

Abstract
The correct interpretation of many molecular biology experiments depends in an essential way on the accuracy and consistency of the existing annotation databases. Such databases are meant to act as repositories for our biological knowledge as we acquire and refine it. Hence, by definition, they are incomplete at any given time. In this paper, we describe a technique that improves our previous method for predicting novel GO annotations by extracting implicit semantic relationships between genes and functions. In this work, we use a vector space model and a number of weighting schemes in addition to our previous latent semantic indexing approach. The technique described here is able to take into consideration the hierarchical structure of the Gene Ontology (GO) and can weight differently GO terms situated at different depths. The prediction abilities of 15 different weighting schemes are compared and evaluated. Nine such schemes were previously used in other problem domains, while six of them are introduced in this paper. The best weighting scheme was a novel scheme, n2tn. Out of the top 50 functional annotations predicted using this weighting scheme, we found support in the literature for 84 percent of them, while 6 percent of the predictions were contradicted by the existing literature. For the remaining 10 percent, we did not find any relevant publications to confirm or contradict the predictions. The n2tn weighting scheme also outperformed the simple binary scheme used in our previous approach.
Index Terms: Gene function prediction, gene annotation, Gene Ontology, vector space model, latent semantic indexing, weighting schemes
1 Introduction
1.1 Background
Gene annotation databases capture the current biological knowledge allowing researchers to interpret the results of life science experiments. In spite of their unquestionable importance, significant problems concerning the annotation databases still exist. One problem is that the annotation databases are currently incomplete. For virtually all sequenced organisms, only a subset of genes is known, and an even smaller subset of genes is functionally annotated [28]. As more knowledge is accumulated, genes and annotations are gradually added to such databases. This means that at any moment in time, it is likely that an annotation database will contain only a subset of all genes of the given organism, and even for those genes that are included, possibly only a subset of their functions is present in the database. In addition to this, most of the annotations are introduced by curators who manually examine the literature. In this process, it is possible that certain confirmed facts reported in existing publications might get overlooked [25]. Another problem is caused by the way these annotations are stored in the structure of the Gene Ontology (GO). There are, for instance, genes that are annotated for a particular molecular function but are not annotated for the corresponding biological process. This is not a problem for a database curator or a life scientist looking for the annotations of a specific gene, since a human can easily make obvious extrapolations. However, this is not how such databases are used most of the time. In a more typical scenario, the researcher will try to interpret the results of a high-throughput experiment using a software that performs an ontological analysis [11], [12], [24], [27], [26], [2], [4], [21], [35], [42], [43]. Such software will query an annotation database in each of the three main branches of the GO graph and calculate a statistical significance based strictly on the data retrieved, making no extrapolations. This type of analysis fails to correctly compute the statistical significance of the genes involved if they are not correctly annotated for each of the three GO categories. We should note here that no matter how thorough the annotators are, as our knowledge improves, new functions will continue to be added, and some of the older ones will be changed or revoked. Thus, due to the intrinsic evolution of scientific knowledge, gene annotations are likely to maintain a dynamic character and hence are unlikely to be considered complete anytime in the near future.
To overcome some of these problems, we previously proposed a method capable of finding gene-function associations that are not explicitly represented in the annotation databases [25]. This technique employs a latent semantic indexing (LSI) approach and was demonstrated using the human genome annotations. This first attempt used a binary representation of the relationships between genes and their functional annotations. However, the binary representation fails to properly capture the hierarchical relationships between various terms. Previous research in information retrieval (IR) has shown that the use of a weighted representation, rather than a binary one, can improve the quality of retrieval operations. Intuitively, IR term weighting attempts to exploit two simple observations: 1) terms that appear repeatedly in a document are better suited to describe the topic of the document than terms that are rarely used, and 2) infrequent terms across the document collection are better differentiators between documents than terms that appear in most or in all documents. Similar relationships might exist between genes and their annotations. Functions that are only associated with few genes carry more information about the genes and can better differentiate between them. Conversely, several closely related functions associated with a given gene will better describe what the gene actually does.
This paper explores the use of vector space model (VSM) weighting schemes in the context of a semantic analysis of biological annotations. The technique described here is able to discover implicit gene-function relationships and propose them to researchers and database curators as novel annotations. We present the results obtained with several weighting schemes on the annotations of the human genome stored in the Onto-Tools database [11], [24], which includes all known annotations from the GO Consortium.
1.2 Related Work
A VSM [5], [6], [16] has been used previously to cluster genes by creating a vector space of genes and MEDLINE abstracts of papers discussing those particular genes [17]. The similarity between genes was assessed by computing a distance between the vectors that were representing them. It was found that weighted vectors improved the results significantly over Boolean vectors [17]. VSM was also used to compute the similarity between GO terms, and the results were compared with two other nonlexical methods for analyzing the GO graph [7]. LSI [5], [6], [9] has recently been utilized for genome-wide expression data analysis [3]. LSI was also employed to identify relations between genes by creating a vector space of genes and MEDLINE abstracts [20]. Earlier IR research has shown that LSI is 30 percent more effective than word matching methods [9]. Ontologies were used in the recent past to overcome the limitations of keyword-based search, especially after the emergence of the Semantic Web [32], [39]. In [39], the authors describe an IR method that combines document annotation and query expansion using ontology terms and results ranking using VSM. Similar techniques are employed by MELISA [1] and Textpresso [30], two medical literature search tools. MELISA uses MEDLINE’s own ontology, MeSH, to semantically enrich the user queries. Textpresso builds an ontology, 80 percent of which is based on GO terms, and uses it for document annotation and query expansion.
Other approaches for predicting functional annotations for a given gene also exist. The most commonly used approach for function prediction uses sequence similarity. This approach is based on the hypothesis that a function can be transferred between similar sequences in different organisms since such similarity has been conserved over long periods of evolution [10]. This method of annotation transfer can result in incorrect function predictions due to reasons such as divergence of function within homologous proteins. Furthermore, this type of inference can also be incorrect because the annotations are only transferred from the closest homolog [23]. In order to overcome these problems, approaches combining sequence similarity data with structural information have been proposed [14], [38]. The guilt by association (GBA) approach [33], [40], [44], based on the observation that functionally related genes tend to share similar mRNA expression profiles, has also been widely applied to predict gene functions [8], [13], [22], [36], [41]. This approach clusters the genes based on their expression profiles in order to predict the gene functions. The GBA approaches are affected by issues such as data transformation [15], [31] and filtering intended to boost the signal-to-noise ratio [19]. An alternative approach uses sequence similarity and protein domain data in order to predict functional annotations [37]. Raychaudhuri et al. [34] proposed a natural language processing approach for automatically extracting gene-function associations from the literature abstracts.
2 Methods
GO maintains an organism-independent ontology of functional annotations that has a directed acyclic graph (DAG) structure. Each node in this graph represents a functional category and groups a number of genes annotated with that category. Researchers and curators endeavor to annotate the genes with the most specific functional category available in each case. For instance, if a gene is known to regulate the cell growth by extracellular stimulus, it is annotated with the specific category “regulation of cell growth by extracellular stimulus (GO:0001560),” instead of a higher level more general category such as “regulation of cell growth (GO:0001558)” or “cell growth (GO:0016049).” However, a gene involved in regulation of cell growth by extracellular stimulus is actually involved in regulation of cell growth, which is indeed part of the cell growth phenomenon. Because of this, we consider that a gene annotated with a specific function f is also associated with the more general functional categories represented by the ancestors of f. In order to represent this in our data, we create a gene-function matrix GF as follows:
| (1) |
The ith row of the matrix GF represents all functions known to be associated with gene gi either directly, as found in the literature, or through its descendants. Similarly, the jth column of the matrix GF represents all genes known to be associated with the function fj or any of its descendants.
Functional categories such as “unknown biological process” are used in GO in order to ensure a consistency of annotations. However, these terms lack any semantic content since they can be used to group completely unrelated genes. Since our goal is to construct a model of the semantic relationships between genes and functions, such terms lacking semantic content are removed from the analysis. Similarly, the top-level nodes, “gene ontology (GO:0003673),” “biological process (GO:0008150),” “molecular function (GO:0003674),” and “cellular component (GO:0005575)” also lack a specific semantic content since all genes will appear related to each of these terms. Therefore, we also remove these GO terms from the GF matrix.
The matrix described by (1) uses a simple binary weighting scheme for the gene-function associations. However, previous work in IR has shown that the performance of a system can be improved in terms of both precision and recall by using more sophisticated weighting schemes instead of a binary scheme [16]. In this paper, we propose a VSM model using different weights for the gene-function associations.
The weighting schemes used in this paper are denoted by three-letter codes, where the first letter refers to the local weight, the second to the global weight, and the third to the normalization method used for the annotation vector. The annotation vectors are the columns in the GF matrix. Each column in the GF matrix contains the weights of the relationships a particular GO term has with each human gene in the GO database.
The local weight is proportional to the number of direct or indirect relationships that exist between a given gene and a given GO term. It is computed using gene frequency gf, which is defined as the number of times a gene is directly or indirectly associated with a function in the GO graph. We use inverse annotation frequency iaf as the global weight, where inverse annotation frequency is defined as the natural log ratio of the total number of GO terms in the GF matrix to the total number of annotations specific to the given gene.
The overall weight for each annotation is computed as a product of the local and the global weights divided by the normalization factor. Because each annotation in the original database is propagated to the higher levels of the GO DAG (1), the GO terms toward the root of the DAG will invariably have higher weights and could bias the semantic contents of the GF matrix. Therefore, the final weights should be normalized so that the terms at higher levels will not dominate the specific terms found close to the leaves of the GO DAG. The local and global weights and the normalization factors used in this paper are presented in Table 2. Maximum and augmented local weights are employed to compensate for high gene frequencies; cosine normalization can be used to compensate for annotations common to a large number of genes.
TABLE 2.
A Comparison between the Annotations Predicted from 2003 GO Data and the Actual 2006 GO Data
| atn | atm | atc | mtn | ntn | ntm | ntc | Its | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| confirmed relations | 53 | 39 | 331 | 16 | 149 | 255 | 339 | 25 |
| relations above threshold | 1552 | 1035 | 29552 | 1576 | 6040 | 14418 | 29033 | 2790 |
| threshold used | 0.17 | 0.34 | 0.04 | 0.04 | 0.02 | 0.12 | 0.04 | 0.04 |
The columns contain the number of confirmed predictions in the May 2006 GO database, the total number of predictions above the threshold, and the threshold used for each of the weighting schemes. atm, atn, ntn, and ntm performed best, but the results were not conclusive.
The hierarchical structure of the GO DAG poses another two problems. The indirect annotations introduced in the GF matrix due to the propagation of the annotations toward the root of the GO DAG are less specific and should be assigned less weight than the annotations that were present in the original database. Also, the annotations near the root of the GO tree are less specific than those close to the leaves and should receive smaller weights. In order to address these problems, depth corrections need to be applied to both local and global weights.
Given a GO term ti and a gene gj directly annotated with a different GO term tj, which is a (possibly distant) descendant of ti, the local weight of the relationship between ti and gj was multiplied with
| (2) |
where d(tj) and d(tj) are the depths of ti and tj, respectively, and α is a factor that indicates how much the weight diminishes between successive levels of the GO tree. For instance, in Fig. 1, the relationship between the gene CASP3 and one of its direct annotations, induction of apoptosis by intracellular signals (GO:0008629), is highly meaningful. However, while CASP3’s relationship with programmed cell death (GO:0012501)—three levels up—is still meaningful even though less informative, its relationship with biological process (GO:0008150)—six levels up—is hardly informative at all. Hence, we would like to gradually diminish the importance of the association between a gene gj and the function ti by the shortest distance in the GO tree between ti and tj. We consider that the association between a given gene and the ancestors of any of its annotation terms exponentially decreases in strength. In most cases, the relationships between genes and GO terms that propagate up more than half of the GO tree’s height are not particularly meaningful anymore. In order to reflect this, α was chosen so that the local weight of an indirect relationship over eight depth levels (half-depth of the GO tree as of May 2003) is approximately 1 percent of the weight of a direct relationship. Hence, α can be calculated from the equation α−8 = 0.01 as approximately 1.7.
Fig. 1.

Depth calculations for local and global weights. The depth used for penalizing the local weight of the indirect relationship between the gene CASP3 and the GO term programmed cell death (GO:0012501) is calculated as the difference between the depth of GO:0008629 (which has a direct relationship with the gene CASP3) and the depth of GO:0012501 (both computed from the root of the GO tree). The depth used for the global weight of the same relationship is the depth of GO:0012501, also computed from the root of the GO tree.
Fig. 1 describes the usage of the depth in calculating the local and global weighting. Gene CASP3 is annotated with the GO term GO:0008629 (induction of apoptosis by intracellular signals), and therefore, it is in an indirect relationship with GO:0012501 (programmed cell death), one of the ancestors for GO:0008629 (shaded in green in Fig. 1). In this figure, d(GO :0008629) = 7, d(GO :0012501) = 4, and the global weight of ti will be
| (3) |
This captures the fact that the association between CASP3 and “programmed cell death” is weaker than the association between the same gene and its direct annotation, “induction of apoptosis by intracellular signals.”
We will illustrate the weighting process by describing the steps required to compute the weight of the relationship between the gene CASP3 and the GO term GO:0012501 (programmed cell death), using the ntn weighting scheme. GO:0012501 has four descendants that have a direct relationship with CASP3: GO:0006915, GO:0006917, GO:0008624, and GO:0008629. These GO terms are located at the following depths from GO:0012501: GO:0006915 at depth 1, GO:0006917 at depth 2, and GO:0008624 and GO:0008629 at depth 3. The ntn scheme uses gene frequency for the local weight; therefore, in order to compute the local weight of the relationship between CASP3 and GO:0012501, we have to sum all depth-penalized weights for each GO:0012501 descendant that has a direct relationship with CASP3. For GO:0006915 at depth 1, we have the weight 1.7−1 = 0.588, for GO:0006917 at depth 2, we have the weight 1.7−2 = 0.346, and for GO:0008624 and GO:0008629 at depth 3, we have the weight 1.7−3 = 0.203. These values sum up to the local weight of 1.341 for this particular relationship. For the global weight, the ntn scheme uses inverse annotation frequency. In May 2003, CASP3 had 25 direct or indirect relationships with GO terms, from a total of 4,683 GO terms that were in the direct or indirect relationships with genes at that date. This allows us to calculate the inverse annotation frequency as ln(4,683/25) = 5.233. We have to penalize this figure for depth. In the same GO release, GO:0012501 was at depth 5 from the root; therefore, the global weight is equal to (1 − (1.7−5) * 5.233 = 4.864. Because no other normalization type is used, in the ntn scheme, the normalization factor is one. This gives us the final ntn weight for the relationship between the gene CASP3 and the GO term GO:0012501: 1.341 * 4.864 = 6.523. The relationships discussed in this paragraph are marked with bold lines in Fig. 1. In-depth descriptions of the classical VSM weighting schemes and the various motivations that inspired them are available in the literature [16].
Eight different weighting schemes, described below, were tested in a first stage: ntn, ntm, ntc, mtn, atn, atm, atc, and lts (note that ntm and ntc are identical with mtm and mtc, respectively). The last scheme, lts, was not described in Table 1. Although lts is a frequently used weighting scheme in IR, in our context, it was outperformed by all the other weighting schemes that we tested. We kept lts in the results section for comparison purposes. The local weight l (logarithmic) used by lts is equal to 1 + ln(gf); its global weight is inverse annotation frequency and the normalization factor used is s (sum), which is equal to sum of weights in the annotation vector.
TABLE 1.
The Description of the Weighting Scheme Codes Used
| Local weighting | ||
|---|---|---|
| Code | Description | |
| n | None: gene frequency, gf, is used as the local weight. | |
| m | Maximum: gene frequency normalized with respect to the maximum gene frequency in an annotation vector is used, i.e., | |
| a | Augmented: gene frequency is augmented as: | |
| Global weighting | ||
| t | Inverse annotation frequency, iaf, is used as the global weight. | |
| Normalization factor | ||
| n | None: normalization factor is not used. | |
| m | Maximum: each weight is normalized with respect to the maximum weight in an annotation vector, i.e., | |
| c |
|
|
The weighting schemes used in this paper are denoted by three-letter codes, where the first letter refers to the local weight, the second to the global weight, and the third to the normalization method used for the annotation vector. For instance, in ntn, the local weighting uses the gene frequency, the global weighting uses the inverse annotation frequency, and no further normalization is performed.
After applying each of the weighting schemes above to the GF matrix, the matrix is decomposed using singular value decomposition (SVD) as (Fig. 2):
| (4) |
where Sm is an m × m diagonal matrix, and m is the rank of GF, i.e., the number of linearly independent rows or columns. The elements of Sm are the singular values of GF. The matrices Gm and are the basis sets of size g × m and m × f, respectively, and are orthogonal, i.e., [18], [25].
Fig. 2.
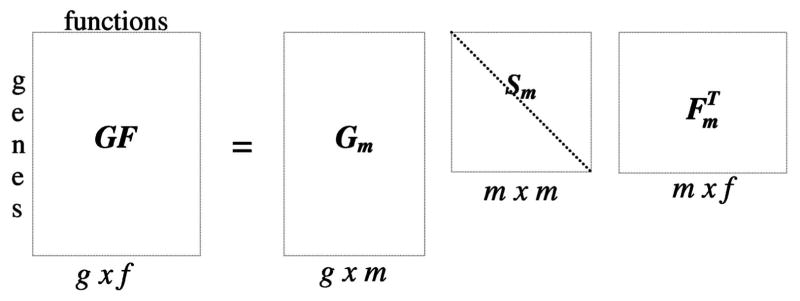
SVD of the gene-function association matrix GF. There are g genes and f functions. Sm is a diagonal matrix such that Sij = 0 if i ≠ j and Sij ≥ 0 if i = j.
SVD rotates the m-dimensional vector space and projects the data into a new vector space, where the highest variation of the data is found along the first dimension, the second highest variation is found along the second dimension, and so on. Reducing the dimensionality of the vector space removes much of the noise from the original data. This is done by selecting only the k largest singular values of Sm and the corresponding vectors in Gm and Fm matrices, creating the matrices Sk, Gk, and Fk (Fig. 3). The product of these matrices, , is the closest rank k approximation of GF in the least squares sense:
| (5) |
Fig. 3.
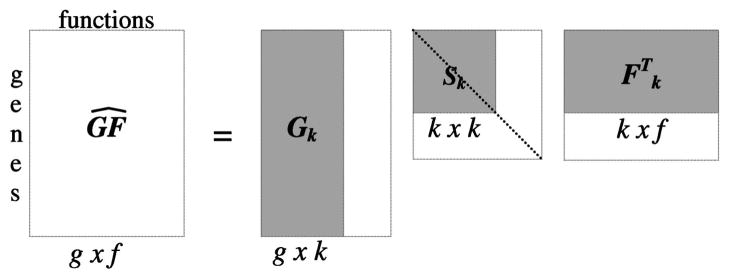
The dimensionality reduction from m to k produces an approximation matrix of the original matrix GF. By reducing the dimensionality, we force the new matrix to capture the latent semantics and filter out the noise. This essentially will capture those interactions that are strongly represented in the data.
The matrix is now expected to contain explicitly all associations that are strongly represented in the data, whether or not such associations were present in the original matrix. Thus, the goal of this process is to reveal those gene-function associations that were not previously known but which are implicitly contained in the data.
After reducing the dimensionality of the system, we then analyze the matrix by comparing its elements with a given threshold T. A value of greater than the threshold T might indicate that gene i has function j. Gene-function relationships with gfij = 0 (i.e., no previously known association between gene g and function f) and correspond to newly discovered associations between genes and functions. Gene-function relationships with gfij ≠ 0 and correspond to known functional annotations that have weak semantic support in the data. Weak semantic support for an annotation does not necessarily mean that the given annotation is incorrect. Such annotation may simply be a novel phenomenon for which not enough information is available and therefore appears inconsistent with the rest of the annotations at the time of the analysis.
3 Results and Discussion
The approach described above was used to examine the entire existing set of GO annotations for the human genome. We were interested in analyzing the GO annotation graph in order to find relationships between genes and functions that are captured in the semantic layer of the graph but are missing from the annotation database itself. In the first step, the performance of eight weighting schemes was investigated on this human annotation data set: ntn, ntm, ntc, mtn, atn, atm, atc, and lts. For each of the eight weighting schemes, the first 50 best scoring relationships were assessed by a human expert. In a second step, the scheme that performed the best, ntn, was altered in another six different weighting schemes, in order to understand what were the terms in the weighting formula that helped it achieve the best performance. We defined one new local weight and two new global weights. The local weight, called n2, had the same local depth factor, but the gene frequency was not used (i.e., it has value 1 for all genes). The global weight nt had the same global depth factor, but the inverse annotation frequency was not used. For the global weight nt2, both the global depth factor and the inverse annotation frequency were not used. The six new weighting schemes derived from ntn were n-nt-n, n2-t-n, n2-t-m, n2-nt-n, n2-nt-m, and n2-nt2-n. The weighting terms, other than the three new terms defined here, have the same meaning as before. For the sake of a simpler notation consistent with the conventions used in IR, the dashes will be omitted henceforth.
The gene-function matrix GF was built using the human annotations contained in the GO database, released in May 2003. The initial GF matrix contained 10,078 genes and 4,693 functional annotations, for a total of 300,204 relations between genes and functions. As discussed, relations that involved the annotations at the root of the GO graph were not included in the GF matrix to prevent these annotations from overwhelming the others. Also, the genes and GO terms that had no associations were not included in the GF matrix, because they do not add semantic information.
We decomposed the matrix GF as in (4) and reduced its dimensionality to the largest 500 eigenvalues. The matrix is constructed as in Fig. 3, by multiplying the reduced matrices that resulted after SVD. The value for the threshold T was calculated as previously described [25]. In essence, the range of values in the initial GF matrix is divided into 100 equal bins. The upper limit of each bin is then used as a threshold (T) to evaluate the number of false positives and false negatives in the data. Gene-function relationships with gfij = 0 (i.e., no previously known association between gene g and function f) and are the false negatives (but also the predictions of our method). Gene-function relationships with gfij ≠ 0 and are the false positives. Assuming that the initial relationships taken from the GO database have the minimum amount of errors, we are selecting as the threshold the value T that minimizes the number of presumed errors (FP + FN).
Initially, we tried to evaluate the accuracy of the weighting schemes by counting the number of confirmed relationships in the annotation database released three years after the data used for input. More specifically, the new associations predicted from the analysis of the annotations from May 2003 were compared with the annotations from May 2006. The thresholds, the number of gene-function relationships that scored above the threshold, and the number of confirmed relationships for each of the weighting schemes investigated in the first stage are shown in Table 2. The number of confirmed predictions in the May 2006 GO database in the first 25, 50, 100, 200, 400, 800, and 1,600 predictions (ordered in the decreasing order of their score) are shown in Fig. 4 for each of the eight weighting schemes. These data show that for few predictions (25–100), atn and atm are able to predict most correct relationships. Beyond 150 predicted relations, ntn and ntm also started to performed well, with ntn yielding the most correct predictions.
Fig. 4.

Automated assessment results: the data points connected with lines represent the number of confirmed predictions in the May 2006 GO database in the first 25, 50, 100, 200, 400, 800, and 1,600 predictions (ordered in the decreasing order of their score) for each of the eight weighting schemes; atm, atn, ntn, and ntm performed best, but the results were not conclusive.
However, by using the existing GO annotations as the gold standard, this type of assessment is somewhat limited. Indeed, this approach only provides a lower bound for the number of true positives because the real number of correctly predicted annotations could be much higher than what is reflected in the current GO annotations used as a reference. In order to address this, we also asked a human expert (A.D.) to assess the top 50 highest scoring relations for each of the weighting schemes. Each predicted annotation was assessed on a scale from −2 to 2 as follows: A score of 2 means that the predicted annotation is very well supported by existing literature (at least two independent papers were found proving that the predicted annotation is correct) or that specific relationship has been included in one of the more recent releases of GO. A score of 1 was given when existing papers suggest that the relationship is correct, without offering indubitable proof. Relationships for which no support was found in the literature able to confirm or contradict them were given a score of 0. A score of −1 was given when papers were found suggesting that the relationship is not correct, and a score of −2 was given when strong literature support was found to prove that the relationship is not correct. The results of this assessment can be seen in Table 3. These data show that ntn is the best performing scheme: among its 50 relationships that were evaluated, 35 are strongly supported in the literature; another five are suggested by various existing research results; on seven of them, there is nothing published yet; and only three were contradicted by the existing knowledge. In the second stage, n2tn performed as good as ntn, despite the fact that it requires fewer steps to compute its weights. The results of the second stage are shown in Table 4.
TABLE 3.
The Results of the Expert Assessment of the Top 50 Predictions
| atn | atm | atc | ntn | ntm | ntc | mtn | Its | bin | |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| 2 | 18 | 21 | 12 | 35 | 26 | 19 | 3 | 4 | 18 |
| 1 | 4 | 9 | 7 | 5 | 6 | 5 | 5 | 7 | 9 |
| 0 | 16 | 17 | 15 | 7 | 12 | 20 | 19 | 34 | 18 |
| −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| −2 | 10 | 1 | 6 | 3 | 6 | 6 | 5 | 4 | 2 |
| obsolete | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 2 |
The most successful weighting scheme, ntn, outperformed the simple binary representation scheme, bin (used in [25]). The last row, “obsolete,” shows the number of relationships involving GO terms that have been retired since the May 2003 release of GO, which was used as the basis for our predictions.
TABLE 4.
The Results of the Manual Assessment in the Second Stage
| nntn | n2tn | n2tm | n2ntn | n2ntm | n2nt2n | ntn | bin | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 2 | 34 | 34 | 29 | 28 | 27 | 25 | 35 | 18 |
| 1 | 5 | 8 | 6 | 8 | 4 | 7 | 5 | 9 |
| 0 | 7 | 5 | 12 | 7 | 13 | 9 | 7 | 18 |
| −1 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 1 |
| −2 | 4 | 2 | 2 | 4 | 3 | 5 | 3 | 2 |
| obsolete | 0 | 0 | 1 | 2 | 2 | 3 | 0 | 2 |
The weighting schemes analyzed in the second stage did not show improved results over the earlier best performing scheme, ntn, but some of them were computationally less demanding.
As examples of predictions made using the n2tn scheme, SLC2A10 and SLC2A9 were predicted to exhibit glucose transporter activity. The human gene SLC2A10 is the solute carrier family 2 (facilitated glucose transporter), member 10 (a validated well-documented structure). Obviously, SLC2A10 has glucose transporter activity. Yet, in spite of the fact that this gene is annotated for the biological process glucose transport, it is not yet annotated for the corresponding molecular function glucose transporter activity. At the same time, the existing annotations for molecular functions are far less specific: sugar porter activity and transporter activity. The human gene SLC2A9, the solute carrier family 2 (facilitated glucose transporter), member 9, is in the same situation. As explained in the introduction, the lack of such apparently simple extrapolations from one GO category to another is frequent in GO and represents a serious problem for the tools that perform an automatic functional profiling.
Probably, the most interesting prediction was beyond a simple extrapolation and involved the human gene aquaporin 1 (Colton blood group) (AQP1). This gene was already annotated in May 2003 for porin activity, transporter activity, and water transporter activity. Hence, it was known that AQP1 is involved in water transport, but AQP1 was not annotated in the GO database for the exact mechanism through which the gene achieves this function. Interestingly, n2tn predicted that AQP1 exhibits water channel activity, which is a very specific and very complex mechanism of water transport. Research proving that aquaporin indeed forms a channel for the water molecules was awarded the Nobel Prize for Chemistry in 2003 [29].
Overall, out of the top 50 functional annotations predicted using the best performing weighting scheme, n2tn, we found support in the literature for 84 percent of them. For 10 percent of our predictions, we did not find any relevant publications, and 6 percent were actually contradicted by existing literature.
4 Conclusion
Gene annotation databases represent an essential resource for modern research in life sciences. Such databases are used on a daily basis by thousands of researchers worldwide. However, it is well known that these annotations are incomplete, and it is likely that some annotations are also incorrect. In this paper, we presented a VSM/LSI approach that can be used in combination with any one of 15 weighting schemes studied here in order to perform a global semantic analysis of the contents of such databases. The technique described here was able to predict novel functional annotations for known human genes. This technique is independent of the organism and can be used to analyze the quality of the data in any public or private annotation database. In our experiments with the human annotations from GO, some the popular IR normalization schemes tested for the local weight and normalization factor actually deteriorated the accuracy. Nevertheless, these normalization schemes may still be useful for other data. On the other hand, our results show that the use of gene frequency, inverse annotation frequency, and depth penalty applied to local and global weights provides better results than the previously proposed binary approach [25].
Acknowledgments
This material is based upon work supported by NSF DBI-0234806, NIH(NCRR) 1S10 RR017857-01, MLSC MEDC-538 and MEDC GR-352, NIH 1R21 CA10074001, 1R21 EB00990-01, and 1R01 NS045207-01. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF, NIH, or any other of the funding agencies.
Biographies
 Bogdan Done is a PhD student at Wayne State University, Detroit, and a member of the Intelligent Systems and Bioinformatics Laboratory in the Department of Computer Science, Wayne State University. His current research interest are focused on applying computational intelligence methods in bioinformatics.
Bogdan Done is a PhD student at Wayne State University, Detroit, and a member of the Intelligent Systems and Bioinformatics Laboratory in the Department of Computer Science, Wayne State University. His current research interest are focused on applying computational intelligence methods in bioinformatics.
 Purvesh Khatri received the PhD degree in computer science from Wayne State University, Detroit. He is currently a postdoctoral fellow in the Intelligent Systems and Bioinformatics Laboratory, Department of Computer Science, Wayne State University. His research interests include bioinformatics, in particular genomics, and ontological analysis of high-throughput gene expression data. He has published 23 peer-reviewed papers. He has served as a reviewer for a number of journals, including Bioinformatics, Nucleic Acids Research, BMC Bioinformatics, and the IEEE/ACM Transactions on Computational Biology and Bioinformatics.
Purvesh Khatri received the PhD degree in computer science from Wayne State University, Detroit. He is currently a postdoctoral fellow in the Intelligent Systems and Bioinformatics Laboratory, Department of Computer Science, Wayne State University. His research interests include bioinformatics, in particular genomics, and ontological analysis of high-throughput gene expression data. He has published 23 peer-reviewed papers. He has served as a reviewer for a number of journals, including Bioinformatics, Nucleic Acids Research, BMC Bioinformatics, and the IEEE/ACM Transactions on Computational Biology and Bioinformatics.
 Arina Done received the Doctor of Medicine degree from Ovidius University, Constanţa, Romania, and the MA degree in biological sciences from Wayne State University, Detroit. She is currently a research associate in the Intelligent Systems and Bioinformatics Laboratory, Department of Computer Science, Wayne State University.
Arina Done received the Doctor of Medicine degree from Ovidius University, Constanţa, Romania, and the MA degree in biological sciences from Wayne State University, Detroit. She is currently a research associate in the Intelligent Systems and Bioinformatics Laboratory, Department of Computer Science, Wayne State University.
 Sorin Drăghici received the PhD degree in computer science from the University of St. Andrews, St. Andrews, United Kingdom. He is the director of the Bioinformatics Core at Karmanos Cancer Institute and an associate professor and the head of the Intelligent Systems and Bioinformatics Laboratory in the Department of Computer Science (http://vortex.cs.wayne.edu), Wayne State University, Detroit. His research interests include bioinformatics, machine learning, and image processing. He has published a best selling book on microarray data analysis entitled Data Analysis Tools for Microarrays (Chapman and Hall/CRC Press, 2003), seven book chapters, and more than 50 peer-reviewed journal and conference papers. He is a senior member of the IEEE.
Sorin Drăghici received the PhD degree in computer science from the University of St. Andrews, St. Andrews, United Kingdom. He is the director of the Bioinformatics Core at Karmanos Cancer Institute and an associate professor and the head of the Intelligent Systems and Bioinformatics Laboratory in the Department of Computer Science (http://vortex.cs.wayne.edu), Wayne State University, Detroit. His research interests include bioinformatics, machine learning, and image processing. He has published a best selling book on microarray data analysis entitled Data Analysis Tools for Microarrays (Chapman and Hall/CRC Press, 2003), seven book chapters, and more than 50 peer-reviewed journal and conference papers. He is a senior member of the IEEE.
Contributor Information
Bogdan Done, Email: bogdandone@wayne.edu.
Purvesh Khatri, Email: purvesh@cs.wayne.edu.
Arina Done, Email: arinadone@yahoo.com.
Sorin Draghici, Email: sod@cs.wayne.edu.
References
- 1.Abasolo JM, Gomez M. MELISA: An Ontology-Based Agent for Information Retrieval in Medicine; Proc First Int’l Workshop on the Semantic Web; 2000. [Google Scholar]
- 2.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: A Web Tool for Finding Significant Associations of Gene Ontology Terms with Groups of Genes. Bioinformatics. 2004;20(4):578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 3.Alter O, Brown PO, Botstein D. Singular Value Decomposition for Genome-Wide Expression Data Processing and Modeling. Proc Nat’l Academy of Sciences USA. 2000;97(18):10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beissbarth T, Speed TP. GOstat: Find Statistically Over-Represented Gene Ontologies within a Group of Genes. Bioinformatics. 2004 Jun;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 5.Berry MW, Drmac Z, Jessup ER. Matrices, Vector Spaces, and Information Retrieval. SIAM Rev. 1999;41(2):335–362. [Google Scholar]
- 6.Berry MW, Dumais ST, O’Brien GW. Using Linear Algebra for Intelligent Information Retrieval. SIAM Rev. 1995;37(4):573–595. [Google Scholar]
- 7.Bodenreider O, Aubry M, Burgun A. Evaluation of the Vector Space Representation in Text-Based Gene Clustering. Proc Pacific Symp Biocomputing (PSB ’05) 2005:91–102. [Google Scholar]
- 8.Brown MPS, Boble Grundy W, Lin D, Cristianini N, Waish Sugnet C, Furgey TS, Ares Manuel M, Haussler D. Knowledge-Based Analysis of Microarray Gene Expression Data by Using Support Vector Machines. Proc Nat’l Academy of Sciences USA. 2000;97(1):262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deerwester S, Dumais ST, Furnas GW, Landauer TK, Harshman R. Indexing by Latent Semantic Analysis. J Am Soc for Information Science. 1990;41(6):391–407. [Google Scholar]
- 10.Devos D, Valencia A. Practical Limits of Function Prediction. PROTEINS: Structure, Function, and Genetics. 2000;41:98–107. [PubMed] [Google Scholar]
- 11.Drăghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, Tainsky MA. Onto-Tools, the Toolkit of the Modern Biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Research. 2003 Jul;31(13):3775–3781. doi: 10.1093/nar/gkg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drăghici S, Khatri P, Martins RP, Charles Ostermeier G, Krawetz SA. Global Functional Profiling of Gene Expression. Genomics. 2003 Feb;81(2):98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- 13.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster Analysis and Display of Genome-Wide Expression Patterns. Proc Nat’l Academy of Sciences USA. 1998 Dec;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fetrow JS, Siew N, Di Gennaro JA, Martinez-Yamout M, Dyson HJ, Skolnick J. Genomic-Scale Comparison of Sequence- and Structure-Based Methods of Function Prediction: Does Structure Provide Additional Insight? Protein Science. 2001;10:1005–1014. doi: 10.1110/ps.49201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geller SC, Gregg JP, Hagerman P, Rocke DM. Transformation and Normalization of Oligonucleotide Microarray Data. Bioinformatics. 2003;19:1817–1823. doi: 10.1093/bioinformatics/btg245. [DOI] [PubMed] [Google Scholar]
- 16.Gerard S. Introduction to Modern Information Retrieval. McGraw-Hill; 1983. [Google Scholar]
- 17.Glenisson P, Antal P, Mathys J, Moreau Y, De Moor B. Evaluation of the Vector Space Representation in Text-Based Gene Clustering. Proc Pacific Symp Biocomputing (PSB ’03) 2003:391–402. doi: 10.1142/9789812776303_0037. [DOI] [PubMed] [Google Scholar]
- 18.Golub G, van Loan CF. Matrix Computations. Johns Hopkins Univ. Press; 1983. [Google Scholar]
- 19.Herrero J, Diaz-Uriarte R, Dopazo J. Gene Expression Data Processing. Bioinformatics. 2003;19:655–656. doi: 10.1093/bioinformatics/btg040. [DOI] [PubMed] [Google Scholar]
- 20.Homayouni R, Heinrich K, Wei L, Berry MW. Gene Clustering by Latent Semantic Indexing of MEDLINE Abstracts. Bioinformatics. 2005;21(1):104–115. doi: 10.1093/bioinformatics/bth464. [DOI] [PubMed] [Google Scholar]
- 21.Hosack DA, Dennis G, Jr, Sherman BT, Clifford Lane H, Lempicki RA. Identifying Biological Themes within Lists of Genes with EASE. Genome Biology. 2003;4(6):P4. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hvidsten TR, Sandvik AK, Laegreid A, Komorowski J. Predictive Gene Function from Gene Expressions and Ontologies. Proc Pacific Symp Biocomputing (PSB) 2001 doi: 10.1142/9789814447362_0030. [DOI] [PubMed] [Google Scholar]
- 23.Karp PD. What We Do Not Know About Sequence Analysis and Sequence Databases. Bioinformatics. 1998;14(9):753–754. doi: 10.1093/bioinformatics/14.9.753. [DOI] [PubMed] [Google Scholar]
- 24.Khatri P, Bhavsar P, Bawa G, Drăghici S. Onto-Tools: An Ensemble of Web-Accessible, Ontology-Based Tools for the Functional Design and Interpretation of High-Throughput Gene Expression Experiments. Nucleic Acids Research. 2004 Jul;32:W449–W456. doi: 10.1093/nar/gkh409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khatri P, Done B, Rao A, Done A, Draghici S. A Semantic Analysis of the Annotations of the Human Genome. Bioinformatics. 2005;21(16):3416–3421. doi: 10.1093/bioinformatics/bti538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khatri P, Draghici S. Ontological Analysis of Gene Expression Data: Current Tools, Limitations, and Open Problems. Bioinformatics. 2005;21(18):3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khatri P, Drăghici S, Charles Ostermeier G, Krawetz SA. Profiling Gene Expression Using Onto-Express. Genomics. 2002 Feb;79(2):266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 28.King OD, Foulger RE, Dwight SS, White JV, Roth FP. Predicting Gene Function from Patterns of Annotation. Genome Research. 2003;13:896–904. doi: 10.1101/gr.440803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozono D, Yasui M, King LS, Agre P. Aquaporin Water Channels: Atomic Structure Molecular Dynamics Meet Clinical Medicine. J Clinical Investigation. 2002;109(11):1395–1399. doi: 10.1172/JCI15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller HM, Kenny EE, Sternberg PW. Textpresso: An Ontology-Based Information Retrieval and Extraction System for Biological Literature. PloS Biology. 2004;2(11):1984–1998. doi: 10.1371/journal.pbio.0020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan W, Lin J, Le C. Model-Based Cluster Analysis of Microarray Gene Expression Data. Genome Biology. 2002;3(2):research0009.1–research0009.8. doi: 10.1186/gb-2002-3-2-research0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popov B, Kiryakov A, Ognyanoff D, Manov D, Kirilov A. Kim—A Semantic Platform for Information Extraction and Retrieval. Natural Language Eng. 2004;10(3–4):375–392. [Google Scholar]
- 33.Quackenbush J. Microarrays—Guilt by Association. Science. 2003;302(5643):240–241. doi: 10.1126/science.1090887. [DOI] [PubMed] [Google Scholar]
- 34.Raychaudhuri S, Chang JT, Sutphin PD, Altman RB. Associating Genes with Gene Ontology Codes Using a Maximum Entropy Analysis of Biomedical Literature. Genome Research. 2002;12:203–214. doi: 10.1101/gr.199701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson J. Vlad: A New GO Tool for Visual Annotation Display. 2007 http://www.informatics.jax.org/~jer/vlad/
- 36.Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, David Haynes J, De la Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA. Discovery of Gene Function by Expression Profiling of the Malaria Parasite Life Cycle. Science. 2003;301(5639):1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- 37.Schug J, Diskin S, Mazzarelli J, Brunk BP, Stoeckert CJ., Jr Predicting Gene Ontology Functions from PromDom and CDD Protein Domains. Genome Research. 2002;12:648–655. doi: 10.1101/gr.222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skolnick J, Fetrow JS. From Genes to Protein Structure and Function: Novel Applications of Computational Approaches in the Genomic Era. Trends in Biotechnology. 2000;18:283–287. doi: 10.1016/s0167-7799(99)01398-0. [DOI] [PubMed] [Google Scholar]
- 39.Vallet D, Fernandez M, Castells P. An Ontology-Based Information Retrieval Model. Proc Second European Semantic Web Conf (ESWC ’05) 2005:455–470. [Google Scholar]
- 40.Walker MG, Volkmuth W, Sprinzak E, Hodgson D, Klingler T. Prediction of Gene Function by Genome-Scale Expression Analysis: Prostate Cancer-Associated Genes. Genome Research. 1999;9(12):1198–1203. doi: 10.1101/gr.9.12.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler SJ. Large-Scale Prediction of Saccharomyces Cerevisiae Gene Function Using Overlapping Transcriptional Clusters. Nature Genetics. 2002 Jul;31(3):255–265. doi: 10.1038/ng906. [DOI] [PubMed] [Google Scholar]
- 42.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Carl Barrett J, Weinstein JN. GoMiner: A Resource for Biological Interpretation of Genomic and Proteomic Data. Genome Biology. 2003 Mar;4(4):R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Schmoyer D, Kirvo S, Snoddy J. GOTree Machine (GOTM): A Web-Based Platform for Interpreting Sets of Interesting Genes Using Gene Ontology Hierarchies. BMC Bioinformatics. 2004 Feb;5:article 16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou GH, Wen XY, Liu H, Schlicht MJ, Hessner MJ, Tonellato PJ, Datta MW. BEAR GeneInfo: A Tool for Identifying Gene-Related Biomedical Publications through User Modifiable Queries. BMC Bioinformatics. 2004 Apr;5:article 46. doi: 10.1186/1471-2105-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]