

Abstract
Summary: At the heart of many modern biotechnological and therapeutic applications lies the need to target specific genomic loci with pinpoint accuracy. Although landmark experiments demonstrate technological maturity in manufacturing and delivering genetic material, the genomic sequence analysis to find suitable targets lags behind. We provide a computational aid for the sophisticated design of sequence-specific ligands and selection of appropriate targets, taking gene location and genomic architecture into account.
Availability: Source code and binaries are downloadable from www.bioinformatics.org.au/triplexator/inspector.
Contact: t.bailey@uq.edu.au
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Targeting genomic loci with high accuracy is essential for modern molecular biology, such as for insertion of genetically engineered material into genomes. Recent publications demonstrate the maturity of techniques using triplex-forming peptide nucleic acids (PNAs) for accurate, sequence-specific gene targeting. Chin et al. (2008) apply PNAs in conjunction with donor DNA to introduce heritable gene modification and Rogers et al. (2012) improve the delivery by linking the antennapedia transport peptide (Antp) to the PNA to enable its transfer into the cell nucleus after systemic administration of the compound.
In contrast to these biotechnological achievements, the determination of genomic targets that can be accurately targeted has been hampered by the lack of dedicated computational tools able to jointly optimize the choice of the target site and the design of the delivery molecule (e.g. PNA) such that the number of possible collateral binding sites (off-targets) is minimized. Although Rogers et al. (2012) were able to demonstrate site specificity by determining that the PNA–Antp conjugate did not alter the nearby control gene cII, which contains a potential triplex target site whose sequence differs from that of the intended target, they acknowledged the possibility of PNA binding occurring at other genomic locations that have sequences similar to the target. The extent to which applications of PNA-based delivery may be hampered by a non-optimal target choice is demonstrated by the fact that the sequence chosen by Rogers et al. (2012) has almost 20 000 identical copies across the mouse genome, making off-target binding a near certainty. The introduction of genetic material at any of these loci, but especially at the 469 sites that are located within exons, would have unpredictable (and probably undesirable) consequences.
This illustrates the need for a computational aid that takes triplex-specific design features into account. Triplex formation of conventional oligonucleotides relies on hydrogen-bonding of the third strand in the major groove of the genomic DNA target and potential in vivo roles of triplexes are reviewed in Buske et al. (2011). In contrast, PNA-based targeting, as used by Chin et al. (2008) and Rogers et al. (2012), depends on strand invasion resulting in a PNA:DNA·PNA triple-helix with a looped-out DNA strand. The peptide backbone provides enhanced resistance to nucleases, and PNAs show an increased affinity for DNA compared with oligonucleotides (Egholm et al., 1993). The target site consists, in either case, of a sequence of consecutive purines that provide the hydrogen donor and acceptor groups that stabilize the triple-helix. Suitable genomic targets thus exhibit a dominant polypurine·polypyrimidine characteristic (poly-R poly-Y).
poly-Y).
2 DESCRIPTION OF TRIPLEX-INSPECTOR
Triplex-Inspector is a computational tool for sequence-specific ligand design and appropriate target selection to increase the accuracy of triplex-based genomic manipulations. The software takes a genomic locus of interest and searches for all putative triplex target sites, leveraging our exhaustive search algorithm Triplexator (Buske et al., 2012). Each of these putative targets is subsequently examined for their uniqueness by searching the genome for any locus with high-sequence similarity to them (illustrated in Fig. 1A and Supplementary Fig. S1). Although we do not present any experimental validation, we demonstrate that Triplex-Inspector greatly facilitates avoiding targets that are clearly sub-optimal because of the presence of (near-) perfect off-targets.
Fig. 1.
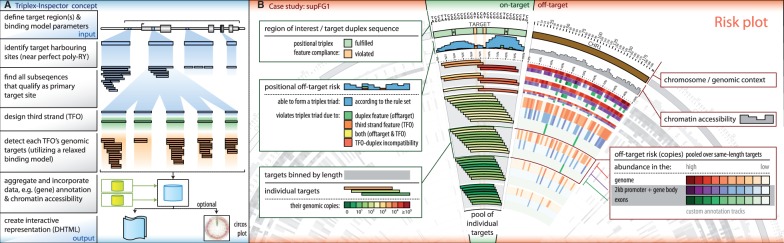
(A) Workflow for automated triplex target-site detection minimizing off-target risk. (B) Interactive target region summary. The centre slice’s histogram shows off-target risk of individual positions for the region of interest (SubFG1), whereas inner tiles show putative target sites and their genome-wide copy number. Feature-annotated (e.g. chromatin accessibility and exons) outer chromosome slices show off-target abundance pooled for all same-length primary targets
The user can separately customize the parameters (e.g. length and mismatch-rate) used in the target site search and uniqueness assessment. Off-targets with high similarity to the target site (high-risk off-targets) can be detected by using the same parameter setting used during the target site search. Relaxing the off-target search criteria allows the detection of less similar (e.g. shorter) off-target sites, giving a more complete picture of the overall risk for cellular toxicity and other unintended consequences.
Off-target sites need to be assessed in their genomic context. For example, off-targets located in exons might be considered more hazardous than occurrences in gene deserts. Triplex-Inspector allows interactive filtering of relevant target sites based on gene annotation data. The target-ligand design can be further optimized by taking chromatin organization into account. Triplex-Inspector allows DNase I hypersensitivity data to be used to highlight genomic regions where off-target binding is more likely (or less likely) because of chromatin accessibility (or inaccessibility).
A single set of criteria for choosing the optimal triplex target site in any given application does not currently exist. Manual, interactive data inspection is, therefore, essential. Triplex-Inspector aggregates all results into an interactive web browser application allowing intuitive data manipulation and visualization that can be shared across different platforms. Triplex-Inspector optionally generates a circos plot (Krzywinski et al., 2009) for each target site cluster that allows the user to visually assess its properties (Fig. 1B). Once the user has identified a target with acceptable off-target risk, Triplex-Inspector outputs the sequences of matching ligands based on the model of Vekhoff et al. (2008). A comparison of Triplex-Inspector to existing tools is available in the Supplementary File.
3 DEMONSTRATING THE BENEFIT OF USING A COMPUTATIONALLY GUIDED DESIGN PROCESS
We identify a target for the supFG1 reporter construct with substantially fewer off-targets than the 18 826 sequence copies of the heuristically chosen 10 nt target site used by Rogers et al. (2012). To find this more suitable target, we use Triplex-Inspector to examine the 30 nt poly-R poly-Y region in the supFG1 construct for any subsequence of at least 10 nt that qualifies as target under the binding model from Rogers et al. (2012). Leveraging the software’s ability to find all off-targets for every candidate target site, we find a 10 nt site located 2 nt downstream of the heuristically chosen target that has about half as many exact copies in the genome.
poly-Y region in the supFG1 construct for any subsequence of at least 10 nt that qualifies as target under the binding model from Rogers et al. (2012). Leveraging the software’s ability to find all off-targets for every candidate target site, we find a 10 nt site located 2 nt downstream of the heuristically chosen target that has about half as many exact copies in the genome.
An optimal target would be unique in the genome and long enough to bind the matching ligand effectively. The ligand should also not contain any subsequences that could bind to off-targets. For example, the 30 nt poly-R poly-Y region in the supFG1 construct is unique in the mouse genome, but Triplex-Inspector finds that it contains subsequences of length
poly-Y region in the supFG1 construct is unique in the mouse genome, but Triplex-Inspector finds that it contains subsequences of length 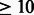 nt that match >100 000 off-target sites. These problematic subsequences are found mainly in the right side of the region (18–30 nt, Fig. 1B).
nt that match >100 000 off-target sites. These problematic subsequences are found mainly in the right side of the region (18–30 nt, Fig. 1B).
Introducing a strategic mismatch at positions that engage the most in off-target binding, e.g. positions 7 and 24 (Fig. 1B, marked with an asterisk), is likely to disrupt the binding stability of shorter subsequences and, therefore, reduces off-target effects. As the optimal balance between uniqueness and off-target risk, Triplex-Inspector identifies a 23 nt subsequence (2–25), which is the shortest unique sequence with no equally stable off-target (as defined by the number of nucleotide triads formed in the triplex). Tolerating non-exonic off-targets reduces the length requirement to 20 nt (4–24) with a total of 38 copies across the genome of which 18 are located in introns. If TFOs can be administered to target cells in a (liver) tissue-specific way, then the number of off-targets, which are accessible based on DNase I hypersensitivity data, drops to two. See the Supplementary File for additional examples targeting biologically important loci.
In conclusion, the achievable specificity of gene targeting is governed by the choice of the primary target site. The ligand design process needs to identify potential off-targets and assess their risk on the basis of their sequence, accessibility and proximity to functional elements. Computational tools are required to process this information optimally and supersede heuristic decision making. Triplex-Inspector addresses this need and aids the design of genome-wide–specific targets for triplex-forming molecules.
Funding: F.A.B. is supported by the UQ Research Scholarship and the UQ International Research Tuition Award; D.C.B. is a research scientist at CSIRO; J.S.M. is funded by the National Health & Medical Research Council Australia Fellowship (631668); T.L.B. is funded by a National Institutes of Health grant (RO-1 RR021692-01).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Buske FA, et al. Potential in vivo roles of nucleic acid triple-helices. RNA Biol. 2011;8:427–439. doi: 10.4161/rna.8.3.14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske FA, et al. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012;22:1372–1381. doi: 10.1101/gr.130237.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JY, et al. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc. Natl Acad. Sci. USA. 2008;105:13514–13519. doi: 10.1073/pnas.0711793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egholm M, et al. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature. 1993;365:566–568. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers FA, et al. Targeted gene modification of hematopoietic progenitor cells in mice following systemic administration of a PNA-peptide conjugate. Mol. Ther. 2012;20:109–118. doi: 10.1038/mt.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekhoff P, et al. Triplex formation on DNA targets: how to choose the oligonucleotide. Biochemistry. 2008;47:12277–12289. doi: 10.1021/bi801087g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.