

Abstract
Diabetes is highly prevalent in India and the proportion of younger patients developing diabetes is on the increase. Apart from the more universally known type 1 diabetes and obesity related type 2 diabetes, monogenic forms of diabetes are also suspected to be prevalent in many young diabetic patients. The identification of the genetic basis of the disease not only guides in therapeutic decision making, but also aids in genetic counselling and prognostication. Genetic testing may establish the occurrence and frequency of early diabetes in our population. This review attempts to explore the utilities and horizons of molecular genetics in the field of maturity onset diabetes of the young (MODY), which include the commoner forms of monogenic diabetes.
Keywords: Consanguinity, epigenetics, maturity onset diabetes of the young, molecular diagnosis, monogenic diabetes, neonatal diabetes
INTRODUCTION
According to the International Diabetes Federation (IDF) Atlas 2011, India stands second in the world with regards to the prevalence of diabetes mellitus. Nearly sixty two million people are affected at present and the number is expected to rise to more than hundred million by 2030.[1] Type 2 diabetes is the commonest cause for this disorder followed by type 1 diabetes; both of which account for more than 95% of cases. Less frequent causes include pancreatic diabetes, monogenic diabetes and secondary forms of diabetes mellitus. The proportion of young people affected with type 2 diabetes is on the increase which parallels the obesity epidemic worldwide with an impact on developing countries as well.[2] There is a shift to the left in the age of onset of type 1 diabetes as shown by studies across the world,[3] though no data has been published from India in this regard. Since diabetes is a silent killer and India being one of the heavily affected countries, more focus needs to be placed on patient care as well as research. Diagnosis of the etiology of young onset diabetes is sometimes a puzzle. Overlapping or atypical features often lead to misdiagnosis. A single clinical characteristic or lab finding may not help reach the diagnosis in such cases. The algorithm [Figure 1] provided below is an attempt to approach a young patient with diabetes.
Figure 1.
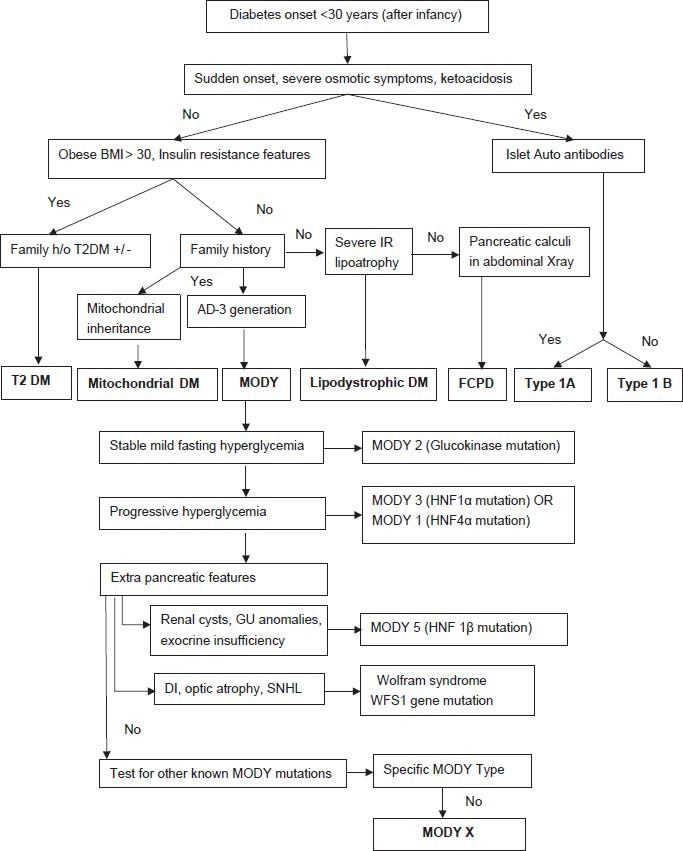
Diagnostic approach to young onset diabetes
Monogenic diabetes
Monogenic diabetes is defined as diabetes that occur due to a mutation of a single gene affecting β-cell function. These mutations involve genes coding for transcription factors involved in β-cell development, differentiation and survival; as well as enzymes like glucokinase (GCK) and carboxy ester lipase (CEL). Neonatal diabetes mellitus (NNDM) and Maturity onset diabetes of the young (MODY) are the 2 main types of monogenic diabetes that result from β-cell dysfunction. Other forms of monogenic diabetes result in defective insulin action or insulin receptor dysfunction.
NEONATAL DIABETES
Neonatal diabetes mellitus (NNDM) is a rare disorder with a reported incidence of 1 in 2 60 000 live births in European population.[4] By definition, neonatal diabetes mellitus, presents within the first 6 months of life with failure to thrive, dehydration and sepsis with or without ketoacidosis. Based on its clinical course, 2 forms have been recognised-transient and permanent. In short, approximately half of these cases have a transient course which resolves in the first few months of life, while others might require lifelong insulin therapy.
Transient neonatal diabetes
Transient neonatal diabetes mellitus (TNDM) results from a developmental defect in β-cell maturation which resolves spontaneously during the postnatal period. It manifests as insulin requiring hyperglycemia within the first week of life. Affected babies are generally growth retarded as insulin is a major foetal growth factor. Ketoacidosis rarely occurs in TNDM. The condition usually resolves within the first year of life. Infants born with TNDM harbour a greater risk of developing type 2 diabetes later in the life. The persistence of the underlying pancreatic defect predisposes to a heightened risk, especially during periods of increased demand like puberty and pregnancy.
The genetics of transient neonatal diabetes
The molecular genetic mechanism resulting in TNDM has been elucidated in a majority of cases. Two third of cases of TNDM results from an imprinting defect on chromosome 6q24.[5] Mutations in the KCNJ11 and ABCC8 genes which encode for Kir6.2 and SUR1 subunits of the ATP sensitive potassium channel, usually result in permanent neonatal diabetes mellitus (PNDM), though transient forms have been reported.[6] Heterozygous mutations in HNF-1β gene have also been shown to result in TNDM though the typical presentation is later in the young adult as MODY5.[7] Transient neonatal diabetes is a well established example of a genomic imprinting disorder. Genomic imprinting is a phenomenon where the phenotypic expression of a particular genetic allele in an individual depends on the parent from which the allele is transmitted. In other words, the allele from one parent is ‘silenced’ through epigenetic mechanisms as methylation and histone modification which results in a monoallelic gene expression. Mutations affecting this single allele have a greater chance of affecting the gene function which results in disease. Abnormalities in the imprinted region of chromosome 6q24 results in the majority of cases of TNDM.[8,9] In the normal foetus only 1 allele of chromosome 6q24 is expressed and this is of paternal origin; the maternal allele being silenced by methylation. Over expression of the paternal allele results from uniparental di-somy or duplication of the 6q24 region of the paternal chromosome. Activation of the maternal allele can occur as a result of hypomethylation of the maternal differentially methylated region (DMR). This results in TNDM as the imprinted region has been shown to contain genes which regulate apoptosis, insulin secretion and foetal growth namely PLAGL1 (Pleomorphic adenoma gene like-1 (also known as ZAC)) and HYMAI- (Hydatidiform mole associated and imprinted).[8] Studies in animal models have proven that over expression of genes in the 6q24 locus adversely affects islet cell development.[10] However, the cellular mechanism of hyperglycemia and its spontaneous resolution in the postnatal period are not well understood. The ZAC gene is also shown to regulate the imprinted region on chromosome 11p15.5 near the Beckwith-Weidmann syndrome locus and 2 other growth regulatory genes namely igfr2 and igfr2r which possibly explain the sharing of associated features such as macroglossia and umbilical hernia in both conditions.[11]
Permanent neonatal diabetes
Permanent neonatal diabetes mellitus (PNDM) is characterised by the early onset of persistent hyperglycemia requiring lifelong treatment. All cases of PNDM result from mutations affecting genes regulating pancreatic development, β-cell function, apoptosis or the insulin molecule as such. Approximately half the cases of PNDM in the Caucasian population have been shown to involve defects in the genes transcribing the 2 subunits of K+ ATP channel Kir6.2 and SUR1, which regulate insulin release from the β-cell.[12,13] These subunit proteins are transcribed by KCNJ11 and ABCC8 genes respectively. Other genes implicated in PNDM include PTF-1α, IPF-1 (Pdx-1), EIF-2AK3, Glucokinase, FOXP3, insulin and GLIS3.
PNDM due to activating mutations in potassium sensitive channels
Glucose sensing by the β-cell and subsequent insulin release is a complex cascade involving multiple receptors and channels. The glucose molecule that is transported into the β-cell through the GLUT receptor undergoes hydrolysis after phosphorylation. The pyruvate molecule that is generated is then metabolised inside the mitochondria to CO2 and H2O to generate ATP molecules. The increased ATP/ADP ratio inside the β-cell closes the potassium channels on the cell membrane resulting in depolarization which subsequently activates the voltage gated calcium channels. The influx of calcium ions causes release of insulin from the secretory granules. The potassium sensitive channel on the β cell membrane has two kinds of subunits-Kir6.2 (Potassium inward rectifying) and SUR1 (sulphonylurea receptor 1) which are encoded by 2 different genes – KCNJ11 (Potassium Channel, subfamily J, member 11) and ABCC8 (ATP Binding Cassette transporter subfamily C, member 8) respectively. Activating mutations in these genes result in persistent opening of the potassium channels preventing insulin release resulting in impaired glucose sensing and hence diabetes. However, the mutant channels retain the ability to bind and respond to sulphonylureas. These potassium channel subunits are expressed in non pancreatic tissues as well; SUR1 being found in neurons while Kir6.2 in the brain, heart and skeletal muscles. This explains the occurrence of associated features like developmental delay, epilepsy and muscle weakness (DEND – Developmental delay, Epilepsy and Neonatal Diabetes- syndrome) in infants with NNDM due to these mutations.[14]
PTF-1α (Pancreatic transcription factor 1, α subunit)
PTF-1α is a transcription factor with critical roles in foetal pancreatic development and maintenance of mature pancreatic function. It has also been found to regulate cerebellar development. Homozygous mutations in gene encoding PTF-1α results in PNDM characterised by cerebellar hypoplasia, pancreatic agenesis, intrauterine growth retardation, respiratory distress and microcephaly as described initially in a consanguineous Pakistani family.[15]
IPF-1 (Insulin promoter factor-1) or PDX-1 (Pancreatic and duodenal homeobox 1)
IPF-1/PDX-1 is one of the early transcription factors regulating the development of both exocrine and endocrine pancreas in the foetus. Homozygous loss of function mutation in the gene encoding this factor results in PNDM with exocrine deficiency due to pancreatic agenesis.[16] Heterozygous mutations in the same gene has been shown to result in early onset diabetes in the young (MODY4) due to impaired β-cell function.
Glucokinase (GCK)
The glucokinase enzyme is the glucose sensor of the β-cell. It phosphorylates the glucose molecules entering the cell and initiates subsequent pathways leading to insulin release. Homozygous mutations in the GCK gene results in complete loss of glycolytic activity in the β-cell resulting in PNDM. In the heterozygous state it results in mild impairment of insulin release manifesting as maturity onset diabetes of the young (MODY2) or as gestational diabetes.[17]
Eukaryotic translation initiation factor-2 kinase-3 (EIF2AK3)-wolcott-rallison syndrome
Wolcott Rallison syndrome is an autosomal recessive disorder characterised by infantile onset diabetes, multiple epiphyseal dysplasia, growth retardation, exocrine pancreatic dysfunction, osteopenia, developmental delay, hypothyroidism, acute liver failure, renal failure, cardiomegaly, cerebellar cortical dysplasia and early death. The molecular defect is localised to the EIF-2AK3 gene on chromosome 2p12 which regulates protein synthesis and folding. The resultant misfolded proteins increase the stress in the endoplasmic reticulum of various tissues leading to cell death by apoptosis which explains the various phenotypic features.[18]
FOXP3 gene mutation (Immune dysregulation, polyendocrinopathy and enteropathy, X-linked-IPEX syndrome)
The IPEX syndrome is an X-linked disorder characterised by the combination of PNDM, intractable diarrhoea with villous atrophy, exfoliative dermatitis, autoimmune hypothyroidism, haemolytic anaemia, recurrent infections due to immune deficiency. The condition is fatal and often results in death before the first birthday. A genetic defect has been identified in the FOXP3 gene on the X chromosome which codes for a forkhead domain-containing protein known as ‘scurfin’ that is required for immune homeostasis.[19] Studies in the mouse model have shown over proliferation of CD4+/CD8- T-lymphocytes with infiltration of multiple organs.[20] There is an increased predilection for autoimmunity in this condition. The diabetes mellitus in this disorder is due to autoimmune destruction of the β-cells as suggested by the presence of a number of islet cell autoantibodies.[19]
Insulin gene mutations
Mutations in the insulin gene have been shown to affect the normal folding of the insulin molecule inside the endoplasmic reticulum resulting in reduced insulin release. The abnormal/misfolded protein increases the intracellular stress leading to apoptosis and cell death.[21] The age of presentation and severity of diabetes in this condition varies widely. It ranges from severe early onset neonatal diabetes to a mild form of MODY and gestational diabetes. Absence of extrapancreatic features differentiates this disorder from that due to the EIF-2AK3 mutation.
GLIS3 – (GLI subfamily of Kruppel-like zinc finger protein-3)
Mutations in GLIS3, yet another transcription factor gene, have been shown to be associated with PNDM with multisystem involvement.[22] The affected infant presents with intrauterine growth retardation, neonatal onset diabetes, congenital hypothyroidism, congenital glaucoma, renal cysts and dysmorphic facies. The protein encoded by the GLIS3 gene plays an important role in various cellular processes during foetal development including that of pancreatic β-cells.
SLC19A2 (Soluble carrier family 19, member 2) – Thiamine transporter mutations
Recessive SLC-19A2 mutations have been shown to result in early onset diabetes, thiamine responsive megaloblastic anaemia and deafness. Neurological deficits, cardiac abnormalities and visual problems are also reported in some of these cases.[23]
Table 1 summarizes the important mutations that cause neonatal diabetes and their characteristics features.[24,25] Identification of the genetic defect in neonatal diabetes helps in tailoring a treatment strategy. Neonatal diabetes due to mutations in the ABCC8 and KCNJ11 genes generally respond well to oral sulfonylureas.[26,27] This allows a shift from insulin therapy to oral drugs which gives immense logistic as well as a psychological advantage to the parents of the patients. Sulfonylurea therapy has been found to ameliorate the associated neurological dysfunction and hence early initiation of the drug is of utmost importance for an improvement in long term prognosis.[28]
Table 1.

So far, mutations in more than ten genes have been identified as a cause for neonatal diabetes. Recently a very high incidence of PNDM is reported from Saudi Arabia from a highly consanguineous population (1 in 21, 196 live births). Fourteen out of seventeen PNDM cases had an identifiable genetic aetiology of which 50% were due to the autosomal recessive Wolcott Rallison syndrome. Interestingly none of the patients had a mutation in KCNJ11, ABCC8 or INS genes.[29] There are case reports on several individual mutations causing NNDM from different parts of India localised on KCNJ11,[30] insulin gene,[31] and a novel mutation in the ABCC8 gene.[32] However no population based study has been published till now. Hence the true prevalence of each mutation is not known in our population. A Neonatal Diabetes Registry has been established in India where facilities are available to sequence KCNJ11, ABCC8 and insulin genes.
MATURITY ONSET DIABETES OF THE YOUNG
The early 1970's witnessed identification of a novel form of diabetes wherein younger people were affected with a familial clustering.[33] These patients had mild or no disease progression and did not require insulin. Subsequently this group came to be called as maturity onset diabetes of the young and was shown to have an autosomal dominant mode of inheritance. Tattersall and Fajans in 1975, proposed the clinical criteria for diagnosing MODY in a diabetic patient which were as follows:[34]
Early onset of Diabetes (<25 years)
Early onset Diabetes in at least 2 or ideally 3 family members
Autosomal dominant mode of inheritance
Non-insulin dependence (not requiring insulin even after 5 years of diagnosis)
Based on these criteria, the reported prevalence of MODY was 1-2% in Europe.[35] A high prevalence of MODY was reported from India also as early as 1985.[36] However this was based on clinical presumption and not substantiated by genetic testing. The molecular genetic basis of this condition was unravelled in the 1990's when 5 of the genetic loci were identified and characterised by various investigators. Thereafter, these conditions were referred to as MODY 1-5 based on the sequence of genes identified. Subsequent years have witnessed an explosion of knowledge in this field with 6 more gene loci harbouring mutations resulting in MODY being identified. It is now clear that the proverbial iceberg effect exists in MODY as well. This was due to a vast majority of diabetic patients being misclassified stereotypically as either type 1 diabetes or type 2 diabetes in the presence of overlapping or atypical features.
Genetic basis of maturity onset diabetes of the young
The MODY group of monogenic diabetes results from heterozygous mutations in the genes coding for various transcription factors involved in the foetal development of the pancreas and the β-cells as well as those regulating the maturation and maintenance of β-cell function after birth. In addition, mutations in enzymes directly or indirectly involved in glucose sensing of the β-cell have also been shown to result in early onset diabetes. There is wide clinical as well as genetic heterogeneity within and among the different MODY subtypes. The following section gives a brief account of the molecular mechanism and clinical peculiarities of each MODY subtype. Table 2 displays a summary of the different MODY mutations described so far.
Table 2.
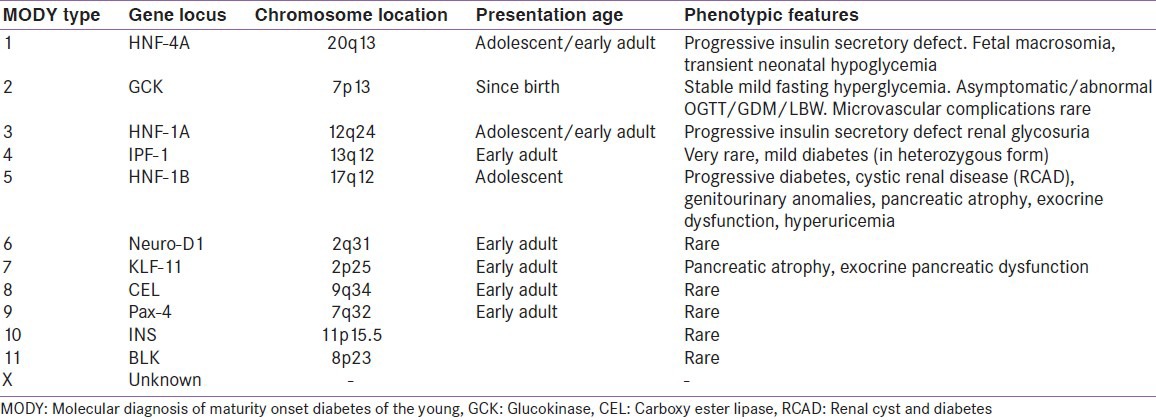
MODY due to mutations in transcription factor genes
The different transcription factors involved in foetal pancreatogenesis include hepatocyte nuclear factor 1-α (HNF-1α), HNF-4α, HNF-1β, IPF-1 (Insulin Promoter Factor-1), Neuro-D1 (Neurogenic Differentiation Factor-1) and Pax 4 and 6 (Paired Homeobox).[37]
HNF-1α mutations (MODY3)
HNF-1α or TCF-1 (Transcription Factor 1) is a homeo-domain containing transcription factor developmentally expressed in the liver, kidney, pancreas and gut. It has a crucial role in regulation of transcription of insulin gene in the mature β-cell as well as the glucose transporter GLUT2.[37,38] The 631aminoacid protein is encoded by the HNF-1α gene located on chromosome 12q. Mutations in the HNF-1α gene leads to progressive insulin secretory defect resulting in a severe form of inherited diabetes that is classically termed MODY3.[39] Recently, it has been established that in HNF-1α mutation carriers β-cell apoptosis is increased. The apoptotic cells induce production of pancreatic stone protein (PSP)/reg1 A from the surviving cells, which serves as a maker of the pathologic process.[40] The reduced β-cell mass results in defective glucose handling which is evidenced by large increments in postprandial 2 hour blood sugar values during a 75 g OGTT. HNF-1α is also functional in the kidneys in the regulation of proximal renal tubular reabsorption of glucose mediated through sodium glucose transporter-2.[41] Hence patients with MODY3 also exhibit low renal threshold for glucose resulting in renal glycosuria. MODY3 patients are sensitive to sulfonylureas, though many patients finally require insulin on a long term basis. Long term microvascular complications occur with the same frequency as in type 2 and type 1 diabetes.[42] Liver adenomas have also been reported to occur in MODY3 subjects, though the association is rare.[43]
The HNF-1α mutation is the most common form of MODY reported in studies from UK and from many European countries.[44,45,46,47] A recent study from Argentina reported a prevalence of 10% in a group of clinically characterised MODY patients of Caucasian ethnicity.[48] However studies in Japanese and Brazilian populations showed a very low prevalence of the HNF-1α gene defects in early onset diabetes.[49,50] In a recent publication, the French Monogenic Diabetes Study group reported a prevalence of 39% of HNF-1α gene mutation in their cohort. 40% of the mutation proven MODY 3 subjects had their age at diagnosis above twenty five years and only about half of these subjects had 3 generations of family history of diabetes. Nearly one third (28%) of the subjects had a BMI of >25kg/m2.[51] Similar figures had been reported from an Irish MODY cohort.[52] This points towards the fact that many cases of MODY3 may be missed if strict criteria are used for patient selection. During a median follow up of twenty years in the French cohort, 45% of the patients required initiation of insulin therapy.
HNF-4α mutation (MODY1)
HNF-4α and HNF-1α transcription factors have been found to exhibit a mutual regulatory function in the mature β-cell.[53] Heterozygous mutations in HNF4α results in a phenotype that resembles MODY3.[54] In addition, it has been observed that such patients are more sensitive to the hypoglycaemic effect of sulfonylureas.[55,56] Mutations in HNF-4α have been reported to result in macrosomia and transient hyperinsulinemic hypoglycaemia in the neonatal period and these subjects subsequently develop an early onset diabetes mellitus (MODY1).[57] The reported prevalence of HNF-4α mutations is less than 5%.[58] However, up to a 30% mutation detection rate for HNF-4α has been reported in those screened negative for HNF-1α mutations in a clinically suggestive MODY cohort.[59]
Insulin Promoter Factor-1/Pancreatoduodenal homeobox-1 (Pdx-1) mutations
IPF-1 is a transcription factor which is important in early pancreatic development, differentiation, maturation and maintenance of β-cells as well as transcriptional regulation of insulin, GLUT2, Glucokinase and Glucagon genes in the mature β-cell. Homozygous mutations in IPF-1 leads to pancreatic agenesis resulting in permanent neonatal diabetes and exocrine pancreatic insufficiency.[60] Heterozygous mutations lead to a variable defect in insulin secretion and diabetes (MODY4).[61]
HNF-1β mutations (MODY5)
HNF-1β, also known as transcription factor-2 (TCF-2), is a member of basic helix-loop-helix proteins. In the embryonic period HNF-1β is involved in the differentiation of the visceral endoderm. Apart from its role in pancreatic development, HNF-1β is required for development of the kidneys and the genital tract. Hence mutations in HNF-1β results in pancreatic insufficiency that includes both exocrine and endocrine dysfunction, cystic renal disease (RCAD-Renal cyst and diabetes syndrome) and anomalies of the vagina and the mullerian structures ranging from uterine aplasia to a bi-cornuate uterus. Early onset slowly progressive non diabetic renal failure may occur in some affected patients.[62,63] A wide spectrum of renal and genitourinary manifestations has been reported in HNF-1β mutation carriers.[64] The renal defect can manifest well before the onset of diabetes.
Neurogenic differentiation factor 1 (MODY6)
Neurogenic differentiation factor 1 (Neuro-D1) is an important transcription factor required for pancreatic development as well as β-cell differentiation. It has also been expressed in the enterocytes and the neurons. Mutations in Neuro-D1 results in an autosomal dominant disorder which resembles type 2 diabetes (MODY6).[65,66]
MODY due to enzymatic mutations
Glucokinase gene mutation (MODY2)
The prime role of Glucokinase (GCK) enzyme in the glucose sensing mechanism of the β-cell is already explained in the section of neonatal diabetes. Heterozygous mutations affecting the GCK gene results in mild fasting hyperglycemia that has been demonstrable from birth but is non progressive.[67] The Glucokinase enzyme is also functional in the liver. As a result of the molecular defect, there is reduced hepatic glucose sensing especially after a meal resulting in reduced glycogen synthesis and stimulation of hepatic gluconeogensis manifesting as postprandial hyperglycemia. However due to a milder degree of hyperglycemia, long term microvascular complications rarely occur in MODY2 compared to other forms of diabetes. GCK mutations may remain asymptomatic in nearly half of the carriers. Pregnancy unmasks the defect in some patients. GCK mutations have been shown to be responsible for gestational diabetes mellitus in 80% of Caucasian pregnant women when strict clinical criteria were applied for patient selection.[68] An affected mother can transmit the genetic defect to the offspring in which case the foetus is born with low birth weight owing to reduced insulin availability in the intrauterine period. However an unaffected foetus can develop macrosomia under the effect of maternal hyperglycemia.[69] Till date more than one hundred twenty different GCK mutations have been identified, each of which causes a variable degree of functional impairment.
Carboxy ester lipase enzyme-CEL (MODY8)
CEL, also known as Bile salt activated lipase, is secreted by the pancreatic acinar cells and is involved in hydrolysis of cholesteryl esters. Genome wide screen detected mutations in the carboxy ester lipase (CEL) gene to be a rare cause for diabetes and exocrine pancreatic insufficiency.[70] Subsequent research has suggested that the underlying functional defect could be due the accumulation of the misfolded protein product of the mutant gene in the pancreas.[71]
Other MODY mutations described
Several other gene mutations have been discovered in recent years as rare causes for early onset diabetes.
The Kruppel like factor 11 is a transcription factor which regulates the transcription of IPF-1 (Pdx-1), one of the crucial transcription factors involved in early pancreatic development.[72] Recently mutations in this gene are identified to be linked with MODY7. Pax 4 and Pax 6 are transcription factors involved in islet cell differentiation. In 2007 two novel mutations in the Pax 4 gene were identified in 2 patients from a Thai population as the cause for diabetes (now termed MODY9).[73]
Homozygous mutations in insulin gene resulting in PNDM have been discussed under the section of neonatal diabetes. The search for a genetic cause for diabetes in children with type 1 diabetes with negative autoantibodies identified mutations in the insulin gene as a rare cause for MODY (MODY10).[74]
The BLK gene codes for a non receptor tyrosine kinase of the src family of proto oncogenes which are involved in cell proliferation and differentiation. It is also found to exert a modulatory role in β-cell development and function. Mutations in BLK gene result in β-cell dysfunction and have been shown to be associated with early onset diabetes (MODY11).[75] Lately, there are reports suggesting mutations in ABCC8 and KCNJ11 genes as causes for MODY (MODY12 and 13 respectively).[76,77]
WHY IS MOLECULAR DIAGNOSIS OF MODY IMPORTANT?
There is extensive genetic heterogeneity with a wide phenotypic spectrum within and among the MODY groups. Table 2 displays the known MODY mutations and the clinical characteristics.[78,79,80] It is therefore important to characterize the different types of MODY and to distinguish it from other types of diabetes, because the treatment protocols for achieving optimal glycaemic control are specific for each type of MODY. Hyperglycemia due to mutations in HNF-1α has been found to be well controlled with Sulfonylureas for a number of years.[81] On the other hand, heterozygous mutations in the GCK gene (MODY2) do not require any pharmacological treatment except in certain physiologically demanding situations like pregnancy.
The clinical spectrum of MODY overlap with the features of both type 1 and type 2 diabetes, thus presenting a diagnostic challenge. Especially in a developing country like India, where there is lack of awareness for a genetic diagnosis of MODY, the probability of MODY being falsely diagnosed as either type 1 or type 2 diabetes is rather high. This may result in patients receiving inappropriate therapy with suboptimal glycaemic control. Identifying specific genes involved in MODY will have potential therapeutic benefit by streamlining the therapy for the particular genotype, better prognostication of the clinical course of the disease and widening our knowledge to give appropriate genetic counselling to the affected individuals.
On a wider perspective, molecular diagnosis of monogenic diabetes is necessary for:
-
Characterising the frequency of disease in the population
Shields et al. has reported that 80% of MODY cases in UK are misdiagnosed as either type 1 or type 2 diabetes.[82] While considering the high prevalence of diabetes in India, these figures are more likely to be higher. An alarming proportion (28%) of adolescents and young adults from Southern India were detected to have impaired fasting glucose during a search for metabolic abnormalities and their anthropometric correlates.[83] Some of these subjects could be harbouring asymptomatic MODY mutations particularly involving the GCK gene.
-
Establishing a direction for applied pharmacogenomics
Patients with mutations in HNF-4α (MODY1) and HNF-1α (MODY3) respond well to sulphonylureas alone, while mutations in GCK (MODY2) are managed with diet alone, and rarely require pharmacotherapy. Mutations in the HNF-1β gene results in severe β-cell dysfunction and these patients require insulin right from a very early stage of the disease. Even though one can diagnostically suspect MODY based on the phenotypical features we have mentioned earlier, one cannot treat the patient without knowing the specific genotype. The specific management protocols for each genetic subtype mandates genetic analysis in patients with clinical suspicion of MODY.
-
Avoiding insulin therapy
Given the fact that MODY is misdiagnosed as either type 1 or type 2 diabetes, many patients end up receiving insulin, along with the complications of insulin therapy and manage to achieve only suboptimal glycaemic control.[84] This can be avoided, and treatment can be streamlined if the genetic subtype is known via molecular genetic methods.
-
Understanding the relationship between MODY mutations and birth weight
It has been proven beyond doubt that a strong relationship exists between maternal malnutrition, low-birth weight and the predisposition to metabolic and cardiovascular disease in later life.[85,86] Studies have demonstrated that exposure to sub-optimal diet during pregnancy leads to gene expression changes resulting in ‘intra uterine programming’, thus helping the foetus in adapting to a suboptimal environment. Overfeeding of these infants in the postnatal period results in the onset of metabolic disease in the early years of life.[87] Epigenetic studies have shown reduced transcription of the HNF-4α gene (HNF-4α is a key transcription factor required for β-cell differentiation and glucose homeostasis) in rat models exposed to low-protein maternal diet during pregnancy and lactation. Such rats also developed diabetes at seventeen months of age showing continued repression of the gene throughout the ageing process.[88] Given the status of maternal malnutrition in our country, molecular studies might help establish the possible association between epigenetic regulation of HNF-4A, low birth weight and early onset diabetes. Genome-wide association scans have further proven the genetic association between low birth weight and type 2 diabetes in early adulthood. Genetic variants in the ADCY5 (Adenylate Cyclase 5) and CCNL1 (Cyclin L1) gene loci have been shown to be associated with low birth weight in Europeans.[89] However, the birth weight lowering effect of the ADCY5 polymorphism could not be replicated in a large Indian birth cohort, though its effect on glucose-insulin homeostasis and the risk of type 2 diabetes was well documented.[90]
-
Identifying the effect of consanguinity on the frequency of MODY
The 1992-1993 National Family Health Survey had shown high frequency of consanguinity and co-efficient of inbreeding in Southern Indian states of Tamil Nadu, Karnataka and Andhra Pradesh.[91] In this context, a higher rate of genetic disorders as MODY may be expected in the population. It is likely that MODY is an under diagnosed subset of diabetes in our country than is thought of.
-
Unravelling the impact of variations in the susceptibility genes on MODY
Genome-wide association studies have identified several susceptibility genes for developing type 2 diabetes.[92] Polymorphisms in the transcription factor 7-like 2 (TCF-7L2) gene have been shown to be strongly associated with increasing risk of developing type 2 diabetes in populations of various ethnicities including Asians.[93,94] Hypothetically speaking, the TCF-7L2 polymorphisms can augment the onset of MODY at an earlier age or modify its clinical expression. It would be interesting to investigate this possible interaction through genetic studies.
What is the status of monogenic diabetes worldwide?
Significant progress in molecular research is taking place worldwide. At the time of writing this paper eleven genes have been identified to harbour MODY related mutations. Epidemiological studies from different ethnic groups have shown varying frequencies of prevalence of these mutations.[95,96,97] Many countries are now offering molecular diagnosis to young people with diabetes before a therapeutic decision can be made. Additionally some of them have established monogenic diabetes registries.
Molecular diagnostic platform used in MODY
Sanger sequencing is a widely used genetic diagnostic platform for identifying MODY and is the gold standard sequencing platform to test single-gene genetic disorders. This sequencing technique has been reported to be >99% sensitive to detect a heterozygous base substitution in MODY genes. Only 3 genes i.e. HNF-1α, HNF-4α and GCK genes are sequenced and are available under as a diagnostic modality in European countries. The turn around time for the genetic diagnosis of MODY for sequencing these 3 genes is around fourty working days for unknown mutations and twenty working days for known mutations.
Based on the 2008 best practice guidelines for genetic diagnosis of MODY, it has been recommended that genetic testing should be performed only for subjects who fit into the clinical criteria of MODY.[80] However it has been reported that >50% of patients with MODY do not present with a classical picture of MODY. The genetic diagnosis of MODY has been recommended by a clinician only when he considers that a genetic diagnosis may make a difference in the clinical management of the patient. Furthermore, owing to issues of affordability, there is a delay from the time of diagnosis of diabetes to the definitive genetic diagnosis of MODY. At present due to limitations in throughput and scalability of the present Sanger sequencing, there are limitations to genetically characterizing patients with MODY, in a fast, comprehensive and cost-efficient manner.
Status of MODY genetics in India
India is making rapid advances in the field of molecular genetics and its application in the field of health and disease. However, few groups of investigators have embarked on the molecular genetic studies in diabetes particularly MODY. The performed studies have been centred on mutations on very few genes which were reported to be relatively common in the western population. Hence, data about MODY in India is basic and preliminary. In 2005, Mohan et al. reported the first single nucleotide polymorphism in HNF-1α gene associated with MODY and early type 2 diabetes from India.[98] They identified that frequency of Ala98Val polymorphism in the HNF-1α gene is significantly higher in MODY patients. The same polymorphism was associated with earlier age of diabetes onset in type 2 diabetes. However, since this study specifically looked at only 1 polymorphism in HNF-1α gene among the several genes involved and there could be several polymorphisms associated with each MODY gene, it cannot be representative of the entire genetic subtypes of MODY.
Another study from Northern India by Sahu et al. showed that A3243G mitochondrial mutation and HNF-1α gene mutations were infrequently associated with early onset type 2 diabetes.[99] However, this study attempted to look at the specific aetiologies of early onset type diabetes and was not uniquely focused on the genetic basis of MODY. In 2009 Radha et al. demonstrated that mutations in the HNF-1α gene comprise about 9% of clinically diagnosed MODY subjects in southern India.[100] They sequenced the coding and promoter regions of HNF-1α gene for mutations in unrelated South Indian subjects, in whom a clinical diagnosis of MODY was made. Nine novel variants comprising 7 mutations (1 novel mutation-538G > C at promoter region and 6 novel coding region mutations) and 2 polymorphisms in the HNF-1α gene were identified. They also observed co-segregation with diabetes of the Arg263His coding region mutation in eight members of one MODY family. In this study, mutations in only 1 gene that was implicated in MODY was looked for, while there are more than ten genes on the list currently. In 2011, the first report on HNF-4α mutation (MODY1) was published by the same group constituting 3.4% of the clinically diagnosed MODY.[101] They identified three novel mutations in the P2 promoter region of HNF-4α. In addition, they also reported single nucleotide polymorphisms of HNF-4α that are both susceptible to and protective against MODY and early onset T2DM.
CONCLUSION
MODY is a clinically and genetically heterogenous group of monogenic disorder causing diabetes in the younger population. Genetic diagnosis of this disorder has huge therapeutic and prognostic benefits. A comprehensive genetic service for the diabetic population particularly for the younger age group may benefit in the long run in terms of treatment as well as genetic counselling. The complexities of the molecular genetic techniques as well the financial requirements for the sophisticated instruments and reagents and the need for expert manpower limits the widespread availability of such a facility in our country at present, however steps in this direction are in the phase of evolution.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Whiting DR, Guariguata L, Weil C, Shaw J. IDF Diabetes Atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94:311–21. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Mohan V, Jaydip R, Deepa R. Type 2 diabetes in Asian Indian youth. Pediatr Diabetes. 2007:28–34. doi: 10.1111/j.1399-5448.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 3.Patterson CC, Dahlquist GG, Gyürüs E, Green A, Soltész G EURODIAB Study Group. Incidence trends for childhood type 1diabetes in Europe during 1989-2003 and predicted new cases 2005-20: A multicentre prospective registration study. Lancet. 2009;373:2027–33. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- 4.Slingerland AS, Shields BM, Flanagan SE, Bruining GJ, Noordam K, Gach A, et al. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia. 2009;52:1683–5. doi: 10.1007/s00125-009-1416-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryan AL, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008;29:265–91. doi: 10.1210/er.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flanagan SE, Patch AM, Mackay DJ, Edghill EL, Gloyn AL, Robinson D, et al. Mutations in ATP-sensitive K+channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56:1930–7. doi: 10.2337/db07-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1β gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89:2905–8. doi: 10.1210/jc.2003-031828. [DOI] [PubMed] [Google Scholar]
- 8.Temple IK, James RS, Crolla JA, Sitch FL, Jacobs PA, Howell WM, et al. An imprinted gene (s) for diabetes? Nat Genet. 1995;9:110–2. doi: 10.1038/ng0295-110. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell BD, Pollin TI. Genomic imprinting in diabetes. Genome Med. 2010;2:55. doi: 10.1186/gm176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma D, Shield JP, Dean W, Leclerc I, Knauf C, Burcelin RR, et al. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114:339–48. doi: 10.1172/JCI19876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel JR, Smallwood A, Harper A, Higgins MJ, Oshimura M, Reik W, et al. Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. J Med Genet. 2000;37:921–6. doi: 10.1136/jmg.37.12.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: Patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004;53:2713–8. doi: 10.2337/diabetes.53.10.2713. [DOI] [PubMed] [Google Scholar]
- 13.Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–66. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 14.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: New clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54:2503–13. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 15.Hoveyda N, Shield JP, Garrett C, Chong WK, Beardsall K, Bentsi-Enchill E, et al. Neonatal diabetes mellitus and cerebellar hypoplasia/agenesis: Report of a new recessive syndrome. J Med Genet. 1999;36:700–4. [PMC free article] [PubMed] [Google Scholar]
- 16.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF-1 gene coding sequence. Nat Genet. 1997;15:106–10. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 17.Njolstad PR, Sovik O, Cuesta-Munoz A, Bjorkhaug L, Massa O, Undlien DE, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588–92. doi: 10.1056/NEJM200105243442104. [DOI] [PubMed] [Google Scholar]
- 18.Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–9. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 19.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 20.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 21.Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA. 2007;104:15040–4. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38:682–7. doi: 10.1038/ng1802. [DOI] [PubMed] [Google Scholar]
- 23.Shaw-Smith C, Flanagan SE, Patch AM, Grulich-Henn J, Habeb AM, Hussain K, et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine-responsive megaloblastic anaemia. Pediatr Diabetes. 2012;13:314–21. doi: 10.1111/j.1399-5448.2012.00855.x. [DOI] [PubMed] [Google Scholar]
- 24.Hattersley A, Bruining J, Shield J, Njolstad P, Donaghue KC. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2009;10:33–42. doi: 10.1111/j.1399-5448.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 25.Greeley SA, Tucker SE, Worrel HI, Skowron KB, Bell GI, Philipson LH. Update in neonatal diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:13–9. doi: 10.1097/MED.0b013e328334f158. [DOI] [PubMed] [Google Scholar]
- 26.Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SE, Larkin B, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355:467–77. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 27.Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31:204–9. doi: 10.2337/dc07-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slingerland AS, Nuboer R, Hadders-Algra M, Hattersley AT, Bruining GJ. Improved motor development and good long-term glycaemic control with sulfonylurea treatment in a patient with the syndrome of intermediate developmental delay, early-onset generalised epilepsy and neonatal diabetes associated with the V59M mutation in the KCNJ11 gene. Diabetologia. 2006;49:2559–63. doi: 10.1007/s00125-006-0407-0. [DOI] [PubMed] [Google Scholar]
- 29.Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K, et al. Incidence, genetics and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatr Diabetes. 2012;13:499–505. doi: 10.1111/j.1399-5448.2011.00828.x. [DOI] [PubMed] [Google Scholar]
- 30.Letha S, Mammen D, Valamparampil JJ. Permanent neonatal diabetes due to KCNJ11 mutation. Indian J Pediatr. 2007;74:947–9. doi: 10.1007/s12098-007-0175-y. [DOI] [PubMed] [Google Scholar]
- 31.Ahamed A, Unnikrishnan AG, Pendsey SS, Nampoothiri S, Bhavani N, Praveen VP, et al. Permanent neonatal diabetes mellitus due to a C96Y heterozygous mutation in the insulin gene. A case report. JOP. 2008;9:715–8. [PubMed] [Google Scholar]
- 32.Jain V, Flanagan SE, Ellard S. Permanent neonatal diabetes caused by a novel mutation. Indian Pediatr. 2012;49:486–8. doi: 10.1007/s13312-012-0093-6. [DOI] [PubMed] [Google Scholar]
- 33.Tattersall RB. Mild familial diabetes with dominant inheritance. Q J Med. 1974;43:339–57. [PubMed] [Google Scholar]
- 34.Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24:44–53. doi: 10.2337/diab.24.1.44. [DOI] [PubMed] [Google Scholar]
- 35.Ledermann HM. Is maturity onset diabetes at young age (MODY) more common in Europe than previously assumed? Lancet. 1995;345:648. doi: 10.1016/s0140-6736(95)90548-0. [DOI] [PubMed] [Google Scholar]
- 36.Mohan V, Ramachandran A, Snehalatha C, Mohan R, Bharani G, Viswanathan M. High prevalence of maturity-onset diabetes of the young (MODY) among Indians. Diabetes care. 1985;8:371–4. doi: 10.2337/diacare.8.4.371. [DOI] [PubMed] [Google Scholar]
- 37.Cerf ME. Transcription factors regulating beta-cell function. Eur J Endocrinol. 2006;155:671–9. doi: 10.1530/eje.1.02277. [DOI] [PubMed] [Google Scholar]
- 38.Galán M, García-Herrero CM, Azriel S, Gargallo M, Durán M, Gorgojo JJ, et al. Differential effects of HNF-1α mutations associated with familial young-onset diabetes on target gene regulation. Mol Med. 2011;17:256–65. doi: 10.2119/molmed.2010.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3) Nature. 1996;384:455–8. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 40.Bacon S, Kyithar MP, Schmid J, Rizvi SR, Bonner C, Graf R, et al. Serum levels of pancreatic stone protein (PSP)/reg1A as an indicator of beta cell apoptosis suggest an increased apoptosis rate in hepatocyte nuclear factor-1 alpha (HNF1A-MODY) carriers from the third decade of life onward. BMC Endocr Disord. 2012;12:13. doi: 10.1186/1472-6823-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pontoglio M, Prie D, Cheret C, Doyen A, Leroy C, Froguel P, et al. HNF-1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep. 2000;1:359–65. doi: 10.1093/embo-reports/kvd071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isomaa B, Henricsson M, Lehto M, Forsblom C, Karanko S, Sarelin L, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia. 1998;41:467–73. doi: 10.1007/s001250050931. [DOI] [PubMed] [Google Scholar]
- 43.Lerario AM, Brito LP, Mariani BM, Fragoso MC, Machado MA, Teixeira R. A mis-sense TCF1 mutation in a patient with mody-3 and liver adenomatosis. Clinics. 2010;65:1059–60. doi: 10.1590/S1807-59322010001000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frayling TM, Bulamn MP, Ellard S, Appleton M, Dronsfield MJ, Mackie AD, et al. Mutations in the hepatocyte nuclear factor-1alpha gene are a common cause of maturity-onset diabetes of the young in the U.K. Diabetes. 1997;46:720–5. doi: 10.2337/diab.46.4.720. [DOI] [PubMed] [Google Scholar]
- 45.Kaisaki PJ, Menzel S, Lindner T, Oda N, Rjasanowski I, Sahm J, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in MODY and early-onset NIDDM: Evidence for a mutational hotspot in exon 4. Diabetes. 1997;46:528–35. doi: 10.2337/diab.46.3.528. [DOI] [PubMed] [Google Scholar]
- 46.Gragnoli C, Cockburn BN, Chiaramonte F, Gorini A, Marietti G, Marozzi G, et al. Early-onset type II diabetes mellitus in Italian families due to mutations in the genes encoding hepatic nuclear factor-1alpha and glucokinase. Diabetologia. 2001;44:1326–9. doi: 10.1007/s001250100644. [DOI] [PubMed] [Google Scholar]
- 47.Estalella I, Rica I, Perez de Nanclares G, Bilbao JR, Vazquez JA, San Pedro JI, et al. Mutations in GCK and HNF-1alpha explain the majority of cases with clinical diagnosis of MODY in Spain. Clin Endocrinol. 2007;67:538–46. doi: 10.1111/j.1365-2265.2007.02921.x. [DOI] [PubMed] [Google Scholar]
- 48.Lopez AP, Foscaldi SA, Perez MS, Rodriguez M, Traversa M, Puchulu FM, et al. HNF-1 alpha gene coding regions mutations screening, in a Caucasian population clinically characterized as MODY from Argentina. Diabetes Res Clin Pract. 2011;91:208–12. doi: 10.1016/j.diabres.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 49.Nishigori H, Yamada S, Kohama T, Utsugi T, Shimizu H, Takeuchi T, et al. Mutations in the hepatocyte nuclear factor-1 alpha gene (MODY3) are not a major cause of early-onset non-insulin-dependent (type 2) diabetes mellitus in Japanese. J Hum Genet. 1998;43:107–10. doi: 10.1007/s100380050049. [DOI] [PubMed] [Google Scholar]
- 50.Furuzawa GK, Giuffrida FM, Oliveira CS, Chacra AR, Dib SA, Reis AF. Low prevalence of MODY2 and MODY3 mutations in Brazilian individuals with clinical MODY phenotype. Diabetes Res Clin Pract. 2008;81:12–4. doi: 10.1016/j.diabres.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 51.Bellanné-Chantelot C, Lévy DJ, Carette C, Saint-Martin C, Riveline JP, Larger E, et al. Clinical characteristics and diagnostic criteria of maturity-onset diabetes of the young (MODY) due to molecular anomalies of the HNF1A gene. J Clin Endocrinol Metab. 2011;96:1346–51. doi: 10.1210/jc.2011-0268. [DOI] [PubMed] [Google Scholar]
- 52.Kyithar MP, Bacon S, Pannu KK, Rizvi SR, Colclough K, Ellard S, et al. Identification of HNF1A-MODY and HNF4A-MODY in Irish families: Phenotypic characteristics and therapeutic implications. Diabetes Metab. 2011;37:512–9. doi: 10.1016/j.diabet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 53.Boj SF, Parrizas M, Maestro MA, Ferrer J. A transcription factor regulatory circuit in differentiated pancreatic cells. Proc Natl Acad Sci USA. 2001;98:14481–6. doi: 10.1073/pnas.241349398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1) Nature. 1996;384:458–60. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 55.Ellard S, Colclough K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor-1alpha (HNF1A) and 4alpha (HNF4A) in maturity-onset diabetes of the young. Hum Mutat. 2006;27:854–69. doi: 10.1002/humu.20357. [DOI] [PubMed] [Google Scholar]
- 56.Pearson ER, Pruhova S, Tack CJ, Johansen A, Castleden HA, Lumb PJ, et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor-4alpha mutations in a large European collection. Diabetologia. 2005;48:878–85. doi: 10.1007/s00125-005-1738-y. [DOI] [PubMed] [Google Scholar]
- 57.Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA. Hepatocyte nuclear factor- 4α gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. J Pediatr. 2011;158:852–4. doi: 10.1016/j.jpeds.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 58.Malecki MT, Yang Y, Antonellis A, Curtis S, Warram JH, Krolewski AS. Identification of new mutations in the hepatocyte nuclear factor-4alpha gene among families with early onset Type 2 diabetes mellitus. Diabet Med. 1999;16:193–200. doi: 10.1046/j.1464-5491.1999.00073.x. [DOI] [PubMed] [Google Scholar]
- 59.Carette C, Dubois-Laforgue D, Saint-Martin C, Clauin S, Beaufils S, Larger E, et al. Familial young-onset forms of diabetes related to HNF4A and rare HNF1A molecular aetiologies. Diabet Med. 2010;27:1454–8. doi: 10.1111/j.1464-5491.2010.03115.x. [DOI] [PubMed] [Google Scholar]
- 60.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–10. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 61.Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-Cell- specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onsetdiabetes. Genes Dev. 1998;12:1763–8. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1beta gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–5. doi: 10.1038/ng1297-384. [DOI] [PubMed] [Google Scholar]
- 63.Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–8. doi: 10.1093/hmg/8.11.2001. [DOI] [PubMed] [Google Scholar]
- 64.Bellanné-Chantelot C, Chauveau D, Gautier JF, Dubois-Laforgue D, Clauin S, Beaufils S, et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med. 2004;140:510–7. doi: 10.7326/0003-4819-140-7-200404060-00009. [DOI] [PubMed] [Google Scholar]
- 65.Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–8. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 66.Kristinsson SY, Thorolfsdottir ET, Talseth B, Steingrimsson E, Thorsson AV, Helgason T, et al. MODY in Iceland is associated with mutations in HNF-1alpha and a novel mutation in Neuro-D1. Diabetologia. 2001;44:2098–103. doi: 10.1007/s001250100016. [DOI] [PubMed] [Google Scholar]
- 67.Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, et al. Familial hyperglycemia due to mutations in glucokinase: Definition of a subtype of diabetes. N Engl J Med. 1993;328:697–702. doi: 10.1056/NEJM199303113281005. [DOI] [PubMed] [Google Scholar]
- 68.Ellard S, Beards F, Allen LI, Shepherd M, Ballantyne E, Harvey R, et al. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia. 2000;43:250–3. doi: 10.1007/s001250050038. [DOI] [PubMed] [Google Scholar]
- 69.Spyer G, Hatersley AT, Sykes JE, Sturley RH, MacLeod KM. Influence of maternal and fetal glucokinase mutations in gestational diabetes. Am J Obstet Gynecol. 2001;185:240–1. doi: 10.1067/mob.2001.113127. [DOI] [PubMed] [Google Scholar]
- 70.Raeder H, Johansson S, Holm PI, Haldorsen IS, Mas E, Sbarra V, et al. Mutations in the CEL-VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38:54–62. doi: 10.1038/ng1708. [DOI] [PubMed] [Google Scholar]
- 71.Johansson BB, Torsvik J, Bjørkhaug L, Vesterhus M, Ragvin A, Tjora E, et al. Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY): A protein misfolding disease. J Biol Chem. 2011;286:34593–605. doi: 10.1074/jbc.M111.222679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fernandez-Zapico ME, van Velkinburgh JC, Gutierrez-Aguilar R, Neve B, Froguel P, Urrutia R, et al. MODY7 gene, KLF-11, is a novel p300-dependent regulator of Pdx1 (MODY4) transcription in pancreatic islet β-cells. J Biol Chem. 2009;284:36482–90. doi: 10.1074/jbc.M109.028852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, Nishi M, et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2007;92:2821–6. doi: 10.1210/jc.2006-1927. [DOI] [PubMed] [Google Scholar]
- 74.Bonfanti R, Colombo C, Nocerino V, Massa O, Lampasona V, Iafusco D, et al. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes auto-antibodies. Diabetes Care. 2009;32:123–5. doi: 10.2337/dc08-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borowiec M, Liew CW, Thompson R, Boonyasrisawat W, Hu J, Mlynarski WM, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and β-cell dysfunction. Proc Natl Acad Sci USA. 2009;106:14460–5. doi: 10.1073/pnas.0906474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. 2012;55:123–7. doi: 10.1007/s00125-011-2319-x. [DOI] [PubMed] [Google Scholar]
- 77.Bonnefond A, Philippe J, Durand E, Dechaume A, Huyvaert M, Montagne L, et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS One. 2012;7:37423. doi: 10.1371/journal.pone.0037423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hattersley AT. Maturity-Onset diabetes of the young: Clinical heterogeneity explained by genetic heterogeneity. Diabet Med. 1998;15:15–24. doi: 10.1002/(SICI)1096-9136(199801)15:1<15::AID-DIA562>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 79.Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:200–13. doi: 10.1038/ncpendmet0778. [DOI] [PubMed] [Google Scholar]
- 80.Ellard S, Bellanné Chantelot C, Hattersley AT. Best practice guidelines for the molecular genetic diagnosis of maturity onset diabetes of the young. Diabetologia. 2008;51:546–53. doi: 10.1007/s00125-008-0942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fajans SS, Brown MB. Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care. 1993;16:1254–61. doi: 10.2337/diacare.16.9.1254. [DOI] [PubMed] [Google Scholar]
- 82.Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): How many cases are we missing? Diabetologia. 2010;53:2504–8. doi: 10.1007/s00125-010-1799-4. [DOI] [PubMed] [Google Scholar]
- 83.Vasan SK, Thomas N, Christopher S, Gitanjali FS, Paul TV, Sanjeevi CB. Anthropometric measurements for the prediction of the metabolic syndrome: A cross-sectional study on adolescents and young adults from Southern India. Heart Asia. 2011;3:2–7. doi: 10.1136/ha.2009.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hathout EH, Cockburn BN, Mace JW, Sharkey J, Chen-Daniel J, Bell GI. A case of hepatocyte nuclear factor-1A diabetes (MODY 3) masquerading as type 1 diabetes in a Mexican-American adolescent and responsive to a low dose of sulphonylurea. Diabetes Care. 1999;22:867–8. doi: 10.2337/diacare.22.5.867. [DOI] [PubMed] [Google Scholar]
- 85.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–41. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 86.Stanner SA, Bulmer K, Andrès C, Lantseva OE, Borodina V, Poteen VV, et al. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ. 1997;315:1342–8. doi: 10.1136/bmj.315.7119.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sandovici I, Smith NH, Nitert MD, Ackers-Johnson M, Uribe-Lewis S, Ito Y, et al. Maternal diet and aging alter the epigenetic control of a promoter–enhancer interaction at the HNF4A gene in rat pancreatic islets. Proc Natl Acad Sci USA. 2011;108:5449–54. doi: 10.1073/pnas.1019007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Freathy RM, Mook-Kanamori DO, Sovio U, Prokopenko I, Timpson NJ, Berry DJ, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat Genet. 2010;42:430–5. doi: 10.1038/ng.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vasan SK, Neville MJ, Antonisamy B, Samuel P, Fall CH, Geethanjali FS, et al. Absence of birth-weight lowering effect of ADCY5 and near CCNL, but association of impaired glucose-insulin homeostasis with ADCY5 in Asian Indians. PLoS One. 2011;6:21331. doi: 10.1371/journal.pone.0021331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krishnamoorthy S, Audinarayana N. Trends in consanguinity in South India. J Biosoc Sci. 2001;33:185–97. doi: 10.1017/s0021932001001857. [DOI] [PubMed] [Google Scholar]
- 92.Janipalli CS, Kumar MV, Vinay DG, Sandeep MN, Bhaskar S, Kulkarni SR, et al. Analysis of 32 common susceptibility genetic variants and their combined effect in predicting risk of type 2 diabetes and related traits in Indians. Diabet Med. 2012;29:121–7. doi: 10.1111/j.1464-5491.2011.03438.x. [DOI] [PubMed] [Google Scholar]
- 93.Tong Y, Lin Y, Zhang Y, Yang J, Liu H, Zhang B. Association between TCF-7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: A large Human Genome Epidemiology (HuGE) review and meta-analysis. BMC Med Genet. 2009;19:10–15. doi: 10.1186/1471-2350-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chandak GR, Janipalli CS, Bhaskar S, Kulkarni SR, Mohankrishna P, Hattersley AT, et al. Common variants in the TCF-7L2 gene are strongly associated with type 2 diabetes mellitus in the Indian population. Diabetologia. 2007;50:63–7. doi: 10.1007/s00125-006-0502-2. [DOI] [PubMed] [Google Scholar]
- 95.Costa A, Bescós M, Velho G, Chêvre J, Vidal J, Sesmilo G, et al. Genetic and clinical characterisation of maturity-onset diabetes of the young in Spanish families. Eur J Endocrinol. 2000;142:380–6. doi: 10.1530/eje.0.1420380. [DOI] [PubMed] [Google Scholar]
- 96.Ehtisham S, Hattersley AT, Dunger DB, Barrett TG. First UK survey of paediatric type 2 diabetes and MODY. Arch Dis Child. 2004;89:526–9. doi: 10.1136/adc.2003.027821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yorifujia T, Fujimarua R, Hosokawaa Y, Tamagawab N, Shiozaki M, Aizu K, et al. Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatric Diabetes. 2012;13:26–32. doi: 10.1111/j.1399-5448.2011.00827.x. [DOI] [PubMed] [Google Scholar]
- 98.Anuradha S, Radha V, Deepa R, Hansen T, Carstensen B, Mohan V, et al. A prevalent amino acid polymorphism at codon 98 (Ala98Val) of the hepatocyte nuclear factor-1 is associated with maturity-onset diabetes of the young and younger age at onset of type 2 diabetes in Asian Indians. Diabetes Care. 2005;28:2430–5. doi: 10.2337/diacare.28.10.2430. [DOI] [PubMed] [Google Scholar]
- 99.Sahu RP, Aggarwal A, Zaidi G, Shah A, Modi K, Kongara S, et al. Etiology of early-onset type 2 diabetes in Indians: islet autoimmunity and mutations in hepatocyte nuclear factor-1α and mitochondrial gene. J Clin Endocrinol Metab. 2007;92:2462–7. doi: 10.1210/jc.2006-2467. [DOI] [PubMed] [Google Scholar]
- 100.Radha V, Ek J, Anuradha S, Hansen T, Pedersen O, Mohan V. Identification of novel variants in hepatocyte nuclear factor-1α gene in south Indian patients with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2009;94:1959–65. doi: 10.1210/jc.2008-2371. [DOI] [PubMed] [Google Scholar]
- 101.Anuradha S, Radha V, Mohan V. Association of novel variants in the hepatocyte nuclear factor-4A gene with maturity onset diabetes of the young and early onset type 2 diabetes. Clin Genet. 2011;80:541–549. doi: 10.1111/j.1399-0004.2010.01577.x. [DOI] [PubMed] [Google Scholar]