

Abstract
Neuroinflammation is a host-defense mechanism associated with restoration of normal structure and function of the brain and neutralization of an insult. Increasing neuropathological and biochemical evidence from the brains of individuals with Parkinson's disease (PD) provides strong evidence for activation of neuroinflammatory pathways. Microglia, the resident innate immune cells, may play a major role in the inflammatory process of the diseased brain of patients with PD. Although microglia forms the first line of defense for the neural parenchyma, uncontrolled activation of microglia may directly affect neurons by releasing various molecular mediators such as inflammatory cytokines (tumor necrosis factor-α, interleukin [IL]-6, and IL-1β), nitric oxide, prostaglandin E2, and reactive oxygen and nitrogen species. Moreover, recent studies have reported that activated microglia phagocytose not only damaged cell debris but also intact neighboring cells. This phenomenon further supports their active participation in self-enduring neuronal damage cycles. As the relationship between PD and neuroinflammation is being studied, there is a realization that both cellular and molecular mediators are most likely assisting pathological processes leading to disease progression. Here, we discuss mediators of neuroinflammation, which are known activators released from damaged parenchyma of the brain and result in neuronal degeneration in patients with PD.
1. Introduction
Parkinson's disease (PD) is a frequent neurological disorder of the basal ganglia characterized by the progressive loss of dopaminergic neurons, mainly in the substantia nigra pars compacta (SNpc) [1], cytoplasmic inclusions of aggregated proteins, and neuroinflammation [2, 3]. Several hypotheses have been postulated regarding the possible causes for neuronal degeneration in patients with PD. These include genetic factors, environmental toxins, mitochondrial dysfunction, and free radical-mediated cell death [4–6]. Although there is less evidence suggesting that neuroinflammation is the primary trigger causing neurodegeneration, preclinical and epidemiological data now strongly suggest that chronic neuroinflammation may be a slow and steady reason for neuronal dysfunction during the asymptomatic stage of PD [7]. Neuroinflammation induced by exposure to either infectious agents or toxicants with proinflammatory characteristics is now increasingly recognized as a major contributor to the pathogenesis of PD [8]. Whitton in 1988 initially suggested the involvement of inflammation in PD by describing upregulation of major histocompatibility complex (MHC) molecules in patients with PD [9]. The hallmarks of neuroinflammation are the presence of activated microglia and reactive astrocytes in the parenchyma (neurons, astrocytes, and endothelial cells) of the central nervous system (CNS), direct participation of the adaptive immune system, increased production of cytokines, chemokines, prostaglandins, a cascade of complement proteins, and reactive oxygen and nitrogen species (ROS/RNS), which in some cases can result in disruption of the blood-brain barrier (BBB) [10]. The extent to which neuroinflammation and peripheral immune responses contribute towards the development of PD or modify its course is not exactly known. In fact, dysregulation of the neuroimmune system has been postulated by many to be one of the underlying causes of the chronic nature of PD.
Several lines of evidence support the hypothesis that glial reactive and inflammatory processes participate in the cascade of events leading to neuronal degeneration [11]. One of the earlier studies reporting neuroinflammation in PD involved a quantitative confirmation of the astroglial reaction using glial fibrillary acidic protein (GFAP) immunostaining in the substantia nigra (SN) of patients with PD [12]. Fundamental work by McGeer et al. [13], over two decades ago, first identified significantly increased levels of human leucocyte antigen-DR-positive microglia in the postmortem brains of patients with PD [14]. Following these reports, an increased number of activated microglial cells had consistently been reported in the neuroinflammatory pathogenesis of PD [11]. Initially, the pathological role played by these glial cells was not fully understood, but activated microglial cells have a deleterious effect on dopaminergic neurons. Microglial cells represent resident brain macrophages that are transformed into activated immunocompetent antigen-presenting cells during the pathological process [15]. Microglia in PD have been observed to grow densely in the striatum and SN with increased expression of proinflammatory mediators, including tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β) [16], IL-2, IL-4, IL-6, transforming growth factor-α (TGF-α), cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS) [14]. TGF-β1 and β2 have also been detected by several investigators in the cerebrospinal fluid (CSF) and brain parenchyma of patients suffering from PD [17]. Taken together, these data indicate that glial cells are one of the most important cells involved in mediating neuroinflammatory events in PD [18].
In addition to glial cells, other cells may also participate in the neuroinflammatory processes in PD. Increasing evidence now demonstrates the involvement of both innate and adaptive immune responses in the pathophysiology of PD [19–21]. Innate immunity does not require the presence of a specific antigen to elicit an immune response, whereas adaptive immunity is activated when specific antigens are presented and recognized by lymphocytes. In contrast, endogenous pathological antigens that normally do not occur in physiological conditions may also initiate adaptive immune responses [22]. Indeed, Hirsch et al., in their various experiments, reported a small number of CD8-positive T lymphocytes in the vicinity of degenerating neurons in the SN of a patient with PD [18]. In line with this observation, an increased density of interferon-(IFN-) γ-positive cells with lymphocytes in the SN of patients with PD has also been reported [18]. Taken together, these data indicate that injured dopaminergic neurons release immunogenic mediators which have the potential to provoke detrimental innate and adaptive immune responses thereby amplifying the neuroinflammatory process in PD. Furthermore, mounting evidence suggests that BBB permeability may be modulated under neuroinflammatory conditions, and trafficking of leukocytes and peripheral macrophages into the brain becomes a normal process that must be tightly regulated to promote brain homeostasis and avoid the neuronal demise [10, 23]. These pathomechanisms not only produce complex cross-talk between the peripheral immune system and CNS but also highlight the interactions between microglial cells and other brain parenchymal cells [18]. Therefore, identifying and understanding the nature and role of the neuroinflammatory mediators involved in the pathogenesis of PD might provide us with various options to target these neuroinflammatory pathways to help curb neuronal death in PD. This review describes various cellular and molecular mediators of neuroinflammation which occur in response to or as part of the ongoing disease progression in PD.
2. Mediators of Neuroinflammation
2.1. Role of Microglia as a Mediator of Neuroinflammation in PD
Glia are composed of three distinct cells types named as microglia, astrocytes, and oligodendrocytes [24]. Several populations of macrophages are present in different compartments of healthy brain tissue, each with a distinct phenotype and morphology. The most abundant of these macrophages are the microglia, the resident macrophages of the brain parenchyma [25]. Microglia maintains homeostasis and performs immune surveillance by continuously examining their environment by extending cellular protrusions [26]. With a plentitude of ion channels, cytokines, Toll-like receptors, and chemokine receptors [21], microglia promptly reacts in response to subtle alterations in their microenvironment such as alterations in ion homeostasis and brain insults, ranging from aggregated proteins to pathogens [27]. Microglial cells are generally quiescent in the normal brain, with their cell bodies barely visible and few detectable fine ramified processes. However, resting microglial cells quickly proliferate, become hypertrophic, and persistently increase expression of a large number of marker molecules such as CD11b, CD68, and MHC-I and II molecules [28] and are further transformed to macrophage-like cells in patients with PD [11, 29, 30]. Microglia may be transformed into M1 or M2 macrophages depending upon the type of stimulus [31, 32]. It is now apparent that microglia occur in many different phenotypes that cannot be readily divided into a small number of discrete subsets following tissue injury [33]. The SN is relatively rich in microglia compared with other brain regions [34, 35]. In addition, a reduced level of intracellular glutathione in the SN dopaminergic neurons makes them much more susceptible to a variety of insults, including activated microglial-mediated injury and oxidative stress [35]. This observation indicates that localization of microglia in the SN predisposes dopaminergic neurons to immunological insult in patients with PD [36].
The selective acute degeneration of dopaminergic cells in the SN can be induced by toxins such as 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and rotenone. In all of these rodent toxin models, dopaminergic cells of the SN degenerate over a period of a few days. A recent study comparing the commonly used medial forebrain or intrastriatal injection of 6-OHDA showed rapid degeneration induced by the toxin, which was accompanied by activation of microglia as assessed by the upregulation of the complement type 3 receptor [37]. Similarly, MPTP-induced neurodegeneration is associated with activated microglia [38, 39]. It has become increasingly apparent that there are various triggers through which microglia are activated to elicit their neurotoxic response. Interestingly, while these diverse toxins exhibit several mechanisms of microglial activation, toxin-induced activation of NADPH oxidase is the most common pathway through which microglia exerts neurotoxicity [36]. Apart from this toxin-induced microglial activation, various other triggers involved in microglial activation include immunological insults such as IFN-γ, lipopolysaccharide (LPS), chemokines (CCL5, CCL2, and CXCL10), neurotransmitters, gangliosides, the CD40 ligand, proteases such as thrombin [40], tissue plasminogen activator [41], matrix metalloproteinase-3 (MMP-3) [21], endogenous disease proteins, and neuronal injury itself [42]. Among these activators, LPS-induced neuroinflammation is one of the most accepted and widely used endotoxin models that induces a strong neuroinflammatory response in BV-2 microglial cells [43, 44] or when injected directly into the vicinity of the SN [45]. Recent findings demonstrate that neurons are not simply passive targets of microglia but rather control microglial activation [3, 46, 47]. A variety of signals that neurons use to modulate microglia can be categorized into excitatory and inhibitory signals. Inhibitory signals from neurons constitutively maintain microglia in their quiescent state and antagonize proinflammatory activity, whereas excitatory signals are inducible and incite activation of microglia under pathological conditions towards a beneficial or detrimental phenotype. Thus, various neuronal signaling molecules actively modulate microglial functions and contribute to the inflammatory milieu of PD [46]. The two subclasses of neuronal inhibitory signals are called released and membrane-bound signals. Released inhibitory signals can be CX3CL1, CD22, TGF-β, brain derived neurotrophic factor (BDNF), neurotrophin-3, nerve growth factor, or neurotransmitters. The release site for neuronal inhibitory signals is not yet known but is thought to be related to synaptic activity [48, 49]. Membrane-bound neuronal inhibitory signals consist of several molecules from the immunoglobulin superfamily including CD200 [50], CD22, and CD47, which are expressed or secreted by neurons that bind to receptors on microglia [48, 51, 52]. These immunoreceptors, so-called tyrosine-based inhibitory motif receptors, contain a cytoplasmic motif that inhibits activation of microglia. This mechanism of maintaining microglia in their silent state also depends upon the density of the ligands expressed by neurons, which are reduced during PD and will thus shift the level of inhibitory tone to activation in microglia [25]. In parallel with the inhibitory signals, neuronal excitatory signals are classified into released and membrane-bound signals. The released excitatory signals control various aspects of microglia function and can be listed as chemokines (CX3CL1, CCL21, and CXCL10) [53–55], glutamate [56], purines (ATP and UTP) [57, 58], or MMP-3 [59, 60] which modulate various aspects of microglia function. Taken together, microglia are maintained in a quiescent state under normal physiological conditions by the orchestrated action of neurons and astrocytes; however, microglia are rapidly activated when integrity of a neuron is disrupted in PD, probably as a result of both direct activation signals from neurons or loss of inhibitory signals by neurons [47].
While it is clear that microglia becomes activated upon neuronal damage, proteases known to modify the extracellular matrix (ECM) may also be a critical mechanism through which damaged neurons activate microglia to produce extracellular superoxide. Earlier reports from Chang et al. emphasized the critical role of ECM proteins in the interactions between microglia and neurons [61]. Later, it was found that MMP-3, a proteinase that degrades ECM components, is released following damage to dopaminergic neurons exposed to 1-methyl-4-phenylpyridinium (MPP+), exerting neurotoxicity to the dopaminergic neurons [59]. It was also observed that exposure of mesencephalic neuron/glia cultures to MPP+ results in a dose-dependent increase in MMP-3 protein expression, both in conditioned media and in cell lysates, indicating that death of dopaminergic neurons upregulates MMP-3 expression. These data suggest that MMP-3 is a crucial mediator released upon damage to dopaminergic neurons and that it activates microglia to further propagate neuronal cell death in PD [42]. Besides MMP-3, damaged or dying dopaminergic neurons release neuromelanin to activate microglia in the SN of patients with PD [62, 63]. Neuromelanin has the potential to be neurotoxic, because excess neuromelanin inhibits the function of dopaminergic neurons, proteasomes and induces the production of toxic factors such as TNF-α, IL-6, and nitric oxide (NO) [64, 65]. Among the proinflammatory mediators released during microglial activation, some act synergistically to produce inflammation-related neuronal damage. Hence, the identification of several potential mediators of microglia activation has allowed a general classification of how microglia respond to stimuli. Microglia can also be activated by products of the classical complement cascade and by chromogranin A [66, 67], which has been reported to occur in the PD SN [68]. Among the array of mediators released, superoxide is necessary for both the induction and amplification of neurotoxicity in PD [69, 70]. NADPH oxidase (PHOX) is the major superoxide-producing enzyme in microglia [71]. Activation of PHOX results in translocation of its cytosolic subunits to the cellular membrane to form a functional enzyme that not only generates superoxide but also controls the levels of other proinflammatory neurotoxic mediators produced by microglia in PD [72]. It has been revealed that PHOX is closely paired with Mac-1 and plays an important role in microglia-mediated neuroinflammation and neurotoxicity [73]. Therefore, coupling between PHOX and Mac-1 might be a central mechanism responsible for the oxidative damage induced by reactive microgliosis that results in progressive neuroinflammation in PD [36].
To date, one of the best elucidated cytotoxic mechanisms induced by proinflammatory cytokines in PD is activation of iNOS. iNOS mediates the synthesis of high levels of NO, which is toxic to dopaminergic neurons [74]. The density of glial cells expressing iNOS increases significantly in the SN of patients with PD compared with that in control subjects [75]. The induction of NOS produces high levels of NO and superoxide radicals for a prolonged period of time. These two ROS can either directly or indirectly promote neuronal death in PD [76]. Prostaglandins and their synthesizing enzymes, such as COX-2, represent a second group of potential culprits in PD. Expression of COX-2 along with the levels of its product, prostaglandin E2 (PGE2), increases significantly in glial cells of the SNpc, which are responsible for many of the cytotoxic effects to dopaminergic neurons in PD [77]. Many reports have demonstrated increased expression of COX-2 in PD [77, 78]. In fact, several studies have observed upregulation of COX-2 in animal models of PD [79–81]. Increased COX-2 expression has also been shown in the SN of postmortem PD specimens compared to that in normal controls [75, 82]. Inhibiting COX-2 [80, 81, 83, 84] and transgenic mice lacking COX-2 expression [85] in models of PD has been demonstrated to increase survival of dopaminergic neurons. Release of α-synuclein (α-syn) from neuronal damage could also incite the production of proinflammatory mediators such as PGE2 from microglia [86], thus, contributing to the progression of nigral neurodegeneration. It has recently been observed that modifying ubiquitin carboxy-terminal hydrolase L1 by cyclopentenone prostaglandins causes protein unfolding and aggregation. Hence, the deleterious effect of COX-2 in PD could be due to the production of cyclopentenone prostaglandins [87]. Although the exact causal link between neuronal injury and microglial activation in PD remains controversial, one of the earliest reported harmful effects demonstrated to cause demise of dopaminergic neurons was microglial-mediated release of proinflammatory cytokines, including IFN-γ [20] IL-1β, TNF-α, IL-2, and IL-6 [88] with elevated levels of TNF-α receptor R1 (p55), bcl-2, soluble Fas, caspase-1 and caspase-3 in postmortem striatum, SN, and CSF of patients with PD [89, 90]. These cytokines, in turn, propagate and intensify neuroinflammation and cause irreversible destruction of SN dopaminergic neurons [91] by a number of mechanisms, including upregulating isoforms of phospholipases A2, generating platelet-activating factor, stimulating NOS, and activating calpain [9, 92]. We have briefly summarized some of the major mediators of neuroinflammation during the pathogenesis of PD in Figure 1.
Figure 1.
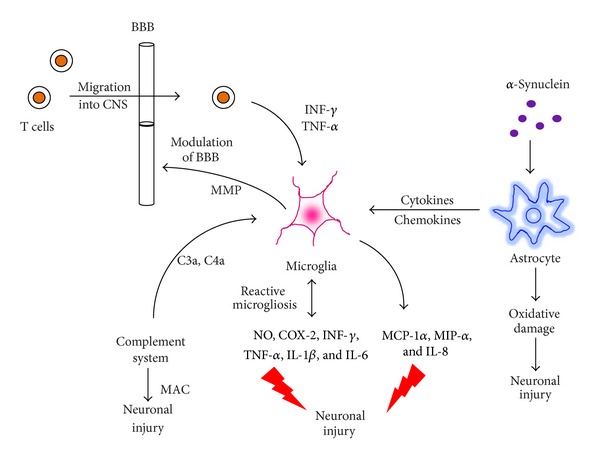
Microglial and astroglial cells become activated during the pathogenesis of Parkinson's disease under the influence of various proinflammatory triggers, including α-synuclein, the complement system, and cytokines released from infiltrated T cells. Activated microglial and astroglial cells further release various neuroinflammatory mediators, including NO, COX-2, IFN-γ, TNF-α, IL-1β & IL-6, chemokines including MCP-1α, MIP-α and CXCL-8, and MAC which have deleterious effect on neuronal survival. Abbreviations: NO: nitric oxide, COX-2: cyclooxygenase, INF-γ: interferon-γ, TNF-α: tumor necrosis factor-α, IL-1β: interleukin-1β, IL-6: interleukin-6, MCP-1α: monocyte chemotactic protein-1, MIP-α: microphage inflammatory protein, IL-8: interleukin-8, MAC: membrane attack complex, α-syn: α-synuclein, MMP: matrix metalloprotein, BBB: blood brain barrier, C3a: complement component 3a, and C4a: complement component 4a.
2.2. Role of T Cells as a Mediator of Neuroinflammation in PD
The brain has long been considered an immune privileged system, as it is protected by the BBB. However, recent findings demonstrate that both the innate and adaptive immune systems play a very critical role disrupting BBB permeability and mediating the pathogenesis of PD via their ability to supply the required signals for antigen presentation and to act as final effectors by T cells [25, 93, 94]. For example, infiltration of T cells has been found in the brains of patients with PD [95] as well as significant infiltration of adoptively transferred immune splenocytes into the brains of MPTP-intoxicated mice and localization within the inflamed SN [93]. Similarly, a recent study of MPTP model demonstrated the necessity of T cells to mediate degeneration of dopaminergic neurons and that dopaminergic neuronal loss is exacerbated by T cells [96]. Increased mutual coexpression of CD4 and CD8 by CD45R0+ T cells with increased expression of CD25, TNF-α receptors, and diminished expression of IFN-γ receptors suggests that subsets of T cell are indeed activated in patients with PD [97]. The influence of infiltrating T cells on dopaminergic neurons has been demonstrated in mice lacking T lymphocytes, wherein death of dopaminergic neurons was significantly attenuated in both types of mice as compared to that in wild-type animals [22]. It was also observed that a subset of CD4+ T cells, rather than CD8+ T cells, mediate the cytotoxic effects on dopaminergic neurons, as survival of dopaminergic neurons after MPTP administration increases in CD4-deficient mice but not in CD8-deficient mice [22]. Further analysis showed that CD4+ T cells exert their cytotoxicity through the Fas-FasL pathway rather than through IFN-γ secretion [98]. Early evidence from the SN by autopsy of patients with PD showed increased numbers of CD8+ T cells in close proximity with activated microglia and degenerating neurons [99]. More recently, both CD4+ and CD8+ T cells have been discovered within the SN of patients with PD [22]. Significant level of unrepaired single-strand DNA breaks and a number of micronuclei are more also observed in lymphocytes and activated T cells from patients with PD due to inflammation and exposure to ROS than those in age-matched controls [100, 101]. CD8+ T cells were the first type of peripheral T lymphocytes to be located in the postmortem brain from a PD patient [13]. Later, infiltration of CD4+ and CD8+ T cells was found in the SN and striatum of MPTP-intoxicated mice [93, 102]. A more recent report provided substantial evidence of significant infiltration of CD4+ and CD8+ T cells in the SN of patients with PD and in MPTP-intoxicated mice [22]. Increased frequencies of activated CD4+ T cells expressing Fas [103], increased IFN-γ-producing Th1 cells, decreased IL-4-producing Th2 cells, and a decrease in CD4+, CD25+ T cells have been found in the peripheral blood of patients with PD [104]. These data suggest complex roles for CD4+ subsets of T cells in mediating the development of PD.
Compelling evidence suggests the possible involvement of the BBB, including changes in lymphocytic subpopulations in the blood and CSF of patients with PD [91, 103]. Moreover, an increased proportion of γδ-positive lymphocytes, thought to play a role in infections and autoimmunity, have also been reported in the CSF and blood of patients with PD [105]. These data suggest that infiltration of immune cells across the BBB into the brain participates in the pathophysiology of PD. This infiltration of peripheral lymphocytes into the brain through the BBB occurs mainly because activated microglia and monocytes in the brains of patients with PD release proinflammatory cytokines and chemokines that act on the vascular endothelium to induce upregulation of cell adhesion molecules, including vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 (ICAM-1) [106] that disrupt the BBB and attract lymphocytes [97] expressing counterreceptors such as leukocyte function antigen-1 (LFA-1) to the neuronal injury site [106]. Activated T cells further release proinflammatory cytokines such as IFN-γ and TNF-α, which positively induce the expression of costimulatory molecules and MHC-II on microglial cells [107]. When these activated microglial molecules bind to their respective receptors on T cells, they are transformed into effector T cells. Among the activated T cells, CD8+ T lymphocytes mediate direct cytotoxic lysis of target cells, recruit and activate accessory cells via proinflammatory mediators, whereas activated CD4+ T lymphocytes induce B cells to produce high-affinity antibodies [91]. Nitrated-α-syn (N-α-syn), a misfolded protein present in intraneuronal inclusions or Lewy bodies in patients with PD, can be released to the extraneuronal space and cross the BBB to enter the cervical lymph nodes, where it can activate antigen-presenting cells [93]. This phenomenon has also been observed in MPTP-intoxicated mice, wherein α-syn drains to cervical lymph nodes [93]. As a novel epitope, N-α-syn can be processed and presented to naive T cells, thus, stimulating them to expand into different subsets of effector T cells. Peripherally activated effector T cells, such as Th1 or Th17 cells, can also cross the BBB and reach inflammatory sites within the brain where they activate microglia and release proinflammatory cytokine IL-17 and secrete cytolytic enzymes such as granzyme B [108]. Although a dysfunctional BBB in patients with PD may show some leakiness [109], it may not be sufficient to allow unrestricted lymphocyte infiltration because CD4/CD8 ratios are 1 : 4.8 [22] compared with the typical 2 : 1 ratio expected for peripheral T cells performing surveillance functions. Thus, the mechanisms by which these T cells gain access to the SN, their activation state, and their functions are questions that remain to be answered [97].
In summary, infiltrating subsets of T cells may induce excessive microglial-mediated inflammation and oxidative stress that exacerbate neuroinflammation in PD. Further study is needed to identify the exact roles of specific subsets of CD4+ T cells on the pathogenic progression of PD [98]. Studies suggest that the adaptive immune system similar to the innate immune system not only responds to but also actively participates in the pathogenesis of PD. However, more work needs to be done to determine if and how they could serve as a potential target for PD therapy [36].
2.3. Role of Astrocytes as a Mediator of Neuroinflammation in PD
The majority of findings from research on PD point toward microglia as the major mediator of neuroinflammation, but the astrocytic reaction is another well-known neuropathological characteristic in the SN of patients with PD [110]. Astrocytes function as supportive cells for neurons and maintain homeostasis and other neuronal functions [111]. Compared to microglia, their role as innate immune cells is somehow less appreciated. Nonetheless, astrocytes form the glia limitans around blood vessels, preventing entry of immune cells via the BBB into the CNS parenchyma [112]. Emerging evidence has focused upon the importance of astrocytes in the regulation of neuroinflammation in PD [113, 114]. The SNpc of many postmortem PD cases had been observed to have an increased number of astrocytes and GFAP immunoreactivity [115] with a decrease in glial-derived neurotrophic factor, BDNF, and ciliary neurotrophic factor [116]. The amount of GFAP-positive astrocytes is inversely proportional to the demise of dopaminergic neurons [12], indicating that dopaminergic neurons are more susceptible to the degenerative process wherein there are fewer astrocytes. Activation of astrocytes is characterized by the formation of hypertrophic and glial scars, which hinder axonal regeneration [42], enlarged cell bodies, and projections into the injured area [117] that seem to be mediated by proteoglycans [118]. During inflammatory conditions, astrocyte-derived granulocyte macrophage-colony stimulating factor, IL-6, CCL2, and CCL5 regulates migration, activation, and proliferation of microglia [119]. Astrocytes may detect neuron-derived α-sys as a degenerative marker released by neurons and get activated to protect neurons. However, such reactive astrocytes are exposed to increasing toxicity from α-sys oligomers and/or protofibrils, until they no longer serve a protective function [120].
Astrocytes play a major role mediating MPTP toxicity, as the active metabolite of MPTP, MPP+ is extruded into the extracellular space from astrocytes and further enters into dopaminergic neurons and induces neurotoxicity by inhibiting complex I in the mitochondrial electron transport chain [113]. Astrocytic activation parallels the development of dopaminergic cell death in the SNpc and striatum, whereas GFAP expression remains high even after most dopaminergic neurons have died due to administration of MPTP. These findings suggest that the astrocytic reaction occurs after neuronal cell death in PD [121]. In a recent study, it was demonstrated that α-syn released from neuronal cells can also be transferred to and accumulate in astrocytes and induce expression of genes associated with immune functions [122]. Proinflammatory cytokines that are differentially expressed in astrocytes in response to extracellular α-syn include IL-1α, IL-1β, IL-6, IL-18, and colony-stimulating factors-1, 2, and 3, suggesting a strong inflammatory response from astrocytes upon exposure to neuron-derived α-syn [122]. Exposure to neuron-derived α-syn also causes dramatic changes in chemokine expression in astrocytes, including CC-type (CCL-3, 4, 5, 12, 20), CXC-type (CXCL-1, 2, 5, 10, 11, 12, 16), and CX3C-type (CX3CL1) chemokines. These chemokines are involved in a variety of functions, such as recruitment of monocytes and macrophages, migration of microglia and neural progenitors, regulation of microglial activity, proliferation and survival of astrocytes, and synaptic plasticity and transmission [123]. These released chemokines, including CXCL12 and CCL5, induce glutamate release and restart the synthesis of cytokines and chemokines in astrocytes, suggesting their role in glia-glia and glia-neuron communication [122].
Other than cytokines, inflammatory oxidants have emerged as key contributors to PD and MPTP-related neurodegeneration. In this context, myeloperoxidase (MPO), a key oxidant-producing enzyme, which is mostly expressed by reactive astrocytes during inflammation, is upregulated in the ventral midbrain of human patients with PD and in MPTP mice [110]. MPO oxidizes nonreactive nitrite whose concentration is increased in parkinsonism [124] to reactive nitrite (NO2 −) and, thus, nitrosylates many proteins [125]. Reactive nitrites also contribute towards the production of the nonradical oxidant, hypochlorous acid (HOCl), which can damage macromolecules indirectly by fuelling hydroxyl freeradicals or directly by converting amines into chloramines, phenols, and unsaturated bond chlorination [126]. Furthermore, MPO also directly produces HOCl from hydrogen peroxide and chloride anion. Thus, HOCl might directly inflict oxidative damage on dopaminergic neurons [110]. Apart from the direct release of proinflammatory cytokines, including TNF-α and IL-6, astrocytes can also be activated by cytokines such as IL-1β and TNF-α released from microglia, thus, producing ROS and RNS [127]. In support of this observation, a recent study has reported that microglial inflammatory responses are enhanced by astrocytes through a nuclear factor-κB-dependent mechanism leading to increased dopaminergic toxicity [127]. Astrocytes also produce mediators which play a vital role mediating the inflammatory reaction that occurs in the SN of patients with PD. For example, ICAM-1-positive astrocytes are seen in the SN in patients with PD and attract reactive microglia to the area because such microglia carries the LFA-1 counterreceptor [95]. In addition, α-syn activates microglia and astrocytes to produce IL-6 and ICAM-1 [128]. This combination further attracts reactive microglia to the site of neuronal injury. The action of α-syn on astrocytes is thought to be through receptors, but the identity of these receptors is currently unknown [3].
Activated CD4+ T cells express and release several inflammatory factors such as the Fas ligand, a cell-surface molecule in the TNF-α family. Fas expression increases in patients with PD and in mice exposed to MPTP [129, 130]. This Fas ligand binds with the Fas receptor expressed on astrocytes and causes a release of various cytokines such as IL-6 and IL-8 and chemokines such as monocyte chemoattractant protein-1 [110]. The detrimental consequence of activating the Fas-Fas ligand pathway in PD has recently been established by several investigators. It has also been reported that mice deficient in Fas are more resistant to MPTP exposure than wild-type controls [131]. The current literature certainly suggests that astrocytes have the ability to modulate the function and survival of dopaminergic neurons in PD.
2.4. Role of the Complement System as a Mediator of Neuroinflammation in PD
The complement system is believed to amplify the effectiveness of both the specific and nonspecific immunological defense system. The complement system destroys invading pathogens, encourages inflammation, and supports phagocytosis of waste materials [132]. The complement system has the full ability to recognize molecular patterns associated with injured tissues and dying cells or molecular patterns on pathogens [133]. Various complement proteins, mostly present in tissue fluids and blood, are in the form of soluble monomers. The complement system can be activated by molecules other than antibodies. One such molecule, which is elevated in the SN of patients with PD, is C-reactive protein [134]. Similar evidence about the involvement with the complement system in PD has also been reported wherein membrane attack complex (MAC) together with all complement protein components has been identified on oligodendroglia of the SN and intracellularly in Lewy bodies of patients with sporadic PD [135, 136] and familial PD [137]. Additionally, elevated levels of MAC [135], C-reactive protein, and complement 3 have been observed in the SN and CSF of patients with PD [138, 139]. Increased mRNA levels of complement components have also been found in affected brain regions of PD models [138]. Activation of the complement system leads to a cascade of events ultimately leading to the destruction of cell surface with three different recognized pathways, which share a common juncture at the level of the C3 protein [140, 141].
Products of the activated complement cascade include opsonizing components (C3b, iC3b, and C4b) [142], which stain material for phagocytosis, MAC, and anaphylatoxins (C3a, C4a, and C5a) [142]. The opsonins perform a clearance function, whereas anaphylatoxins are involved in generation of the neuroinflammatory response [143]. In contrast, MAC induces cell death by entering cell membranes and causing organelles to leak. Although MAC destroys foreign cells and viruses, nearby host cells are at a significant risk of lysis if they are not protected by MAC [135, 140]. The complement system also contributes to the secretion of inflammatory cytokines from activated microglial cells [144]. Very recent evidence has demonstrated involvement of the complement system in the pathogenesis of PD, wherein the only cells in the SN and other brain areas that express C1q are microglia [145]. One of the important features of PD is that degeneration of dopaminergic neurons is accompanied by the deposition of extracellular neuromelanin. Degenerating neurons along with neuromelanin granules are opsonized by C1q and phagocytosed by C1q-positive microglia and macrophages in the perivascular spaces and parenchyma. Furthermore, the luminal surfaces of blood vessels in the SN of patients with PD have attached neuromelanin-laden C1q-positive cells. Thus, microglia are capable of clearing cellular debris from degenerating neurons of the SN and phagocytosing cells through the C1q-mediated pathway in PD [145]. Pentraxin is one of the mediators which activates the complement system by binding to the collagen tail of C1q. Pentraxin is an acute-phase protein that is involved in innate immunity and inflammatory response. Glial cells may be the major cellular source of this protein in the CNS. Under the inflammatory milieu of PD, pentraxin proteins secreted by reactive glial cells are detected in the plasma and CSF of patients with PD [146, 147]. Hence, pentraxin could serve as an inflammatory biomarker for PD. Overall, it seems clear that there is a role for the complement system in inflammation-mediated neurodegeneration in PD [138, 140]; hence, research aiming at developing effective inhibitors targeting these sites appears to be worthwhile.
3. Conclusion
PD is one of the most common neurodegenerative diseases with a well-established group of symptoms. Although a number of different mechanisms have been considered responsible for the development of PD, none are absolute. Growing evidence from patients and experimental models of PD has indicated that neuroinflammation is one of the driving forces in the pathogenesis of PD. The CNS had been thought to be an immunologically protected organ, but this notion has now undergone considerable reassessment. It has become apparent from a number of reports that various neuronal injury signals from different neuronal cell types in response to environmental insults, involving many mediators, incite and disseminate the ongoing neuroinflammation in PD. We have summarized the evidence wherein neuroinflammatory mediators play a key role in the pathogenesis of PD. Neuroinflammatory mediators have a profound action on CNS cells that differently affect the progress of inflammation and neuronal death. Therefore, regulating the production of neuroinflammatory mediators or their action on respective receptors would be an effective approach to mitigate the inflammatory processes in PD. Thus, further studies are required to form a more comprehensive idea about the role of these neuroinflammatory mediators in PD. Furthermore, it is of significant interest for ongoing research to identify and target various neuroinflammatory mediators released in response to various toxins to help explain how neuronal damage can signal inflammation and propagate neuronal cell death. This knowledge might serve to develop pharmacological strategies for treating the neuroinflammation in PD.
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2011-0014923) and was also supported by the Regional Innovation Center (RIC) Program of the Ministry of Knowledge Economy through the Bio-Food and Drug Research Center at Konkuk University, Republic of Korea.
References
- 1.Nolan YM, Sullivan AM, Toulouse A. Parkinson's disease in the nuclear age of neuroinflammation. Trends in Molecular Medicine. 2013;19(3):187–196. doi: 10.1016/j.molmed.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annual Review of Neuroscience. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 3.McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Movement Disorders. 2008;23(4):474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 4.Schapira AHV. Evidence for mitochondrial dysfunction in Parkinson’s disease—a critical appraisal. Movement Disorders. 1994;9(2):125–138. doi: 10.1002/mds.870090202. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Shachar D, Zuk R, Glinka Y. Dopamine neurotoxicity: inhibition of mitochondrial respiration. Journal of Neurochemistry. 1995;64(2):718–723. doi: 10.1046/j.1471-4159.1995.64020718.x. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg RN. Mitochondrial therapy for Parkinson disease. Archives of Neurology. 2002;59(10):p. 1523. doi: 10.1001/archneur.59.10.1523. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Tran T, Tansey MG. Neuroinflammation in Parkinson’s disease. Journal of Neuroimmune Pharmacology. 2009;4(4):419–429. doi: 10.1007/s11481-009-9176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson’s disease. Advances in Neurology. 2001;86:83–89. [PubMed] [Google Scholar]
- 9.Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson’s disease. British Journal of Pharmacology. 2007;150(8):963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 11.Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Movement Disorders. 1998;13(2):221–227. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- 12.Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience. 1993;52(1):1–6. doi: 10.1016/0306-4522(93)90175-f. [DOI] [PubMed] [Google Scholar]
- 13.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Annals of Neurology. 1988;24(4):574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- 14.Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Experimental Neurology. 2007;208(1):1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cross AK, Woodroofe MN. Immunoregulation of microglial functional properties. Microscopy Research and Technique. 2001;54(1):10–17. doi: 10.1002/jemt.1115. [DOI] [PubMed] [Google Scholar]
- 16.Hunot S, Dugas N, Faucheux B, et al. FcεRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. The Journal of Neuroscience. 1999;19(9):3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. Journal of Neural Transmission, Supplement. 2000;(58):143–151. [PubMed] [Google Scholar]
- 18.Hirsch EC, Hunot S, Hartmann A. Neuroinflammatory processes in Parkinson’s disease. Parkinsonism and Related Disorders. 2005;11(supplement 1):S9–S15. doi: 10.1016/j.parkreldis.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism and Related Disorders. 2012;18(supplement 1):S210–S212. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- 20.Lee Mosley R, Benner EJ, Kadiu I, et al. Neuroinflammation, oxidative stress, and the pathogenesis of Parkinson’s disease. Clinical Neuroscience Research. 2006;6(5):261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone DK, Reynolds AD, Mosley RL, Gendelman HE. Innate and adaptive immunity for the pathobiology of Parkinson’s disease. Antioxidants and Redox Signaling. 2009;11(9):2151–2166. doi: 10.1089/ars.2009.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brochard V, Combadière B, Prigent A, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. The Journal of Clinical Investigation. 2009;119(1):182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rezai-Zadeh K, Gate D, Town T. CNS infiltration of peripheral immune cells: D-day for neurodegenerative disease? Journal of Neuroimmune Pharmacology. 2009;4(4):462–475. doi: 10.1007/s11481-009-9166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teismann P, Schulz JB. Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation. Cell and Tissue Research. 2004;318(1):149–161. doi: 10.1007/s00441-004-0944-0. [DOI] [PubMed] [Google Scholar]
- 25.Perry VH. Innate inflammation in Parkinson's disease. Cold Spring Harbor Perspectives in Medicine. 2012;2(9) doi: 10.1101/cshperspect.a009373.a009373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nimmerjahn A, Kirchhoff F, Helmchen F. Neuroscience: resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 27.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 28.Hunot S, Hirsch EC, Isacson I, et al. Neuroinflammatory processes in Parkinson’s disease. Annals of Neurology. 2003;53(supplement 3):S49–S60. doi: 10.1002/ana.10481. [DOI] [PubMed] [Google Scholar]
- 29.McGeer PL, McGeer E. Innate immunity, local inflammation, and degenerative disease. Science of Aging Knowledge Environment. 2002;2002(29, article re3) doi: 10.1126/sageke.2002.29.re3. [DOI] [PubMed] [Google Scholar]
- 30.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology of Disease. 2006;21(2):404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature Reviews Immunology. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 32.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 35.Kim W, Mohney RP, Wilson B, Jeohn G, Liu B, Hong J. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. The Journal of Neuroscience. 2000;20(16):6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian L, Flood PM, Hong J. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. Journal of Neural Transmission. 2010;117(8):971–979. doi: 10.1007/s00702-010-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh S, Finn DP, Dowd E. Time-course of nigrostriatal neurodegeneration and neuroinflammation in the 6-hydroxydopamine-induced axonal and terminal lesion models of Parkinson’s disease in the rat. Neuroscience. 2011;175:251–261. doi: 10.1016/j.neuroscience.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 38.McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine administration. Annals of Neurology. 2003;54(5):599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- 39.Sugama S, Yang L, Cho BP, et al. Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Research. 2003;964(2):288–294. doi: 10.1016/s0006-8993(02)04085-4. [DOI] [PubMed] [Google Scholar]
- 40.Möller T, Hanisch U, Ransom BR. Thrombin-induced activation of cultured rodent microglia. Journal of Neurochemistry. 2000;75(4):1539–1547. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- 41.Tsirka SE. Clinical implications of the involvement of tPA in neuronal cell death. Journal of Molecular Medicine. 1997;75(5):341–347. doi: 10.1007/s001090050119. [DOI] [PubMed] [Google Scholar]
- 42.Block ML, Hong J. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Progress in Neurobiology. 2005;76(2):77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Ko HM, Koppula S, Kim B, et al. Inflexin attenuates proinflammatory responses and nuclear factor-ΚB activation in LPS-treated microglia. European Journal of Pharmacology. 2010;633(1–3):98–106. doi: 10.1016/j.ejphar.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Chow Y, Lee K, Vidyadaran S, et al. Cardamonin from Alpinia rafflesiana inhibits inflammatory responses in IFN-γ/LPS-stimulated BV2 microglia via NF-κB signalling pathway. International Immunopharmacology. 2012;12(4):657–665. doi: 10.1016/j.intimp.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Herrera AJ, Castaño A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiology of Disease. 2000;7(4):429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- 46.Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal “On” and “Off” signals control microglia. Trends in Neurosciences. 2007;30(11):596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Experimental and Molecular Medicine. 2006;38(4):333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- 48.Hoek RH, Ruuls SR, Murphy CA, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290(5497):1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 49.Wright GJ, Jones M, Puklavec MJ, Brown MH, Barclay AN. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology. 2001;102(2):173–179. doi: 10.1046/j.1365-2567.2001.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neumann H. Control of glial immune function by neurons. Glia. 2001;36(2):191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- 51.Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nature Reviews Immunology. 2006;6(6):457–464. doi: 10.1038/nri1859. [DOI] [PubMed] [Google Scholar]
- 52.Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 53.Limatola C, Lauro C, Catalano M, et al. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. Journal of Neuroimmunology. 2005;166(1-2):19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 54.de Jong EK, Dijkstra IM, Hensens M, et al. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. The Journal of Neuroscience. 2005;25(33):7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein RS, Lin E, Zhang B, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. Journal of Virology. 2005;79(17):11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. The Journal of Neuroscience. 2003;23(6):2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Arcuino G, Takano T, et al. P2X7 receptor inhibition improves recovery after spinal cord injury. Nature Medicine. 2004;10(8):821–827. doi: 10.1038/nm1082. [DOI] [PubMed] [Google Scholar]
- 58.Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446(7139):1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim YS, Kim SS, Cho JJ, et al. Matrix metalloproteinase-3: a novel signaling proteinase from apoptotic neuronal cells that activates microglia. The Journal of Neuroscience. 2005;25(14):3701–3711. doi: 10.1523/JNEUROSCI.4346-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoon SK, Dong HC, Block ML, et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. The FASEB Journal. 2007;21(1):179–187. doi: 10.1096/fj.06-5865com. [DOI] [PubMed] [Google Scholar]
- 61.Chang RCC, Hudson P, Wilson B, Haddon L, Hong J. Influence of neurons on lipopolysaccharide-stimulated production of nitric oxide and tumor necrosis factor-α by cultured glia. Brain Research. 2000;853(2):236–244. doi: 10.1016/s0006-8993(99)02255-6. [DOI] [PubMed] [Google Scholar]
- 62.Zecca L, Zucca FA, Wilms H, Sulzer D. Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends in Neurosciences. 2003;26(11):578–580. doi: 10.1016/j.tins.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 63.Calabrese VP, Hadfield MG. Parkinsonism and extraocular motor abnormalities with unusual neuropathological findings. Movement Disorders. 1991;6(3):257–260. doi: 10.1002/mds.870060311. [DOI] [PubMed] [Google Scholar]
- 64.Shamoto-Nagai M, Maruyama W, Akao Y, et al. Neuromelanin inhibits enzymatic activity of 26S proteasome in human dopaminergic SH-SY5Y cells. Journal of Neural Transmission. 2004;111(10-11):1253–1265. doi: 10.1007/s00702-004-0211-2. [DOI] [PubMed] [Google Scholar]
- 65.Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, Lucius R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson’s disease. The FASEB Journal. 2003;17(3):500–502. doi: 10.1096/fj.02-0314fje. [DOI] [PubMed] [Google Scholar]
- 66.Ciesielski-Treska J, Ulrich G, Taupenot L, et al. Chromogranin a induces a neurotoxic phenotype in brain microglial cells. The Journal of Biological Chemistry. 1998;273(23):14339–14346. doi: 10.1074/jbc.273.23.14339. [DOI] [PubMed] [Google Scholar]
- 67.Taupenot L, Ciesielski-Treska J, Ulrich G, Chasserot-Golaz S, Aunis D, Bader M-F. Chromogranin A triggers a phenotypic transformation and the generation of nitric oxide in brain microglial cells. Neuroscience. 1996;72(2):377–389. doi: 10.1016/0306-4522(96)83172-1. [DOI] [PubMed] [Google Scholar]
- 68.Yasuhara O, Kawamata T, Aimi Y, McGeer EG, McGeer PL. Expression of chromogranin A in lesions in the central nervous system from patients with neurological diseases. Neuroscience Letters. 1994;170(1):13–16. doi: 10.1016/0304-3940(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 69.Wu X, Block ML, Zhang W, et al. The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxidants and Redox Signaling. 2005;7(5-6):654–661. doi: 10.1089/ars.2005.7.654. [DOI] [PubMed] [Google Scholar]
- 70.Block ML, Zecca L, Hong J. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 71.Choi DK, Koppula S, Choi M, Suk K. Recent developments in the inhibitors of neuroinflammation and neurodegeneration: inflammatory oxidative enzymes as a drug target. Expert Opinion on Therapeutic Patents. 2010;20(11):1531–1546. doi: 10.1517/13543776.2010.525220. [DOI] [PubMed] [Google Scholar]
- 72.Qian L, Block ML, Wei S, et al. Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. Journal of Pharmacology and Experimental Therapeutics. 2006;319(1):44–52. doi: 10.1124/jpet.106.106351. [DOI] [PubMed] [Google Scholar]
- 73.Zhang W, Dallas S, Zhang D, et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia. 2007;55(11):1178–1188. doi: 10.1002/glia.20532. [DOI] [PubMed] [Google Scholar]
- 74.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. The Journal of Neuroscience. 1993;13(6):2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Molecular and Cellular Neuroscience. 2000;16(6):724–739. doi: 10.1006/mcne.2000.0914. [DOI] [PubMed] [Google Scholar]
- 76.Koppula S, Kumar H, Kim IS, Choi DK. Reactive oxygen species and inhibitors of inflammatory enzymes, NADPH oxidase, and iNOS in experimental models of Parkinson's disease. Mediators of Inflammation. 2012;2012:16 pages. doi: 10.1155/2012/823902.823902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Teismann P. COX-2 in the neurodegenerative process of Parkinson's disease. Biofactors. 2012;38(6):395–397. doi: 10.1002/biof.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsai S, Chao C, Yin M. Preventive and therapeutic effects of caffeic acid against inflammatory injury in striatum of MPTP-treated mice. European Journal of Pharmacology. 2011;670(2-3):441–447. doi: 10.1016/j.ejphar.2011.09.171. [DOI] [PubMed] [Google Scholar]
- 79.Przybylkowski A, Kurkowska-Jastrzebska I, Joniec I, Ciesielska A, Czlonkowska A, Czlonkowski A. Cyclooxygenases mRNA and protein expression in striata in the experimental mouse model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine administration to mouse. Brain Research. 2004;1019(1-2):144–151. doi: 10.1016/j.brainres.2004.05.095. [DOI] [PubMed] [Google Scholar]
- 80.Teismann P, Tieu K, Choi D, et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang T, Pei Z, Zhang W, et al. MPP+-induced COX-2 activation and subsequent dopaminergic neurodegeneration. The FASEB Journal. 2005;19(9):1134–1136. doi: 10.1096/fj.04-2457fje. [DOI] [PubMed] [Google Scholar]
- 82.Teismann P, Vila M, Choi D-K, et al. COX-2 and neurodegeneration in Parkinson’s disease. Annals of the New York Academy of Sciences. 2003;991:272–277. doi: 10.1111/j.1749-6632.2003.tb07482.x. [DOI] [PubMed] [Google Scholar]
- 83.Sánchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell A, Isacson O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. Journal of Neuroinflammation. 2004;1(1, article 6) doi: 10.1186/1742-2094-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teismann P, Ferger B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson's disease. Synapse. 2001;39(2):167–174. doi: 10.1002/1098-2396(200102)39:2<167::AID-SYN8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 85.Feng Z, Li D, Fung PCW, Pei Z, Ramsden DB, Ho S. COX-2-deficient mice are less prone to MPTP-neurotoxicity than wild-type mice. NeuroReport. 2003;14(15):1927–1929. doi: 10.1097/00001756-200310270-00009. [DOI] [PubMed] [Google Scholar]
- 86.Zhang W, Wang T, Pei Z, et al. Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. The FASEB Journal. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 87.Koharudin LMI, Liu H, Di Maio R, Kodali RB, Graham SH, Gronenborn AM. Cyclopentenone prostaglandin-induced unfolding and aggregation of the Parkinson disease-associated UCH-L1. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(15):6835–6840. doi: 10.1073/pnas.1002295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Lella Ezcurra AL, Chertoff M, Ferrari C, Graciarena M, Pitossi F. Chronic expression of low levels of tumor necrosis factor-α in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiology of Disease. 2010;37(3):630–640. doi: 10.1016/j.nbd.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 89.Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. Journal of Neuroinflammation. 2012;9, article 82 doi: 10.1186/1742-2094-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson’s disease. Journal of Neural Transmission, Supplement. 2000;(60):277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 91.Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson’s disease. Progress in Neurobiology. 2002;68(5):325–340. doi: 10.1016/s0301-0082(02)00127-2. [DOI] [PubMed] [Google Scholar]
- 92.Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. Journal of Neurochemistry. 2007;101(3):577–599. doi: 10.1111/j.1471-4159.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- 93.Benner EJ, Banerjee R, Reynolds AD, et al. Nitrated α-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001376.e1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. Journal of Immunology. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 95.Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Experimental Neurology. 2006;197(2):275–283. doi: 10.1016/j.expneurol.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 96.Depboylu C, Stricker S, Ghobril JP, Oertel WH, Priller J, Hoglinger GU. Brain-resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T-lymphocytes in the MPTP mouse model of Parkinson disease. Experimental Neurology. 2012;238(2):183–191. doi: 10.1016/j.expneurol.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 97.Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson's disease. Cold Spring Harbor Perspectives in Medicine. 2012;2(1) doi: 10.1101/cshperspect.a009381.a009381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cao J, Li K, Shen Y. Activated immune cells in Parkinson’s disease. Journal of Neuroimmune Pharmacology. 2011;6(3):323–329. doi: 10.1007/s11481-011-9280-9. [DOI] [PubMed] [Google Scholar]
- 99.McGeer EG, Singh EA, McGeer PL. Peripheral-type benzodiazepine binding in Alzheimer disease. Alzheimer Disease and Associated Disorders. 1988;2(4):331–336. doi: 10.1097/00002093-198802040-00001. [DOI] [PubMed] [Google Scholar]
- 100.Petrozzi L, Lucetti C, Scarpato R, et al. Cytogenetic alterations in lymphocytes of Alzheimer’s disease and Parkinson’s disease patients. Neurological Sciences. 2002;23(supplement 2):S97–S98. doi: 10.1007/s100720200087. [DOI] [PubMed] [Google Scholar]
- 101.Migliore L, Petrozzi L, Lucetti C, et al. Oxidative damage and cytogenetic analysis in leukocytes of Parkinson’s disease patients. Neurology. 2002;58(12):1809–1815. doi: 10.1212/wnl.58.12.1809. [DOI] [PubMed] [Google Scholar]
- 102.Kurkowska-Jastrzȩbska I, Wrońska A, Kohutnicka M, Członkowski A, Członkowska A. MHC class II positive microglia and lymphocytic infiltration are present in the substantia nigra and striatum in mouse model of Parkinson’s disease. Acta Neurobiologiae Experimentalis. 1999;59(1):1–8. doi: 10.55782/ane-1999-1289. [DOI] [PubMed] [Google Scholar]
- 103.Hisanaga K, Asagi M, Itoyama Y, Iwasaki Y. Increase in peripheral CD4 bright+ CD8 dull+ T cells in Parkinson disease. Archives of Neurology. 2001;58(10):1580–1583. doi: 10.1001/archneur.58.10.1580. [DOI] [PubMed] [Google Scholar]
- 104.Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism and Related Disorders. 2005;11(8):493–498. doi: 10.1016/j.parkreldis.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 105.Fiszer U, Mix E, Fredrikson S, Kostulas V, Olsson T, Link H. γδ + T cells are increased in patients with Parkinson’s disease. Journal of the Neurological Sciences. 1994;121(1):39–45. doi: 10.1016/0022-510x(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 106.Neumann H, Wekerle H. Neuronal control of the immune response in the central nervous system: linking brain immunity to neurodegeneration. Journal of Neuropathology and Experimental Neurology. 1998;57(1):1–9. doi: 10.1097/00005072-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 107.Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microscopy Research and Technique. 2001;54(1):47–58. doi: 10.1002/jemt.1120. [DOI] [PubMed] [Google Scholar]
- 108.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nature Medicine. 2007;13(10):1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kortekaas R, Leenders KL, Van Oostrom JCH, et al. Blood-brain barrier dysfunction in Parkinsonian midbrain in vivo. Annals of Neurology. 2005;57(2):176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 110.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 111.Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews. 2006;86(3):1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- 112.Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends in Immunology. 2007;28(1):5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 113.Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson’s disease. Neurotherapeutics. 2010;7(4):413–423. doi: 10.1016/j.nurt.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nutt JG, Wooten GF. Diagnosis and initial management of Parkinson’s disease. The New England Journal of Medicine. 2005;353(10):1021–1027. doi: 10.1056/NEJMcp043908. [DOI] [PubMed] [Google Scholar]
- 115.Wu DC, Tieu K, Cohen O, et al. Glial cell response: a pathogenic factor in Parkinson’s disease. Journal of NeuroVirology. 2002;8(6):551–558. doi: 10.1080/13550280290100905. [DOI] [PubMed] [Google Scholar]
- 116.Sellers RS, Antman M, Phillips J, Khan KN, Furst SM. Effects of Miglyol 812 on rats after 4 weeks of gavage as compared with methylcellulose/Tween 80. Drug and Chemical Toxicology. 2005;28(4):423–432. doi: 10.1080/01480540500262839. [DOI] [PubMed] [Google Scholar]
- 117.Eddelston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54(1):15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hirsch S, Bähr M. Growth promoting and inhibitory effects of glial cells in the mammalian nervous system. Advances in Experimental Medicine and Biology. 1999;468:199–205. doi: 10.1007/978-1-4615-4685-6_16. [DOI] [PubMed] [Google Scholar]
- 119.Lee H, Kim C, Lee S. Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxidative Medicine and Cellular Longevity. 2010;3(4):283–287. doi: 10.4161/oxim.3.4.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sekiyama K, Sugama S, Fujita M, et al. Neuroinflammation in Parkinson’s disease and related disorders: a lesson from genetically manipulated mouse models of α-synucleinopathies. Parkinson’s Disease. 2012;2012:8 pages. doi: 10.1155/2012/271732.271732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Przedborski S, Jackson-Lewis V, Djaldetti R, et al. The parkinsonian toxin MPTP: action and mechanism. Restorative Neurology and Neuroscience. 2000;16(2):135–142. [PubMed] [Google Scholar]
- 122.Lee H, Suk J, Patrick C, et al. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. The Journal of Biological Chemistry. 2010;285(12):9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in Immunology. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 124.Qureshi GA, Baig S, Bednar I, Sodersten P, Forsberg G, Siden A. Increased cerebrospinal fluid concentration of nitrite ire Parkinson’s disease. NeuroReport. 1995;6(12):1642–1644. doi: 10.1097/00001756-199508000-00013. [DOI] [PubMed] [Google Scholar]
- 125.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite: a potential additional mechanism of nitric oxide-dependent toxicity. The Journal of Biological Chemistry. 1997;272(12):7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 126.Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92(9):3007–3017. [PubMed] [Google Scholar]
- 127.Saijo K, Winner B, Carson CT, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137(1):47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klegeris A, Pelech S, Giasson BI, et al. α-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiology of Aging. 2008;29(5):739–752. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 129.Ferrer I, Blanco R, Cutillas B, Ambrosio S. Fas and Fas-L expression in Huntington’s disease and Parkinson’s disease. Neuropathology and Applied Neurobiology. 2000;26(5):424–433. doi: 10.1046/j.1365-2990.2000.00267.x. [DOI] [PubMed] [Google Scholar]
- 130.Hayley S, Crocker SJ, Smith PD, et al. Regulation of dopaminergic loss by Fas in a 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine model of Parkinson's disease. The Journal of Neuroscience. 2004;24(8):2045–2053. doi: 10.1523/JNEUROSCI.4564-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Karunakaran S, Saeed U, Mishra M, et al. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. The Journal of Neuroscience. 2008;28(47):12500–12509. doi: 10.1523/JNEUROSCI.4511-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rus H, Cudrici C, David S, Niculescu F. The complement system in central nervous system diseases. Autoimmunity. 2006;39(5):395–402. doi: 10.1080/08916930600739605. [DOI] [PubMed] [Google Scholar]
- 133.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease—a brief review of the basic science and clinical literature. Cold Spring Harbor Perspectives in Medicine. 2012;2(1) doi: 10.1101/cshperspect.a006346.a006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McGeer PE, Yasojima K, McGeer PL. Inflammation in the pathogenesis of Parkinson's disease. British Columbia Medical Journal. 2001;43(3):138–141. [Google Scholar]
- 135.McGeer EG, McGeer PL. Pharmacologic approaches to the treatment of amyotrophic lateral sclerosis. BioDrugs. 2005;19(1):31–37. doi: 10.2165/00063030-200519010-00004. [DOI] [PubMed] [Google Scholar]
- 136.Loeffler DA, Camp DM, Conant SB. Complement activation in the Parkinson’s disease substantia nigra: an immunocytochemical study. Journal of Neuroinflammation. 2006;3, article 29 doi: 10.1186/1742-2094-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yamada T, McGeer EG, Schelper RL, et al. Histological and biochemical pathology in a family with autosomal dominant Parkinsonism and dementia. Neurology Psychiatry and Brain Research. 1993;2(1):26–35. [Google Scholar]
- 138.McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism and Related Disorders. 2004;10(supplement 1):S3–S7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 139.Wang Y, Hancock AM, Bradner J, et al. Complement 3 and factor H in human cerebrospinal fluid in Parkinson’s disease, Alzheimer’s disease, and multiple-system atrophy. American Journal of Pathology. 2011;178(4):1509–1516. doi: 10.1016/j.ajpath.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Molecular Immunology. 2007;44(5):999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 141.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nature Immunology. 2010;11(9):785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Annals of the New York Academy of Sciences. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- 143.Shen Y, Meri S. Yin and Yang: complement activation and regulation in Alzheimer’s disease. Progress in Neurobiology. 2003;70(6):463–472. doi: 10.1016/j.pneurobio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 144.O’Barr S, Cooper NR. The C5a complement activation peptide increases IL-1β and IL-6 release from amyloid-β primed human monocytes: implications for Alzheimer’s disease. Journal of Neuroimmunology. 2000;109(2):87–94. doi: 10.1016/s0165-5728(00)00291-5. [DOI] [PubMed] [Google Scholar]
- 145.Depboylu C, Schäfer MK-H, Arias-Carrión O, Oertel WH, Weihe E, Höglinger GU. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. Journal of Neuropathology and Experimental Neurology. 2011;70(2):125–132. doi: 10.1097/NEN.0b013e31820805b9. [DOI] [PubMed] [Google Scholar]
- 146.Lee H, Choi J, Suk K. Increases of pentraxin 3 plasma levels in patients with Parkinson’s disease. Movement Disorders. 2011;26(13):2364–2370. doi: 10.1002/mds.23871. [DOI] [PubMed] [Google Scholar]
- 147.Yin GN, Lee HW, Cho J, Suk K. Neuronal pentraxin receptor in cerebrospinal fluid as a potential biomarker for neurodegenerative diseases. Brain Research. 2009;1265:158–170. doi: 10.1016/j.brainres.2009.01.058. [DOI] [PubMed] [Google Scholar]