcalister et al., 2005). Given these relatively successful parallel efforts associated with culturing and expanding endothelial cells in vitro, the endothelium does not appear to present a “significant” hurdle to corneal tissue engineering effort at this time. Ultimately, continued efforts to expand primary endothelial cells in culture will be critical to the success of engineered corneas.
Stroma
The adult human stroma is approximately 500 microns thick, relatively acellular (3–10% quiescent corneal keratocytes by volume), and comprises aligned arrays of hydrated type I/V heterotypic collagen fibrils (15% wet weight) of uniform diameter (32±0.7 nm) (Meek and Leonard, 1993); glycosaminoglycans (GAGs) keratan sulfate and dermatan sulfate (1% wet weight (Anseth, 1961)); various proteoglycan (PG) core proteins (Axelsson and Heinegard, 1975) and other protein constituents including fibronectin, laminin and type VI collagen (among other collagens). The collagen fibrils are packed in 300 to 500 parallel arrays (lamellae) which are generally parallel to the corneal surface (Hamada et al., 1972) and are principally responsible for the observed tensile mechanical properties of the cornea (reviewed in (Ethier et al., 2004)). The PGs and their associated GAGs contribute to the cornea’s compressive and swelling material properties (Hedbys, 1961) and to the uniform spacing of the collagen fibrils (Scott, 1991). We believe that the corneal stroma should be the current focus of researchers attempting to produce a corneal tissue analog and we have concentrated on this part of the cornea for a number of reasons: First, there have been very few concerted efforts aimed at reproducing the architecture of a natural corneal stroma (Orwin et al., 2003; Crabb et al., 2006; Crabb et al., 2006; Guo et al., 2007), while there have been multiple and relatively successful attempts to culture the limiting cell layers of the cornea (on a stromal scaffolding)(Minami et al., 1993; Zieske et al., 1994; Germain et al., 1999; Griffith et al., 1999; Griffith et al., 2002; Li et al., 2003; Germain et al., 2004; Li et al., 2005) fully reviewed in (Ruberti et al., 2007). Second, the corneal stroma provides the majority of the principal functions of the corneal tissue. No corneal analog will work without a mechanically strong and clear, properly shaped stroma. Third, the stroma is extremely organized on the nanoscale making it a very difficult and interesting basic materials engineering problem. Finally, because of the highly-organized nature of the stromal matrix, successful reproduction of the architecture requires detailed examination of in vivo/in vitro matrix assembly (from development to scar resolution) and the development of novel engineering methods to control collagen fibrillogenesis. Such methods have broader implications for connective tissue remodelling, homeostasis and pathology.
Note on the tear film
The tear film is a complex, multicomponent structured ocular surface coating which provides both a smooth optical interface and protection from pathogens. It is the secretary product of three-separate systems (conjunctival goblet cells, meibomian glands and lacrimal glands) none of which are part of the cornea proper. Therefore we will not consider the tear film in this Chapter; although we note that it is a critical component of the ocular surface, without which neither donor corneal grafts, nor tissue engineered replacements would survive.
1.3.2 Corneal Function
The native cornea provides three fundamental functional attributes (which we consider essential design requirements for an artificial corneal construct) to the ocular optical system: protection, transmission and refraction of the incident light to the retina. The mechanisms by which the tissue simultaneously meets all three of these requirements are discussed below.
Protection
The cornea provides both transport protection (in the form of a barrier) and mechanical protection.
Transport protection
The transport of deleterious chemicals and pathogens is impeded by the tight junctions of superficial squamous cells of the corneal epithelium (Sugrue and Zieske, 1997). Thus, any natural, stromal analog, should be inductive for the migration of epithelial cells from the host peripheral corneal tissue and support the formation of a mulitlaminar, adherent epithelium with complete tight junctions (as donor corneas do (Boot et al., 1991)).
Mechanical protection
Protection of the fragile intraocular contents is provided by the tensile properties of the stromal extracellular matrix. The tensile mechanical strength of the tough ocular tunic (of which the cornea is a continuous part) must be high enough to withstand chronic tensile stress induce by the intraocular pressue (IOP) and permit survival of significant traumatic impacts without rupture. The overall biomechanical properties of the cornea proper are complex because the stromal tissue is highly anisotropic, heterogeneous and viscoelastic. A full review of corneal mechanics is provided in Ethier et al 2004 ((Ethier et al., 2004). Biomechanical properties are typically derivative of the orientation of collagen fibrils in load-bearing connective tissue. This is also true for the cornea when one is considering the tensile modulus in the direction of tangential loads. As might be expected, in the human corneal stroma, the preferred collagen fibril orientation as determined by X-ray (Aghamohammadzadeh et al., 2004; Meek and Boote, 2004) and SEM (Radner et al., 1998; Radner and Mallinger, 2002) (Figure 2a) is reflected in the measured tensile strength of excised test specimens (Figure 2b). It is also reflected clinically by the tendency of astigmatic axes to be aligned with either tangential or sagittal meridians. It is important to note that the fibril organization model of Meek and Boote (Aghamohammadzadeh et al., 2004) is very recent (2004) and is the result of full thickness integrations or averages of X-Ray interactions with the collagen fibrils. We thus are only beginning to understand the complex arrangement of fibrils in the corneal stroma, which begs the question: How can we recreate what we do not truly know?
Figure 2.
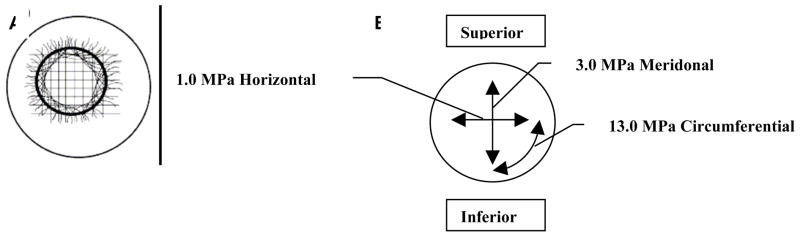
A. Proposed model of fibril orientation in the cornea based on X-Ray synchrotron data. B. Variation in corneal tensile strength as a function of direction. Though the cornea is typically considered a simple nematic stack of lamellae comprising aligned collagen which alternate in orientation by 90 degrees, recent investigations demonstrate an array of fibrils which run circumferentially around the periphery. Preferred fibril orientation and aligned fibril concentration is reflected in the tensile strength or modulus. The figure shows the tensile modulus found in test strips excised and loaded in the direction of the arrows. (A) with permission from (Meek and Boote, 2004) and B with permission from Ruberti et al. (Ruberti et al., 2007).
The compressive behavior of the cornea is dominated not by collagen, but by its associated proteoglycans. More specifically, compressive corneal mechanics are dependent upon hydration and are primarily the result of the 40 mEq of fixed negative charges (at normal hydration) bound to the stromal proteoglycans (Hedbys, 1961) (Kostyuk et al., 2002). Thus the cornea typically exhibits a net negative swelling pressure of approximately 60 mmHg (Hedbys et al., 1963), which is the result of the combined action of the epithelial barrier and the endothelial active pump (reviewed in Ruberti and Klyce (Ruberti and Klyce, 2002)). It has been known for over 100 years that stromas will swell and become opaque if excised and placed hypotonic fluid (Leber, 1873). We assert that the compressive properties of the cornea are not critical to protect the globe, but are a consequence of the presence of the proteoglycans which are thought to maintain the collagen fibrillar spacing (Scott, 1991). Nonetheless, any accurate reproduction of a natural collagen/GAG based stroma will also require the same sort of hydration control system (due to the fixed charges on the GAGs) to maintain transparency. It is important to note that if a corneal analog can be produced which does not swell and remains clear, a transport system which deturgesces the cornea may not be required. This is the ostensibly valid argument of researchers pursuing synthetic hydrogel-based corneas which promote epithelialization (Sweeney et al., 2003). In the case of these synthetic porous implants, the epithelium would still be required to provide a barrier function and to maintain the tear/air interface. Research has demonstrated however that endothelial cells may still be necessary to aid the formation of healthy epithelium (Zieske et al., 1994; Orwin and Hubel, 2000).
Transmission of light
The native cornea is an extremely efficient transmitter of incident visible light (Cox et al., 1970), with such efficiency dependent on the relative content and distribution of water in the stroma (Goldman et al., 1968). The transmission of light is critical to the function of the tissue. Thus the transparency of the stromal matrix, which constitutes the majority of the tissue thickness (and the light interaction) is also critical to corneal function. To understand the optics of corneal matrix transparency, it is necessary to examine the stromal collagen architecture at the nanoscale and to examine the role of the keratocytes.
Keratocytes and Corneal Crystallins
Within the last decade, it has been shown that keratocytes contribute actively to corneal transparency (Jester et al., 1999). Following corneal wounding, such as PRK surgery, the de-differentiation of keratocytes to fibroblast and/or myofibroblasts can lead to a clinically relevant corneal haze (Jester et al., 1999). The haze has been attributed to poor optical properties of the cells which have undergone de-differentiation. Their ability to “index match” to allow transmission of light appears to be a function of the content of soluble enzyme crystallins expressed in the cytoplasm of cells with a keratocyte phenotype (Karring et al., 2004). Thus, the cell population in corneal constructs should be of the appropriate phenotype to reduce cell-induced optical haze. For a review of factors controlling corneal stromal cell phenotype see West-Mays and Dwivedi (West-Mays and Dwivedi, 2006).
Stromal Fibril Organization and Transparency
In Maurice’s 1957 landmark paper on corneal transparency, he concluded that a regular crystalline arrangement of the monodisperse diameter collagen fibrils in the cornea was required to maintain transparency to incident light (Maurice, 1957). Later Hart and Farrell and then Benedek showed theoretically that corneas could be transparent even if there was only limited correlation in the spacing of the collagen (Hart and Farrell, 1969; Benedek, 1971). Benedek demonstrated theoretically that for transparency, it was important that the collagen fibrils should not pack together and that areas of collagen depletion (lakes) larger than the wavelength of light must not exist (Benedek, 1971). The fundamental argument against the requirement of extremely ordered fibrils was derived from examination of Bowman’s membrane in the shark which is both transparent and disordered (Figure 3) (Goldman and Benedek, 1967). Thus, while it is not necessary that the collagen fibrils in an engineered stroma be uniformly spaced, they should be much smaller than the wavelength of light, have a reasonably monodisperse diameter distribution and exhibit uniform center-to-center spacing. Further, there should not be large regions (on the order of the wavelength of light) devoid of fibrils.
Figure 3.

Bowman’s layer in the dogfish. There is no discernible “organization” to the the fibrils in this layer yet this portion of the cornea is more transparent than the underlying more “organized” stroma proper. With permission from (Goldman and Benedek, 1967)
Refraction of light
The combination of the nearly perfectly spherical corneal anterior surface and the index of refraction change at the air/tear film interface generate approximately 80% (42 diopters) of the total refractive power of the human ocular system. Precisely how the cornea forms such an effective refractive surface remains a matter of speculation, but is likely to reside in the details of the stromal collagen nanoscale arrangement coupled with the distribution of the mechanical tensile load (which is a consequence of IOP via Pascal’s Principle and Laplace’s Law). For instance, the fact that there are (putatively) no covalent crosslinks binding collagen fibrils to one another, fibrils may move relatively freely to distribute the load induced by the IOP. The precise pattern of in vivo corneal collagen load-bearing remains difficult to determine. After a detailed mechanical analysis of corneal transport, Friedman concluded that the anteriormost lamellae carry the load from the IOP (Friedman, 1972). In his paper, he cited the clinical manifestations of sub-epithelial edema as evidence supporting this proposed load distribution. More recently however, evidence has been provided that indicates that in the rabbit (Mcphee et al., 1985) and Henninghausen (Hennighausen et al., 1998) and in the human (Hjortdal, 1996) that the IOP load is distributed evenly through the thickness of the corneal stroma. In areas were the stromal lamellae are parallel to the corneal surface and given the monodispersity in the fibril diameters, this finding implies that the collagen fibrils should “feel” approximately the same strain (or stretch). How this load sharing, which requires precise control of fibril length and prestrain, is accomplished is an excellent research question. Some mechanism must ensure during both development and growth, that the corneal curvature remains reasonably spherical. This requires that the load be distributed with precision among the collagen fibrils. The solution to this question may involve a matrix remodeling control system such as that putatively responsible for ocular globe length control (Troilo and Wallman, 1991; Troilo, 1992).
1.3.3 Linking Corneal Form and Function
The stroma comprises 90% of the total corneal thickness, thus it is not surprising that it plays a major role in providing the principal functions of the cornea. We suggest that the stroma owes its success in simultaneously meeting the three corneal design requirements (protection, transmission and refraction) to its exquisite nanoscale organization. Figure 4 shows the organization of the stromal collagen fibrils, which have a virtually monodisperse diameter distribution (transparency), reasonably uniform local interfibrillar spacing (transparency), no interfibrillar covalent crosslinks (refraction?) and are arranged in parallel arrays which are generally tangential to the corneal surface (mechanical strength).
Figure 4.

Transmission Electron Micrograph of collagen fibril arrangement in cornea. Corneal collagen fibrils have a highly monodisperse diameter distribution and are arranged in oriented arrays within lamellae which are approximately 1–2 microns thick. (Section is normal to tangent plane). Image courtesy of Dr. Haiyan Gong.
We assert that this remarkable and persistent nanoscale arrangement of fibrils which persists throughout the cornea (with the exception of Bowman’s membrane) is directly responsible for corneal function and thus links corneal form and function at the level of the nanoscale. The stromal organization appears to be the principal reason why attempts to engineer a viable cornea have been unsuccessful to date. Any corneal analog comprising a natural collagenous extracellular matrix (ECM), must closely mimic this arrangement or risk being too weak, too opaque or too irregular to form an appropriate refractive surface. It is for this reason that we believe that efforts to produce a natural cornea via tissue engineering should focus on reproducing the stroma and in particular on reproducing the nanoscale stromal architecture.
1.4 In Vitro Culture of Epithelium and Endothelium
There has already been extensive and reasonably successful effort expended on reproducing the epithelium and endothelium in vitro from primary human cells. This suggests that if a viable stroma is produced, methods are already available to populate it with limiting functional cell layers.
1.4.1 Culturing the Corneal Epithelium Ex Vivo
The epithelium is responsible for the protection of the stroma and the maintenance of the tear film. Though it is not critical to produce corneal constructs with adherent epithelia to replace failed corneas (donor corneas are debrided of their epithelium prior to implantation), it is important that constructs induce the attachment, spreading and ultimately growth to confluence of epithelial cells to form a patent anterior corneal barrier. A healthy corneal epithelium should form a confluent multilaminar (5–7 cell layers) stratified structure with complete tight junctions surrounding the surface cells. Desmosomes should be present between the cells and hemi-desmosomes with anchoring fibrils projecting into an adherent basement membrane. Culture of epithelial cells onto various substrates has been achieved by numerous investigators: rabbit epithelial cells on plastic (Sundar-Raj et al., 1980); on denuded rabbit corneal stroma (Friend et al., 1982) and on collagen gels (Geggel et al., 1985). It was realized early in this effort that culture technique and cell response to substrates were critical to epithelial morphology and protein expression, leading Trinkaus-Randall to demonstrate that epithelial cells responded differently to different substrates (Trinkaus-Randall et al., 1988) (Trinkaus-Randall and Gipson, 1985) and Minami et al to show that airlifted cultures produced appropriately stratified epithelial layers (Minami et al., 1993). In 1994, Zieske et al. demonstrated that rabbit epithelium basement membrane and differentiation was profoundly influenced by the presence of endothelial cells in an airlifted culture system (Zieske et al., 1994). Figure 5 shows the morphology of the epithelium grown at the moist air interface and in the presence of the endothelial cells.
Figure 5.
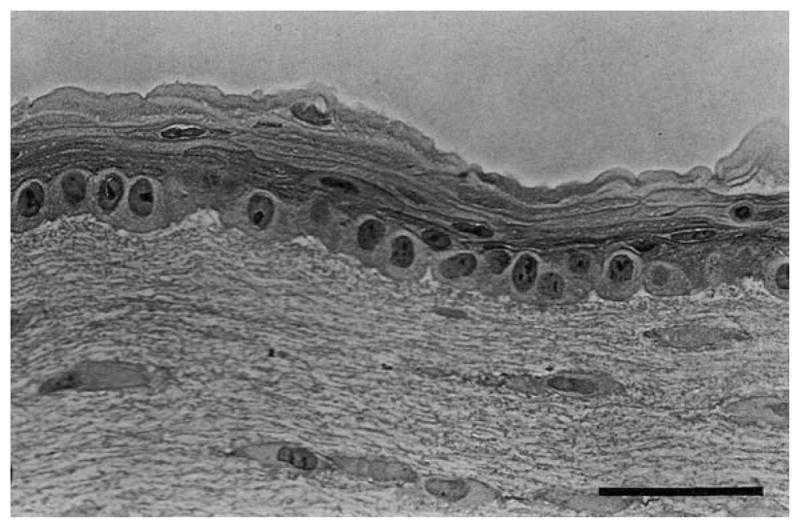
Light micrograph of stratified corneal epithelium grown using air-lift technique and in organotypic, co-culture with endothelium and keratocytes. Bar is 50 microns. with permission from Zieske et al 1994 (Zieske et al., 1994)
Culture of human corneal epithelial cells onto collagen-based substrates for the purposes of corneal tissue engineering has been performed with reasonable success by a number of investigators: primary cells on collagen gels (and on stromal blocks) (Ohji et al., 1994), immortalized cells on glutaraldehyde fixed collagen/chondroitin sulfate gels, (Griffith et al., 1999) primary limbal epithelial cells on collagen populated with keratocytes (Germain et al., 1999), primary epithelial cells on dehydrothermally crosslinked collagen sponges populated with keratocytes and in the presence of endothelial cells (Orwin and Hubel, 2000). One of the more important results of this body of work is the fact that careful attention should be paid to the location of cell harvesting because there are spatial differences in the proliferative capacity of human corneal epithelial cells, with limbally-derived cells showing the greatest ability to undergo multiple passages in culture (Lindberg et al., 1993). In general, progress in this area has been exceptional and it appears certain that once a more appropriate stromal analog has been produced, attempts to epithelialize it will be well-informed.
1.4.2 Culturing the Corneal Endothelium Ex Vivo
Corneal tissue has a tendency to imbibe fluid and become opaque as a consequence of the large imbibition pressure imparted by the stromal GAGs (Hedbys et al., 1963). In 1972, David Maurice (Maurice, 1972) determined that the mechanism responsible for countering the swelling pressure and keeping the cornea in a state of relative deturgescence (and thus transparent) is an active transporter located in the corneal endothelium. For tissue engineered corneas with a natural stromal analog, a functioning confluent endothelial layer would provide a critical component of the corneal transport system. In addition, it has also been shown that endothelial cell co-culture plays a role in generation of appropriate structure and differentiation behavior of corneal epithelium (Zieske et al., 1994; Orwin and Hubel, 2000). Thus, even if the stromal analog comprises a material which does not require “pumping down” to become transparent, unknown signaling molecules derived from a functioning endothelium may still be required to guarantee the patency of the corneal epithelium. Unfortunately, unlike those of other mammals such as rabbit and pig, human corneal endothelial cells do not proliferate in vivo, instead, they undergo polymegathism and pleomorphism to cover exposed Descemet’s membrane following the loss of neighboring cells (Yee et al., 1985). Human corneal endothelial cells (HCECs), which are contact inhibited and arrested in the G1 phase of the cell cycle (Joyce et al., 1996; Joyce et al., 1996), are perniciously non-proliferative and unique culture methods and cell selection protocols have been evolved specifically to encourage proliferation (Engelmann et al., 1999; Chen et al., 2001). The recent progress in endothelial culture techniques has made the difficult task of supplying enough vital, differentiated and functional untransformed human corneal endothelial cells to populate corneal constructs a distinct possibility(Bohnke et al., 1999; Engelmann et al., 1999; Chen et al., 2001; Joyce and Zhu, 2004). The laboratory of Dr. Joyce has been particularly successful in inducing the proliferation of HCECs in culture (Chen et al., 2001; Joyce and Zhu, 2004; Zhu and Joyce, 2004; Konomi et al., 2005). In 2001, they successfully cultured HCECs from donors 50 to 80 years old onto denuded Descemet’s membrane of recipient corneas and achieved remarkably high cell densities. The methods are not trivial and the Joyce laboratory employs donor selection criteria to increase the probability of obtaining highly dense cell cultures. These extremely encouraging results have, for the first time, engendered the possibility of transplanting untransformed HCECs onto a patient’s denuded Descemet’s membrane or of seeding HCECs in high density on artificial corneal constructs. The ability to culture untransformed HCECs is a major advance toward the goal of producing a tissue engineered cornea.
2.0 Stromal cell behavior in vivo
The corneal stroma is the secretary product of mesenchymal fibroblastic cells which are thought to ultimately differentiate into corneal keratocytes. At the heart of classical tissue engineering approaches to producing a stroma-like tissue is the primary corneal fibroblast (usually a de-differentiated corneal keratocyte extracted from donor tissue). Understanding how these cells behave during development, in the normal adult and during wound-healing is critical to the effective production of functional, cell-derived stromal constructs. The distinction between fibroblast behavior during development and during wound healing is particularly important to ascertain.
21. Stromal Cells in Development
Development of the cornea stroma is fundamentally different from wound healing (Cintron et al., 1973; Cintron et al., 1981).The distinction is a critical one for tissue engineers to grasp given the fact that normal stromal architecture arises during development only and not during wound healing (Cintron et al., 1981). Since classical tissue engineering follows a scar model (replacement of a provisional matrix or scaffold) and has enjoyed limited success, it is possible that we may discover a better way forward through the examination of the process of corneal development which appears to be the only way normal corneal tissue can be produced rapidly and in bulk. Much of what we have learned about the developing cornea has been derived from the incisive work of Hay, Coulombre, Linsenmayer, Birk, Trelstad and others from investigations into the developmental biology of the chicken (Hay and Revel, 1969; Trelstad and Coulombre, 1971; Bard and Higginson, 1977; Birk and Trelstad, 1984; Linsenmayer et al., 1998)}. Chick corneal embryogenesis is an intricately orchestrated and complex phenomenon. Beginning at day 3 (stage 18), from superficial epithelial cells derived from an offshoot of the developing brain, an orthogonally-organized primary corneal stroma of type I collagen is secreted layer-by-layer (Hay and Revel, 1969) The confined geometry of developing cornea at day 5 (stage 27) with the initially deposited 10 micron thick, acellular, primary stroma is shown in Figure 6.
Figure 6.
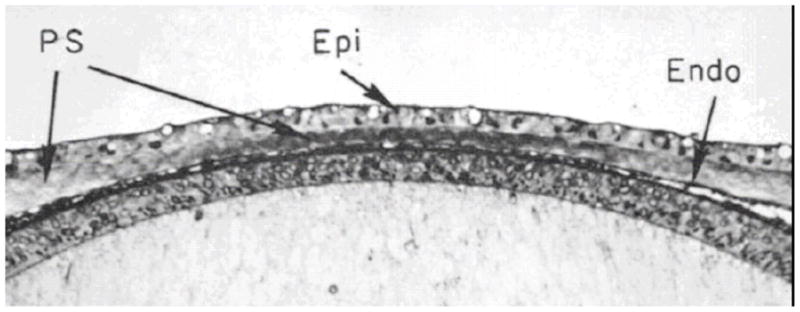
Developing chick cornea. Epithelium, primary stroma and endothelium which is continuous with the lens are depicted. The acellular primary stroma is 10 microns thick at this stage of development (stage 27; day 5). With permission from Trelstad and Coulombre (Trelstad and Coulombre, 1971)
During primary stroma deposition, the younger fiber layers appear anteriorly beneath the basal lamina of the superficial epithelium, pushing older layers towards the nascent lens. Peripheral neural crest cells migrate (the first wave) between the developing stroma and the lens to form the corneal endothelium. The orthogonal arrangement of collagen fibrils in the primary stroma reaches about 10 microns thick at day 5 and then expands rapidly to 60 microns (due to endothelial secretion of hyaluronan into the stroma (Toole and Trelstad, 1971)) Figure 7 shows the exquisite organization of the primary stroma in the developing chick. Understanding the mechanism by which a sheet of epithelial cells controls collagen fibrillogenesis and organization could inform future attempts to produce stromal tissue de novo and in vitro. The expansion of the primary stroma permits the second wave of neural crest cells to colonize the tissue and then differentiate into fibroblasts (Bard and Hay, 1975). These fibroblasts are thought to utilize the template, possibly via a contact guidance mechanism, provided by the “primary” stroma to generate “secondary stroma” (Bard and Higginson, 1977; Birk and Silver, 1984) in register with the primary stromal template (Trelstad and Coulombre, 1971). To make the cornea convex, the endothelium secretes proteoglycans into the anterior chamber. These charged molecules swell to push the cornea forward (Bard and Abbott, 1979), beginning the establishment of the constant curvature necessary for effective refraction. The swelling may also serve to mechanically load the primary stroma and to make it a more effective organizing substrate for the production of secondary stroma as fibroblasts are known to migrate along lines of higher strain/stress (Lo et al., 2000). Subsequent to innervation and the stromal collapse to transparency, collagen is continually laid down, apparently to stabilize corneal shape (Linsenmayer et al., 1986).
Figure 7.

Primary stroma in developing chick cornea. A) TEM of primary stromal collagen showing orthogonal and discontinuous fibrils. B) QFDE image of orthogonal primary stromal collagen. With permission from Trelstad and Coulombre (Trelstad and Coulombre, 1971) and Hirsch et al. (Hirsch et al., 1999).
The preceding review, which owes its fidelity to the excellent developing chick cornea model, serves to demonstrate that embryological development of the chick corneal stroma is an extremely complex, controlled, time and spatially dependent process that may utilize contact guidance, geometrical confinement and possibly mechanical strain to influence secondary stromal secretion and organization by the neural crest-derived mesenchymal corneal fibroblasts. Unfortunately, much less is known about the development of the mammalian corneal stroma and still less is known about human stromal development. However, it is apparent that the development of the mammalian stroma proceeds quite differently than the avian cornea (Cintron et al., 1983). There are also substantial differences in corneal development among mammals. Figure 9 shows the rabbit cornea during development and the human developing cornea prior to stromal deposition. Note that the endothelial cell layer is not present during the invasion of the stromal mesenchymal cells in rabbits or human (the endothelium is produced by primary neural-crest cell waves in both avian and in some non-human primate developing corneas (Hay and Revel, 1969; Ozanics et al., 1977; Sevel and Isaacs, 1988). One of the most striking differences between mammalian and avian corneal development is that in mammals, there is no evidence of the secretion or presence of a primary stroma (Ozanics et al., 1977; Cintron et al., 1983; Sevel and Isaacs, 1988). Prior to mesenchymal cell invasion in mammalian stroma synthesis, a “flocculent” fibrillar material does occupy the prospective stroma, but it does not comprise an orthogonal array of fibrils. Thus, there appears not to be a need for a secondary stromal template in mammalian stromal synthesis. The lens still appears to influence the shape of the posterior cornea and may play a mechanical (and/or chemical) role in guiding collagen fibrillogenesis and stromal organization. Another important distinction is the early organization and flattening of the posterior stromal cells in mammals (prior to deturgescence) as opposed to chick (post deturgescence). Figure 10 demonstrates the geometry of mammalian stromal developmental process.
Figure 9.
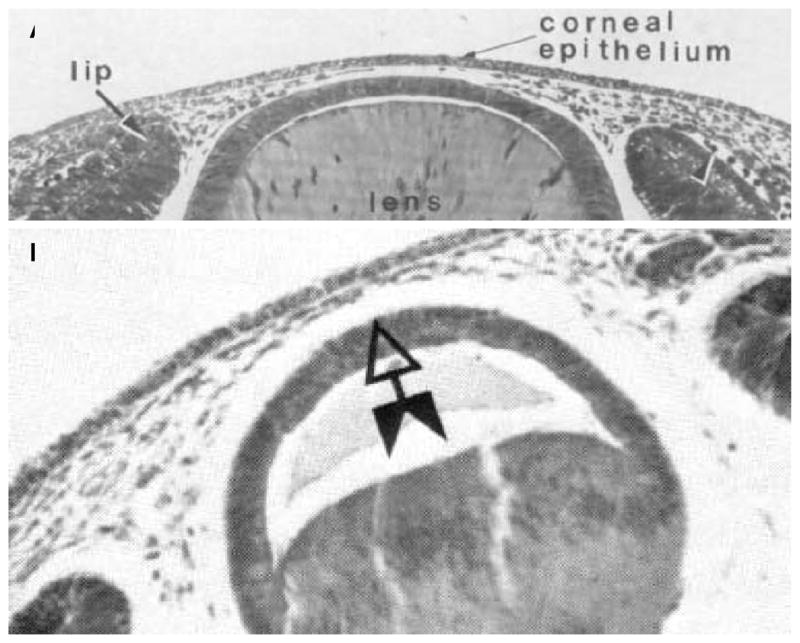
Light micrographs of developing (A) rabbit and (B) human cornea at the initiation of mesenchymal invasion. Note the similarity in the geometry. Both rabbit and human do not have discernible endothelium at the time of mesenchymal invasion, while some primates and avians do. With permission from (Cintron et al., 1983; Sevel and Isaacs, 1988).
Figure 10.

Light micrographic sequence of the developing rabbit stroma from Day 14 through birth to post-natal day 21. The depicted progression is generally representative of mammalian stromal development (sans Bowman’s). The sequence demonstrates prospective stroma formation (day 14), mesenchymal cell invasion (day 15), strom0l synthesis and expansion (days 16 to 23), increase in posterior stromal organization cell flattening (days 16–21), stromal compaction/deturgescence (days 23-birth), Anterior stromal cell flattening (days 26-birth). With permission from Cintron et al (Cintron et al., 1983).
It is also important to consider the fact that during development, there is typically a very high cell density (relative to ECM) with apparent cell-cell communication guiding the process (Cintron et al., 1983). In the rabbit, Cintron measured the ratio of DNA to hydroxyproline and determined that during development there is an initial rapid period of proliferation followed by synthesis of matrix. Thus the ratio of DNA to hydroxyproline is high during development (as opposed to the adult cornea and during adult wound healing). Figure 11 shows the packed parallel arrangement of cells in the central stroma during corneal development in the rabbit.
Figure 11.
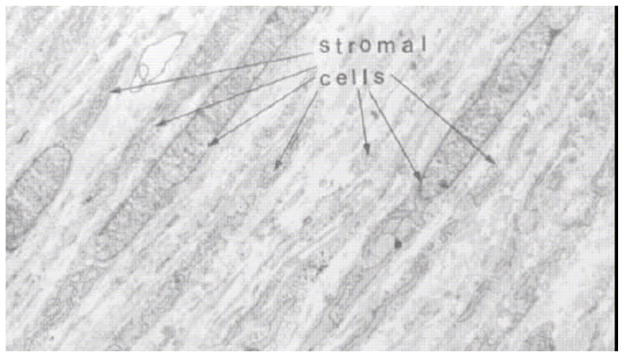
Transmission electron micrograph of 21 day rabbit fetal cornea demonstrating the density of cells in the developing tissue. With permission from (Cintron et al., 1983)
In addition to high cell density, developing tissues are initially very thin and supported by vascular membranes (Cintron et al., 1983). Diffusion limitations, which present significant limitations to tissue engineers (Vacanti 2006), are thus minimized.
2.1.1 Lessons for tissue engineers
There are numerous lessons for tissue engineers to extract from examination of the development of the vertebrate stroma. 1) The process is obviously complex and is both temporally and spatially constrained in that it depends on the interaction of multiple, dynamically changing structures (lens, lens epithelium, corneal epithelium etc). 2) Organization of stromal collagen fibrils into orthogonal arrays can arise by more than one route (templated in chick, non-templated in mammals) indicating some laxity in the developmental constraints imposed on the process (good news for tissue engineers), 3) an intact functioning endothelium is not necessary for stromal development in humans, 4) geometrical constraint or molding of the cornea (either by the lens apposition or by the transudate from receding vasculature (Sevel and Isaacs, 1988)) appears to be a common factor across avian and mammalian taxa, 5) Anterior-posterior expansion (and contraction) of the stroma during matrix synthesis appears to be a common factor across avian and mammalian taxa, 6) the ratio of cells-to-matrix is very high at the outset of development (Figure 11) and 7) diffusion limitations are minimized by the initially compact developing tissue and the support of a vascular pannus.
Though examination of the developing cornea has slowed over the last decade, more specific information on the process is required to inform the effort of tissue engineers who, too often, begin with an approach adopted from a completely different tissue with different design requirements (e.g. skin). A recent investigation on the rapidly developing and experimentally accessible zebrafish cornea suggests that this model exhibits a primary stroma and a Bowman’s membrane (Zhao et al., 2006). The use of such models with modern immunohistochemical methods and live imaging could provide a new level of temporal fidelity to engineers involved in the design of replacement corneas.
2.2 Stromal Cells in the Adult Quiescent Cornea
In the healthy adult human cornea, the stroma contains a sparse network of dendritic (see Figure 12), refractive index-matched (due to cytoplasmic crystallin proteins (Jester et al., 1999)), “keratocytes” which are thought to be quiescent, although they have been observed to contain synthetic cellular apparatus (Muller et al., 1995). Keratocytes maintain the stroma via homeostatic remodeling principally through the production of proteoglycans (decorin, lumican, osteoglycin, keratocan, and biglycan have been detected in keratocyte cultures) (Hassell et al., 1992; Funderburgh et al., 1996; Berryhill et al., 2001; Funderburgh et al., 2003)}. They comprise approximately 5%–10% of the stromal volume and are distributed in a helical network from the anterior to the posterior surface (Muller et al., 1995). The keratocyte network is likely to function as a syncitium as patent gap junctions and cell-cell contacts have been demonstrated between the cells (Bard and Hay, 1975; Hasty and Hay, 1977; Muller et al., 1995; Watsky, 1995). Identification of the keratocyte phenotype in cultures can be accomplished through detection of cytoplasmic aldehyde dehydrogenase (Jester et al., 1999), integrin receptor blockade (Masur et al., 1993; Masur et al., 1999) and the proteoglycan synthesis signature with keratocan a particularly reliable marker (Berryhill et al., 2002; Carlson et al., 2005).
Figure 12.

(A) SEM of an adult human corneal keratocyte (K) showing the complex dendritc morphology of the cell body and processes. (B) Light micrographs of keratocyte network in the feline. Keratocytes are connected by broad cellular processes (open arrows) extending from main cell body which contains nucleus (arrows) With permission from Muller et al 1995 (Muller et al., 1995) (A) and Jester et al 1994 (Jester et al., 1994) (B)
In general, keratocytes are quiescent (with exception of proteoglycan production) and exhibit only wispy organization of filamentous actin in the vicinity of the cell cortex. Their mobility is likely limited given their expression of integrins specific for type I collagen (Masur et al., 1993) and lack of organized, cytoplasmic f-actin or smooth muscle actin (SMA). Following insult, keratocytes demonstrate their ability to change phenotype to “address” the injury, but often produce conditions which reduce the transparency of the cornea (Fini, 1999; Jester et al., 1999; Jester et al., 1999; Zieske, 2001; Zieske et al., 2001; West-Mays and Dwivedi, 2006). The change in phenotype results in substantial changes in cytoplasmic constituent molecules (f-actin, aldehyde dehydrogenase (ALDH), etc). The loss of ALDH in particular can result in the development of corneal haziness because the cells lose their ability to index-match with the surrounding matrix (Jester et al., 1999; Jester et al., 1999). For corneal tissue engineers, it is preferable that mature stromal constructs provide an environment which promotes and maintains the quiescent, index-matching keratocytic phenotype.
2.3 Stromal Cells in Wound Healing
The fine details of the stromal response to injury have been established by numerous investigations, many of the latter studies were motivated principally by the increased popularity of refractive surgery. Corneal stromal wound response is mediated by the keratocytes (and their de-differentiated repair phenotypes) following injury and has been extensively reviewed by Fini (Fini, 1999). It is essential for tissue engineers to understand the difference between wound healing and development. Both processes produce extracellular matrix, however, the nature of the matrix and the process by which it is formed in each case is very different. Wound healing is an event that occurs following injury in an adult animal. It is typically designed to mitigate the immediate effect of the injury and save the life of the animal, however, “it can never restore function to the damage organ.” (Fini, 1999). This statement is particularly true for the human cornea. Upon wounding, keratocytes can dedifferentiate into repair phenotypes, fibroblasts or myofibroblasts, which either participate in the normal wound healing process or undergo apoptosis (Dohlman et al., 1968; Matsuda and Smelser, 1973; Garana et al., 1992; Fini, 1999; Jester et al., 1999; Zieske, 2001; Zieske et al., 2001; West-Mays and Dwivedi, 2006). Keratocytes subjacent (within about 200–300 microns from wound edge) to an injury to the epithelium undergo apoptosis, while kerotocytes distal to the injury dedifferentiate into repair phenotypes.
2.3.1 Repair Fibroblasts
Repair fibroblasts are distinct from quiescent keratocytes in that they are actively motile, synthesize collagen and produce proteoglycans with a different synthetic signature than those found in normal or developing cornea (Figure 13) (Funderburgh and Chandler, 1989; Cintron et al., 1990; Garana et al., 1992; Jester et al., 1994; Funderburgh et al., 2003). Repair fibroblasts arise from the phenotypic transformation of keratocytes at the edge of the acellular zone following wounding (Dohlman et al., 1968; Matsuda and Smelser, 1973). The cells display the classical fusiform fibroblastic morphology and infiltrate the wound within 2 days. The repair fibroblasts exhibit f-actin, α-actinin and non-muscle myosin which has been transferred from the cortex to stress fibers in the cytoplasm in the form of stress fibers (similar to cultured fibroblasts)(Jester et al., 1994). Repair fibroblasts also express α5β1which binds fibronectin, a fibrillar wound matrix component synthesized by repair fibroblasts and thought to facilitate wound invasion and possibly contraction (Welch et al 1990, Garana et al 1992, Masur 1993). An important hallmark of fibroblast mediated repair is associated with the production of matrix degrading molecules including members of the matrix metalloproteinase family (see Fini (Fini, 1999)for an extensive review of the topic). It is well-known that remodeling of ECM requires the synthesis of new matrix components as well as the synthesis of matrix degrading molecules. It is thought that the paracrine signal stimulatory for the production of collagenases by fibroblasts is IL-1α while TGF-β exerts an ECM component synthetic effect (Strissel et al., 1997), Zeiske recent data). TGF-β also appears to induce the transition of the fibroblast to the myofibroblast phenotype in skin (Desmouliere et al., 1993) and in corneal fibroblast in culture (Jester et al., 1996).
Figure 13.
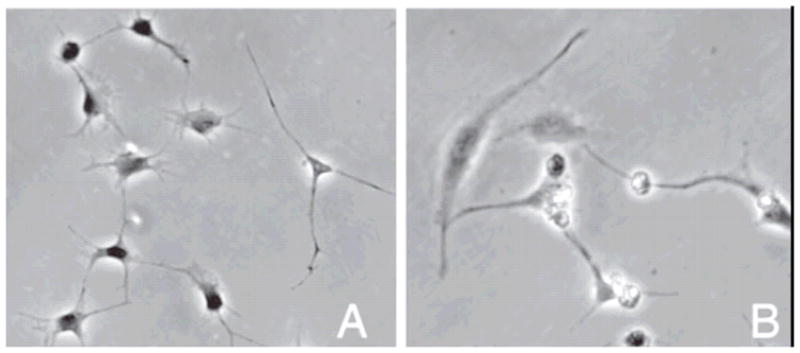
Phase contrast optical micrographs of dendritic keratocytes (A) versus more fibroblastic (B-repair type fibroblasts) cells in culture. With permission from (Musselmann et al., 2005).
2.3.2 Myofibroblasts
An exhaustive review on the role of myofibroblasts in the cornea has been provided by the pre-eminent expert in the field (Jester et al., 1999).We will briefly review some of the more relevant aspects of myofibroblast behavior in corneal wound repair. Concomitant with the invasion of the corneal wound with repair fibroblasts, another type of cell, the corneal myofibroblast, also appears, particularly when the wound ruptures the basement membrane of the epithelium. It has been suggested that corneal myofibroblasts play an important role in wound contraction (Jester et al., 1987; Garana et al., 1992) while corneal fibroblasts synthesize and remodel the wound matrix through the production of matrix components and matrix degrading proteins (Cintron et al., 1981; Fini, 1999). The idea that myofibroblasts are principally involved in contraction is supported by the data which show that cells expressing α-SMA are confined to the wound “plug” (Jester et al., 1999). Further, the expression of α-SMA is found to be temporally transient, appearing with the onset of contraction and disappearing when contraction is complete (Jester et al., 1995). The process of wound contraction in linear incision wounds in corneas exhibited an atypical pattern where myofibroblastic cells organized themselves, collagen type I and fibronectin along the incision boundary (but somehow adherent to both wound borders-Figure 14a) (Petroll et al., 1993). Wound contracture is thought occur via a mechanism similar to tightening a shoestring (Figure 14b). Jester points out that this mechanism could only be appreciated by live, 3-D + time confocal microscopy (Jester et al., 1999). Thus the myofibroblast, whose expression can be induced by TGF-β, is a transient cell responsible for closing wounds. The orientation of the cells and organization of matrix associated with the myofibroblasts appears to be dictated by wound position and direction.
Figure 14.
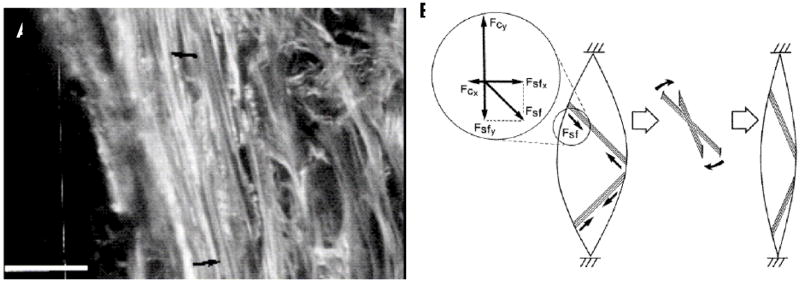
(A) Confocal Scanning Laser Microscope image of myofibroblast stress fibers aligned adjacent to the wound margin (dark area) in a linear incision wound in a rabbit cornea. (B) Schematic of the possible “shoestring” wound contracture model in the rabbit. In vivo confocal 3-D imaging revealed that wound contracture in corneal wounds my occur in a manner similar to pulling closed the top of a shoe with shoestings. With permission from Petroll et al (Petroll et al., 1993)).
2.3.2 Lessons for the tissue engineer
Though the wound healing response can save the eye, the tissue that is produced is not the same as that generated during development even after two years of remodeling (Cintron et al., 1981). This is possibly because there are clear disadvantages in corneal wound healing over development, 1) In wound healing, there are limited supporting structures/cell types (i.e. lens, lens epithelium, vascular pannus), with only the epithelium and endothelium in close proximity, 2) the diffusion distances are large and limit access to nutrients, proteins and oxygen, 3) the ratio of cells-to-matrix is lower in wound healing which means the cells must exert organizing influence over longer distances, 4) in wound healing, the potential space is occupied by a fibrin plug which potentially “imprints” disorganization on the matrix produced by repair fibroblasts, 5) In scar models, the geometry depends on the wound type (linear or trephination), which is likely to further constrain the cell’s ability to self-organize in a predictable manner, particularly in the presence of the resorbing fibrin matrix within the wound.
It is important for tissue engineers to consider what conditions promote the production of organized matrix in vivo. Development produces a unique, complex, spatially and temporally varying environment in a 3-D bioreactor well suited to produce organized matrix while wound healing is a stop gap measure which then relies on inefficient remodeling over long time periods with poor nutrient and metabolic supply to resolve the matrix organization. We postulate that to produce highly-organized matrix similar to a corneal stroma, it is better to do so under conditions similar to development, which is the only time significant quantities of organized stromal matrix are produced rapidly. One characteristic that is constantly exhibited by corneal mesenchymal cells, keratocytes, repair fibroblasts and myofibroblasts is the presence of cell-cell contacts, which has been observed during development (Hasty and Hay, 1977; Cintron et al., 1983), quiescence (Muller et al., 1995) and wound healing (Jester et al., 1995). These stromal cell-cell contacts have been shown to be physiologically functional in both quiescent and wounded corneas (Watsky, 1995). It is possible that the organization of the cells (and thus any deposited matrix) may depend on their ability to communicate. When developing constructs, the ability of the cells to form connections should be considered, particularly when initially dense scaffoldings are going to be utilized.
3.0 Stromal Cell Behavior In Vitro
Now that a reasonably thorough review of the behavior of corneal stromal cells in vivo has been undertaken, tissue engineers should also understand the behavior of these cells following their extraction from living tissue. There has been a long history of manipulation of corneal stromal cell lines, primary corneal stromal cells, and primary human corneal stromal cells (pHCSC) in vitro (see Ruberti et al (Ruberti et al., 2007) for extensive review). In the remainder of this chapter, we will attempt to focus principally on primary (or low passage) human corneal stromal cells which currently hold the most promise of producing clinically viable and ethically acceptable tissue engineered constructs (in the future directions section we will mention tissue engineering with stem-like cells recently isolated from corneas by Du et al. (Du et al., 2007)). However, where there has been substantial supporting data added from cells isolated from other species, the species will be identified.
3.1 Primary corneal stromal cell culture systems
Work with pHCSCs has its roots in the early seventies when Newsome determined that pHCSCs are more proliferative under certain culture conditions (which typically include the use of FBS) than either the corneal epithelium or endothelium (Newsome et al., 1974). The cultured cells, which were likely more akin to the repair fibroblasts (Fini, 1999), migrated from explants after 4 days and then overran the epithelial cell layers which had emerged from the explant earlier. This was one of the first indications that human corneal keratocytes could be grown successfully and in large numbers in culture. Interestingly, if the cultures were allowed to proliferate, the fibroblasts would overgrow with the second layer of cells roughly perpendicular to the first layer (Figure 15). This phenomenon has been observed by multiple investigators using human and chick fibroblasts (Newsome et al., 1974; Bard and Hay, 1975; Guo et al., 2007).
Figure 15.
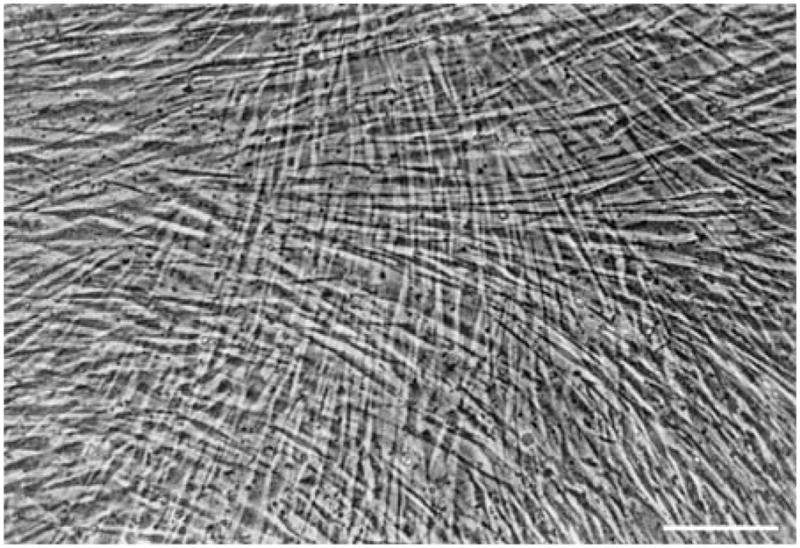
Phase contrast image of a multilayered primary human corneal fibroblast culture after one week in culture. Modified from Guo et al (Guo et al., 2007) with permission. Bar is 20 microns
3.1.1 Stimulated secretion of matrix components
Stoesser et al. (Stoesser et al., 1978) followed the work of Newsome with the idea that cultured pHCSCs might be induced to produce collagen in culture if they are stimulated with ascorbic acid (a co-factor for the hydroxylation of proline). They demonstrated that type I collagen was synthesized in large quantities by the cultured cells. In 1996, slightly longer term cultures of pHCSCs stimulated with ascorbic acid were shown to secrete and assemble collagen of the types found in the normal cornea (I,V and VI) in the appropriate ratios (Ruggiero et al., 1996). These two studies along with earlier work on isolation and successful culturing of corneal fibroblasts suggest that it may be possible to stimulate pHCSCs in scaffold-free systems to produce an organized 3-D ECM that is similar to the normal corneal architecture. The results of our attempt to do this in long-term culture will be discussed in the section on in vitro corneal tissue engineering.
3.1.2 Corneal stromal cell differentiation control
Corneal stromal cells possess the ability to de-differentiate (and re-differentiate). It is thus important for tissue engineers to maintain control over the mutable phenotype of the corneal keratocyte. Fetal bovine serum (FBS) induces the conversion of keratocytes from their quiescent phenotype to a fibroblastic phenotype (Beales et al., 1999)(bovine). Further differentiation to a myofibroblastic phenotype can be induced by adding TGF-β (Jester et al., 1996) (rabbit). Partial reversibility of fibroblasts to a keratocyte-like morphology is possible by removal of the serum in the medium (Berryhill et al., 2002)(bovine). Typically, the stromal analogs in tissue engineered corneas include FBS in the medium (Germain et al., 1999; Griffith et al., 1999; Orwin and Hubel, 2000), which should induce the transformation to a fibroblastic synthetic phenotype. Ultimately however, the ability to redifferentiate fibroblasts to a keratocyte phenotype, which possess corneal crystallins (ALDH) in large quantities, will be important for restoration of transparency to stromas in corneal constructs (Jester et al., 1999).
3.1.3 Stromal cells in 3-D culture
There is only limited information available on the behavior of pHCSCs in 3-D culture systems. However, the natural environment of mesenchymal fibroblasts is 3-D and their behavior in 3-dimensions is more relevant to both basic science and tissue engineering (Hay, 2005). In 1992, Doane et al. seeded chick corneal fibroblasts into collagen gels and observed that they oriented orthogonally and produced limited amounts of organized collagen (Doane et al., 1992). Thus it is quite possible that corneal fibroblasts possess a persistent ex vivo “memory” of their developmental history. In an excellent demonstration of this idea across fibroblasts with different developmental origins, Doane and Birk (Doane and Birk, 1991) cultured chick corneal, skin and tendon fibroblasts into 3-d collagen gels. The fibroblasts self-organized in a manner consistent with their developmental origins (corneal cells into orthogonal sheets, tendon cells in parallel bundles and dermal cells with no particular orientation). This behavior is particularly encouraging for tissue engineers as it suggests that the cells do possess intrinsic organizational ability derivative of their developmental lineage.
Tissue engineers have seeded fibroblasts into collagen gels to produce “thick” substrates to which epithelium and endothelium will adhere. Griffith et al (Griffith et al., 1999) demonstrated that immortalized human keratocytes populated glutaraldehyde cross-linked collagen-chondroitin sulfate gels. However, little information about the resulting cell-matrix interaction was provided. Orwin and Hubel (Orwin and Hubel, 2000) demonstrated that primary human keratocytes migrated into, proliferated, aligned with and synthesized collagen (and GAGs) in collagen-based dehydrothermally cross-linked sponges.
3.1.4 Effect of mechanical loads - Mechanobiology
The effect of gravity and the need to transmit, produce and absorb mechanical forces constantly during normal activity suggests that response to mechanical stimulus may be an important component of fibroblast behavior. Indeed, it has long been known that there are real effects on ECM structure which are related to the applied mechanical environment (Wolff, 1891).The importance of mechanical stimulation during the development of the musculoskeletal system is reviewed extensively by Carter and Beaupre (Carter and Beaupre, 2001). Numerous examples of the effect of disrupting normal mechanical stimulation have been documented and include bone fragility which is observed in infants who are hypokinetic during gestation (Rodriguez et al., 1988; Rodriguez et al., 1988) and loss of bone tissue during space flight (Zerath, 1998). The ability of other connective tissue to remodel in response to mechanical forces is dramatically demonstrated by the behavior of vascular tissue when subjected to elevated shear stress or elevated internal pressure. In general, increasing the flow rate through blood vessels induces a remodeling response which increases vessel caliber (presumably to lower wall shear stress to a target value) (Zarins et al., 1987). Decreasing the pressure in a blood vessel induces a remodeling response which thins the vessel wall (presumably to keep wall tensile stress at a target value) (Bayer et al., 1999). Tissue engineers have already been attempting to use mechanical stimulation to produce stronger cell synthesized constructs and have recently demonstrated that mechanical force alone can induce the differentiation of fibroblast cells in a tissue engineered scaffolding (Altman et al., 2002). The specific molecular events which control cellular response to mechanical stimuli are currently the subject of intense research in the field of mechanotransduction. Recently the term “mechanome” was coined and the topic was formally introduced at the World Congress of Biomechanics in a plenary presentation given by Professor Roger Kamm of MIT. The mechanome refers to the constellation of molecules and systems of molecules that are mechanoresponsive. In the last few decades, there has been extensive investigation into the response of fibroblasts/tissue to mechanical stimulation (Chamay and Tschantz, 1972; Cowin, 1983; Akeson et al., 1987; Michna and Hartmann, 1989; Bishop and Lindahl, 1999; Arokoski et al., 2000; Cowin, 2000; Humphrey, 2001; Altman et al., 2002; Cowin, 2004). However, there are very few investigations into the behavior of corneal fibroblasts in mechanically stimulated 3-D networks. Roy et al (Roy et al., 1997) performed some of the first quantitative assessments of corneal fibroblast (rabbit) force generation in collagen gels and Karmichos and Petroll (Karamichos et al., 2007) have recently investigated the interplay between human corneal fibroblasts and mechanical constraint in 3-D cultures. Their conclusions suggest that mechanical constraint significantly impacts the behavior of the cells (spreading, alignment and contractile force) leading to enhanced matrix reorganization particularly between the ends of cells which are aligned with the cell-generated stress field (Figure 16).
Figure 16.
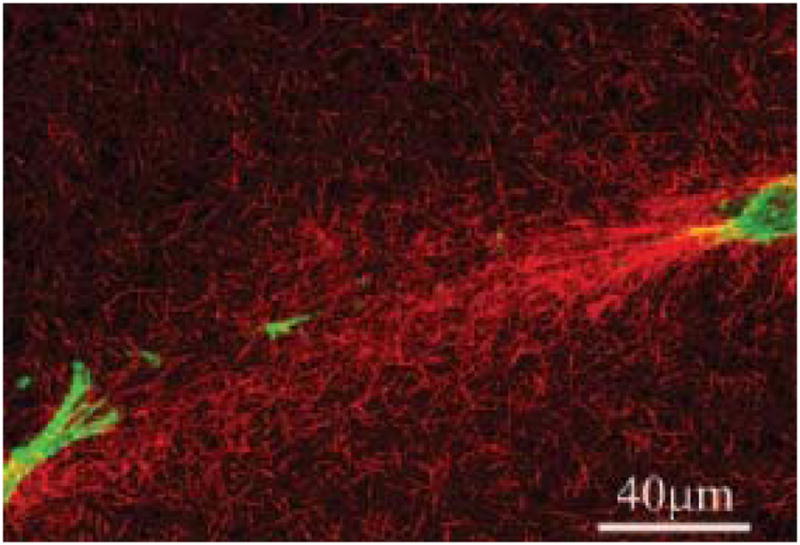
Confocal reflectance microscopy of human corneal fibroblasts in a reconstituted type I collagen gel that has been mechanically loaded by fibroblast contractile force. The cells produce organization in bundles of collagen fibrils between their “ends” when they are induced to align by the mechanical constraints on the matrix. With permission from Karmichos et al (Karamichos et al., 2007)
Mechanical stimulation is likely to be important to the production of mechanically stable engineered corneal tissue. Mechanical loads have been demonstrated to enhance both organization and mechanical strength in other engineered connective tissues (Juncosa-Melvin et al., 2006; Stoddart et al., 2006; Duty et al., 2007). Mechanical forces are also known to alter the migration patterns of cells via a mechanism termed “durotaxis” (Lo et al., 2000). Finally, tissue loading is also a putative controller of matrix development/growth in the in vivo developing cornea as demonstrated by the effect of IOP reduction (which leads to loss of matrix stress) on developing chick eyes (Neath et al., 1991).
3.1.5 Contact guidance
Early in the study of chick corneal development, it was proposed that the primary corneal stroma acted as a template to guide the deposition of the secondary stroma secreted by the invading mesenchymal cells (Trelstad and Coulombre, 1971). Experimental demonstration of the phenomenon of contact guidance for fibroblasts came in 1972 when Elsdale and Bard cultured lung fibroblasts on shear-aligned collagen gels (Elsdale and Bard, 1972). The result of contact with aligned collagen produced oriented fibroblasts in the direction of the fibrils.
Since that time, there have been a few attempts to contact guide pHCSCs using anisotropic substrates (Teixeira et al., 2004) and on templated, anisotropic type I collagen gels (Vrana et al., 2008). An important adjunct to the process of contact guided locomotion and induced cell orientation is the fact that fibroblastic cells are known to produce collagen in alignment with the long axis of the cell body (Wang et al., 2003). Thus it may follow that by orienting the pHCSCs it is possible to orient their synthesized ECM. In the field of corneal tissue engineering, this phenomenon could prove to be particularly useful.
4.0 Stromal Tissue Engineering
At the present time, no clinically viable human corneal equivalents have been produced by tissue engineering methods. In fact, to our knowledge, no full-thickness tissue engineered corneal constructs have been implanted successfully into trephinated test animals to simulate corneal transplantation. However, there have been some partial thickness lamellar keratoplasties attempted in animals (Li et al., 2003). It is our view that the major obstacle to the production of a successful engineered cornea is the difficulty associated with reproducing (or at least simulating) the stromal architecture. Examination of the literature quickly reveals that reproducing the corneal stroma in vitro is challenging. To be fair, it cannot be claimed that significant effort has been expended with the specific goal of reproducing the stromal architecture. This is possibly because “classical” tissue engineering has focused primarily on developing advanced cell culture methods and on the development of a dizzying array of controllable, degradable scaffoldings (see Yarlagadda et al (Yarlagadda et al., 2005) for a general review of tissue scaffolds). Corneal tissue engineering efforts to date have generally been derivative of this “classical” approach.
4.1 General approaches to corneal tissue engineering
If one performs a review of the literature and also examines the current research trends in leading laboratories, it appears that the tissue “engineering” of a corneal stroma can be undertaken a number of ways: 1) Classical tissue engineering: Seeding fibroblastic cells into degradable matrices for subsequent remodeling in vitro and/or in vivo, 2) Developmental tissue engineering: Stimulating fibroblastic cells to produce a functional stroma-like matrix in vitro prior to implantation, 3) De Novo Tissue Engineering: Engineering the assembly of collagenous matrix to produce, a priori, an organized, functional, stroma-like structure into which fibroblasts may then be seeded and 4) Hybrid approaches: a combination of the latter approaches (e.g. seeding activated corneal fibroblasts onto/into an aligned template (which may or may not be collagenous) similar to the mechanism of primary/secondary stromal secretion found during chick corneal development (Trelstad and Coulombre, 1971).
The vast majority of attempts to reproduce stromas can be categorized as “classical” tissue engineering, while developmental and de novo stromal tissue engineering are emerging from the “concept” stage. Interestingly some hybrid approaches to the problem have just begun to appear in the literature. In the remainder of this chapter we will assess what has been accomplished in the classical and developmental approaches to tissue engineering, examine how hybrid approaches are being conducted and finish with a look into newer methods which may be leveraged to produce stroma-like materials de novo.
An underappreciated problem associated with in vivo remodeling of degradable scaffoldings for load-bearing tissue
Regardless of the method by which corneal constructs are produced, if they are to supplant donors as the graft material of choice, they will need to be functional at the time of implantation. At the very least, the implant and the method of graft fixation will have to withstand the stresses associated with minor traumatic events immediately following surgery. Significant in vivo remodeling of the stromal matrix is a potentially serious pitfall for many engineered load-bearing constructs under prospective design (for tissues other than cornea as well). The difficulty arises from the fact that the in vivo remodeling of a degradable scaffolding amounts to an intentionally produced mechanical instability with numerous uncontrolled variables (i.e. individual immune response, individual wound healing and fibroblastic response). If the scaffolding degrades and is not properly rebuilt in situ, the tissue will undergo mechanical failure, possibly resulting in loss of the globe. It is difficult to imagine that medical device governing bodies (such as the FDA) would allow implantation of constructs intentionally designed to undergo substantial in vivo degradation and remodeling. This puts significant constraints on tissue engineers because it suggests that the construct must be both functional and highly stable at the time of implantation.
4.1.1 Classical tissue engineering of the human corneal stroma
The majority of stromal analogs for tissue engineered corneas have been created by seeding human corneal stromal cells into collagen-based scaffoldings which are apparently designed to be remodelled (Borderie et al., 1999; Germain et al., 1999; Griffith et al., 1999; Orwin and Hubel, 2000; Li et al., 2003). Simple, reconstituted type I collagen derived from bovine skin or from rat tail tendon has been used by many investigators interested in constructing stromal analogs (Geggel et al., 1985; Minami et al., 1993; Ohji et al., 1994; Zieske et al., 1994; Germain et al., 1999). Acid-soluble atelo-collagen monomers such as Vitrogen-100® or Purecol® can be purchased in quantity and forms gels reliably by neutralization with NaOH followed by warming to 37°C. The resulting gels are low-density, weak, disorganized and comprise long, native-looking type I collagen fibrils. Type I collagen gels are highly biocompatible and can be assembled in the presence of corneal fibroblasts without significant loss of cells (Germain et al., 1999). Unfortunately, reconstituted collagen gels are not known to be particularly stable in vivo or in vitro without some form of crosslinking (Charulatha and Rajaram, 1997). One of the most notable corneal constructs was built around a stromal analog which was produced by exposing the type I collagen gels to 0.03–0.04% glutaraldehyde followed by treatment with glycine (to scavenge the unbound glutaraldehyde) (Griffith et al., 1999).
Because of the known importance of glycosaminoglycans in the control of stromal properties (Hedbys, 1961), Griffith added chondroitin sulfate to her stromal analogs (Griffith et al., 1999). The net result of the crosslinking and addition of GAGs to the simple type I collagen gel, followed by human fibroblast seeding, produced a clear, populated corneal stroma (Figure 18). The details of the mechanical properties, quantitative values for transparency (corrected for thickness) and in vivo remodeling behavior of the cornea of Griffith et al (Griffith et al., 1999) are not available. Recall that mechanical strength and transparency of the stroma are two of the three primary functions of the cornea. These initial constructs were subsequently replaced by a superior collagen-synthetic polymer hybrid four years later. Figure 19 shows a collagen-based stroma next to a collagen-TERP5 construct (Li et al., 2003). The copolymer [poly(N-isopropylacrylamide-coacrylic acid-coacryloxysuccinimide), PNiPAAmcoAAc-coASI or TERP] was used because it spontaneously crosslinks proteins and anchors peptides through primary amines. The TERP and a second formulation, TERP5, in which some of the acrylosuccinimide groups were reacted with YIGSR (to facilitate neurite extension and epithelial cell growth) were combined with bovine atelo-collagen and molded to produce a corneal stromal analog. The resulting stromal analogs were transparent and for the TERP5 (with YIGSR) they supported up to five layers of human immortalized epithelial cells and promoted rapid neurite invasion in vitro.
Figure 18.
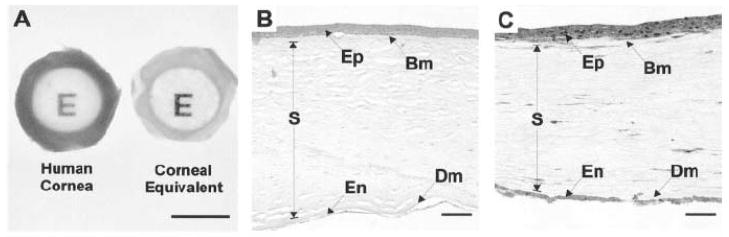
Corneal construct of Griffith et al. (A) Demonstration of construct clarity compared to post-mortem human cornea. Scale bar 10 mm (B) Cultured eye bank cornea (C) cultured corneal equivalent. Scale bar 100 microns. Epithelium – EP; Bowman’s membrane – Bm; Stroma – S; Descemet’s – Dm; Endothelium – En. With permission from Griffith et al., 1999
Figure 19.
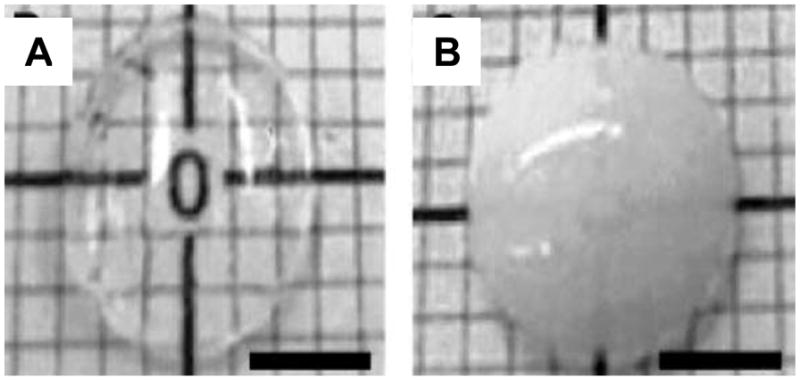
Photographic comparison of the TERP5-collagen composite of Li et al.,2003 (A) and a reconstituted type I collagen gel (B). With permission from Li et al. (Li et al., 2003))
The TERP5 collagen-synthetic-YIGSR based stromas were strong enough to be sutured (unlike the collagen only gels) and were implanted into micropigs (by LKP leaving the posterior cornea and endothelium intact). The implant copolymer matrix was quietly integrated, elicited rapid epithelialization (including the deposition of type VII collagen at epithelial matrix interface) and an enhanced rate of reinnervation (compared to allografts). However, though the stromal constructs were suturable, care had to be taken to prevent cracking and damage during implantation. The authors state outright that their stromal construct is weak (but did not quantify the strength directly) compared to native corneal stroma. This is probably due to the random arrangement of fibrils and low density (3.4% wt/vol) of the collagen-polymer matrix. In 2005, Li et al (Li et al., 2005) further characterized a series of similar collagen-synthetic gels by numerous methods including suture pull-out tests which demonstrated that these materials are quite weak. This is possibly why only partial thickness keratoplasties were performed with these materials. In general, this approach to stromal tissue engineering possesses some significant advantages over reconstituted atelo-collagen gels including higher strength, better clarity and improved cell-inductive behavior (for epithelium and neurites). However, remodeling of the material to a clear, strong stroma-like tissue analog was not demonstrated in vivo and they appeared to be too weak to provide a full-thickness graft at the time of implantation.
In an effort to manipulate the mechanical properties and the pore size of collagen constructs, Orwin and Hubel (Orwin and Hubel, 2000) constructed stromal analogs which began with collagen fibrils eluted from bovine corium and were formed into “sponges” by grinding, lyophilization and dehydrothermal (DHT) crosslinking. The sponges supported colonization by human primary cells (epithelium, endothelium and pHCSCs) either alone or in combinations (epithelium/endothelium or epithelium/pHCSCs) in vitro. The fine details of the stromal matrix/cell interaction were not extensively investigated, however the keratocytes appeared to align with fibrils and synthesize matrix components (collagen and proteoglycans) Figure 20.
Figure 20.

H&E stained stromal analog of Orwin and Hubel. (A) By day 10 of culture the cells have migrated through the stromal sponge. (B) By day 22 of culture cells have elongated along the collagen fibers. With permission from Orwin and Hubel (Orwin and Hubel, 2000).
In 2003, the same laboratory (Orwin et al., 2003) added exogenous chondroitin sulfate and/or hyaluronic acid to DHT cross-linked collagen sponges and assessed the evolving mechanical and optical properties as a function of time in culture with pHCSCs (Orwin et al., 2003). Ultimately, the DHT collagen sponges were mechanically and optically “better” than reconstituted collagen gels, but were substantially inferior to native corneas in the critical areas of strength and clarity (equilibrium compressive modulus was 3 orders of magnitude smaller than native cornea, transparency was, at best, half that of the native cornea). Of particular concern was the mechanical instability of the sponge in culture, which exhibited a sharp decline in modulus followed by a slow return to about ½ the starting modulus at day 21. In an attempt to further improve the mechanical properties of collagen-based scaffoldings, dense collagen films were produced via a modified DHT-crosslinking method and seeded with pHCSCs (Crabb et al., 2006). In this case, the cells did not penetrate the films but remained on the surface and produced variable diameter collagen fibrils (35–75 nm as measured by SEM). The mechanical properties of the films (which were at least an order of magnitude lower than the native cornea upon fabrication), remained unchanged during the culture period (up to 4 weeks).
In a parallel study, Crabb et al. (Crabb et al., 2006) demonstrated that the ECM synthesized by the pHCSCs on DHT cross-linked collagen substrates was not particularly well-organized. In addition, there were two distinct distributions of fibril diameters produced by the cells on the films. One set was about 100 nm in diameter while the other set was 47 ± 17 nm. The average and distribution of the diameters for the smaller set indicates that the synthesized fibrils were not normal for the cornea. Details of the fibril composition are lacking but it was assumed that they were collagen (though there is evidence that fibronectin was also present in the culture). It is unclear how these “stromal constructs” would be used to reconstruct a cornea as the cells did not penetrate the dense scaffold. Nonetheless, Crabb et al. (Crabb et al., 2006) are beginning to examine the results of the synthetic behavior of pHCSCs in culture and on collagen. This is a significant step towards what we term “developmental” tissue engineering.
The net result of attempts to produce a stromal analog via “classical” approaches has fallen far short of an implantable, clear, strong cornea. The qualified success of the classical tissue engineering approach for skin has produced similar expectations for many other tissues, including the cornea. A simple illustration (Figure 23) may demonstrate why engineering a cornea is much more difficult than engineering skin.
Figure 23.

Scanning electron micrographs of a reconstituted type I collagen gel (A), cell-assembled collagen in native human corneal stroma (B), cell-assembled collagen in human hip skin (C). In the native cornea, arrays of highly-organized fibrils split and pass above and below lamellae with different orientations. In skin, the collagen is more loosely organized and does not have to be transparent. It appears simpler to transform the structure from A to C than it is to transform the structure from A to B. Image in (B) with permission from Radner et al., 1998 (Radner et al., 1998). Image in (C) with permission from Fligiel et al (Fligiel et al., 2003).
Degradable reconstituted type I collagen scaffoldings such as the one pictured in Figure 23a, would need to be resorbed and replaced with something mimetic of the image in Figure 23b to produce a cornea. This is unlikely on short time-scales, as organized stroma-like ECM is synthesized in quantity in the human cornea only during development, where there is no observable degradable matrix and where there exists a very high cell density. We speculate that disorganized degradable scaffoldings could impair the ability of pHCSCs to coordinate the construction of organized collagen by prevention of cell-cell communication and/or by “imprinting” disorganization on the cell arrangement and subsequently on the newly synthesized matrix. In general, the process of pHCSC remodelling of a degradable scaffolding (either in vitro or in vivo) resembles stromal scar resolution which, even under ideal conditions in vivo, can take years (Cintron et al., 1982). If this fact is coupled with the difficult requirement that an implanted resorbing scaffold retain its clarity and mechanical strength during the remodeling process, the prospect of a developing a suitable corneal construct via “classical” tissue engineering appears to be unlikely at the moment. However, if the resorbing scaffolding could enhance the organization of the fibroblasts, promote synthesis of new organized matrix and promote cell-cell communication during remodeling in vitro, this approach could become more tractable (Baker and Mauck, 2007).
4.1.2 Developmental tissue engineering
The development of the human corneal stroma takes place under specialized conditions in a very thin layer between the epithelium and the nascent lens (Cintron et al., 1983). It appears, at least for the cornea, that the rapid production of highly-organized matrix occurs under conditions which include a high density of synthetic cells (Cintron et al., 1983) in a confined space. Corneal tissue engineers should closely examine stromal development, because this is where (and when) the problems associated with tissue construction have already been solved. To our knowledge only one corneal tissue engineering investigation has derived an approach that can be described as partially “mimetic” of development. Of course, there is little chance that all of the events in corneal development can be duplicated in culture. Nonetheless, there are some features of development that can be replicated in culture which may strongly influence the organization of synthesized matrix. In addition, human keratocytes appear to possess some intriguing characteristics which may facilitate “recapitulation of development” in vitro. First, we have already reviewed the fact that primary human keratocytes possess a mutable phenotype and can de-differentiate (and possibly re-differentiate) under investigator control in culture (Fini, 1999; Jester et al., 1999; Berryhill et al., 2002). Second, primary human corneal stromal cells cultured on plates at high-density stratify to form multiple layers of cells which alternate in the orientation of their long axes by about 90 degrees in adjacent layers (Newsome et al., 1974, Guo, 2007 #294) Third, pHCSCs can be stimulated to produce collagen by the addition of ascorbic acid (AA) to the fibroblast culture medium (Stoesser et al., 1978). Fourth, the type and relative quantities of collagens produced by AA stimulated pHCSCs in culture was found to be consistent with that of the normal corneal stroma (Ruggiero et al., 1996). In a recent investigation which attempts to characterize the organization and morphology of ECM synthesized by pHCSCs, we took some simple steps which deviated from classical tissue engineering in a deliberate attempt to make the culture method somewhat mimic development (Guo et al., 2007). Specifically, we removed the degradable scaffolding, seeded the pHCSCs at relatively high density (to promote stratification), stimulated the cells to produce collagen with stabilized AA and kept the cells in culture for extended periods (up to 12 weeks). We did not provide any orienting structures, just bare polycarbonate transwell membranes. Under these conditions, the cells produced significant ECM which possessed large areas of organized small diameter collagen fibrils (Figure 24).
Figure 24.

Standard TEM micrographs of pHCSCs stimulated to produce collagen. (A) low magnification image of cells and matrix in the constructs. The collagen fibrils clearly alternate in direction in neighboring lamellae. (B) Higher magnification showing at least 9 direction changes in collagen lamellar orientation. (C) Higher magnification image showing detailed fibril organization. Bars (A,B) 2 μm and (C) 1μm. With permission from Guo et al. (Guo et al., 2007)
Adjacent lamellae alternated in direction, were parallel to each other and parallel to the transwell membrane. The fibril diameters were slightly polydisperse and slightly large compared to the normal adult cornea (38±7.0 nm vs. 32±0.7 nm). The constructs were evaluated at 1, 4, 8 and 12 weeks and the thickness peaked at 8 weeks at approximately 50 microns as measured by optical Differential Interference Contrast (DIC) imaging. Within the constructs, organization of the fibrils (as detected by z-scan DIC) was limited to the 25 microns closest to the transwell membrane. Figure 25 shows an example of organized matrix (en face optical section) as seen by DIC optical imaging.
Figure 25.

Contrast-Enhanced Optical DIC image of cell-assembled collagenous ECM. Location of image section is approximately 8 microns above and parallel to the transwell membrane. Image texture is suggestive of alternating arrays of fibrillar structures which persist over large length scales (order 10s of microns). The construct in this image is 4 weeks old. Bar is 10 microns. Image courtesy of Nima Saeidi. (unpublished data)
Immunohistochemistry demonstrated that the cell-assembled constructs possessed both type V and type VI collagen, both of which are present in the normal cornea. The most striking aspect of the cell-assembled constructs was their resemblance to the developing mammalian cornea (Figure 26). In particular, the alternating arrays of collagen, the stratification of the clearly active fibroblast cells and the high cellular density are all found in developing stroma (Cintron et al., 1983). The ability of untransformed human corneal fibroblasts to produce organized arrays of reasonably monodisperse collagen when stimulated is a very tantalizing development in the pursuit of an engineered cornea. Though it has not been proven definitively, we believe that stratification of the cells is critical to the development of the stromal organization. It is likely that cellular organization is necessary to promote organization of subsequently synthesized collagen (fibroblast cells have been shown to synthesize collagen in the direction of their long axis (Wang et al., 2003)). We also believe that the close proximity of the cells to one another could be an important and necessary factor in controlling both the fibrillogenesis and the direction of the assembled collagen, particularly if collagen organization is achieved by cholesteric means (cholesteric collagen assembly will be discussed in the section on de novo corneal tissue engineering). One of our key hypotheses is that degradable disorganized scaffoldings (into which fibroblasts are seeded) interfere with the production of organized matrix by preventing the fibroblasts from self-ordering, from communicating and from controlling the local space into which collagen will be secreted and assembled. The converse of this hypothesis is also likely true: degradable scaffoldings which promote fibroblast organization, cell-cell communication and allow control of the local space may enhance the formation of organized matrix. For example, the use of a surface that promotes the alignment of pHCSCs prior to the deposition of matrix may result in the production of aligned matrix similar to the normal stroma (Vrana et al 2006).
Figure 26.

Transmission electron micrographs of 4 week old stromal construct produced by untransformed human corneal fibroblasts in culture. Note synthetically active stromal cells with prominent RER and orthogonal arrangement of fibrils in lamellae. The architecture of the construct organization is very similar to that found in the developing mammalian corneal stroma. Arrows indicate prominent dilated RER. Bar is 2 microns. With permission from Guo et al. (Guo et al., 2007)
Note on stem cells
We have not covered work with embryonic stem cells in this chapter partly because of the ethical concerns associated with their use. However, there are other cells which exhibit the pluripotential behavior of stem cells which have not been derived by destruction of an embryo. In a recent paper, Du et al (Du et al., 2007) reports the use of adult stromal stem cells extracted from human corneas to make stroma-like matrix in vitro. These adult human corneal stromal stem cells (HCSSCs) produced organized ECM most effectively at the edges of 3-D pellet culture (they did little in the center of the pellet or when cultured in fibrin gels at low density). Interestingly, Du et al (Du et al., 2007) demonstrated that cell-cell junctions were more prominent in the areas of the pellet culture where the matrix was most-organized and concluded that communication plays an important role in the synthesis of organized tissue.
Naturally, the ability to utilize cells which may possess more plasticity and synthetic ability is exciting. Such cells would not require differentiation to fibroblast phenotype followed by multiple expansions, but would need to differentiate into keratocytes eventually. They also represent an approach that is more similar to development than using pHCSCs. However, these cells may also produce quite challenging phenotype control issues in culture. We await further refinement of this methodology for use in corneal stromal tissue engineering.
4.1.3 De novo tissue engineering
The insoluble components of the normal adult corneal stromal matrix comprises a complex milieu of collagens (I,V,VI,XII,XIII) and proteoglycans (decorin, lumican, biglycan, mimecan, keratocan). These molecules appear to be arranged in specific patterns both over both short and long length scales. Short-range organization includes the incorporation of type I/V collagen in small diameter heterotypic fibrils (Birk et al., 1988), the association of collagen VI along the collagen I/V fibrils (Zimmermann et al., 1986), the association of KSPGs with the collagen I/V “a” and “c” bands (Scott and Haigh, 1985) etc). Over long length scales collagen I/V fibrils are arranged in parallel in lamellae which alternate in direction but form a very specific network structure (Meek and Boote, 2004). The development of this arrangement of fibrils is derivative of a complex series of cell-mediated and self-assembly events which take place in a genetically controlled 3-D cell culture system in which the enhancements associated with millions of years of evolution are embedded. No one believes that a corneal stroma can be precisely reproduced (or even poorly mimicked) at the current time on the bench top. However, it may be possible to take advantage of the intelligence that is encoded into ECM molecules to produce, de novo, a matrix that is at least partially “functional” at the time of implantation and which undergoes only limited remodeling/enhancement. The process of producing such constructs is what we mean by the term “De novo tissue engineering”. Our focus will be on reproducing collagen organization because we believe that collagen is the “template” molecule in connective tissues (this is true for bone in particular). De novo tissue engineering of collagenous matrices is clearly in its infancy, however, it is rooted in seminal investigations which date back to the 1950’s when the ability of collagen monomers extracted from tissue to self-assemble into native type fibrils was extensively examined (Highberger et al., 1951; Gross et al., 1954; Gross, 1958; Gross, 1958). This first series of quantitative and morphologically advanced investigations into the in vitro self-assembly of extracted collagens has provided the basis for numerous investigations which have since probed the kinetics and resulting morphology of collagen assembly de novo. The results of this body of work are beautifully summarized by Prockop and Hulmes 1994 (Prockop and Hulmes, 1994). Some of the more important results of these investigations demonstrate that : 1) collagen monomers are made soluble by the presence of C- and N-terminal propeptides at relatively high concentrations (up to 1.5 mg/ml in physiological buffer (Mould et al., 1990), 2) fibril diameter depends on the temperature during assembly (Kadler et al., 1990), 3) assembling fibrils possess an inherent polarity (Kadler et al., 1990), 4) assembled fibril morphology is influenced by numerous factors including the momomer concentration (Mould and Hulmes, 1987), pH (Yuan and Veis, 1973), terminal propeptides (Mould et al., 1990), other collagen monomers (Birk et al., 1990; Wenstrup et al., 2004) and proteoglycan core proteins (Brown et al., 2002). In spite of this large body of work investigating factors which control collagen fibril assembly, methods designed to control the organization of the resulting fibrils have received little attention. One of the earliest references to a method designed to control collagen fibril organization is in a paper by Elsdale and Bard (Elsdale and Bard, 1972), where collagen fibrils in films were aligned by inclining the surface during polymerization. In addition to this drainage method, collagen alignment has also been attempted by applying a magnetic field (Guido and Tranquillo, 1993; Torbet et al., 2007), dip pen lithography (Wilson et al., 2001), cholesteric methods (Giraud-Guille et al., 2003), electrospinning (Matthews et al., 2002) and even by freezing and thawing (Faraj et al., 2007). Our efforts have focused on three different methods designed to control the organization of collagen fibrils specifically for the purposes of corneal stromal reconstruction.
Shear alignment of collagen
Our first approach utilizes a shearing flow to produce organized collagen fibrils on substrates. With this method, cold collagen solution (Allergan, Irvine, CA) can be driven through a warmed shear chamber. The kinetics of collagen assembly are readily observed (via optical DIC microscopy) as well as the organization of the polymerizing fibrils (Figure 28).
Figure 28.
DIC optical micrograph. Collagen fibrils assembled on clean borosilicate glass in the shear chamber. The fibrils were assembled in the presence of a 18 sec−1 shear rate (arrow indicates flow direction). The kinetics of the assembly process suggest that collagen begins aggregating very rapidly on the glass surface (within 2 minutes) and that the alignment of the fibrils quickly becomes compromised by the formation of fibillar loops (inset in figure). (Bar is 10 microns). Image courtesy of Nima Saeidi (unpublished data – manuscript in preparation)
These loops are likely the result of an instablility caused by the attempted upstream growth of the collagen fibrils which ultimately are “turned” back downstream. It is thus difficult to produce completely aligned collagen layers via a pure shear-influence. In addition, the morphology of the fibrils in these shear-aligned substrates does not appear to be properly D-banded (Figure 29).
Figure 29.
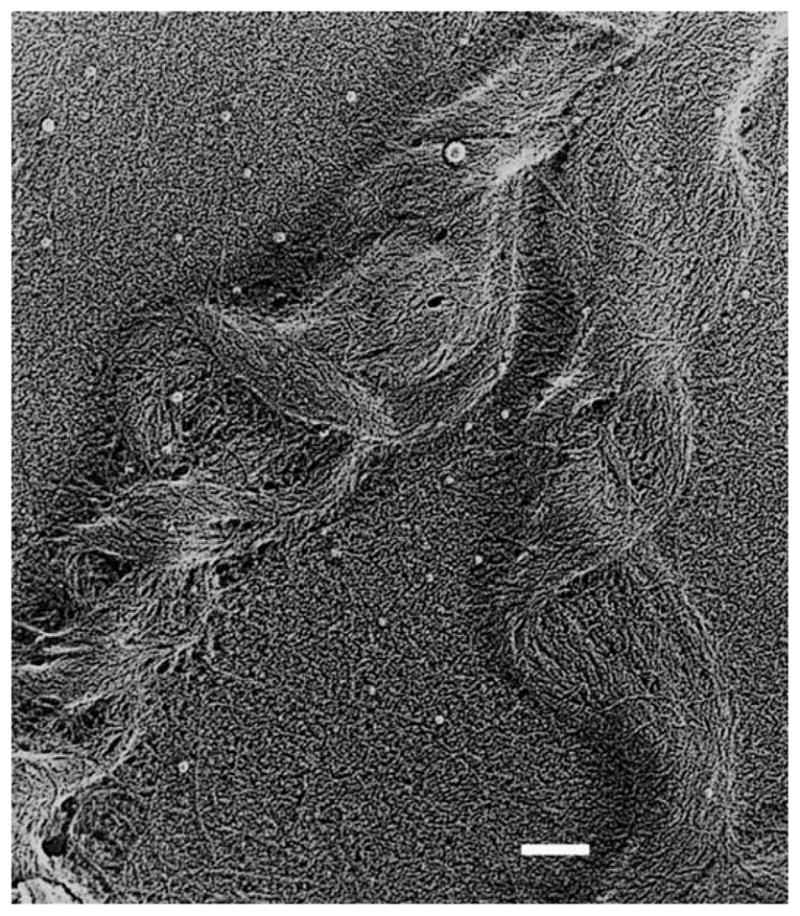
Quick Freeze Deep Etch image of extracted collagen, aligned and re-assembled in shear chamber. Morphology of the fibrils does not match native D-periodic banding. Individual monomers can be observed interacting with both the fibril and the substrate surface. This multiple interaction is likely the cause of the disrupted structure. Bar is 50 nm. Photograph courtesy of Nima Saeidi (unpublished data – manuscript in preparation)
It appears that the assembly of the collagen is disrupted by the adhesion to the glass substrate.
The use of the shear chamber was illuminating but the alignment force can be further enhanced by confinement which can be achieved when collagen is assembled in a thin film via spin coating (Braithwaite and Ruberti US patent 7,048,963).
Since the patent application by Braithwaite and Ruberti, the spin-coating method has been refined and the improved alignment induced by the combination of shear stress and confinement can be readily seen in the Figure 30. In their patent Braithwaite and Ruberti demonstrate that spin-coated collagen fibrils may be crossed at any arbitrary angle merely by employing an offset glass coverslip into the spinning disk (Figure 31a). The glass substrate can be rotated through any angle between coating runs to produce 3-D, alternating arrays of aligned collagen fibrils (Figure 31b).
Figure 30.

Aligned collagen layer on glass substrate produced by introducing cold collagen solution onto warm spinning disk. Note the degree of alignment is excellent. However, even during spincoating, there is upstream polymerization instability which causes fibrils to turn, disrupting the alignment (highlighted circle). Black arrows indicate flow direction. Spin rate 1000 rpm; collagen flow rate 0.5 ml/min (3.0 mg/ml Purecol); 12 minute run. Bar is 10 microns. With permission from (Ruberti et al., 2007) Photo courtesy of Nima Saeidi (unpublished data – manuscript in preparation)
Figure 31.
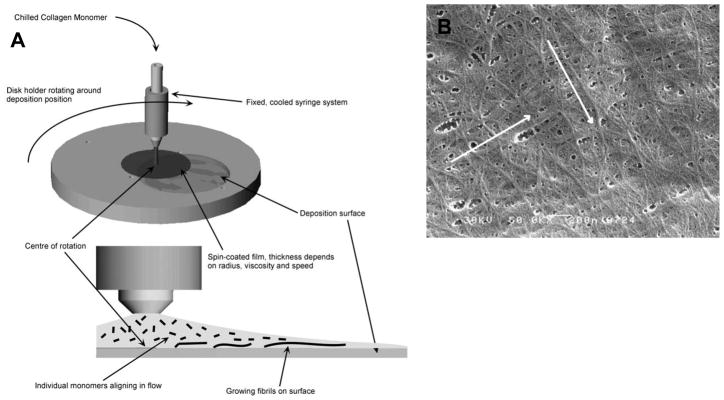
(A) Offset disk concept where collagen is deposited onto glass substrate offset in spinning disk. The collagen flows with a slight radial spread over substrate surface. Substrate may be rotated between coating runs to produce layered aligned structures (above). (B) SEM of collagen orthogonal layers produced in successive runs. Flow rate 0.25 ml/min, 1000 rpm, 15 min per run. Modified from Braithwaite and Ruberti US Pat 7,048,963
Though the organization of collagen via shear assembly both with and without confinement has been reasonably successful, there are some very challenging technical hurdles. In the shear chamber, the difficulty associated with the upstream growth instability will make it tough to produce a good stromal analog where all of the collagen in a particular layer is aligned. It is also difficult to modify the shear chamber to produce multiple alternating layers of aligned fibrils. Nonetheless, the aligned collagen that can be produced should be good enough to use as a “contact guidance” substrate for influencing pHCSCs to grow in a particular orientation (which may be all that is required to induce formation of a stroma – this is a hybrid, de novo/developmental approach).
In the spin coating apparatus, in addition to the upstream growth instability, there are significant problems maintaining the integrity of the very thin films required to confine the growing collagen (while shearing them). This problem is particularly difficult if multiple orthogonal layers are to be grown to produce a stromal analog. In spite of the difficulties associated with shear-alignment of collagen, the resulting collagen film (from a spin coating run) has produced collagen that was aligned well enough to contact guide human corneal fibroblasts within six hours in a pilot experiment (Figure 32). Human corneal fibroblasts also aligned on denuded rabbit stroma, however within a longer period of of time (24 h).
Figure 32.
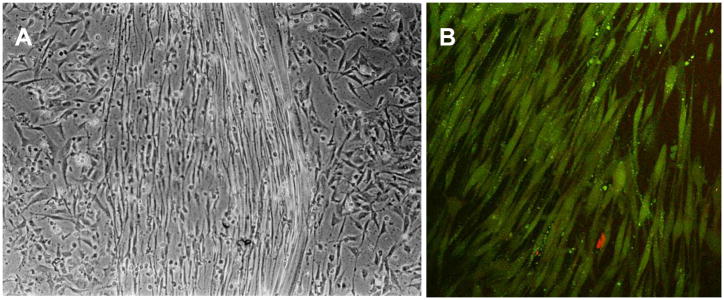
(A) Phase contrast optical microscopy of human corneal fibroblasts cultured on shear-aligned collagen from spin coater. Within six hours, fibroblasts had aligned in the direction of the collagen on the glass (center region). Fibroblasts on glass which was scraped clean of shear-aligned collagen were arranged randomly. (B) Confocal image of human corneal fibroblasts grown on denuded rabbit stroma after 24 hours in culture. Fibroblasts align (most likely with the fibril orientation in the exposed stroma). Images courtesy of Xiaoqing Guo.
Biomimetic approaches to collagen organization
Our collective experience with shear-influenced collagen self-assembly have lead us to conclude that the difficulty of producing multiple layers of aligned, alternating collagen fibrils is much higher than producing a reasonably aligned single layer. In addition, shear-alignment requires the use of large amounts of collagen solution, most of which ends up in a waste jar because only a fraction of the monomers populate the sheared interface. Thus, producing a full-thickness functional stroma via shear alignment of collagen is both unlikely and expensive. Competing methods designed to organize collagen suffer from a host of difficult technical problems as well. Electrospinning is a violent process typically done in non-aqueous solvents which is unlikely to permit the use of morphology-controlling molecules in addition to the collagen. Dip-pen lithography is currently limited to very small areas. Magnetic alignment requires very strong field strengths and complex solution handling to produce alternating layers. Lastly, freeze-thawing offers little control of organization. To address the problem of creating organized collagenous arrays, we reexamined stromal development and the development of other tissues where highly-aligned collagen is produced. Because we asked ourselves a fundamentally different question than is typically asked by biologists: “How can we do this ourselves?” vs. “How is this done?” we were lead down what we believe is a very promising path toward de novo corneal tissue engineering.
There appear to be two competing hypotheses regarding the assembly and organization of collagen in vivo. The first and most substantially documented by experimental data is the notion that fibroblast cells produce and expel monomers into “fibropositors” or cell “surface crypts” where they are assembled into fibrils or intermediate filaments and discharged “vectorially” into the extracellular space (Birk and Trelstad, 1984) (Canty et al., 2004). The second possibility is that the cells mediate the fibrillogenesis of collagen by controlling the local concentration and orientation of monomers to produce cholesteric patterns which then are converted into organized matrix (Giraud-Guille, 1992). We have been working on reproducing both of these methods in benchtop systems designed to generate organized stroma-like collagen arrays de novo.
Fibripositors and the collagen printer concept
Figure 33 shows a descriptive annotated image of how a mesenchymal fibroblast might produce the collagen of the corneal stroma during development. In his model, Birk suggests that fibroblastic cells locally control the organization of the collagen in “surface crypts”. The collagen fibrils are formed and aligned in the crypts and then discharged into the extracellular space where they possibly “fuse” with other fibrils which are aligned in the same direction. The concept of the fibripositor appeared to be amenable to reproduction using available membrane technology (polycarbonate track-etched membranes), pepsin-extracted collagen and transport theory. Figure 34 demonstrates the concept of the fibripositor analog or “nanoloom”.
Figure 33.

Model of Birk and Trelstad (Birk and Trelstad, 1984) in which collagen fibrils are produced and aligned in compartments or surface crypts. In this model the cells are responsible for both the synthesis and organization of each fibril at the local level. Note that the same cell secretes collagen fibrillar arrays at right angles to one another. Thus the cell cannot move while depositing the fibils (and still maintain orthogonal fibril orientation). 1, 2, 3 types of surface invaginations producing collagen forming extracellular spaces. SV - secretory vesicle, With permission from (Birk and Trelstad, 1984)
Figure 34.
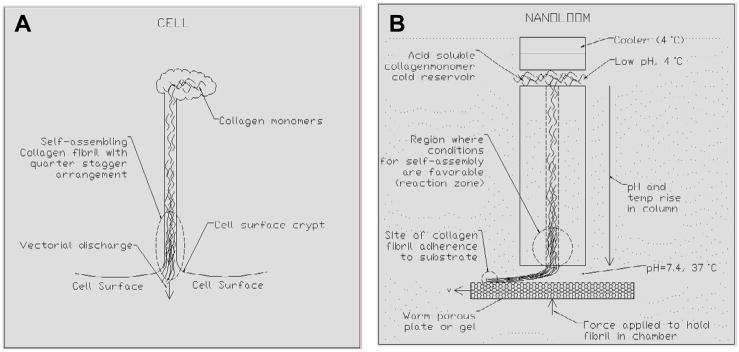
(A) Model of fibripositor in cell. Procollagen is released from the secretory vesicle into the surface crypt where monomer orientation is controlled. One or both of the propeptides are cleaved to allow fibrillogenesis and the resulting collagen fibrils are “vectorially discharged” into the ECM. (B) Single reactor (fibripositor analog) in collagen nanoloom. Atelo, acid soluble collagen monomers (2.8 mg/ml) are kept cold (4°C) and at low pH on one side of polycarbonate track-etched membrane populated with 80 nm pores. The collagen is driven down the pore with a pressure gradient where conditions at the outlet promote fibrillogenesis (high pH, 37°C, high NaCl concentration). Fibrils are bound to a substrate which moves past the membrane, drawing assembling collagen from the pore. Image courtesy Northeastern University Capstone Design Course.
The nanoloom has been under development for approximately three years. The device pictured in Figure 34 is the subject of international patent PCT application 60/720,696 application and was constructed by undergraduate students as part of their Capstone Design Course at Northeastern University. The nanoloom was subsequently refined via adjustable parameters (i.e. temperature, concentration, ionic strength etc). However, printing collagen in this manner has proven to be terribly difficult so far. Our best result was obtained by driving (via a hydrostatic pressure head) 2.8 mg/ml, cold, atelo-collagen through a polycarbonate track-etched membrane (80 nm pore size) into a warm PBS bath and onto a silanized glass plate. Figure 35 shows a broken strip of what we suspect is aligned collagen printed by the nanoloom, although this has not yet been proven or reproduced.
Figure 35.
(A) Collagen Nanoloom was constructed in 2004 and comprises a 3-D nanomotion gantry, a peltier cooled collagen printer head (highlighted circle) and a heated copper substrate on which to print. At the heart of the design is a biomimetic array of fibripositors which are 80 nm in diameter and 6 microns long and contained in a polycarbonate track-etched membrane. (B) DIC optical image of a single broken strip of aligned “collagen” found after an attempted printer run. Three years of effort on the device have raised some very sophisticated engineering problems. Nonetheless it will likely provide some level of control over collagen organization when optimized. Bar is 20 microns. Images courtesy Kathryn Portale (unpublished data-manuscript in preparation).
Self-organizing collagen fibrils: Cholesteric effect
In 1982, Trelstad postulated that the stromal organization of submammalian corneas was the result of the cholesteric liquid crystal behavior of collagen (Trelstad, 1982). He based this argument on the fact that the handedness of the small angle shift in collagen fibril alignment as one progresses from the anterior to the posterior stroma was the same in both eyes. There was no bilateral symmetry, suggesting that the behavior is likely physicochemical. There have been numerous papers which address the possibility that collagen in load-bearing connective tissue is organized by liquid-crystal behavior but the most prolific work has come from the laboratory of Marie Giraud-Guille (Giraud-Guille, 1988; Giraud Guille et al., 2005). Their investigation of the liquid crystal behavior of collagen monomers has demonstrated that fibrillar organizational patterns similar to those found in native connective tissue can be produced purely by concentrating collagen monomers and inducing fibrillogenesis. Figure 36 shows the results of one study in which collagen was assembled de novo resulting in fibrillar patterns which mimic the collagen structure in ligament and in demineralized compact bone.
Figure 36.

Collagen fibrils formed by neutralizing concentrated collagen monomer solutions. Patterns of the resulting fibrils mimic patterns found in ligament (a) and in compact bone (b). Bars are 1 micron (a) and 500 nm (b). With permission from (Giraud-Guille et al., 2003)
Upon review of corneal development and examination of the stromal constructs that we have grown in a scaffold-free model, it seemed quite plausible that the organization of collagen fibrils could be the result of a similar mechanism. In the case of the cornea, collagen monomers would have to be concentrated enough to produce nematic organization (in the neighborhood of 50–100 mg/ml (Gobeaux et al., 2008)). This could be accomplished if the cells form stratified layers and secrete large amounts of collagen monomers into the confined space between the organized layers of cells. The most troubling part of this hypothesis is the fact that collagen should form aggregates above 1.5 mg/ml, even with intact propeptides (Mould et al., 1990). Assuming higher concentrations can be stabilized, the direction of the monomeric alignment may be controlled over short distances (and potentially long distances as well) from the cell surface via geometric features or by adhesion complexes. This idea is fundamentally different from the fibripositor concept as it requires that the fibrillogenesis occur en mass outside the cells. In essence, the organization of the monomers would come first and the fibrils would “precipitate” from the aligned monomers. Interestingly, if the fibrillogenesis was produced by a wave of cell synthesized collagen propeptidases, it would appear as if the cell was “vectorially” discharging the fibrils. Instead, the fibrils would be condensing from the concentrated and organized monomers (which would not appear in standard TEM). There is some evidence that this is a plausible alternative theory. Figure 37 shows aligned collagen fibrils near a mesenchymal cell in developing chick corneal stroma at day 14 (Young et al., 2007) where it appears as if the fibrils are being vectorially discharged from the cell (although it is hard to imagine collagen being discharged without buckling – one cannot “push” a rope). Note that the fibrils are discontinuous and that there is quite a bit of “white space” in the micrograph. The second plate shows aligned collagen fibrils in the vicinity of a pHCSC in our scaffold free stromal development model. Again, the fibrils are discontinuous and there appears to be a lot of “white space”. In the third plate in Figure 37, a quick-freeze deep-etch (QFDE) micrograph shows just how dense the matrix is adjacent to the cell in the same scaffold-free model of development. It is possible that the “white space” could be collagen monomer or another molecule which does not stain in the TEM prep.
Figure 37.

Transmission Electron Micrographs of cell-synthesized organized collagen. (A) Standard TEM of embryonic chicken stroma at developmental day 14. Collagen fibrils appear to be “extruded” from cell. Note the large electron lucent areas. (B) standard TEM of pHCSC in scaffold-free development model at 11 weeks. Note discontinuous collagen and “white” space. (C) QFDE of 11 week pHCSC in development model. Aligned fibrils are present (lower right). Note that there is no “white” space detected by QFDE which does not depend on stains. Image in (A) with permssion from (Young et al., 2007). Images in B and C courtesy Drs. Ruberti and Zieske (manuscript under review at Developmental Dynamics)
A well-designed experiment will be able to determine if embryonic stromas contain enough collagen monomer to reach nematic concentrations in the spaces adjacent to the fibroblasts. From these observations and a review of the fine work of Giraud-Guille, we surmised that a similar approach could be taken on the benchtop which might lead to reproduction of the aligned planar collagen layers found in the normal human corneal stroma. The idea was to simply concentrate and confine collagen monomers in the thin space between two coverslips and then “trip” fibrillogenesis. Figure 38A is a schematic depicting the general methodology (which is trivially simple). The results (also shown in Figure 38) indicate that a very thin layer of completely aligned collagen fibrillar structures could be produced using this liquid crystal approach combined with confinement. These results were obtained in less than a few days (compare to three years for the nanoloom). It is clear that collagen (even the atelo, pepsin extracted, type I collagen monomers we use) are coded with the information necessary to produce organized hierarchical structures over long-distances.
Figure 38.

(A) Schematic of the general approach to concentrate and confine collagen monomers. Step 1 – place low concentration collagen between two coverslips inside dialysis tube and immersed in osmicant (PEG in this case). Step 2 - Allow dialysis to continue until desired concentration is achieved. Step 3 – induce fibrillogenesis of the concentrated monomer solution. (B) DIC optical micrograph of the results of similar experiment. Concentration and confinement of monomers between two featureless coverslips produced an organized array of collagen fibrils. Image in (B) courtesy of Nima Saeidi (unpublished data – manuscript in preparation)
Further experimentation concentrating collagen monomers to induce liquid crystalline behavior prior to fibrillogenesis has yielded thicker constructs in which arrays of collagen change direction (Figure 39). Thus, not only can liquid crystalline collagen monomers polymerize into aligned arrays of fibrils, the aligned collagen arrays naturally alternate in direction to form structures which appear suitable to carry loads. The morphology of collagen fibrils produced from neutralizing and warming cholesteric collagen monomers was examined by standard TEM preparation. The fibrils, which result from the fibrillogenesis of type I atelo-collagen monomers at very high concentrations, appear to be distinct, small and highly-organized (Figure 40).
Figure 39.

DIC optical image of collagen fibrillar arrays. The method of production of this collagen construct is similar to that of Figure 38. Low concentration collagen monomer solution was dialyzed against PEG and placed between two coverslips. The resulting constructs contained multiple layers of organized collagen alternate in direction. Image courtesy of Nima Saeidi. Bar is 20 microns (unpublished data – manuscript in preparation)
Figure 40.

TEM of collagen fibrils produced from liquid crystalline type I atelo-collagen monomers. The fibrils are small (about 20 nm) and well-aligned. Banding is not entirely clear, but can be seen in some areas. Bar 100 nm. Image courtesy of Nima Saeidi (unpublished data – manuscript in preparation)
The concerning aspect of the TEM images is the lack of clear banding (though some banding can be observed). This could be the result of the usual difficulty of observing bands in small diameter fibrils or the fibrils may truly be unbanded. Even if the fibrils are not banded, it is quite likely that the addition of proteoglycans (decorin/lumican) or other matrix molecules (type V collagen) known to alter fibril morphology will enhance the banding and diameter distribution (Birk et al., 1990; Brown et al., 2002). Another difficulty with most of the work on organizing collagen is the fact that it is typically done with readily available atelo-type I collagen monomers. Constructs derived from this material will always lack lysyl oxidase cross-links and will thus not be strong unless cross-linked in some other manner. Nonetheless, it appears that collagen has adequate information encoded into the triple helical domain to produce native-like lamellae which are persistent over long distances.
De novo tissue engineering of load-bearing constructs (including the cornea) will ultimately rely on the ability to control collagen alignment, long-range structure, fibril spacing and morphology. The former two objectives are likely to be aided considerably by the fact that collagen appears to be a “smart” engineering material (which fibroblastic cells have learned to manipulate with amazing precision over 800 million years of co-evolution). The liquid crystalline behavior, the ability to self-assemble (both radially and laterally) and recently demonstrated increase in fibril stability with mechanical load (Nabeshima et al., 1996; Bass et al., 2004; Ruberti and Hallab, 2005) make control over this polymerizing molecule potentially quite simple. Fibril spacing and morphology control is more of a challenge but may quite possibly be affected by addition of the same class of components: proteoglycans (Chakravarti et al., 1998; Chakravarti et al., 2000; Brown et al., 2002; Chakravarti et al., 2006).
5.0 Future directions
Much of the previous section addressed nascent efforts which are likely to mature into solid approaches to engineering a functional corneal stroma. Of the four types of tissue engineering defined (classical, developmental, de novo and hybrid), developmental and hybrid appear to hold the most promise. This is because pHSSCs and corneal stromal stem cells appear to possess the intrinsic ability to reproduce stroma-like matrix particularly when the cells can organize and proliferate to high density. It may be a simple matter of providing them the appropriate geometric/chemical/mechanical environment to induce formation of a functional stroma (including providing an aligned substrate or pre-organized scaffolding). The principal difficulties will be associated with phenotype control (pHSSCs being too differentiated; CSSCs being too undifferentiated) and controlling the resulting cell-synthesized construct long-range organization. Ultimately, whichever cells produce the tissue will have to be replaced with quiescent keratocytes (presumable by phenotypic switching). We cautiously discount classical tissue engineering particularly if the degradable scaffolding is disorganized as we believe that this will “imprint” disorganization on the cell patterning and ultimately the cell-derived matrix. Our inclination is to avoid the in vivo remodeling typically associated with classical tissue engineering approaches because of the uncertainty of individual host responses to implants. De novo tissue engineering is very nascent and will require a significant number of basic science investigations before a useful construct could be produced in this manner. Thus in the near term, we will likely continue to depend on the cells (with heavy support) to make what we need, although the empiricism in this process makes rationale design impossible. Ultimately, producing functional tissue de novo is desirable because it would allow full control over the process and the final construct morphology while enabling a rationale approach to optimize the tissue. However, the ability to employ rationale design will have to wait until the corneal tissue engineering tool box is full.
In closing, it is important to mention the often overlooked role of mechanical force in shaping structural tissues such as the cornea. In addition to the now well-established role of mechanical force in determining cell behavior (the effect of mechanical forces on corneal fibroblasts has been explored (Petroll et al., 2003; Karamichos et al., 2007)), there have been recent developments which suggest that collagen itself is a “smart” load-adaptive biomaterial (Nabeshima et al., 1996; Ruberti and Hallab, 2005). In their paper, Ruberti and Hallab demonstrated that mechanical force (or strain) alone can determine whether a collagen fibril is cleaved or retained when strips of devitalized cornea are exposed to collagenase. This suggests that collagen organization and remodeling may be under direct mechanical control (as well as cellular control). Thus it is likely that in addition to the particular tissue engineering approach chosen to produce a stromal construct, judicious application of mechanical force will play a key role in the structural development of engineered corneas (as it probably does in vivo).
Figure 8.

Critical stages in corneal development. Images depicts corneal collagen organization in transverse section (large rectangles) and en face section (small circles). At stage 27 (day 5) the epithelium has generated a primary stroma that strictly orthogonal and approximately 10 microns thick. Following a rapid expansion at the end of day 5, secondary stroma is produced posteriorly by invading mesenchymal cells. Note the shift in the register of the orthogonal collagen in the anterior uninvaded primary stroma. At the end of this stage (stage 33 (day 7)) the nascent stroma is 170 microns thick with the anterior 10 microns uninvaded. By stage 35 (day 9) the stroma is 190 microns thick with the posterior 180 microns invaded my mesenchymal cells. The anteriormost 40 microns demonstrate an angular shift between orthogonal layers of collagen, while the posterior 150 microns are in register. With permission from Trelstad and Coulombre (Trelstad and Coulombre, 1971)
Figure 17.

Photomicrograph of Giemsa-stained preparation illustrating the alignment of fibroblasts with orthogonal layers of shear-aligned collagen. Collagen was aligned during polymerization by draining collagen monomer solution on inclined plate. Cells were introduced onto first layer of aligned collagen. A second layer of aligned collagen was produced orthogonal to the first followed by plating a second population of cells. Note resulting orthogonal arrangement of cells. With permission from Elsdale and Bard (Elsdale and Bard, 1972).
Figure 21.
SEM micrographs of primary human corneal cells seeded onto dense collagen films. A) Rounded cells after one day on the film. B) Confluent cells after one week on the film. Asterisk indicates film top surface. With permission From Crabb et al. (Crabb et al., 2006)
Figure 22.
SEM micrographs of ECM synthesized by primary human corneal cells seeded onto dense collagen films after 5 weeks in culture. Arrows indicate smooth “fibrils” which are approximately 100 nm in diameter while the bracket indicates smaller diameter fibrils (47 nm) with a relatively large dispersity (± 17 nm). With permission from Crabb et al (Crabb et al., 2006)
Figure 27.
Immunofluorescence and TEM micrographs of HCSSCs in pellet culture. (A) Fluorescence image of connexin and cadherin staining in pellet culture showing prominent staining for these junctional proteins in the periphery. (B) Electron Micrograph demonstrating organized arrangement of fibrils in the area with the increased staining for junctional proteins. With permission from Du et al. (Du et al., 2007)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jeffrey W. Ruberti, Department of Mechanical and Industrial Engineering, Northeastern University, Boston, MA
James D. Zieske, Schepens Eye Research Institute and Department of Ophthalmology, Harvard Medical School, Boston, MA
References
- Aghamohammadzadeh H, Newton RH, Meek KM. X-ray scattering used to map the preferred collagen orientation in the human cornea and limbus. Structure. 2004;12:249–256. doi: 10.1016/j.str.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Aiken-O’Neill P, Mannis MJ. Summary of comeal transplant activity Eye Bank Association of America. Cornea. 2002;21:1–3. doi: 10.1097/00003226-200201000-00001. [DOI] [PubMed] [Google Scholar]
- Akeson WH, Amiel D, Abel MF, Garfin SR, Woo SL. Effects of immobilization on joints. Clin Orthop. 1987:28–37. [PubMed] [Google Scholar]
- Alper MG. The anesthetic eye: an investigation of changes in the anterior ocular segment of the monkey caused by interrupting the trigeminal nerve at various levels along its course. Trans Am Ophthalmol Soc. 1975;73:323–365. [PMC free article] [PubMed] [Google Scholar]
- Altman GH, Horan RL, Martin I, Farhadi J, Stark PR, Volloch V, Richmond JC, Vunjak-Novakovic G, Kaplan DL. Cell differentiation by mechanical stress. Faseb J. 2002;16:270–272. doi: 10.1096/fj.01-0656fje. [DOI] [PubMed] [Google Scholar]
- Anseth A. Studies on corneal polysaccharides. III. Topographic and comparative biochemistry. Exp Eye Res. 1961;1:106–115. doi: 10.1016/s0014-4835(61)80015-8. [DOI] [PubMed] [Google Scholar]
- Arokoski JP, Jurvelin JS, Vaatainen U, Helminen HJ. Normal and pathological adaptations of articular cartilage to joint loading. Scand J Med Sci Sports. 2000;10:186–198. doi: 10.1034/j.1600-0838.2000.010004186.x. [DOI] [PubMed] [Google Scholar]
- Axelsson I, Heinegard D. Fractionation of proteoglycans from bovine corneal stroma. Biochem J. 1975;145:491–500. doi: 10.1042/bj1450491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Mauck RL. The effect of nanofiber alignment on the maturation of engineered meniscus constructs. Biomaterials. 2007;28:1967–1977. doi: 10.1016/j.biomaterials.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JB, Abbott AS. Matrices containing glycosaminoglycans in the developing anterior chambers of chick and Xenopus embryonic eyes. Dev Biol. 1979;68:472–486. doi: 10.1016/0012-1606(79)90219-7. [DOI] [PubMed] [Google Scholar]
- Bard JB, Hay ED. The behavior of fibroblasts from the developing avian cornea. Morphology and movement in situ and in vitro. J Cell Biol. 1975;67:400–418. doi: 10.1083/jcb.67.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JB, Higginson K. Fibroblast-collagen interactions in the formation of the secondary stroma of the chick cornea. J Cell Biol. 1977;74:816–827. doi: 10.1083/jcb.74.3.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass EC, Wistrom EV, Diederich CJ, Nau WH, Pellegrino R, Ruberti J, Lotz JC. Heat-induced changes in porcine annulus fibrosus biomechanics. J Biomech. 2004;37:233–240. doi: 10.1016/j.jbiomech.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Bayer IM, Adamson SL, Langille BL. Atrophic remodeling of the artery-cuffed artery. Arterioscler Thromb Vasc Biol. 1999;19:1499–1505. doi: 10.1161/01.atv.19.6.1499. [DOI] [PubMed] [Google Scholar]
- Beales MP, Funderburgh JL, Jester JV, Hassell JR. Proteoglycan synthesis by bovine keratocytes and corneal fibroblasts: maintenance of the keratocyte phenotype in culture. Invest Ophthalmol Vis Sci. 1999;40:1658–1663. [PubMed] [Google Scholar]
- Benedek GB. Theory of the transparency of the eye. Applied Optics. 1971;10:459–473. doi: 10.1364/AO.10.000459. [DOI] [PubMed] [Google Scholar]
- Berryhill BL, Beales MP, Hassell JR. Production of prostaglandin D synthase as a keratan sulfate proteoglycan by cultured bovine keratocytes. Invest Ophthalmol Vis Sci. 2001;42:1201–1207. [PubMed] [Google Scholar]
- Berryhill BL, Kader R, Kane B, Birk DE, Feng J, Hassell JR. Partial restoration of the keratocyte phenotype to bovine keratocytes made fibroblastic by serum. Invest Ophthalmol Vis Sci. 2002;43:3416–3421. [PubMed] [Google Scholar]
- Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J Cell Sci. 1990;95(Pt 4):649–657. doi: 10.1242/jcs.95.4.649. [DOI] [PubMed] [Google Scholar]
- Birk DE, Fitch JM, Babiarz JP, Linsenmayer TF. Collagen type I and type V are present in the same fibril in the avian corneal stroma. J Cell Biol. 1988;106:999–1008. doi: 10.1083/jcb.106.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk DE, Silver FH. Kinetic analysis of collagen fibrillogenesis: II. Corneal and scleral type I collagen. Collagen Related Research. 1984 doi: 10.1016/s0174-173x(84)80034-5. [DOI] [PubMed] [Google Scholar]
- Birk DE, Trelstad RL. Extracellular compartments in matrix morphogenesis: collagen fibril, bundle, and lamellar formation by corneal fibroblasts. J Cell Biol. 1984;99:2024–2033. doi: 10.1083/jcb.99.6.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JE, Lindahl G. Regulation of cardiovascular collagen synthesis by mechanical load. Cardiovasc Res. 1999;42:27–44. doi: 10.1016/s0008-6363(99)00021-8. [DOI] [PubMed] [Google Scholar]
- Bohnke M, Eggli P, Engelmann K. Transplantation of cultured adult human or porcine corneal endothelial cells onto human recipients in vitro. Part II: Evaluation in the scanning electron microscope. Cornea. 1999;18:207–213. doi: 10.1097/00003226-199903000-00011. [DOI] [PubMed] [Google Scholar]
- Bonanno JA. Identity and regulation of ion transport mechanisms in the corneal endothelium. Prog Retin Eye Res. 2003;22:69–94. doi: 10.1016/s1350-9462(02)00059-9. [DOI] [PubMed] [Google Scholar]
- Boot JP, van Best JA, Stolwijk TR, Sterk CC. Epithelial permeability in corneal grafts by fluorophotometry. Graefes Arch Clin Exp Ophthalmol. 1991;229:533–535. doi: 10.1007/BF00203316. [DOI] [PubMed] [Google Scholar]
- Borderie VM, Mourra N, Laroche L. Influence of fetal calf serum, fibroblast growth factors, and hepatocyte growth factor on three-dimensional cultures of human keratocytes in collagen gel matrix. Graefes Arch Clin Exp Ophthalmol. 1999;237:861–869. doi: 10.1007/s004170050324. [DOI] [PubMed] [Google Scholar]
- Brown CT, Lin P, Walsh MT, Gantz D, Nugent MA, Trinkaus-Randall V. Extraction and purification of decorin from corneal stroma retain structure and biological activity. Protein Expr Purif. 2002;25:389–399. doi: 10.1016/s1046-5928(02)00025-6. [DOI] [PubMed] [Google Scholar]
- Canty EG, Lu Y, Meadows RS, Shaw MK, Holmes DF, Kadler KE. Coalignment of plasma membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J Cell Biol. 2004;165:553–563. doi: 10.1083/jcb.200312071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson EC, Liu CY, Chikama T, Hayashi Y, Kao CW, Birk DE, Funderburgh JL, Jester JV, Kao WW. Keratocan, a cornea-specific keratan sulfate proteoglycan, is regulated by lumican. J Biol Chem. 2005;280:25541–25547. doi: 10.1074/jbc.M500249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter D, Beaupre G. Skeletal form and function: Mechanobiology of skeletal development, aging and regeneration. Cambridge, Press Syndicate of the University of Cambridge; 2001. [Google Scholar]
- Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H. Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol. 1998;141:1277–1286. doi: 10.1083/jcb.141.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti S, Petroll WM, Hassell JR, Jester JV, Lass JH, Paul J, Birk DE. Corneal opacity in lumican-null mice: defects in collagen fibril structure and packing in the posterior stroma. Invest Ophthalmol Vis Sci. 2000;41:3365–3373. [PMC free article] [PubMed] [Google Scholar]
- Chakravarti S, Zhang G, Chervoneva I, Roberts L, Birk DE. Collagen fibril assembly during postnatal development and dysfunctional regulation in the lumican-deficient murine cornea. Dev Dyn. 2006;235:2493–2506. doi: 10.1002/dvdy.20868. [DOI] [PubMed] [Google Scholar]
- Chamay A, Tschantz P. Mechanical influences in bone remodeling. Experimental research on Wolff’s law. J Biomech. 1972;5:173–180. doi: 10.1016/0021-9290(72)90053-x. [DOI] [PubMed] [Google Scholar]
- Charulatha V, Rajaram A. Crosslinking density and resorption of dimethyl suberimidate-treated collagen. J Biomed Mater Res. 1997;36:478–486. doi: 10.1002/(sici)1097-4636(19970915)36:4<478::aid-jbm5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Chen KH, Azar D, Joyce NC. Transplantation of adult human corneal endothelium ex vivo: a morphologic study. Cornea. 2001;20:731–737. doi: 10.1097/00003226-200110000-00012. [DOI] [PubMed] [Google Scholar]
- Cintron C, Covington H, Kublin CL. Morphogenesis of rabbit corneal stroma. Invest Ophthalmol Vis Sci. 1983;24:543–556. [PubMed] [Google Scholar]
- Cintron C, Gregory JD, Damle SP, Kublin CL. Biochemical analyses of proteoglycans in rabbit corneal scars. Invest Ophthalmol Vis Sci. 1990;31:1975–1981. [PubMed] [Google Scholar]
- Cintron C, Hong BS, Kublin CL. Quantitative analysis of collagen from normal developing corneas and corneal scars. Curr Eye Res. 1981;1:1–8. doi: 10.3109/02713688109019966. [DOI] [PubMed] [Google Scholar]
- Cintron C, Schneider H, Kublin C. Corneal scar formation. Exp Eye Res. 1973;17:251–259. doi: 10.1016/0014-4835(73)90176-0. [DOI] [PubMed] [Google Scholar]
- Cintron C, Szamier RB, Hassinger LC, Kublin CL. Scanning electron microscopy of rabbit corneal scars. Invest Ophthalmol Vis Sci. 1982;23:50–63. [PubMed] [Google Scholar]
- Cowin SC. The mechanical and stress adaptive properties of bone. Ann Biomed Eng. 1983;11:263–295. doi: 10.1007/BF02363288. [DOI] [PubMed] [Google Scholar]
- Cowin SC. How is a tissue built? J Biomech Eng. 2000;122:553–569. doi: 10.1115/1.1324665. [DOI] [PubMed] [Google Scholar]
- Cowin SC. Tissue growth and remodeling. Annu Rev Biomed Eng. 2004;6:77–107. doi: 10.1146/annurev.bioeng.6.040803.140250. [DOI] [PubMed] [Google Scholar]
- Cox JL, Farrell RA, Hart RW, Langham ME. The transparency of the mammalian cornea. J Physiol. 1970;210:601–616. doi: 10.1113/jphysiol.1970.sp009230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb RA, Chau EP, Decoteau DM, Hubel A. Microstructural characteristics of extracellular matrix produced by stromal fibroblasts. Ann Biomed Eng. 2006;34:1615–1627. doi: 10.1007/s10439-006-9181-x. [DOI] [PubMed] [Google Scholar]
- Crabb RA, Chau EP, Evans MC, Barocas VH, Hubel A. Biomechanical and microstructural characteristics of a collagen film-based corneal stroma equivalent. Tissue Eng. 2006;12:1565–1575. doi: 10.1089/ten.2006.12.1565. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doane KJ, Babiarz JP, Fitch JM, Linsenmayer TF, Birk DE. Collagen fibril assembly by corneal fibroblasts in three-dimensional collagen gel cultures: small-diameter heterotypic fibrils are deposited in the absence of keratan sulfate proteoglycan. Exp Cell Res. 1992;202:113–124. doi: 10.1016/0014-4827(92)90410-a. [DOI] [PubMed] [Google Scholar]
- Doane KJ, Birk DE. Fibroblasts retain their tissue phenotype when grown in three-dimensional collagen gels. Exp Cell Res. 1991;195:432–442. doi: 10.1016/0014-4827(91)90394-a. [DOI] [PubMed] [Google Scholar]
- Dohlman CH, Gasset AR, Rose J. The effect of the absence of corneal epithelium or endothelium on the stromal keratocytes. Invest Ophthalmol. 1968;7:520–534. [PubMed] [Google Scholar]
- Du Y, Sundarraj N, Funderburgh ML, Harvey SA, Birk DE, Funderburgh JL. Secretion and organization of a cornea-like tissue in vitro by stem cells from human corneal stroma. Invest Ophthalmol Vis Sci. 2007;48:5038–5045. doi: 10.1167/iovs.07-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duty AO, Oest ME, Guldberg RE. Cyclic mechanical compression increases mineralization of cell-seeded polymer scaffolds in vivo. J Biomech Eng. 2007;129:531–539. doi: 10.1115/1.2746375. [DOI] [PubMed] [Google Scholar]
- Elsdale T, Bard J. Collagen substrata for studies on cell behavior. J Cell Biol. 1972;54:626–637. doi: 10.1083/jcb.54.3.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann K, Bednarz J, Valtink M. Prospects for endothelial transplantation. Exp Eye Res. 2004;78:573–578. doi: 10.1016/s0014-4835(03)00209-4. [DOI] [PubMed] [Google Scholar]
- Engelmann K, Drexler D, Bohnke M. Transplantation of adult human or porcine corneal endothelial cells onto human recipients in vitro. Part I: Cell culturing and transplantation procedure. Cornea. 1999;18:199–206. doi: 10.1097/00003226-199903000-00010. [DOI] [PubMed] [Google Scholar]
- Ethier CR, Johnson M, Ruberti J. Ocular biomechanics and biotransport. Annu Rev Biomed Eng. 2004;6:249–273. doi: 10.1146/annurev.bioeng.6.040803.140055. [DOI] [PubMed] [Google Scholar]
- Faraj KA, van Kuppevelt TH, Daamen WF. Construction of collagen scaffolds that mimic the three-dimensional architecture of specific tissues. Tissue Eng. 2007;13:2387–2394. doi: 10.1089/ten.2006.0320. [DOI] [PubMed] [Google Scholar]
- Fini ME. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res. 1999;18:529–551. doi: 10.1016/s1350-9462(98)00033-0. [DOI] [PubMed] [Google Scholar]
- Fligiel SE, Varani J, Datta SC, Kang S, Fisher GJ, Voorhees JJ. Collagen degradation in aged/photodamaged skin in vivo and after exposure to matrix metalloproteinase-1 in vitro. J Invest Dermatol. 2003;120:842–848. doi: 10.1046/j.1523-1747.2003.12148.x. [DOI] [PubMed] [Google Scholar]
- Friedman MH. A quantitative description of equilibrium and homeostatic thickness regulation in the in vivo cornea. I. Normal cornea. Biophys J. 1972;12:648–665. doi: 10.1016/S0006-3495(72)86110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend J, Kinoshita S, Thoft RA, Eliason JA. Corneal epithelial cell cultures on stromal carriers. Invest Ophthalmol Vis Sci. 1982;23:41–49. [PubMed] [Google Scholar]
- Funderburgh JL, Chandler JW. Proteoglycans of rabbit corneas with nonperforating wounds. Invest Ophthalmol Vis Sci. 1989;30:435–442. [PubMed] [Google Scholar]
- Funderburgh JL, Funderburgh ML, Mann MM, Prakash S, Conrad GW. Synthesis of corneal keratan sulfate proteoglycans by bovine keratocytes in vitro. J Biol Chem. 1996;271:31431–31436. doi: 10.1074/jbc.271.49.31431. [DOI] [PubMed] [Google Scholar]
- Funderburgh JL, Mann MM, Funderburgh ML. Keratocyte phenotype mediates proteoglycan structure: a role for fibroblasts in corneal fibrosis. J Biol Chem. 2003;278:45629–45637. doi: 10.1074/jbc.M303292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garana RM, Petroll WM, Chen WT, Herman IM, Barry P, Andrews P, Cavanagh HD, Jester JV. Radial keratotomy. II. Role of the myofibroblast in corneal wound contraction. Invest Ophthalmol Vis Sci. 1992;33:3271–3282. [PubMed] [Google Scholar]
- Geggel HS, Friend J, Thoft RA. Collagen gel for ocular surface. Invest Ophthalmol Vis Sci. 1985;26:901–905. [PubMed] [Google Scholar]
- Germain L, Auger FA, Grandbois E, Guignard R, Giasson M, Boisjoly H, Guerin SL. Reconstructed human cornea produced in vitro by tissue engineering. Pathobiology. 1999;67:140–147. doi: 10.1159/000028064. [DOI] [PubMed] [Google Scholar]
- Germain L, Carrier P, Deschambeault A, Talbot M, Guerin SL, Auger FA. Fibroblasts modulate differentiation and stratification of epithelial cells on reconstructed human cornea through the production of soluble factors. ARVO, Fort Lauderdale 2004 [Google Scholar]
- Giraud-Guille MM. Twisted plywood architecture of collagen fibrils in human compact bone osteons. Calcif Tissue Int. 1988;42:167–180. doi: 10.1007/BF02556330. [DOI] [PubMed] [Google Scholar]
- Giraud-Guille MM. Liquid crystallinity in condensed type I collagen solutions. A clue to the packing of collagen in extracellular matrices. J Mol Biol. 1992;224:861–873. doi: 10.1016/0022-2836(92)90567-4. [DOI] [PubMed] [Google Scholar]
- Giraud-Guille MM, Besseau L, Martin R. Liquid crystalline assemblies of collagen in bone and in vitro systems. J Biomech. 2003;36:1571–1579. doi: 10.1016/s0021-9290(03)00134-9. [DOI] [PubMed] [Google Scholar]
- Giraud Guille MM, Mosser G, Helary C, Eglin D. Bone matrix like assemblies of collagen: from liquid crystals to gels and biomimetic materials. Micron. 2005;36:602–608. doi: 10.1016/j.micron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Gobeaux F, Mosser G, Anglo A, Panine P, Davidson P, Giraud-Guille MM, Belamie E. Fibrillogenesis in Dense Collagen Solutions: A Physicochemical Study. J Mol Biol. 2008 doi: 10.1016/j.jmb.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Goldman JN, Benedek GB. The relationship between morphology and transparency in the nonswelling corneal stroma of the shark. Invest Ophthalmol. 1967;6:574–600. [PubMed] [Google Scholar]
- Goldman JN, Benedek GB, Dohlman CH, Kravitt B. Structural alterations affecting transparency in swollen human corneas. Invest Ophthalmol. 1968;7:501–519. [PubMed] [Google Scholar]
- Griffith M, Hakim M, Shimmura S, Watsky MA, Li F, Carlsson D, Doillon CJ, Nakamura M, Suuronen E, Shinozaki N, Nakata K, Sheardown H. Artificial human corneas: scaffolds for transplantation and host regeneration. Cornea. 2002;21:S54–61. doi: 10.1097/01.ico.0000263120.68768.f8. [DOI] [PubMed] [Google Scholar]
- Griffith M, Osborne R, Munger R, Xiong X, Doillon CJ, Laycock NL, Hakim M, Song Y, Watsky MA. Functional human corneal equivalents constructed from cell lines. Science. 1999;286:2169–2172. doi: 10.1126/science.286.5447.2169. [DOI] [PubMed] [Google Scholar]
- Gross J. Studies on the formation of collagen. I. Properties and fractionation of neutral salt extracts of normal guinea pig connective tissue. J Exp Med. 1958;107:247–263. doi: 10.1084/jem.107.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross J. Studies on the formation of collagen. II. The influence of growth rate on neutral salt extracts of guinea pig dermis. J Exp Med. 1958;107:265–277. doi: 10.1084/jem.107.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross J, Highberger JH, Schmitt FO. Collagen Structures Considered as States of Aggregation of a Kinetic Unit. the Tropocollagen Particle. Proc Natl Acad Sci U S A. 1954;40:679–688. doi: 10.1073/pnas.40.8.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guido S, Tranquillo RT. A methodology for the systematic and quantitative study of cell contact guidance in oriented collagen gels. Correlation of fibroblast orientation and gel birefringence. J Cell Sci. 1993;105(Pt 2):317–331. doi: 10.1242/jcs.105.2.317. [DOI] [PubMed] [Google Scholar]
- Guo X, Hutcheon AE, Melotti SA, Zieske JD, Trinkaus-Randall V, Ruberti JW. Morphologic characterization of organized extracellular matrix deposition by ascorbic acid-stimulated human corneal fibroblasts. Invest Ophthalmol Vis Sci. 2007;48:4050–4060. doi: 10.1167/iovs.06-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada R, Giraud JP, Graf B, Pouliquen Y. Etude analytique et statistique des lamelles, des keratocytes, des fibrillesdel collagene de la region centrale de la cornee humaine normale. Arch Ophtalmol. 1972;32:563–570. [PubMed] [Google Scholar]
- Hanna C, O’Brien JE. Cell production and migration in the epithelial layer of the cornea. Arch Ophthalmol. 1960;64:536–539. doi: 10.1001/archopht.1960.01840010538009. [DOI] [PubMed] [Google Scholar]
- Hart RW, Farrell RA. Light scattering in the cornea. J Opt Soc Am. 1969;59:766–774. doi: 10.1364/josa.59.000766. [DOI] [PubMed] [Google Scholar]
- Hassell JR, Schrecengost PK, Rada JA, SundarRaj N, Sossi G, Thoft RA. Biosynthesis of stromal matrix proteoglycans and basement membrane components by human corneal fibroblasts. Invest Ophthalmol Vis Sci. 1992;33:547–557. [PubMed] [Google Scholar]
- Hasty DL, Hay ED. Freeze-fracture studies of the developing cell surface. I. The plasmalemma of the corneal fibroblast. J Cell Biol. 1977;72:667–686. doi: 10.1083/jcb.72.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn. 2005;233:706–720. doi: 10.1002/dvdy.20345. [DOI] [PubMed] [Google Scholar]
- Hay ED, Revel JP. Fine structure of the developing avian cornea. New York, Basel: 1969. [PubMed] [Google Scholar]
- Hedbys BO. The role of polysaccharides in corneal swelling. Experimental Eye Research. 1961;1:81. doi: 10.1016/s0014-4835(61)80012-2. [DOI] [PubMed] [Google Scholar]
- Hedbys BO, Mishima S, Maurice DM. The imbibition pressure of the corneal stroma. Exp Eye Res. 1963;2:99–111. doi: 10.1016/s0014-4835(63)80001-9. [DOI] [PubMed] [Google Scholar]
- Hennighausen H, Feldman ST, Bille JF, McCulloch AD. Anterior-posterior strain variation in normally hydrated and swollen rabbit cornea. Invest Ophthalmol Vis Sci. 1998;39:253–262. [PubMed] [Google Scholar]
- Highberger JH, Gross J, Schmitt FO. The interaction of mucoprotein with soluble collagen; an electron microscope study. Proc Natl Acad Sci U S A. 1951;37:286–291. doi: 10.1073/pnas.37.5.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch M, Noske W, Prenant G, Renard G. Fine structure of the developing avian corneal stroma as revealed by quick-freeze, deep-etch electron microscopy. Exp Eye Res. 1999;69:267–277. doi: 10.1006/exer.1999.0695. [DOI] [PubMed] [Google Scholar]
- Hjortdal JO. Regional elastic performance of the human cornea. J Biomech. 1996;29:931–942. doi: 10.1016/0021-9290(95)00152-2. [DOI] [PubMed] [Google Scholar]
- Humphrey JD. Stress, strain, and mechanotransduction in cells. J Biomech Eng. 2001;123:638–641. doi: 10.1115/1.1406131. [DOI] [PubMed] [Google Scholar]
- Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM. Induction of alpha-smooth muscle actin expression and myofibroblast transformation in cultured corneal keratocytes. Cornea. 1996;15:505–516. [PubMed] [Google Scholar]
- Jester JV, Barry PA, Lind GJ, Petroll WM, Garana R, Cavanagh HD. Corneal keratocytes: in situ and in vitro organization of cytoskeletal contractile proteins. Invest Ophthalmol Vis Sci. 1994;35:730–743. [PubMed] [Google Scholar]
- Jester JV, Moller-Pedersen T, Huang J, Sax CM, Kays WT, Cavangh HD, Petroll WM, Piatigorsky J. The cellular basis of corneal transparency: evidence for ‘corneal crystallins’. J Cell Sci. 1999;112(Pt 5):613–622. doi: 10.1242/jcs.112.5.613. [DOI] [PubMed] [Google Scholar]
- Jester JV, Petroll WM, Barry PA, Cavanagh HD. Expression of alpha-smooth muscle (alpha-SM) actin during corneal stromal wound healing. Invest Ophthalmol Vis Sci. 1995;36:809–819. [PubMed] [Google Scholar]
- Jester JV, Petroll WM, Barry PA, Cavanagh HD. Temporal, 3-dimensional, cellular anatomy of corneal wound tissue. J Anat. 1995;186(Pt 2):301–311. [PMC free article] [PubMed] [Google Scholar]
- Jester JV, Petroll WM, Cavanagh HD. Corneal stromal wound healing in refractive surgery: the role of myofibroblasts. Prog Retin Eye Res. 1999;18:311–356. doi: 10.1016/s1350-9462(98)00021-4. [DOI] [PubMed] [Google Scholar]
- Jester JV, Rodrigues MM, Herman IM. Characterization of avascular corneal wound healing fibroblasts. New insights into the myofibroblast. Am J Pathol. 1987;127:140–148. [PMC free article] [PubMed] [Google Scholar]
- Joyce NC, Meklir B, Joyce SJ, Zieske JD. Cell cycle protein expression and proliferative status in human corneal cells. Invest Ophthalmol Vis Sci. 1996;37:645–655. [PubMed] [Google Scholar]
- Joyce NC, Navon SE, Roy S, Zieske JD. Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest Ophthalmol Vis Sci. 1996;37:1566–1575. [PubMed] [Google Scholar]
- Joyce NC, Zhu CC. Human corneal endothelial cell proliferation: potential for use in regenerative medicine. Cornea. 2004;23:S8–S19. doi: 10.1097/01.ico.0000136666.63870.18. [DOI] [PubMed] [Google Scholar]
- Juncosa-Melvin N, Shearn JT, Boivin GP, Gooch C, Galloway MT, West JR, Nirmalanandhan VS, Bradica G, Butler DL. Effects of mechanical stimulation on the biomechanics and histology of stem cell-collagen sponge constructs for rabbit patellar tendon repair. Tissue Eng. 2006;12:2291–2300. doi: 10.1089/ten.2006.12.2291. [DOI] [PubMed] [Google Scholar]
- Kadler KE, Hojima Y, Prockop DJ. Collagen fibrils in vitro grow from pointed tips in the C- to N-terminal direction. Biochem J. 1990;268:339–343. doi: 10.1042/bj2680339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadler KE, Hulmes DJ, Hojima Y, Prockop DJ. Assembly of type I collagen fibrils de novo by the specific enzymic cleavage of pC collagen. The fibrils formed at about 37 degrees C are similar in diameter, roundness, and apparent flexibility to the collagen fibrils seen in connective tissue. Ann N Y Acad Sci. 1990;580:214–224. doi: 10.1111/j.1749-6632.1990.tb17930.x. [DOI] [PubMed] [Google Scholar]
- Karamichos D, Lakshman N, Petroll WM. Regulation of corneal fibroblast morphology and collagen reorganization by extracellular matrix mechanical properties. Invest Ophthalmol Vis Sci. 2007;48:5030–5037. doi: 10.1167/iovs.07-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karring H, Thogersen IB, Klintworth GK, Enghild JJ, Moller-Pedersen T. Proteomic analysis of the soluble fraction from human corneal fibroblasts with reference to ocular transparency. Mol Cell Proteomics. 2004;3:660–674. doi: 10.1074/mcp.M400016-MCP200. [DOI] [PubMed] [Google Scholar]
- Ko HC, Milthorpe BK, McFarland CD. Engineering thick tissues--the vascularisation problem. Eur Cell Mater. 2007;14:1–18. doi: 10.22203/ecm.v014a01. discussion 18–19. [DOI] [PubMed] [Google Scholar]
- Konomi K, Zhu C, Harris D, Joyce NC. Comparison of the proliferative capacity of human corneal endothelial cells from the central and peripheral areas. Invest Ophthalmol Vis Sci. 2005;46:4086–4091. doi: 10.1167/iovs.05-0245. [DOI] [PubMed] [Google Scholar]
- Kostyuk O, Nalovina O, Mubard TM, Regini JW, Meek KM, Quantock AJ, Elliott GF, Hodson SA. Transparency of the bovine corneal stroma at physiological hydration and its dependence on concentration of the ambient anion. J Physiol. 2002;543:633–642. doi: 10.1113/jphysiol.2002.021527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer R, Vacanti JP. Tissue engineering. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- Leber T. Studien uber den Flussigkeitswechsel im Auge. Graefes Arch Ophthalmol. 1873;19:87. [Google Scholar]
- Li F, Carlsson D, Lohmann C, Suuronen E, Vascotto S, Kobuch K, Sheardown H, Munger R, Nakamura M, Griffith M. Cellular and nerve regeneration within a biosynthetic extracellular matrix for corneal transplantation. Proc Natl Acad Sci U S A. 2003;100:15346–15351. doi: 10.1073/pnas.2536767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Griffith M, Li Z, Tanodekaew S, Sheardown H, Hakim M, Carlsson DJ. Recruitment of multiple cell lines by collagen-synthetic copolymer matrices in corneal regeneration. Biomaterials. 2005;26:3093–3104. doi: 10.1016/j.biomaterials.2004.07.063. [DOI] [PubMed] [Google Scholar]
- Lindberg K, Brown ME, Chaves HV, Kenyon KR, Rheinwald JG. In vitro propagation of human ocular surface epithelial cells for transplantation. Invest Ophthalmol Vis Sci. 1993;34:2672–2679. [PubMed] [Google Scholar]
- Linsenmayer TF, Fitch JM, Gordon MK, Cai CX, Igoe F, Marchant JK, Birk DE. Development and roles of collagenous matrices in the embryonic avian cornea. Prog Retin Eye Res. 1998;17:231–265. doi: 10.1016/s1350-9462(97)00010-4. [DOI] [PubMed] [Google Scholar]
- Linsenmayer TF, Gibney E, Fitch JM. Embryonic avian cornea contains layers of collagen with greater than average stability. J Cell Biol. 1986;103:1587–1593. doi: 10.1083/jcb.103.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masur SK, Cheung JK, Antohi S. Identification of integrins in cultured corneal fibroblasts and in isolated keratocytes. Invest Ophthalmol Vis Sci. 1993;34:2690–2698. [PubMed] [Google Scholar]
- Masur SK, Conors RJ, Jr, Cheung JK, Antohi S. Matrix adhesion characteristics of corneal myofibroblasts. Invest Ophthalmol Vis Sci. 1999;40:904–910. [PubMed] [Google Scholar]
- Matsuda H, Smelser GK. Electron microscopy of corneal wound healing. Exp Eye Res. 1973;16:427–442. doi: 10.1016/0014-4835(73)90100-0. [DOI] [PubMed] [Google Scholar]
- Matthews JA, Wnek GE, Simpson DG, Bowlin GL. Electrospinning of collagen nanofibers. Biomacromolecules. 2002;3:232–238. doi: 10.1021/bm015533u. [DOI] [PubMed] [Google Scholar]
- Maurice DM. The structure and transparency of the cornea. Journal of Physiology. 1957;136:263–286. doi: 10.1113/jphysiol.1957.sp005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DM. The location of the fluid pump in the cornea. Journal of Physiology. 1972;221:43–54. doi: 10.1113/jphysiol.1972.sp009737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister JC, Joyce NC, Harris DL, Ali RR, Larkin DF. Induction of replication in human corneal endothelial cells by E2F2 transcription factor cDNA transfer. Invest Ophthalmol Vis Sci. 2005;46:3597–3603. doi: 10.1167/iovs.04-0551. [DOI] [PubMed] [Google Scholar]
- McPhee TJ, Bourne WM, Brubaker RF. Location of the stress-bearing layers of the cornea. Invest Ophthalmol Vis Sci. 1985;26:869–872. [PubMed] [Google Scholar]
- Meek KM, Boote C. The organization of collagen in the corneal stroma. Exp Eye Res. 2004;78:503–512. doi: 10.1016/j.exer.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Meek KM, Leonard DW. Ultrastructure of the corneal stroma: a comparative study. Biophys J. 1993;64:273–280. doi: 10.1016/S0006-3495(93)81364-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michna H, Hartmann G. Adaptation of tendon collagen to exercise. Int Orthop. 1989;13:161–165. doi: 10.1007/BF00268040. [DOI] [PubMed] [Google Scholar]
- Minami Y, Sugihara H, Oono S. Reconstruction of cornea in three-dimensional collagen gel matrix culture. Invest Ophthalmol Vis Sci. 1993;34:2316–2324. [PubMed] [Google Scholar]
- Mould AP, Hulmes DJ. Surface-induced aggregation of type I procollagen. J Mol Biol. 1987;195:543–553. doi: 10.1016/0022-2836(87)90182-3. [DOI] [PubMed] [Google Scholar]
- Mould AP, Hulmes DJ, Holmes DF, Cummings C, Sear CH, Chapman JA. D-periodic assemblies of type I procollagen. J Mol Biol. 1990;211:581–594. doi: 10.1016/0022-2836(90)90267-P. [DOI] [PubMed] [Google Scholar]
- Muller LJ, Pels L, Vrensen GF. Novel aspects of the ultrastructural organization of human corneal keratocytes. Invest Ophthalmol Vis Sci. 1995;36:2557–2567. [PubMed] [Google Scholar]
- Musselmann K, Alexandrou B, Kane B, Hassell JR. Maintenance of the keratocyte phenotype during cell proliferation stimulated by insulin. J Biol Chem. 2005;280:32634–32639. doi: 10.1074/jbc.M504724200. [DOI] [PubMed] [Google Scholar]
- Nabeshima Y, Grood ES, Sakurai A, Herman JH. Uniaxial tension inhibits tendon collagen degradation by collagenase in vitro. J Orthop Res. 1996;14:123–130. doi: 10.1002/jor.1100140120. [DOI] [PubMed] [Google Scholar]
- Neath P, Roche SM, Bee JA. Intraocular pressure-dependent and -independent phases of growth of the embryonic chick eye and cornea. Invest Ophthalmol Vis Sci. 1991;32:2483–2491. [PubMed] [Google Scholar]
- Nerem RM. Tissue engineering: the hope, the hype, and the future. Tissue Eng. 2006;12:1143–1150. doi: 10.1089/ten.2006.12.1143. [DOI] [PubMed] [Google Scholar]
- Newsome DA, Takasugi M, Kenyon KR, Stark WF, Opelz G. Human corneal cells in vitro: morphology and histocompatibility (HL-A) antigens of pure cell populations. Invest Ophthalmol. 1974;13:23–32. [PubMed] [Google Scholar]
- Ohji M, SundarRaj N, Hassell JR, Thoft RA. Basement membrane synthesis by human corneal epithelial cells in vitro. Invest Ophthalmol Vis Sci. 1994;35:479–485. [PubMed] [Google Scholar]
- Orwin EJ, Borene ML, Hubel A. Biomechanical and optical characteristics of a corneal stromal equivalent. J Biomech Eng. 2003;125:439–444. doi: 10.1115/1.1589773. [DOI] [PubMed] [Google Scholar]
- Orwin EJ, Hubel A. In vitro culture characteristics of corneal epithelial, endothelial, and keratocyte cells in a native collagen matrix. Tissue Eng. 2000;6:307–319. doi: 10.1089/107632700418038. [DOI] [PubMed] [Google Scholar]
- Ozanics V, Rayborn M, Sagun D. Observations on the morphology of the developing primate cornea: epithelium, its innervation and anterior stroma. J Morphol. 1977;153:263–297. doi: 10.1002/jmor.1051530207. [DOI] [PubMed] [Google Scholar]
- Petroll WM, Cavanagh HD, Barry P, Andrews P, Jester JV. Quantitative analysis of stress fiber orientation during corneal wound contraction. J Cell Sci. 1993;104(Pt 2):353–363. doi: 10.1242/jcs.104.2.353. [DOI] [PubMed] [Google Scholar]
- Petroll WM, Ma L, Jester JV. Direct correlation of collagen matrix deformation with focal adhesion dynamics in living corneal fibroblasts. J Cell Sci. 2003;116:1481–1491. doi: 10.1242/jcs.00357. [DOI] [PubMed] [Google Scholar]
- Prockop D, Hulmes D. Assembly of collagen fibrils de novo from soluble precursors: Polymerization and copolymerization of procollagen, pN-collagen, and mutated collagens. In: Yurchenco BDPD, Mecham RP, editors. Extracellular Matrix Assembly and Structure. San Diego: Academic Press, Inc; 1994. pp. 47–90. [Google Scholar]
- Radner W, Mallinger R. Interlacing of collagen lamellae in the midstroma of the human cornea. Cornea. 2002;21:598–601. doi: 10.1097/00003226-200208000-00013. [DOI] [PubMed] [Google Scholar]
- Radner W, Zehetmayer M, Aufreiter R, Mallinger R. Interlacing and cross-angle distribution of collagen lamellae in the human cornea. Cornea. 1998;17:537–543. doi: 10.1097/00003226-199809000-00012. [DOI] [PubMed] [Google Scholar]
- Rodriguez JI, Garcia-Alix A, Palacios J, Paniagua R. Changes in the long bones due to fetal immobility caused by neuromuscular disease. A radiographic and histological study. J Bone Joint Surg Am. 1988;70:1052–1060. [PubMed] [Google Scholar]
- Rodriguez JI, Palacios J, Garcia-Alix A, Pastor I, Paniagua R. Effects of immobilization on fetal bone development. A morphometric study in newborns with congenital neuromuscular diseases with intrauterine onset. Calcif Tissue Int. 1988;43:335–339. doi: 10.1007/BF02553275. [DOI] [PubMed] [Google Scholar]
- Roy P, Petroll WM, Cavanagh HD, Chuong CJ, Jester JV. An in vitro force measurement assay to study the early mechanical interaction between corneal fibroblasts and collagen matrix. Exp Cell Res. 1997;232:106–117. doi: 10.1006/excr.1997.3511. [DOI] [PubMed] [Google Scholar]
- Ruberti J, Zieske J, Trinkaus-Randall V. In: Corneal Tissue Replacement. Principles of TIssue Engineering. Lanza R, Langer R, Vacanti J, editors. Chap 68 Elsevier/Academic Press; 2007. [Google Scholar]
- Ruberti JW, Hallab NJ. Strain-controlled enzymatic cleavage of collagen in loaded matrix. Biochem Biophys Res Commun. 2005;336:483–489. doi: 10.1016/j.bbrc.2005.08.128. [DOI] [PubMed] [Google Scholar]
- Ruberti JW, Klyce SD. Physiological System Models of the Cornea. In: Hung GK, Ciuffreda KJ, editors. Models of the visual system. New York: Kluwer Academic/Plenum Publishers; 2002. pp. 3–55. [Google Scholar]
- Ruggiero F, Burillon C, Garrone R. Human corneal fibrillogenesis. Collagen V structural analysis and fibrillar assembly by stromal fibroblasts in culture. Invest Ophthalmol Vis Sci. 1996;37:1749–1760. [PubMed] [Google Scholar]
- Schermer A, Galvin S, Sun TT. Differentiation-related expression of a major 64K corneal keratin in vivo and in culture suggests limbal location of corneal epithelial stem cells. J Cell Biol. 1986;103:49–62. doi: 10.1083/jcb.103.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JE. Proteoglycan: collagen interactions and corneal ultrastructure. Biochem Soc Trans. 1991;19:877–881. doi: 10.1042/bst0190877. [DOI] [PubMed] [Google Scholar]
- Scott JE, Haigh M. ‘Small’-proteoglycan:collagen interactions: keratan sulphate proteoglycan associates with rabbit corneal collagen fibrils at the ‘a’ and ‘c’ bands. Biosci Rep. 1985;5:765–774. doi: 10.1007/BF01119875. [DOI] [PubMed] [Google Scholar]
- Sevel D, Isaacs R. A re-evaluation of corneal development. Trans Am Ophthalmol Soc. 1988;86:178–207. [PMC free article] [PubMed] [Google Scholar]
- Smelser GK. Corneal hydration. Comparative physiology of fish and mammals. Invest Ophthalmol. 1962;1:11–32. [PubMed] [Google Scholar]
- Stoddart MJ, Ettinger L, Hauselmann HJ. Enhanced matrix synthesis in de novo, scaffold free cartilage-like tissue subjected to compression and shear. Biotechnol Bioeng. 2006;95:1043–1051. doi: 10.1002/bit.21052. [DOI] [PubMed] [Google Scholar]
- Stoesser TR, Church RL, Brown SI. Partial characterization of human collagen and procollagen secreted by human corneal stromal fibroblasts in cell culture. Invest Ophthalmol Vis Sci. 1978;17:264–271. [PubMed] [Google Scholar]
- Strissel KJ, Rinehart WB, Fini ME. Regulation of paracrine cytokine balance controlling collagenase synthesis by corneal cells. Invest Ophthalmol Vis Sci. 1997;38:546–552. [PubMed] [Google Scholar]
- Sugrue SP, Zieske JD. ZO1 in corneal epithelium: association to the zonula occludens and adherens junctions. Exp Eye Res. 1997;64:11–20. doi: 10.1006/exer.1996.0175. [DOI] [PubMed] [Google Scholar]
- Sundar-Raj CV, Freeman IL, Brown SI. Selective growth of rabbit corneal epithelial cells in culture and basement membrane collagen synthesis. Invest Ophthalmol Vis Sci. 1980;19:1222–1230. [PubMed] [Google Scholar]
- Sweeney DF, Xie RZ, Evans MD, Vannas A, Tout SD, Griesser HJ, Johnson G, Steele JG. A comparison of biological coatings for the promotion of corneal epithelialization of synthetic surface in vivo. Invest Ophthalmol Vis Sci. 2003;44:3301–3309. doi: 10.1167/iovs.02-0561. [DOI] [PubMed] [Google Scholar]
- Teixeira AI, Nealey PF, Murphy CJ. Responses of human keratocytes to micro- and nanostructured substrates. J Biomed Mater Res A. 2004;71:369–376. doi: 10.1002/jbm.a.30089. [DOI] [PubMed] [Google Scholar]
- Thompson RW, Jr, Price MO, Bowers PJ, Price FW., Jr Long-term graft survival after penetrating keratoplasty. Ophthalmology. 2003;110:1396–1402. doi: 10.1016/S0161-6420(03)00463-9. [DOI] [PubMed] [Google Scholar]
- Toole BP, Trelstad RL. Hyaluronate production and removal during corneal development in the chick. Dev Biol. 1971;26:28–35. doi: 10.1016/0012-1606(71)90104-7. [DOI] [PubMed] [Google Scholar]
- Torbet J, Malbouyres M, Builles N, Justin V, Roulet M, Damour O, Oldberg A, Ruggiero F, Hulmes DJ. Orthogonal scaffold of magnetically aligned collagen lamellae for corneal stroma reconstruction. Biomaterials. 2007;28:4268–4276. doi: 10.1016/j.biomaterials.2007.05.024. [DOI] [PubMed] [Google Scholar]
- Trelstad RL. The bilaterally asymmetrical architecture of the submammalian corneal stroma resembles a cholesteric liquid crystal. Dev Biol. 1982;92:133–134. doi: 10.1016/0012-1606(82)90157-9. [DOI] [PubMed] [Google Scholar]
- Trelstad RL, Coulombre AJ. Morphogenesis of the collagenous stroma in the chick cornea. J Cell Biol. 1971;50:840–858. doi: 10.1083/jcb.50.3.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkaus-Randall V, Gipson IK. A technique for obtaining basal corneal epithelial cells. Invest Ophthalmol Vis Sci. 1985;26:233–237. [PubMed] [Google Scholar]
- Trinkaus-Randall V, Newton A, Franzblau C. Influence of substratum on corneal epithelial cell growth and protein synthesis. Invest Ophthalmol Vis Sci. 1988;29:1800–1809. [PubMed] [Google Scholar]
- Troilo D. Neonatal eye growth and emmetropisation--a literature review. Eye. 1992;6(Pt 2):154–160. doi: 10.1038/eye.1992.31. [DOI] [PubMed] [Google Scholar]
- Troilo D, Wallman J. The regulation of eye growth and refractive state: an experimental study of emmetropization. Vision Res. 1991;31:1237–1250. doi: 10.1016/0042-6989(91)90048-a. [DOI] [PubMed] [Google Scholar]
- Vacanti CA. History of tissue engineering and a glimpse into its future. Tissue Eng. 2006;12:1137–1142. doi: 10.1089/ten.2006.12.1137. [DOI] [PubMed] [Google Scholar]
- Viola J, Bhavya L, Oren G. Emergence of tissue engineeing as a research field. Cambridge, MA: Abt Associates Inc for National Science Foundation; 2003. pp. 3–129. [Google Scholar]
- Vrana E, Builles N, Hindie M, Damour O, Aydinli A, Hasirci V. Contact guidance enhances the quality of a tissue engineered corneal stroma. J Biomed Mater Res A. 2008;84:454–463. doi: 10.1002/jbm.a.31442. [DOI] [PubMed] [Google Scholar]
- Wang JH, Jia F, Gilbert TW, Woo SL. Cell orientation determines the alignment of cell-produced collagenous matrix. J Biomech. 2003;36:97–102. doi: 10.1016/s0021-9290(02)00233-6. [DOI] [PubMed] [Google Scholar]
- Watsky MA. Keratocyte gap junctional communication in normal and wounded rabbit corneas and human corneas. Invest Ophthalmol Vis Sci. 1995;36:2568–2576. [PubMed] [Google Scholar]
- Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- West-Mays JA, Dwivedi DJ. The keratocyte: corneal stromal cell with variable repair phenotypes. Int J Biochem Cell Biol. 2006;38:1625–1631. doi: 10.1016/j.biocel.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness: a global perspective. Bull World Health Organ. 2001;79:214–221. [PMC free article] [PubMed] [Google Scholar]
- Wilson DL, Martin R, Hong S, Cronin-Golomb M, Mirkin CA, Kaplan DL. Surface organization and nanopatterning of collagen by dip-pen nanolithography. Proc Natl Acad Sci U S A. 2001;98:13660–13664. doi: 10.1073/pnas.241323198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J. Das Gesetz Der Transformation Der Knochen. Berlin: A Hirschwald; 1891. [Google Scholar]
- Yarlagadda PK, Chandrasekharan M, Shyan JY. Recent advances and current developments in tissue scaffolding. Biomed Mater Eng. 2005;15:159–177. [PubMed] [Google Scholar]
- Yee RW, Matsuda M, Schultz RO, Edelhauser HF. Changes in the normal corneal endothelial cellular pattern as a function of age. Curr Eye Res. 1985;4:671–678. doi: 10.3109/02713688509017661. [DOI] [PubMed] [Google Scholar]
- Young RD, Gealy EC, Liles M, Caterson B, Ralphs JR, Quantock AJ. Keratan sulfate glycosaminoglycan and the association with collagen fibrils in rudimentary lamellae in the developing avian cornea. Invest Ophthalmol Vis Sci. 2007;48:3083–3088. doi: 10.1167/iovs.06-1323. [DOI] [PubMed] [Google Scholar]
- Yuan L, Veis A. The self-assembly of collagen molecules. Biopolymers. 1973;12:1437–1444. doi: 10.1002/bip.1973.360120618. [DOI] [PubMed] [Google Scholar]
- Zarins CK, Zatina MA, Giddens DP, Ku DN, Glagov S. Shear stress regulation of artery lumen diameter in experimental atherogenesis. J Vasc Surg. 1987;5:413–420. [PubMed] [Google Scholar]
- Zerath E. Effects of microgravity on bone and calcium homeostasis. Adv Space Res. 1998;21:1049–1058. doi: 10.1016/s0273-1177(98)00026-x. [DOI] [PubMed] [Google Scholar]
- Zhao XC, Yee RW, Norcom E, Burgess H, Avanesov AS, Barrish JP, Malicki J. The zebrafish cornea: structure and development. Invest Ophthalmol Vis Sci. 2006;47:4341–4348. doi: 10.1167/iovs.05-1611. [DOI] [PubMed] [Google Scholar]
- Zhu C, Joyce NC. Proliferative response of corneal endothelial cells from young and older donors. Invest Ophthalmol Vis Sci. 2004;45:1743–1751. doi: 10.1167/iovs.03-0814. [DOI] [PubMed] [Google Scholar]
- Zieske JD. Extracellular matrix and wound healing. Curr Opin Ophthalmol. 2001;12:237–241. doi: 10.1097/00055735-200108000-00001. [DOI] [PubMed] [Google Scholar]
- Zieske JD, Guimaraes SR, Hutcheon AE. Kinetics of keratocyte proliferation in response to epithelial debridement. Exp Eye Res. 2001;72:33–39. doi: 10.1006/exer.2000.0926. [DOI] [PubMed] [Google Scholar]
- Zieske JD, Mason VS, Wasson ME, Meunier SF, Nolte CJ, Fukai N, Olsen BR, Parenteau NL. Basement membrane assembly and differentiation of cultured corneal cells: importance of culture environment and endothelial cell interaction. Exp Cell Res. 1994;214:621–633. doi: 10.1006/excr.1994.1300. [DOI] [PubMed] [Google Scholar]
- Zimmermann DR, Trueb B, Winterhalter KH, Witmer R, Fischer RW. Type VI collagen is a major component of the human cornea. FEBS Lett. 1986;197:55–58. doi: 10.1016/0014-5793(86)80297-6. [DOI] [PubMed] [Google Scholar]