

Abstract
Early-onset generalized torsion dystonia, also known as DYT1 dystonia, is a childhood onset heritable neurological movement disorder involving painful, involuntary muscle contractions, sustained abnormal postures, and repetitive movements. It is caused by a GAG deletion in the Tor1A gene located on chromosome 9. TorsinA, the product of the Tor1A gene, is expressed throughout the brain beginning early in embryonic development. It plays a role in the regulation of nuclear envelope-cytoskeletal interactions, and presumably nuclear translocation. Since nuclear translocation, powered by cytoskeletal traction, is critical for cell proliferation and migration, we examined whether neurogenesis and neuronal migration are affected in Tor1A/−/− mouse brain. Our data show that interkinetic nuclear migration and the pattern of migration of newly generated neurons are impaired in the dorsal forebrain of the Tor1A/−/− embryo. However, neurogenesis is not altered significantly. The rate of migration of cells from explants of the medial ganglionic eminence is also impaired in the Tor1A−/− embryo. Thus, loss of torsinA results in subtle but significant alterations in cell proliferation and migration in the embryonic forebrain. These subtle developmental changes are consistent with a lack of significant changes in neuronal numbers, neuronal positioning or size of brain regions in DYT1 dystonia patients.
Keywords: Dystonia, DYT1, Medial ganglionic eminence, Nkx2-1
Introduction
Early-onset generalized torsion dystonia is a heritable neurological movement disorder caused by a three base-pair GAG deletion (delGAG) in the Tor1A gene [1, 2]. The mutation exhibits relatively low penetrance, with only about 30–40% of the individuals exhibiting the symptoms. This type of dystonia is also known as DYT1 dystonia [1, 2]. Generally, the disease onset is before the age of 20 years, and individuals who carry the mutation but do not develop symptoms by the age of 30 years tend to escape the illness completely [3–5]. These intriguing characteristics of the disease pose significant challenges for defining the neurobiological mechanisms underlying DYT1 dystonia. Post mortem examination of brains of DYT1 dystonia patients does not reveal significant neuronal loss, neuronal malpositioning, or gross abnormalities in gray or white matter in any brain region. However, enlargement of neuronal somata in the substantia nigra pars compacta [6] as well as altered microstructure or fractional anisotropy of the white matter [7] was reported in individuals carrying the DYT1 genotype with or without dystonic symptoms. Mouse models with deletion of the entire Tor1A gene, or knockin of the human or mouse delGAG mutation do not display overt signs of dystonia [8–16]. However, electrophysiological and neurochemical analyses in some of the mouse models reveal changes in dopamine release, dopamine D2 receptor signaling and impaired synaptic plasticity in the basal ganglia [8, 17–22].
Functional brain imaging studies of human subjects show that asymptomatic carriers of the delGAG mutation display abnormal motor function, implicating underlying functional impairment, which is suggested to be of developmental origin [23–26]. Other lines of evidence also support a developmental origin for DYT1 dystonia. For example, torsinA, the product of the Tor1A gene is expressed throughout the brain starting early in embryonic development in mice and humans [27–29]. TorsinA expression in the mouse forebrain peaks during the prenatal and early postnatal periods when neurogenesis and neuronal migration are robustly underway [27]. Moreover, torsinA is believed to play a role in cytoskeletal dynamics and in anchoring the nuclear envelope to the cytoskeleton [30]. Since the cytoskeleton plays critical roles in nuclear and cellular translocation during neurogenesis and neuronal migration, we hypothesized a developmental role for torsinA in these two processes. We tested our hypothesis using a Tor1A knockout mouse model [10]. Our data show that the loss of Tor1A produces subtle but significant changes in both interkinetic nuclear migration in the precursor cells and migration of newborn neurons in the embryonic telencephalon. These data suggest a role for torsinA in brain development and offer a neurobiological basis for the developmental origins of DYT1 dystonia.
Materials and Methods
Animals
Heterozygous Tor1A knockout mice (Tor1A+/−) [10] were received from Dr. Yuqing Li (University of Florida, Gainesville, Fla., USA). We crossed Tor1A+/− mice to generate timed pregnancies. The day of vaginal plug detection was considered embryonic day 0 (E0). The pregnant dams were housed individually, in a temperature- and humidity-controlled environment, on a 12-hour light/dark cycle with food and water available ad libitum. All of the experimental procedures were in full compliance with institutional guidelines at the Massachusetts General Hospital and the NIH Guide for the Care and Use of Laboratory Animals.
Bromodeoxyuridine Administration
For studies of cell proliferation and neurogenesis, we used the S-phase marker bromodeoxyuridine (BrdU; Sigma Chemical Company, St. Louis, Mo., USA). BrdU was injected intraperitone-ally at a dose of 50 mg/kg body weight.
Embryo Collection and Processing
The studies described here were performed on embryonic mice because Tor1A−/− mice die around the time of birth [10], and experiments on postnatal Tor1A−/− mice are not feasible. For collection of embryos, the dams were anesthetized (ketamine, 50 mg/kg body weight and xylazine, 10 mg/kg body weight, i.p.) and the embryos were removed by hysterotomy. The age of each embryo was ascertained by examination of the external morphological features [31, 32]. Embryos (or entire litters) that did not fulfill the criteria for a given age were discarded from analysis. The embryos were decapitated immediately upon removal from the dam and embryonic heads or brains were fixed by immersion in 4% paraformaldehyde. Following at least 24 h in the fixative solution at 4°C, the tissue samples were cryoprotected in 20% sucrose and frozen using powdered dry ice [33]. The samples were stored at −80°C until further processing.
Cryosectioning and Immunohistochemistry
The paraformaldehyde-fixed, frozen embryonic heads or brains were cut in the coronal plane on a cryostat at 20 μm thickness. The sections were mounted on Superfrost+ slides (Fisher) and the slides were stored at −80°C. For immunohistochemistry, the slides were thawed and an antigen retrieval step was performed by microwaving the slides in warm antigen retrieval buffer (Retrievagen, BD Pharmingen, San Jose, Calif., USA). Non-specific binding was blocked with 10% normal donkey serum, containing 0.3% Triton X-100 (to improve permeability) for 1 h at room temperature. Sections were then incubated overnight at 4°C with the following primary antibodies at the dilutions indicated: BrdU (rat monoclonal; Abcam-ab6326), Tbr1, Tbr2, Ki67 (rabbit polyclonals; Abcam ab31940, ab23345 and ab15580, respectively; dilutions for all 1:200), and phosphohistone H3 (pH3; rabbit polyclonal, Millipore 06–570, 1:1,000). Sections were washed with PBS and incubated at room temperature for 1 h with anti-rat Cy3 or anti-rabbit Alexa Fluor 488 (1:200 Jackson Immunoresearch, West Grove, Pa., USA; 1:200 Invitrogen, Carlsbad, Calif., USA, respectively). Following additional washes with PBS, all slides were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, Calif., USA).
Microscopic Analysis
Following immunohistochemistry, the sections were examined using a Zeiss PASCAL confocal microscope. Uniform penetration of the antibodies throughout the section was ascertained by splitting the image of the 20-μm-thick cryostat section into 2.5-μm-thick digital sections and examining the quality of the immunofluorescence in each digital section. Only those cryostat sections in which each of the 2.5-μm-thick sections showed uniform labeling were included in further analysis.
Counting Labeled Cells
We counted immunohistochemically labeled cells in sections of the dorsal cerebral wall (DCW; future frontal cortex) and the medial ganglionic eminence (MGE) of the basal forebrain for analysis of cell proliferation and migration. From each 20-μm-thick section of the DCW or MGE, two 2.5-μm-thick digital sections were randomly chosen for analysis. A two-dimensional grid of known size was superimposed on the images. The grid was further subdivided into rectangular bins [33]. Labeled cells were counted and the numerical density of labeled cells was expressed as numbers per square micrometer.
Analysis of Interkinetic Nuclear Migration
We used pH3 immunohistochemistry to identify nuclei in M-phase of the cell cycle and analyze interkinetic nuclear migration in radial precursor cells located in the ventricular zone [34, 35]. A characteristic feature of precursor cells organized as pseudostratified epithelia, such as the ventricular neuroepithelium of the embryonic forebrain, is the to-and-fro movement of the nuclei between the apical and basal poles of the cell in step with the stages of the cell cycle [36–38]. During this process, the precursor cell per se remains stationary: it does not migrate. However, the nucleus migrates or translocates within the precursor cell, such that it is near the basal pole, farthest from the apical surface of the cell (which is at the lateral ventricular border) during S-phase and at the basal pole during M-phase. During G1- and G2-phases, the nucleus assumes intermediate positions. Nuclei in M-phase of the cell cycle express pH3 [39]. Therefore, pH3 immunohistochemistry labels nuclei that lie at the apical pole of the cells near the ventricular border. To determine whether the interkinetic nuclear migration was impaired in the forebrain of the Tor1A−/− embryos, we analyzed the proportion of pH3+ (M-phase) nuclei located at or near the lateral ventricular border (expected or normal location of M-phase nuclei) versus further away from the lateral ventricular border (and toward the basal pole of the precursor cell).
Analysis of Cell Output
We analyzed cell output by using a BrdU−Ki67 double-labeling paradigm [40, 41]. In this paradigm, cells in S-phase are labeled with a single administration of the thymidine analog BrdU to the pregnant dam. Following a 24-hour survival period, the embryos are removed and histological sections of the embryonic brain are processed for double immunohistochemistry to reveal the BrdU and an endogenous marker of proliferating cells: Ki67. The latter labels cells in all phases of the cell cycle (S-, G2-, M-, and G1-phases). In this paradigm, cells labeled only with BrdU (BrdU+Ki67− cells) represent cells that were in S-phase during a 3-hour period following the BrdU injection (24 h earlier) and that had left the cell cycle to become postmitotic (ceased cell division) by the time the embryo was sacrificed. In other words, the BrdU+Ki67− cells represent the cell output from the precursor cell pool and provide us with an index of neurogenesis. We injected BrdU on E14 and the embryos were collected for immunohistochemical labeling for BrdU and Ki67 on E15. We counted the numbers of BrdU+Ki67− cells in a 100 × 350 μm2 sector of the neuroepithelium of the DCW and the MGE [41].
Analysis of Neuronal Migration in MGE Explants
We collected embryos at E13 and E15, as described above. The embryonic brains were removed and placed in 8% agarose (type VII, Sigma). Coronal sections of the brains were cut at a thickness of 250 μm on a Vibratome. A sterile biopsy punch (Miltex Inc., York, Pa., USA) was used to collect disks of MGE from the Vibratome sections [33]. The MGE is a component of the ganglionic eminence, a transient embryonic neuroepithelial structure surrounding the lateral ventricles in the basal forebrain. The other components of the ganglionic eminence are the lateral and caudal ganglionic eminences. The majority of the neurons produced in the MGE are migratory neurons destined to become cortical GABA neurons [42–46]. On the other hand, neurons produced in the lateral ganglionic eminence are a mixed population: some destined to the olfactory bulb, some to the striatum and the others to the cortex [44, 47]. Thus, MGE explants provide us with a relatively homogeneous population of cells, which aids interpretation of the data. The caudal ganglionic eminence is too small a structure for the type of explant assays used here, at least in our hands. Finally, we selected the MGE for analysis because MGE cells express Nkx2-1, a transcription factor that we have examined in this study. For all these reasons, we believe that the MGE was the appropriate choice for the experiments described here.
The tissue disks obtained from the Vibratome sections were cultured in Matrigel (BD Biosciences) and neurobasal medium containing 2% B-27 supplement, penicillin, streptomycin and glutamine. Following 24 h in culture, the explants were fixed with 4% paraformaldehyde for 2 h, washed with PBS and examined under a microscope using phase-contrast optics. An ocular grid (200 × 400 μm2) was superimposed on the microscopic image of the explant and the migrating cells). The grid was further subdivided into 200 × 20 μm bins. For each explant, cells in 2 such grids were counted. The position of the first grid was determined at random while the second grid was always positioned 180° from the first. For each genotype, we used 4 or 5 explants taken from 3 separate litters.
In additional experiments, some of the explants were processed for tubulin immunohistochemistry to reveal the processes of the migrating cells (β-III tubulin, mouse monoclonal, Millipore MAB55441, 1:1,000). Nuclei were labeled with ToPro3 (Invitrogen T3605, 1:1,000).
Real-Time Quantitative PCR
We used laser capture microdissection (LCM) to collect samples of postmitotic cells from the basal forebrain (striatal differentiating fields) of cryostat sections of E15 brains, as described previously [48]. Briefly, the frozen heads of the embryos were sectioned at 12 μm thickness in the coronal plane on a cryostat and mounted on RNase-free, MembraneSlides® (Molecular Machines and Industries, Glattbrugg, Switzerland), dried at room temperature and stored at −80°C. The sections were viewed under a Nikon TE2000 inverted microscope connected to CellCut® LCM equipment (Molecular Machines and Industries). The striatal differentiating fields were identified based on anatomical location and dissected (UVCut software package; Molecular Machines and Industries). Immediately upon dissection, the sample was picked up on adhesive lids of 500-μl mini IsolationCaps® (Molecular Machines and Industries). Samples were collected from both hemispheres and pooled. We used 2 or 3 embryos from each genotype. Because the amount of tissue available in these samples is very small, we used a RNAqueous-Micro kit to extract RNA. We converted 2 μg RNA to cDNA by RT reaction using SuperScript III and random hexamer primers (Invitrogen). We analyzed expression of Nkx2-1 (Mm004747558-m1) and 18s rRNA (Life Technologies 4333760T) using proprietary primers and fluorescent probes designed by Applied Biosystems. TaqMan Universal master PCR mix was used for all PCRs (Applied Biosystems). RT PCR was performed using Prism 7500 (Applied Biosystems) and the following thermal settings: 1 cycle of 2 min at 50°C, 1 cycle of 10 min at 95°C, 40 cycle of 15 s at 95°C and 1 min at 60°C. Expression of Nkx2-1 mRNA was normalized to endogenous 18s mRNA control using the 2ΔΔCt method [49] and analyzed using Q-gene software [50].
Nkx2-1 is expressed in MGE precursor cells as well as MGE-derived postmitotic cells that accumulate in the striatal differentiating fields [51–54]. We used LCM for collecting samples of striatal differentiating fields (as opposed to using free-hand micro-dissection) to permit analysis of Nkx2-1 mRNA only in postmitotic cells. The need for distinction between postmitotic and proliferating cells arises because the Nkx2-1 expression in the postmitotic cells (and not precursor cells) influences migration of MGE-derived neurons [52], which is the focus of analyses in our study. Free-hand micro-dissection will not permit reliable collection of exclusively the postmitotic region of the basal forebrain. The striatal differentiating fields of the E15 mouse contain predominantly young neurons. The radial glial cell bodies are essentially restricted to the ganglionic eminences [55–57]. Therefore, we did not use cell type-specific stains to discriminate between neurons and glial cells.
Statistical Analysis
Differences between genotype were analyzed for statistical significance by using the two-tailed Student t test.
Results
Cell Proliferation and Output
We analyzed cell proliferation by examining the interkinetic nuclear migration using pH3 immunohistochemistry and cell output using the BrdU−Ki67 double-labeling paradigm. The numerical density of pH3+ profiles was not significantly different in the E15 DCW or the E15 MGE between Tor1A+/+ and Tor1A−/− mice (fig. 1, DCW: mean ± SEM; Tor1A+/+ 0.00088 ± 0.00011; Tor1A−/− 0.00060 ± 0.00009; t = 1.915, d.f. = 6, p > 0.05; MGE: mean ± SEM; Tor1A+/+ 0.00053 ± 0.00004; Tor1A−/− 0.00073 ± 0.00012; t = 1.611, d.f. = 6, p > 0.05), suggesting that the loss of Tor1A did not significantly affect cell proliferation in either forebrain region. However, the numerical density of pH3+ profiles near the ventricular border in the DCW of the E15 Tor1A−/− mice was significantly lower compared to that in the Tor1A+/+ mice (fig. 1d, t = 5.292, d.f. = 2, p > 0.05; fig. 1e, mean ± SEM; Tor1A+/+ 0.00078 ± 0.00009; Tor1A−/− 0.00044 ± 0.00004; t = 3.189, d.f. = 6, p < 0.05). In contrast, there was no significant difference in the numerical density of pH3+ profiles located near the ventricular border in the MGE between the two genotypes (data not shown, t = 0.2182, d.f. = 2, p > 0.05; mean ± SEM; Tor1A+/+ 0.00031 ± 0.00001; Tor1A−/− 0.00035 ± 0.00006; t = 0.6, d.f. = 6, p > 0.05).
Fig. 1.
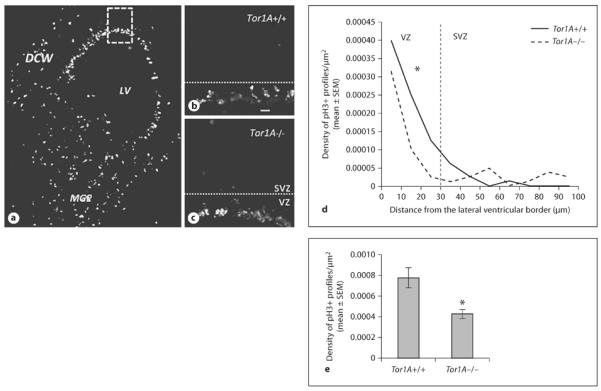
Photomicrographs of pH3-labeled profiles in coronal sections of E15 Tor1A+/+ and Tor1A−/− mice. a Low-magnification image from an E15 Tor1A+/+ forebrain, showing pH3+ profiles in the DCW and the MGE encircling the LV. The boxed area in a outlines the presumptive prefrontal cortex, and is shown at higher power for representative Tor1A+/+ (b) and Tor1A−/− (c) sections. The dotted lines in b and c represent the approximate border between the VZ and SVZ. The numerical density of pH3+ profiles was calculated at 10-μm intervals between the ventricular border and the pial surface (d). The numerical density in the VZ of the DCW was lower in Tor1A−/− mice compared to Tor1A+/+ littermates (e). * p < 0.05, error bars indicate SEM.
Analysis of cell output using the BrdU−Ki67 double-labeling paradigm did not reveal significant differences in the numerical density of BrdU+Ki67− profiles in the E15 DCW or MGE between Tor1A+/+ and Tor1A−/− mice (fig. 2a–d, DCW: mean ± SEM; Tor1A+/+ 0.0022 ± 0.0003; Tor1A−/− 0.0025 ± 0.0002; t = 0.9137, d.f. = 6, p > 0.05; MGE: mean ± SEM; Tor1A+/+ 0.0014 ± 0.0001; Tor1A−/− 0.0015 ± 0.0001; t = 0.7732, d.f. = 8, p > 0.05). In additional studies, we compared the numerical density of Tbr1+ and Tbr2+ cells in the DCW of E15 Tor1A+/+ and Tor1A−/− mice (fig. 3a–d). Tbr1 is expressed in newly generated cortical excitatory neurons whereas Tbr2 is expressed in progenitor cells of the dorsal cerebral wall [58, 59]. Neither Tbr1 nor Tbr2 expression showed significant differences between Tor1A+/+ and Tor1A−/− mice (fig. 3e,f, Tbr2+: mean ± SEM; Tor1A+/+ 0.0065 ± 0.0005; Tor1A−/− 0.0068 ± 0.0006; t = 0.4247, d.f. = 9, p > 0.05; Tbr1+: mean ± SEM; Tor1A+/+ 0.0048 ± 0.0001; Tor1A−/− 0.0047 ± 0.0005; t = 0.2643, d.f. = 4, p > 0.05).
Fig. 2.

Quantification of cell output from the DCW (a, b (c, d) of E15 Tor1A+/+ and Tor1A−/− mice using the BrdU-Ki67 double-labeling method. BrdU+Ki67− cells (red) are postmitotic and represent the cell output fraction. BrdU−Ki67+ cells (green) and BrdU+Ki67+ cells (orange/yellow) are proliferating precursor cells. Number of postmitotic cells (red, black in the printed version) per bin as a percentage of total is plotted against the distance from the ventricular border at 10-μm intervals in the DCW and MGE (e). Arrows in e point to the two peaks in the distribution of postmitotic cells in the Tor1A+/+ embryo. The two peaks are not apparent in the distribution of postmitotic cells in the Tor1A−/− embryo. Numerical density (per 1,000 μm2) of postmitotic cells within the different zones of the cerebral wall is shown for DCW and MGE (f). The broken lines in e represent approximate boundaries between VZ and SVZ, and SVZ and CP. * p < 0.05, error bars indicate SEM. Colors refer to the online version only.
Fig. 3.
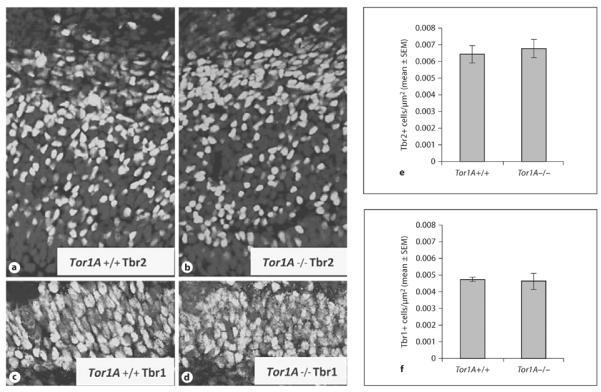
Immunohistochemistry was performed to identify Tbr2+ intermediate progenitor cells (a, b) and Tbr1+ newborn postmitotic cells (c, d) within the presumptive prefrontal cortex of E15 Tor1A+/+ and Tor1A−/− mice. Numerical density of Tbr2+ (e) and Tbr1+ (f) cells was compared between Tor1A+/+ and Tor1A−/− embryos. Error bars indicate SEM.
When we compared the distribution of BrdU+Ki67− cells within the ventricular zone (VZ), subventricular zone (SVZ) and cortical plate (CP) of the E15 DCW, we found a significant increase in the density of cells in the SVZ of the Tor1A−/− mice compared to the Tor1A+/+ mice (fig. 2f, SVZ: mean ± SEM; Tor1A+/+ 0.00143 ± 0.00010; Tor1A−/− 0.00172 ± 0.00003; t = 2.665, d.f. = 6, p < 0.05). Neither the VZ nor the CP showed significant differences (fig. 2f, VZ: mean ± SEM; Tor1A+/+ 0.00011 ± 0.00003; Tor1A−/− 0.00019 ± 0.00005; t = 1.360, d.f. = 6, p > 0.05; CP: Tor1A+/+ 0.00068 ± 0.00022; Tor1A−/− 0.00062 ± 0.00026; t = 0.1691, d.f. = 6, p > 0.05). The bimodal distribution of the BrdU+Ki67− cells, which is characteristic of cell output from the DCW [60, 61], was evident in the Tor1A+/+ but not the Tor1A−/− cerebral wall (fig. 2e, arrows). However, the distribution of BrdU+Ki67− cells within the VZ or SVZ of the E15 MGE did not reveal significant differences between Tor1A−/− and Tor1A+/+ mice (fig. 2e,f, VZ: mean 8 SEM; Tor1A+/+ 0.00025 ± 0.00004; Tor1A−/− 0.00018 ± 0.00003; t = 1.476, d.f. = 8, p > 0.05; SVZ: mean ± SEM; Tor1A+/+ 0.0011 ± 0.0001; Tor1A−/− 0.0013 ± 0.0001; t = 1.476, d.f. = 8, p > 0.05).
Neuronal Migration
In explants of the MGE cultured in Matrigel for 24 h, we did not find significant differences between E13 or E15 Tor1A+/+ and Tor1A−/− mice in the numerical density of cells migrating away from the explant (E13: mean ± SEM; Tor1A+/+ 0.019 ± 0.001; Tor1A−/− 0.0019 ± 0.003; t = 0.1678, d.f. = 8, p > 0.05; E15: mean ± SEM; Tor1A+/+ 0.0013 ± 0.0000; Tor1A−/− 0.0013 ± 0.0001; t = 0.3129, d.f. = 8, p > 0.05). However, when we compared the distribution of cells as a function of distance from the edge of the explant, there were significantly fewer cells in each of the 10 bins closest to explant edge (a total distance of 200 μm) in the Tor1A−/− mice compared to the Tor1A+/+ mice (fig. 4e, E13: t = 4.686, d.f. = 9, p < 0.01; E15: t = 4.601, d.f. = 9, p < 0.01).
Fig. 4.
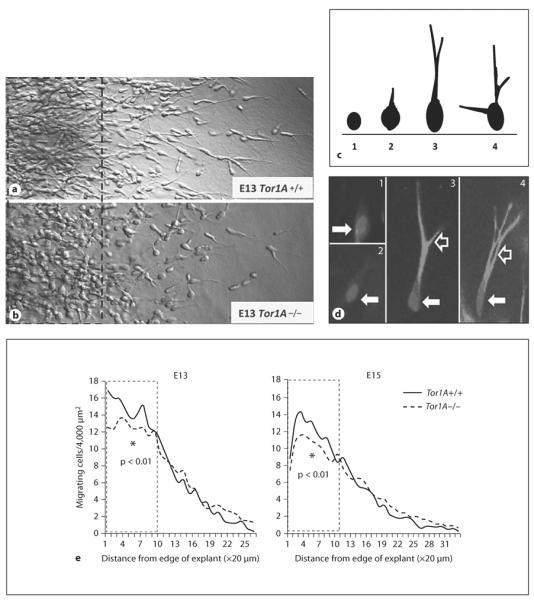
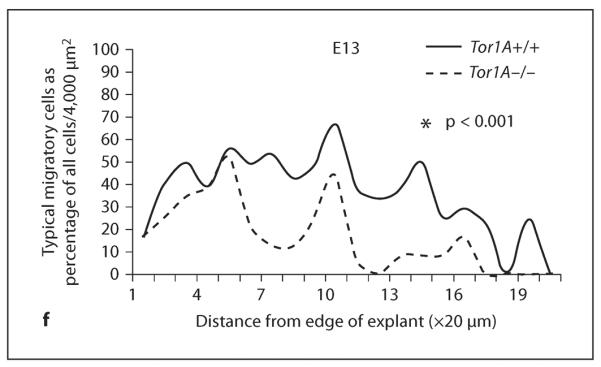
Cell migration in explants of the medial ganglionic eminence of the basal forebrain obtained from E13 and E15 Tor1A+/+ and Tor1A−/− embryos. Explants were cultured in Matrigel for about 24 h. Cells have migrated away from the edge of the explant (on the left of the images) taken from Tor1A+/+ (a) and Tor1A−/− (b) embryos. Broken lines in a and b represent 200 μm distance from the explant edge. c A diagrammatic illustration of the diverse morphologies of migrating cells. The morphologies ranged from rounded cells without a process (1) to cells with elongated cell bodies and multiple well-defined leading processes (4). Intermediate morphologies (2, 3) were also observed. For analysis of migrating cell morphology, cells with morphologies represented in 2, 3 and 4 were considered cells with typical migratory cell morphology, whereas cells with the morphology shown in 1 were considered cells without the typical morphology. Examples of the different cell types are shown in d, where 1 and 2 show cells with morphology depicted in c1, and 3 and 4 show cells with morphologies depicted in c3 and c4. The solid white arrows in d point to cell bodies and the open white arrows point to processes. e The number of migrating cells and (cells with morphological features shown in d2, 3 and 4) as a percentage of all cells (i.e. cell types shown in d1–4) were plotted as a function of distance from the edge of the explant (in 20 μm increments). There are fewer migrating cells within 200 μm from the explant edge in the Tor1A−/− explants compared to Tor1A+/+ explants at E13 and E15 (e). The distribution of cells with typical migrating cell morphology as a percentage of all cells was significantly different between E13 Tor1A−/− and Tor1A+/+ explants. * p < 0.01 Tor1A+/+. f The percentage of cells with migrating cell morphologies.
Since many of the migrating cells in the Tor1A−/− explants appeared to be rounded in morphology compared to the cells in the Tor1A+/+ explants, which appeared to have a typical migrating neuron morphology (elongated cell body and at least one leading process; fig. 4c, d), we quantified the relative proportions of cells with rounded versus typical migrating cell morphology in the E13 explants. The rationale for this analysis was twofold. First, there is the possibility that cells with and without the typical migratory morphology may migrate at different rates of speed. Second, the different morphologies may indicate differences in the ability of the cells to reorganize the cytoskeleton to achieve a morphology that is optimal for initiation and/or continuation of normal migration. We expressed the number of cells with typical migrating neuron morphology as a percentage of total number of cells exiting the explant (fig. 4f). The data were collected separately for each bin. When we compared the distribution of the cells as a function of distance from the edge of the explant, there was a statistically significant difference in the distributions between the Tor1A+/+ and Tor1A−/− explants (fig. 4f, t = 6.359, d.f. = 19, p < 0.001), with the Tor1A−/− explants showing smaller percentages of cells with migrating neuron morphology in virtually every bin through the entire migration field.
Expression of Nkx2-1
As mentioned previously, Nkx2-1 is a transcription factor expressed in proliferating and postmitotic cells of the MGE. Upregulation of Nkx2-1 in postmitotic cells is associated with decreased ability to migrate away from the basal forebrain and into the dorsal forebrain [52]. Therefore, we analyzed Nkx2-1 mRNA expression in the striatal differentiating fields, which contain postmitot -ic cells derived from the MGE in E15 Tor1A+/+ and Tor1A−/− mice. There was a significant increase in the expression of Nkx2-1 mRNA in the striatal differentiating fields of the Tor1A−/− mice compared to the Tor1A+/+ littermates (fig. 5, mean ± SEM; Tor1A−/− 5.1 × 10−7 ± 4.7 × 10−7; Tor1A+/+ 3.6 × 10−6 ± 4.4 × 10−6; t = 4.809, d.f. = 4, p < 0.01).
Fig. 5.
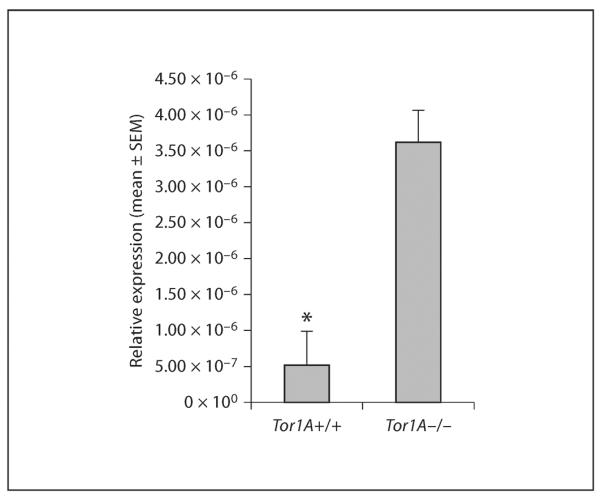
Analysis of Nkx2-1 mRNA expression in postmitotic cells of the E15 striatal differentiating fields isolated using laser capture microdissection. Nkx2-1 mRNA expression was normalized to expression of the endogenous 18s mRNA. The Nkx2-1 mRNA expression is significantly increased in Tor1A−/− embryos compared to Tor1A+/+ embryos. * p < 0.01.
Discussion
We used a Tor1A−/− mouse model to study the role of torsinA in neurogenesis and neuronal migration in the embryonic forebrain. Our in vivo data show that subtle but significant changes occur in the interkinetic nuclear migration of precursor cells and the pattern of exit of postmitotic cells from the neuroepithelium of the DCW in Tor1A−/− mouse embryos (fig 1, 2). Cell output or neurogenesis was not significantly affected either in the DCW or the MGE. Our in vitro studies showed significant changes in MGE cell migration in Tor1A−/− mouse embryos. Finally, expression of the mRNA for the homeobox transcription factor Nkx2-1 was significantly higher in the Tor1A−/−basal forebrain. Physiological levels of expression of Nkx2-1 are critical for MGE cell migration. Thus, interkinetic nuclear migration in precursor cells, migration of postmitotic neurons and expression of Nkx2-1 expression were altered by the loss of the Tor1A gene in the embryonic mouse forebrain.
A principal goal of this study was to test the hypothesis that torsinA, the product of the Tor1A gene, influences brain development. Multiple lines of evidence form the background to this hypothesis. First, torsinA is expressed at its highest level during embryonic and early postnatal development in the mouse forebrain [27]. It is also expressed robustly in the human fetal brain [28, 29]. Second, mice lacking torsinA or mice homozygous for the delGAG mutation die around the time of birth [8, 12, 62], suggesting critical roles for torsinA in development. Studies in nematodes suggest that torsinA is critical for precursor cell proliferation [63, 64]. Finally, DYT1 dystonia has a childhood onset, and individuals carrying the DYT1 mutation show structural and functional changes in the sensorimotor cortex even without displaying overt signs of dystonia [7, 24, 65]. Thus, it is unlikely that the onset of symptoms in the second decade of life temporally coincides exactly with the onset of structural changes underlying the symptoms. Rather, the structural changes likely begin early, during the process of brain development and maturation, and are precursors to the development of dystonia. Collectively, these findings suggest that torsinA has a role in brain development. The data presented here lend support to this possibility.
Interkinetic Nuclear Migration
The only phenomenon associated with cell proliferation that was altered in the Tor1A−/− embryos was the interkinetic nuclear migration of precursor cells in the VZ of the DCW (fig. 1). The overall cell output or neurogenesis was not altered. How the loss of torsinA resulted in altered interkinetic nuclear migration is not yet clear. One possibility is that torsinA may be involved in the regulation of cytoskeletal dynamics critical for the normal to-and-fro movement of the nucleus during cell division in step with the cell cycle stages and, in the absence of torsinA, the interkinetic nuclear migration may be affected. TorsinA is reported to be involved in tethering the nucleus to the cytoskeleton [12, 30] as well as maintaining the structural integrity of the nuclear envelope [66]. However, these suggestions must remain speculative for the present.
Neuronal Migration in the DCW
Newly postmitotic cells in the embryonic DCW rapidly exit the neuroepithelium and migrate through the different laminae of the developing cerebral wall to settle in the CP [67, 68]. Upon exit from the neuroepithelium in the VZ, the newly born cells segregate into two populations: one exiting rapidly and the other slowly [60, 61, 69]. As a result, when the position of a cohort of the newly postmitotic cells is plotted against distance from the VZ in the developing cerebral wall, a bimodal distribution becomes evident with a taller peak in the SVZ (slowly migrating) and a shorter peak in the CP (rapidly migrating) [60, 70]. Our data show such a bimodal distribution in the DCW of the Tor1A+/+ embryos (fig. 2e). However, in the Tor1A−/− embryo, the newly postmitotic cells did not show a bimodal distribution (fig. 2e).
We suggest that the loss of the bimodal distribution of the postmitotic cells in the Tor1A−/− DCW is due to changes in the speed of cell migration. The rationale for our suggestion is the following. First, there were no changes in the total numbers of postmitotic cells produced by the precursor cells (i.e. cell output was not significantly altered by the loss of torsinA). Therefore, changes in the numbers of newly generated cells could not have contributed to the changes in the pattern of cell distribution, namely the loss of the bimodal distribution. Second, a greater number of postmitotic cells (BrdU+Ki67− cells) were found in the SVZ in the Tor1A−/− embryo compared to its Tor1A+/+ counterparts (fig. 2f). The increase in the SVZ BrdU+Ki67− cells is unlikely to be due to increased SVZ precursor cell proliferation because Tbr2 labeling (which marks SVZ precursor cells) did not show significant differences between Tor1A+/+ and Tor1A−/− embryos (fig. 3). Therefore, we suggest that the only plausible explanation for the loss of the bimodal distribution of postmitotic cells (BrdU+ Ki67− cells) in the DCW of the Tor1A−/− embryo is altered pattern of cell exit or cell migration in the DCW.
Although the significance of the slow versus rapid rate of migration of the newly generated cells is not known fully, it is believed that the different migratory rates are associated with different transcriptional profiles of the newly generated cells, and that the two subpopulations of cells correspond to distinct functional classes of cortical projection neurons defined by their laminar position and connectivity [71–73]. Therefore, we speculate that the loss of the normal pattern of migration of cells in the DCW of the Tor1A−/− DCW may underscore a role for torsinA in cortical neuron differentiation and acquisition of functional properties.
MGE Neuronal Migration
Our data show that the loss of torsinA also alters migration of neurons in the basal forebrain – specifically the neurons generated in the MGE. The distribution of the postmitotic BrdU+Ki67− cells was different in the MGE of Tor1A+/+ versus Tor1A−/− embryos (fig. 2). However, the differences were not statistically significant. One reason for the lack of statistical significance may be that the MGE cell migration rate is greater than that of the DCW cells [74–76]. Therefore, our in vivo BrdU-Ki67 labeling paradigm may not have permitted sufficiently long periods for capturing any differences in MGE cell migration between the two genotypes. To overcome this potential problem, we used an in vitro explant culture system to study MGE cell migration [33]. We found significant differences in the distribution of migrating cells within 200 μm from the edge of the MGE explant taken from Tor1A−/− embryos compared to the Tor1A+/+ embryos (Tor1A−/− < Tor1A+/+; fig. 4). However, there was no significant difference at distances greater than 200 μm. There are likely multiple explanations for these findings. For example, Tor1A−/− cells may migrate faster initially and then migrate slowly at distances > 200 μm over the 24-hour culture period. Alternatively, an early wave of migrating cells may migrate at comparable speeds in the Tor1A−/− and Tor1A+/+ explants and reach the farthest points (> 200 μm) of the matrix, while a second wave may migrate more quickly in the initial 200 μm of matrix and then migrate more slowly at distances > 200 μm in Tor1A−/− explants. We cannot distinguish between these possibilities. However, the lack of bimodal distribution of BrdU+Ki67− cells (fig. 2) in the DCW of the Tor1A−/− embryo suggests that different rates of migration of two populations of cells may be a likely explanation in the MGE explants as well.
TorsinA and Nkx2-1 Expression
Our data show that loss of torsinA upregulates Nkx2-1 mRNA expression in the basal forebrain. Upregulation of Nkx2-1 is sufficient to decrease the ability of MGE-derived cells to migrate from the basal to the dorsal forebrain [52] . Although Nkx2-1 is expressed in proliferating and postmitotic cells in the basal forebrain, our RT PCR analysis used samples of postmitotic cells isolated using LCM. Therefore, the upregulation of Nkx2-1 mRNA reported here occurred in postmitotic cells, many of which would have migrated away from the MGE. The effects of TorsinA on Nkx2-1 expression will not be relevant to the dorsal forebrain because Nkx2-1 is not expressed there. In other words, the loss of torsinA may affect multiple mechanisms in a region-specific manner.
Additional Mechanisms for Impaired Neuronal Migration
The data from the in vivo Brdu-Ki67 labeling studies and the in vitro explant cultures both indicate that loss of torsinA may contribute to changes in neuronal migration. However, the in vivo and in vitro experimental paradigms may highlight different mechanisms. In the in vitro explant studies, cells from Tor1A−/− and Tor1A+/+ explants both migrate in a highly permissive environment provided by the Matrigel whereas in the in vivo paradigm, the Tor1A−/− and Tor1A+/+ migratory environments may be different, and may differentially influence the rate of migration. Thus, the in vivo data are influenced by cell-intrinsic and cell-extrinsic factors whereas the in vitro explant data provide identical and favorable cell-extrinsic environments, with genotype-dependent cell-intrinsic factors being the only variable. Our MGE explant studies showed that fewer cells in the Tor1A−/− MGE explants displayed the characteristic morphology of migrating neurons (elongated cell body, and a well-developed leading process). Therefore, it is possible that the loss of torsinA somehow impaired the normal reorganization of the cytoskeleton and organelles associated with cell migration [74, 76] . Thus, the in vitro MGE explant data underscored cell-intrinsic differences between Tor1A−/− and Tor1A+/+ embryos.
Although the present study was not designed to address cell biological mechanisms associated with the influence of torsinA on neuronal migration, the literature on the role of torsinA in nuclear envelope-cytoskeletal interactions warrants some speculation on this subject. For example, Tor1A−/− fibroblasts show mislocalization of nesprin, an outer nuclear membrane protein, and impaired interaction between the nuclear envelope and the cytoskeleton as well as delayed fibroblast migration in a wound-healing assay [30] . Other reports show changes in nuclear envelope structure in the Tor1A−/− embryonic brain [66]. Therefore, we speculate that impaired nuclear-cytoskeletal interactions in the Tor1A−/− embryonic forebrain may be responsible for changes in interkinetic nuclear migration as well as neuronal migration, processes which require efficient nuclear-cytoskeletal interactions [74].
Conclusions and Significance
Our data suggest that torsinA may play a role in the regulation of interkinetic nuclear migration in progenitor cells and migration of newly generated neurons in the dorsal and basal forebrain. The rate of migration of MGE-derived cells was also significantly altered in the Tor1A−/− embryo. The long-term consequences of impaired neuronal migration cannot be evaluated directly because postnatal Tor1A−/− mice are not viable. However, even a transient delay in the migration of MGE-derived neurons, most of which are GABA neurons of the dorsal cerebral wall, leads to long-term impairment of cortical inhibitory-excitatory balance [41], which in turn produces significant functional consequences [77, 78]. Therefore, we speculate that the subtle changes in forebrain neuronal migration observed in the Tor1A−/− embryos in the present study may contribute to long-term functional changes in forebrain neurons.
Acknowledgements
This work was supported by USPHS grants P50 NS037409 and P30NS045776. We are grateful to Igor Bagayev of the Massachusetts General Hospital confocal microscope facility for expert assistance with confocal microscopy and to the staff of the Massachusetts General Hospital's Center for Comparative Medicine for expert assistance with mouse colony management.
References
- 1.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 2.Ozelius LJ, Page CE, Klein C, Hewett JW, Mineta M, Leung J, Shalish C, Bressman SB, de Leon D, Brin MF, Fahn S, Corey DP, Breakefield XO. The TOR1A (DYT1) gene family and its role in early onset torsion dystonia. Genomics. 1999;62:377–384. doi: 10.1006/geno.1999.6039. [DOI] [PubMed] [Google Scholar]
- 3.Bressman SB, Heiman GA, Nygaard TG, Ozelius LJ, Hunt AL, Brin MF, Gordon MF, Moskowitz CB, de Leon D, Burke RE, et al. A study of idiopathic torsion dystonia in a non-jewish family: Evidence for genetic heterogeneity. Neurology. 1994;44:283–287. doi: 10.1212/wnl.44.2.283. [DOI] [PubMed] [Google Scholar]
- 4.Bressman SB, Sabatti C, Raymond D, de Leon D, Klein C, Kramer PL, Brin MF, Fahn S, Breakefield X, Ozelius LJ, Risch NJ. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. 2000;54:1746–1752. doi: 10.1212/wnl.54.9.1746. [DOI] [PubMed] [Google Scholar]
- 5.Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- 6.Rostasy K, Augood SJ, Hewett JW, Leung JC, Sasaki H, Ozelius LJ, Ramesh V, Standaert DG, Breakefield XO, Hedreen JC. TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol Dis. 2003;12:11–24. doi: 10.1016/s0969-9961(02)00010-4. [DOI] [PubMed] [Google Scholar]
- 7.Carbon M, Kingsley PB, Su S, Smith GS, Spetsieris P, Bressman S, Eidelberg D. Micro-structural white matter changes in carriers of the DYT1 gene mutation. Ann Neurol. 2004;56:283–286. doi: 10.1002/ana.20177. [DOI] [PubMed] [Google Scholar]
- 8.Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y. Generation and characterization of Dyt1 deltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 9.Dang MT, Yokoi F, Pence MA, Li Y. Motor deficits and hyperactivity in Dyt1 knockdown mice. Neurosci Res. 2006;56:470–474. doi: 10.1016/j.neures.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoi F, Dang MT, Mitsui S, Li J, Li Y. Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice. J Biochem. 2008;143:39–47. doi: 10.1093/jb/mvm191. [DOI] [PubMed] [Google Scholar]
- 11.Yokoi F, Dang MT, Li Y. Improved motor performance in Dyt1 ΔGAG heterozygous knock-in mice by cerebellar Purkinje-cell specific Dyt1 conditional knocking-out. Behav Brain Res. 2012;130:389–398. doi: 10.1016/j.bbr.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanabe LM, Martin C, Dauer WT. Genetic background modulates the phenotype of a mouse model of DYT1 dystonia. PLoS One. 2012;7:e32245. doi: 10.1371/journal.pone.0032245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulug AM, Vo A, Argyelan M, Tanabe L, Schiffer WK, Dewey S, Dauer WT, Eidelberg D. Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc Natl Acad Sci USA. 2011;108:6638–6643. doi: 10.1073/pnas.1016445108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, Breakefield XO. Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci. 2005;25:5351–5355. doi: 10.1523/JNEUROSCI.0855-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shashidharan P, Sandu D, Potla U, Armata IA, Walker RH, McNaught KS, Weisz D, Sreenath T, Brin MF, Olanow CW. Transgenic mouse model of early-onset DYT1 dystonia. Hum Mol Genet. 2005;14:125–133. doi: 10.1093/hmg/ddi012. [DOI] [PubMed] [Google Scholar]
- 17.Balcioglu A, Kim MO, Sharma N, Cha JH, Breakefield XO, Standaert DG. Dopamine release is impaired in a mouse model of DYT1 dystonia. J Neurochem. 2007;102:783–788. doi: 10.1111/j.1471-4159.2007.04590.x. [DOI] [PubMed] [Google Scholar]
- 18.Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–2349. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Sciamanna G, Tassone A, Martella G, Mandolesi G, Puglisi F, Cuomo D, Madeo G, Ponterio G, Standaert DG, Bonsi P, Pisani A. Developmental profile of the aberrant dopa-mine D2 receptor response in striatal cholinergic interneurons in DYT1 dystonia. PLoS One. 2011;6:e24261. doi: 10.1371/journal.pone.0024261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoi F, Dang MT, Li J, Standaert DG, Li Y. Motor deficits and decreased striatal dopa-mine receptor 2 binding activity in the striatum-specific DYT1 conditional knockout mice. PLoS One. 2011;6:e24539. doi: 10.1371/journal.pone.0024539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grundmann K. Primary torsion dystonia. Arch Neurol. 2005;62:682–685. doi: 10.1001/archneur.62.4.682. [DOI] [PubMed] [Google Scholar]
- 23.Carbon M, Argyelan M, Eidelberg D. Functional imaging in hereditary dystonia. Eur J Neurol. 2010;17(suppl 1):58–64. doi: 10.1111/j.1468-1331.2010.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carbon M, Argyelan M, Habeck C, Ghilardi MF, Fitzpatrick T, Dhawan V, Pourfar M, Bressman SB, Eidelberg D. Increased sensorimotor network activity in DYT1 dystonia: a functional imaging study. Brain. 2010;133:690–700. doi: 10.1093/brain/awq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghilardi MF, Carbon M, Silvestri G, Dhawan V, Tagliati M, Bressman S, Ghez C, Eidelberg D. Impaired sequence learning in carriers of the DYT1 dystonia mutation. Ann Neurol. 2003;54:102–109. doi: 10.1002/ana.10610. [DOI] [PubMed] [Google Scholar]
- 26.Eidelberg D. Abnormal brain networks in DYT1 dystonia. Adv Neurol. 1998;78:127–133. [PubMed] [Google Scholar]
- 27.Vasudevan A, Breakefield XO, Bhide PG. Developmental patterns of torsinA and torsinB expression. Brain Res. 2006;1073-1074:139–145. doi: 10.1016/j.brainres.2005.12.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siegert S, Bahn E, Kramer ML, Schulz-Schaeffer WJ, Hewett JW, Breakefield XO, Hedreen JC, Rostasy KM. TorsinA expression is detectable in human infants as young as 4 weeks old. Brain Res Dev Brain Res. 2005;157:19–26. doi: 10.1016/j.devbrainres.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 29.Shashidharan P, Kramer BC, Walker RH, Olanow CW, Brin MF. Immunohistochemical localization and distribution of torsinA in normal human and rat brain. Brain Res. 2000;853:197–206. doi: 10.1016/s0006-8993(99)02232-5. [DOI] [PubMed] [Google Scholar]
- 30.Nery FC, Zeng J, Niland BP, Hewett J, Farley J, Irimia D, Li Y, Wiche G, Sonnenberg A, Breakefield XO. TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J Cell Sci. 2008;121:3476–3486. doi: 10.1242/jcs.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Theiler K. Development and Normal Stages from Fertilization to 4 Weeks of Age. Springer; Berlin: 1972. The House Mouse. [Google Scholar]
- 32.Kaufman MH. The Atlas of Mouse Development. ed 2 Academic Press; New York: 1992. [Google Scholar]
- 33.McCarthy DM, Zhang X, Darnell SB, Sangrey GR, Yanagawa Y, Sadri-Vakili G, Bhide PG. Cocaine alters BDNF expression and neuronal migration in the embryonic mouse forebrain. J Neurosci. 2011;31:13400–13411. doi: 10.1523/JNEUROSCI.2944-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noctor SC, Martinez-Cerdeno V, Kriegstein AR. Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol. 2008;508:28–44. doi: 10.1002/cne.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauer ME, Walker BE. Radioautographic study of interkinetic nuclear migration in the neural tube. Proc Soc Exp Biol Med. 1959;101:557–560. doi: 10.3181/00379727-101-25014. [DOI] [PubMed] [Google Scholar]
- 37.Smart IH. Proliferative characteristics of the ependymal layer during the early development of the mouse neocortex: a pilot study based on recording the number, location and plane of cleavage of mitotic figures. J Anat. 1973;116:67–91. [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi T, Nowakowski RS, Caviness VS., Jr. BUdR as an S-phase marker for quantitative studies of cytokinetic behaviour in the murine cerebral ventricular zone. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 39.Preuss U, Landsberg G, Scheidtmann KH. Novel mitosis-specific phosphorylation of histone H3 at Thr11 mediated by Dlk/Zip kinase. Nucleic Acids Res. 2003;31:878–885. doi: 10.1093/nar/gkg176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 41.McCarthy D, Bhide PG. Prenatal cocaine exposure decreases parvalbumin immunore-active neurons and GABA-to-projection neuron ratio in the medial prefrontal cortex. Dev Neurosci. 2012 doi: 10.1159/000337172. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 42.Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 43.Rallu M, Corbin JG, Fishell G. Parsing the prosencephalon. Nat Rev Neurosci. 2002;3:943–951. doi: 10.1038/nrn989. [DOI] [PubMed] [Google Scholar]
- 44.Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- 45.Parnavelas JG, Anderson SA, Lavdas AA, Grigoriou M, Pachnis V, Rubenstein JL. The contribution of the ganglionic eminence to the neuronal cell types of the cerebral cortex. Novartis Found Symp. 2000;228:129–139. doi: 10.1002/0470846631.ch10. discussion 139–147. [DOI] [PubMed] [Google Scholar]
- 46.Rubenstein JL, Anderson S, Shi L, Miyashita-Lin E, Bulfone A, Hevner R. Genetic control of cortical regionalization and connectivity. Cereb Cortex. 1999;9:524–532. doi: 10.1093/cercor/9.6.524. [DOI] [PubMed] [Google Scholar]
- 47.Stenman J, Toresson H, Campbell K. Identification of two distinct progenitor populations in the lateral ganglionic eminence: Implications for striatal and olfactory bulb neurogenesis. J Neurosci. 2003;23:167–174. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Araki KY, Sims JR, Bhide PG. Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and postnatal development. Brain Res. 2007;1156:31–45. doi: 10.1016/j.brainres.2007.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 50.Simon P. Q-gene: Processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 51.Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nobrega-Pereira S, Kessaris N, Du T, Kimura S, Anderson SA, Marin O. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron. 2008;59:733–745. doi: 10.1016/j.neuron.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular re-specification within the basal telencephalon: Evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 54.Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2622. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Misson J-P, Austin CP, Takahashi T, Cepko CL, Caviness VS., Jr. The alignment of migrating neural cells in relation to the murine neopallial radial glial fiber system. Cereb Cortex. 1991;1:221–229. doi: 10.1093/cercor/1.3.221. [DOI] [PubMed] [Google Scholar]
- 56.Halliday AL, Cepko CL. Generation and migration of cells in the developing striatum. Neuron. 1992;9:15–26. doi: 10.1016/0896-6273(92)90216-z. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi T, Misson J-P, Caviness VS., Jr Glial process elongation and branching in the developing murine neocortex: a qualitative and quantitative immunohistochemical analysis. JComp Neurol. 1990;302:15–28. doi: 10.1002/cne.903020103. [DOI] [PubMed] [Google Scholar]
- 58.Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hevner RF, Daza RA, Rubenstein JL, Stun-nenberg H, Olavarria JF, Englund C. Beyond laminar fate: toward a molecular classification of cortical projection/pyramidal neurons. Dev Neurosci. 2003;25:139–151. doi: 10.1159/000072263. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi T, Nowakowski RS, Caviness VS., Jr Mode of cell proliferation in the developing mouse neocortex. Proc Natl Acad Sci USA. 1994;91:375–379. doi: 10.1073/pnas.91.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bayer SA, Altman J. Neocortical Development. Raven Press; New York: 1991. [Google Scholar]
- 62.Dauer W, Goodchild R. Mouse models of torsinA dysfunction. Adv Neurol. 2004;94:67–72. [PubMed] [Google Scholar]
- 63.Basham SE, Rose LS. Mutations in ooc-5 and ooc-3 disrupt oocyte formation and the reestablishment of asymmetric par protein localization in two-cell Caenorhabditis elegans embryos. Dev Biol. 1999;215:253–263. doi: 10.1006/dbio.1999.9447. [DOI] [PubMed] [Google Scholar]
- 64.Basham SE, Rose LS. The Caenorhabditis elegans polarity gene ooc-5 encodes a torsin-related protein of the AAA ATPase super-family. Development. 2001;128:4645–4656. doi: 10.1242/dev.128.22.4645. [DOI] [PubMed] [Google Scholar]
- 65.Ghilardi M-F, Carbon M, Silvestri G, Dhawan V, Tagliati M, Bressman S, Ghez C, Eidelberg D. Impaired sequence learning in carriers of the DYT1 dystonia mutation. Ann Neurol. 2003;54:102–109. doi: 10.1002/ana.10610. [DOI] [PubMed] [Google Scholar]
- 66.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 67.Takahashi T, Nowakowski RS, Caviness VS., Jr Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone of the fetal mouse. J Neurosci. 1993;13:820–833. doi: 10.1523/JNEUROSCI.13-02-00820.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995;15:6046–6057. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McConnell SK, Kaznowski CE. Cell cycle dependence of laminar determination in developing neocortex. Science. 1991;254:282–285. doi: 10.1126/science.254.5029.282. [DOI] [PubMed] [Google Scholar]
- 70.Takahashi T, Nowakowski RS, Caviness VS., Jr The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J Neurosci. 1996;16:6183–6196. doi: 10.1523/JNEUROSCI.16-19-06183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caviness VS, Jr, Takahashi T, Miyama S, Nowakowski RS, Delalle I. Regulation of normal proliferation in the developing cerebrum potential actions of trophic factors. Exp Neurol. 1996;137:357–366. doi: 10.1006/exnr.1996.0037. [DOI] [PubMed] [Google Scholar]
- 72.Caviness VS, Jr, Goto T, Tarui T, Takahashi T, Bhide PG, Nowakowski RS. Cell output, cell cycle duration and neuronal specification: a model of integrated mechanisms of the neo-cortical proliferative process. Cereb Cortex. 2003;13:592–598. doi: 10.1093/cercor/13.6.592. [DOI] [PubMed] [Google Scholar]
- 73.Caviness VS, Jr., Nowakowski RS, Bhide PG. Neocortical neurogenesis: morphogenetic gradients and beyond. Trends Neurosci. 2009;32:443–450. doi: 10.1016/j.tins.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Metin C, Vallee RB, Rakic P, Bhide PG. Modes and mishaps of neuronal migration in the mammalian brain. J Neurosci. 2008;28:11746–11752. doi: 10.1523/JNEUROSCI.3860-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- 76.Bellion A, Baudoin JP, Alvarez C, Bornens M, Metin C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/ centrosome associated with centrosome splitting and myosin contraction at the rear. J Neurosci. 2005;25:5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu H, Lim B, Poo MM. Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J Neurosci. 2009;29:12664–12674. doi: 10.1523/JNEUROSCI.1984-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang CC, Liang YC, Hsu KS. Prenatal cocaine exposure enhances long-term potentiation induction in rat medial prefrontal cortex. Int J Neuropsychopharmacol. 2011:1–13. doi: 10.1017/S1461145710000258. [DOI] [PubMed] [Google Scholar]