

Abstract
Neuropsychiatric disease frequently presents with an underlying hyporeactivity or hyperreactivity of the HPA stress axis, suggesting an exceptional vulnerability of this circuitry to external perturbations. Parental lifetime exposures to environmental challenges are associated with increased offspring neuropsychiatric disease risk, and likely contribute to stress dysregulation. While maternal influences have been extensively examined, much less is known regarding the specific role of paternal factors. To investigate the potential mechanisms by which paternal stress may contribute to offspring hypothalamic–pituitary–adrenal (HPA) axis dysregulation, we exposed mice to 6 weeks of chronic stress before breeding. As epidemiological studies support variation in paternal germ cell susceptibility to reprogramming across the lifespan, male stress exposure occurred either throughout puberty or in adulthood. Remarkably, offspring of sires from both paternal stress groups displayed significantly reduced HPA stress axis responsivity. Gene set enrichment analyses in offspring stress regulating brain regions, the paraventricular nucleus (PVN) and the bed nucleus of stria terminalis, revealed global pattern changes in transcription suggestive of epigenetic reprogramming and consistent with altered offspring stress responsivity, including increased expression of glucocorticoid-responsive genes in the PVN. In examining potential epigenetic mechanisms of germ cell transmission, we found robust changes in sperm microRNA (miR) content, where nine specific miRs were significantly increased in both paternal stress groups. Overall, these results demonstrate that paternal experience across the lifespan can induce germ cell epigenetic reprogramming and impact offspring HPA stress axis regulation, and may therefore offer novel insight into factors influencing neuropsychiatric disease risk.
Introduction
Stress dysregulation is a common underlying feature in neurodevelopmental and affective disorders, with hyperreactivity and hyporeactivity of the HPA stress axis reported in schizophrenia, autism, and depression (Arborelius et al., 1999; Moghaddam, 2002; Nestler et al., 2002; Walker et al., 2008; Corbett et al., 2009). Parental exposures to stress, infection, malnutrition, and advanced age have been linked with increased offspring presentation of neuropsychiatric diseases (Kinney et al., 2008; Rosenfield et al., 2010; Brown, 2011; Hultman et al., 2011; Patterson, 2011). The HPA stress axis is remarkably vulnerable to environmental perturbations, and parental experiences likely influence disease risk by altering stress pathway regulation (Matthews, 2002). While maternal insults experienced during pregnancy can directly impact fetal development, the mechanisms by which lifelong maternal or paternal exposures can affect offspring neurodevelopment are less well understood (Bale et al., 2010).
Epigenetic marks in germ cells are a proposed mechanism by which parental environmental exposures shape offspring neurodevelopment (Jirtle and Skinner, 2007; Guerrero-Bosagna and Skinner, 2012). When examining mechanisms of transgenerational transmission, studies of maternal experience must consider the complex maternal–fetal/neonatal interaction, including changes in the intrauterine environment, lactation, and maternal behaviors (Champagne, 2008). However, paternal studies in rodent models, where the sire does not participate in rearing of the offspring, allow for relative exclusion of factors outside of the germ cell contribution, providing a homogenous and easily accessible tissue for examining epigenetic marks (Rando, 2012).
Research from rodent models of male exposure to dietary challenge, drugs of abuse, or social defeat support the transmission of paternal experiences to offspring through epigenetic marks in sperm (Carone et al., 2010; Guerrero-Bosagna et al., 2010; Ng et al., 2010; Dietz et al., 2011; Vassoler et al., 2013). Although mature sperm lack the machinery necessary for chromatin remodeling, spermatozoa RNA populations, histone modifications, and methylation patterns reflect heritable responses to environmental challenges that may occur during spermatogenesis (Godmann et al., 2009; Skinner and Guerrero-Bosagna, 2009). Of specific interest is the sperm microRNA (miR) content, as miRs are a mode of dynamic epigenetic regulation that rapidly respond to environmental perturbations and likely regulate gene expression postfertilization (Giraldez et al., 2006; Rassoulzadegan et al., 2006; Grandjean et al., 2009; Leung and Sharp, 2010). Epidemiological evidence from Swedish famine years suggests that germ cell susceptibility to reprogramming, and its subsequent effects on future generations' disease risk, is greatest during the slow growth period of preadolescence (Kaati et al., 2002, 2007; Pembrey et al., 2006).
Therefore, to examine the effect of paternal stress exposure across the lifespan on offspring HPA axis regulation, male mice were exposed to 6 weeks of chronic stress, before breeding, either throughout puberty or only in adulthood. Offspring stress reactivity and gene array analysis of offspring stress regulatory brain regions, the the paraventricular nucleus (PVN) and bed nucleus of stria terminalis (BNST), were subsequently examined. Paternal sperm miR content was assessed as an indication of potential epigenetic marks associated with the reprogramming of offspring physiology or behavior.
Materials and Methods
Animals
All mice bred for paternal stress studies were virgin, in-house C57BL/6:129 F1 hybrid mice. The hybrid vigor of this background strain provides a reproducible balance of stress responsivity, behavioral performance, and maternal care (Mueller and Bale, 2006, 2007, 2008; Morgan and Bale, 2011). All mice were housed in a 12 h light/dark cycle with ambient temperature 22°C and relative humidity of 42%. Food (Purina Rodent Chow; 28.1% protein, 59.8% carbohydrate, 12.1% fat) and water were provided ad libitum. All studies were performed according to experimental protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee, and all procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Chronic variable stress
Administration of chronic variable stress was performed as described previously (Mueller and Bale, 2006). Briefly, males were randomly assigned to a control nonstressed group (n = 13) or to one of two experimental groups, receiving 42 d of chronic variable stress throughout puberty (PN28-70; n = 13) or in adulthood (PN56-98; n = 13). Seven different stressors were randomized and administered one per day. The order of seven stressors varied across weeks. The duration of stress (42 d) was selected to encompass a complete round of spermatogenesis, exposing sperm to stress at all stages of maturation (Oakberg, 1956). Stressors, selected because they are nonhabituating, do not induce pain, and do not affect food or water intake, included the following: 36 h constant light, 15 min exposure to fox odor (1:5000, 2,4,5-trimethylthiazole; Acros Organics), novel object (marbles) overnight, 15 min restraint in a 50 ml conical tube, multiple cage changes, novel 100 dB white noise (Sleep Machine; Brookstone) overnight, and saturated bedding overnight.
Breeding scheme
Following completion of chronic variable stress, all males were housed with a single female for 14 d to remove the acute effects of stress on male behavior and mature sperm stored in the epididymis. Males were then bred with naive females for 1–3 nights. To minimize male–female interactions, which may influence maternal investment or care and subsequently affect offspring development (Curley et al., 2011), pregnancy was established by confirmation of a copulation plug (identified each morning within 1 h after lights on) and signaled immediate removal of the female to her own cage containing a nestlet. Breeding resulted in litters sired from control (n = 8), pubertally stressed (n = 10), and adult stressed (n = 9) males.
Characterization of offspring physiological and behavioral phenotypes
Same-sex littermates were split into three groups for physiological and behavioral characterization (maximum of one male and female per litter per group). Groups were examined for either HPA stress axis responsivity and prepulse inhibition (PPI), performance on the tail suspension test (TST) and Barnes maze, or performance in the light–dark (LD) box and postmortem molecular measures. A minimum 10 d recovery period separated tests.
HPA axis responsivity.
Plasma corticosterone was measured following an acute 15 min restraint in a 50 ml conical tube. Testing occurred 2–5 h after lights on. Tail blood from adult mice (n = 6–9 per group) was collected at onset and completion of restraint (0 and 15 min, respectively) and 15 and 75 min after the end of restraint (30 and 90 min, respectively). Tail blood collection requires <30 s to complete. Samples were immediately mixed with 50 mm EDTA and centrifuged 10 min at 5000 rpm. Plasma was collected and stored at −80°C until analysis. Corticosterone levels were determined by 125I-corticosterone radioimmunoassay (MP Biomedicals) following kit instructions. The minimum detection limit of the assay was 7.7 ng/ml, and the intraassay coefficient of variation was 7.1%.
Prepulse inhibition.
PPI of acoustic startle was recorded in SR-LAB startle chambers (San Diego Instruments) as described previously (Geyer and Dulawa, 2003). All test sessions were conducted 2–6 h after lights off. A PPI test session began with 5 min acclimation to background noise (65 dB) followed by five consecutive acute habituation tones (40 ms duration; 120 dB). Ten repetitions of each of the following trial types were then presented in pseudorandomized blocks: startle pulse only (40 ms; 120 dB), no stimulus (65 dB; background), and prepulses +4, +8, or +16 dB above background (20 ms; 69, 73, or 81 dB) preceding the startle pulse (40 ms; 120 dB) by 100 ms. Intertrial intervals averaged 15 s. Acoustic startle response was defined as the peak startle magnitude recorded during 65 consecutive 1 ms readings following the startle pulse onset. The percentage PPI for each prepulse intensity was calculated for each animal (n = 5–6 per group) as [1 − (average response to prepulse + startle)/(average response to startle only)] × 100 before group averages were compared. Animals with average response in startle only trials of ≤50 mV were excluded from analysis.
Tail suspension test.
The TST was performed as described previously (Steru et al., 1985). Briefly, adult mice (n = 7–10 per group) were secured to a rod by adhesive tape placed 1 cm from the tip of the tail and suspended 50 cm above the bench top. Testing occurred 2–5 h after lights on. Total immobility time, defined as the absence of movement except whisker twitches and respiration, was recorded over the 6 min test session. Testing was recorded digitally and analyzed manually using ANY-maze v4.75 software (Stoelting).
Barnes maze.
A modified Barnes maze was performed as described previously (Mueller and Bale, 2007). Briefly, adult mice (n = 7–10 per group) were trained on two trials per day for 3 d (4 h separation between trials) to find a target escape box. The location of the target escape box under 1 of 24 holes evenly spaced around the perimeter of a 90 cm circular disk remained constant throughout training. Latency to identify the target escape box by nose poke or head deflection was determined for each trial. Each trial ended when the mouse identified and entered the target box or after 4 min of elapsed time. If the target box was not successfully entered within 4 min, the investigator guided the mouse into the target box, and a latency of 240 s was assigned. Testing was digitally recorded and analyzed manually using ANY-maze v4.75 software (Stoelting).
Light–dark box.
The light–dark box test was performed as described previously (Bale et al., 2000; McEuen et al., 2009). Adult mice (n = 6–8 per group) were placed in the light compartment at the beginning of the 10 min test session. Light intensities were set at 5 lux in the dark compartment and 300 lux in the light compartment. All testing occurred 2–5 h after lights off. Total time spent in the light compartment and the number of light-to-dark transitions were analyzed with ANY-maze v4.75 software (Stoelting).
Mechanistic evaluation of blunted stress responsivity
Acute selective serotonin reuptake inhibitor.
Activation of the HPA axis by citalopram, in the absence of any additional stressor was examined as described previously (Goel et al., 2011). Plasma corticosterone was measured in offspring (n = 5–8 per group) as described above, except that animals were injected with 15 mg/kg citalopram (Sigma-Aldrich) or vehicle (0.9% saline) at time 0 min and were not restrained.
Adult tissue collection.
Adult male and female offspring of paternal stress or control sires (n = 6–8 per group) were rapidly decapitated under isoflurane anesthesia at 22–24 weeks of age. Whole brains were removed, immediately frozen on dry ice, and stored at −80°C. The pituitary gland and left adrenal gland were removed, flash frozen in liquid nitrogen, and stored at −80°C. Approximately 5 × 106 sperm were isolated from the caudal epididymis of adult males into PBS with 1% BSA at 37°C through a swim-up assay. After settling for 30 min, sperm-containing supernatant was centrifuged for 5 min at 4000 rpm. Sperm pellets were stored −80°C.
Peripheral gene expression.
Whole pituitary and left adrenal glands (n = 6–8 per group) were bath sonicated for 7 min (30 s on/off cycles) in TRIzol reagent (Invitrogen), and total RNA was isolated according to manufacturer's protocol. cDNA was transcribed using the High-Capacity cDNA reverse transcriptase kit (Applied Biosystems). Changes in pituitary corticotrophin-releasing factor receptor 1 (CRFr1; NM_007762.4, Mm00432670_m1), pituitary proopiomelanocortin (POMC; NM_008895.3, Mm00435874_m1), adrenal melanocortin 2 receptor (Mc2r; NM_008560.2, Mm00434865_s1), and adrenal 11-β hydroxysteriod dehydrogenase 1 (11βHSD-1; NM_001044751.1, Mm00476182_m1) were measured by quantitative real-time PCR using TaqMan gene expression assays (Applied Biosystems). Samples were run in triplicate for the target genes and endogenous controls (pituitary, Gapdh, NM_0080842.2, Mm99999915_g1; adrenal, Actb, NM_007393.3, Mm00607939_s1) on the same 96-well plate. Analysis was performed by the comparative Ct method, and expression levels were normalized to control male offspring (Schmittgen and Livak, 2008).
Brain gene expression.
Whole brains were cryosectioned at −20°C. Using a hollow needle (Ted Pella), brain regions were micropunched according to the atlas of Paxinos and Franklin (2001) as follows: BNST, 0.75 mm bilateral punches from one 300 μm slice +0.25 to −0.05 relative to bregma (atlas Figures 25–32); PVN, 1.00 mm punch along the midline from two successive 300 μm slices −0.50 to −0.80 and −0.80 to −1.10 relative to bregma (atlas Figures 36–40). Punches were bath sonicated for 2.5 min (30 s on/off cycles) in TRIzol reagent (Invitrogen). RNA was isolated by RNeasy kit (Qiagen) and suspended in RNase-free water. Total RNA (n = 5–6 per group) was sent to the University of Pennsylvania Path BioResource Molecular Profiling Core for Affymetrix GeneChip Mouse Gene 1.0 ST analysis. Gene set enrichment analysis (GSEAv2.0.7; Broad Institute, Cambridge, MA) was used to interpret patterns of gene expression (Mootha et al., 2003; Subramanian et al., 2005). Collections of c2 (curated: Kegg, Biocarta, and Reactome), c3 (motif), and c5 [gene ontology (GO)] annotated gene sets were obtained from the Molecular Signature Database (MSigDBv3.0.1; Broad Institute) available for use with GSEA software.
Analysis of sperm miR environment
Total RNA (n = 4 per group) was extracted from sperm pellets using TRIzol reagent (Invitrogen). Total RNA (100 ng) was reverse transcribed to cDNA using Megaplex RT pool A and B primers and Multiscribe reverse transcriptase, and then quantified using the TaqMan Array MicroRNA card A and B Set v3.0 (Applied Biosystems). Analysis was performed using the comparative Ct method (Schmittgen and Livak, 2008). For each sample, the average of the Ct values of mammu6 and sno202 was used as an endogenous loading control. Expression levels of each sample were normalized to the average expression level of male controls. miRs with expression in <50% of samples or Ct values >35 were excluded, such that analysis of significant differences in expression of specific miRs was conducted with n = 2–4 per group. The miRWalk database, which identifies putative miR target sequences in mRNA 3′-UTR commonly predicted by miRWalk, miRDB.org, miRanda, and TargetScan algorithms, was used to determine predicted targets of significantly changed miRs (Dweep et al., 2011).
Statistics
An investigator blind to animal treatment group conducted all studies and analyses. Total corticosterone was analyzed by multivariate ANOVA (sex by paternal stress for restraint experiment; sex by paternal stress by drug for citalopram experiment) with time as a repeated measure. PPI was analyzed by two-way ANOVA (sex by paternal stress) with prepulse intensity as a repeated measure. Baseline acoustic startle, TST immobility time, and LD box measures were analyzed by two-way ANOVA (sex by paternal stress). Latency to find target on the Barnes maze was analyzed by two-way ANOVA (sex by paternal stress) with trial as a repeated measure. Adrenal and pituitary gene expression analysis was analyzed by two-way ANOVA (sex by paternal stress). Data greater than two SDs away from each group mean were considered outliers and excluded from analysis. Main effects and interactions were further analyzed with Tukey's HSD post hoc test. Significant differences were identified at p ≤ 0.05. All statistics above were performed using JMP8 (SAS) software, and all data are reported as mean ± SEM. Microarray data were modeled as “control” versus “paternal stress” in accordance with behavioral and physiological data. Thresholds for multiple comparisons were set at false discovery rate (FDR) ≤ 0.05. Phenotype permutations (1000) were computed in GSEA to determine the FDR, nominal p value, and normalized enrichment score (NES) of each gene set. Gene sets with FDRs of ≤0.25, p ≤ 0.01, and NESs of ≥1.6 met significance thresholds. For miR analysis, heat maps and hierarchical clustering were performed using MuliExperiment Viewer (TM4.org) with Pearson's correlation, and miRs significantly altered in both pubertal and adult stress groups were identified by one-way ANOVA with post hoc Student's t test (p < 0.05).
Results
Analysis of offspring physiology and behavior
Offspring HPA axis
To determine whether paternal exposure to chronic stress during pubertal or adult windows altered offspring stress responsivity, we examined plasma corticosterone levels following a 15 min restraint stress. There were significant effects of paternal stress (F(2,35) = 9.3, p = 0.0006) and sex (F(1,35) = 22.68, p < 0.0001) on corticosterone levels that did not interact (F(2,35) = 0.58, p = 0.56), and a significant effect of time (F(3,33) = 107.4, p < 0.0001) (Fig. 1A,B). Similarly, there were significant effects of paternal stress (F(2,35) = 5.9, p = 0.006) and sex (F(1,35) = 18.7, p = 0.0001) on total corticosterone that did not interact (F(2,35) = 0.2, p = 0.86). Total corticosterone was lower in paternal stress conditions compared to controls and in males compared to females (p < 0.05) (Fig. 1A,B, inset). The following statistical outliers in corticosterone levels at each of the four time points were removed: one pubertal male and one pubertal female at time 0, one pubertal female at time 15, none at time 30, and one adult male and one adult female at time 120.
Figure 1.

Paternal stress experienced throughout puberty or in adulthood elicited stress dysregulation in offspring. A, B, Both male (A) and female (B) offspring of sires that had been exposed to chronic stress throughout puberty or only in adulthood produced less corticosterone relative to offspring of control sires following a 15 min restraint (shaded column). Total AUC of corticosterone production (shown in the inset) showed a significant decrease in both stress groups relative to controls. An expected sex difference in corticosterone production was observed, with females showing a greater response than males. Data are presented as mean ± SEM. n = 6–9 litters per group. *p < 0.05.
Offspring behavior
To examine the degree to which blunted stress axis responsivity was representative of global stress sensitivity in offspring of stressed sires, we assessed offspring PPI, stress-coping behavior in the TST, spatial learning in an adverse environment on the Barnes maze, and anxiety-like behavior in the LD box. PPI did not differ between males and females (F(1,23) = 0.54, p = 0.47) or between paternal stress and control groups (F(2,23) = 0.28, p = 0.75), though an expected within-subject effect of prepulse intensity was observed, with higher prepulses eliciting greater prepulse inhibition (F(2,22) = 64.1, p < 0.0001) (Fig. 2A,B). Max startle to 120 dB tone was also unaffected by sex (F(1,23) = 0.55, p = 0.46) or paternal stress (F(2,23) = 0.07, p = 0.93) (Fig. 2C). In the TST, there was no effect of sex (F(1,40) = 2.2, p = 0.12) or paternal stress (F(2,40) = 0.01, p = 0.89) on total time spent immobile (Fig. 2D). Analysis of latency to find the target on the Barnes maze similarly revealed no effect of sex (F(1,29) = 0.66, p = 0.78) or paternal stress (F(2,29) = 0.41, p = 0.66), and latency to find target decreased across trials for all groups (F(5,25) = 44.83, p < 0.0001) (Fig. 2E,F). In the LD box, females spent significantly more time in the light than males (F(1,32) = 5.9, p = 0.02), but there was no effect of paternal stress (F(2,32) = 1.5, p = 0.21) (Fig. 2G). To determine whether Barnes maze and LD box results were affected by locomotor ability, light-to-dark transitions were measured in the LD box. There was no effect of sex (F(1,32) = 3.44, p = 0.07) or paternal stress on the number of transitions (F(2,32) = 0.20, p = 0.81) (Fig. 2H). Statistical outliers were removed only from analysis of maximum startle and prepulse inhibition (one adult female) (Fig. 2B,C) and from analysis of total time in light (one pubertal male and one control female) (Fig. 2G).
Figure 2.
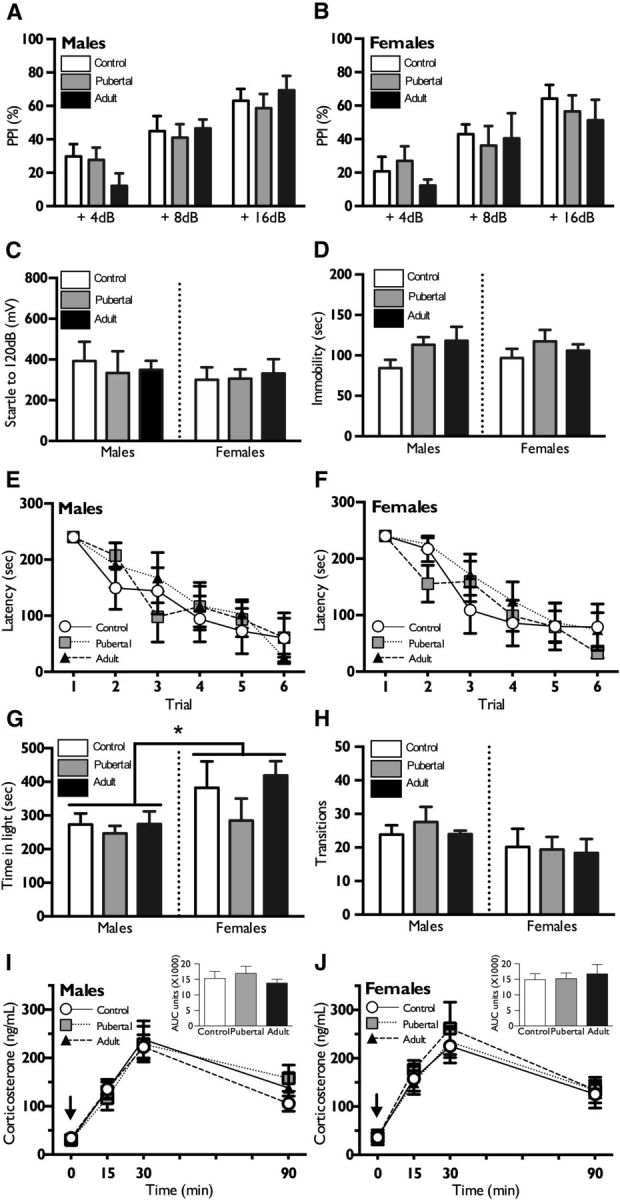
Paternal stress does not alter offspring performance in behavioral assessments or HPA response to an SSRI. A, B, Neither male (A) nor female (B) offspring of stressed sires showed altered PPI of the acoustic startle response compared to control offspring. C, PPI max startle was also unchanged by paternal stress in both sexes. D, No significant differences in time spent immobile on the tail suspension test were observed between control and paternally stressed male or female offspring. E, F, In the Barnes maze spatial learning and memory task, no differences were detected in the latency to learn the task. G, H, In the light–dark box, no treatment group differences were detected for time spent in the light or transitions between the compartments. There was an overall effect of females spending more time in the light compartment than males. I, J, Following administration of an SSRI, citalopram, at time 0 (black arrow), male and female offspring of stressed or control sires exhibited similar stress reactivity. Total AUC of corticosterone production (shown in the inset) did not vary with sex or paternal stress group. Data are presented as mean ± SEM. n = 5–9 litters per group. *p < 0.05.
Mechanistic evaluation of blunted stress reactivity
Acute selective serotonin reuptake inhibitor treatment
To investigate the potential role of serotonin in mediating changes in offspring stress axis responsivity, we evaluated offspring corticosterone levels in response to acute selective serotonin reuptake inhibitor (SSRI) administration. Citalopram administration elicited similar HPA activation in all groups, as there was a main effect of drug (F(1,52) = 14.6, p = 0.0004) on corticosterone level that did not interact with sex (F(1,52) = 0.003, p = 0.96) or paternal stress (F(2,52) = 0.88, p = 0.42) (Fig. 2I,J). The following statistical outliers in corticosterone levels at each of the four time points were removed: at time 0, one pubertal vehicle male, one citalopram control female, one pubertal vehicle female, and one adult vehicle female; none at time 15 or time 30; and at time 120, 1 pubertal vehicle female and one adult citalopram female.
Peripheral gene expression analysis
To determine whether changes in peripheral control of the HPA axis explain offspring blunted stress responsivity following paternal stress exposure, we evaluated gene expression in offspring pituitary and adrenal glands. Neither pubertal nor adult paternal stress significantly altered the expression of genes involved in the offspring HPA axis response, including pituitary CRFr1 (F(2,27) = 1.30, p = 0.29), pituitary POMC (F(2,27) = 0.13, p = 0.90), adrenal Mc2r (F(2,26) = 2.32, p = 0.12), or adrenal 11βHSD-1 (F(2,26) = 0.55, p = 0.58). Consistent with sex differences in the expression of these genes reported previously (Goel and Bale, 2010), significant effects of sex were observed: CRFr1 was higher in males (F(1,27) = 21.42, p < 0.0001), POMC was higher in females (F(1,27) = 14.39, p = 0.001), Mc2r was higher in females (F(1,26) = 24.28, p < 0.0001), and 11βHSD-1 was higher in males (F(1,26) = 16.92, p = 0.003). There were no interactions of paternal stress and sex for CRFr1 (F(2,27) = 0.24, p = 0.79), POMC (F(2,27) = 0.01, p = 0.98), MC2R (F(2,26) = 2.0, p = 0.16), or 11βHSD-1 (F(2,26) = 0.05, p = 0.95) (Fig. 3A–D).
Figure 3.

Stress axis-related gene expression in offspring pituitary and adrenal glands was not significantly different between control (C), pubertal stress (P), or adult stress (A) groups. A–D, Real-time PCR analysis revealed no effect of paternal stress on pituitary CRFr1 (A), POMC (B), adrenal Mc2r (C), or 11βHSD-1 (D). Sex differences were observed, with males showing higher expression of CRFr1 and 11βHSD-1 and lower expression of Mc2r and POMC relative to females. Data are presented as mean ± SEM. n = 6–8 litters per group. **p < 0.001; ***p < 0.0001.
Brain gene expression analysis
To examine changes in central regulators of the HPA axis following paternal stress, we evaluated patterns of gene expression in the PVN and BNST with a microarray. By multiple comparison analysis, no single gene met both threshold and significance criteria. By GSEA analyses, c3 gene sets in the PVN and c3 and c5 gene sets in the BNST were enriched following stress (Table 1). Gene sets enriched in offspring PVN following paternal stress were sorted according to the degree of enrichment (Table 2). Due to the large number of significant gene sets in the BNST, enriched gene sets were organized according to predicted function or miR family (Table 3). Members of c3 gene sets share known or likely regulatory elements in their promoters or 3′-UTRs, and c3 gene sets are named according to the transcription factor or miR that binds that conserved sequence. c3 gene sets sharing the same name represent distinct binding sites for the given transcription factor or miR. Members of c5 gene sets share a common gene ontology annotation, and c5 gene sets are named for that GO annotation.
Table 1.
Summary of gene sets meeting GSEA enrichment criteria of FDR ≤ 0.25, p ≤ 0.01, and NES ≥ 1.6
| Total | PVN (enriched) | BNST (enriched) | |
|---|---|---|---|
| c2 (curated) | 305 | 0 (0.0%) | 0 (0.0%) |
| c3 (motif) | 776 | 16 (2.1%) | 114 (14.7%) |
| c5 (gene ontology) | 969 | 0 (0.0%) | 6 (<1%) |
Table 2.
Gene sets enriched in offspring PVN following paternal stress
| Gene set | Type | Size | NES | p | FDR |
|---|---|---|---|---|---|
| NR3CI (GR) | c3 | 107 | −1.683 | 0.008 | 0.235 |
| CEBPdelta | c3 | 212 | −1.682 | 0.001 | 0.220 |
| FOXD3 | c3 | 178 | −1.682 | 0.006 | 0.207 |
| miR-522 | c3 | 144 | −1.678 | 0.008 | 0.203 |
| NKX2–5 | c3 | 109 | −1.665 | 0.008 | 0.196 |
| PAX8 | c3 | 88 | −1.661 | 0.004 | 0.194 |
| TITF1 | c3 | 212 | −1.657 | 0.002 | 0.194 |
| PIT1 | c3 | 195 | −1.649 | 0.010 | 0.201 |
| DDIT3 | c3 | 209 | −1.641 | 0.004 | 0.200 |
| NFI | c3 | 226 | −1.633 | 0.001 | 0.193 |
| FOXI3 | c3 | 165 | −1.625 | 0.010 | 0.193 |
| IPFI | c3 | 220 | −1.617 | 0.001 | 0.190 |
| AR | c3 | 227 | −1.606 | 0.002 | 0.203 |
Members of c3 gene sets share a binding motif for the designated transcription factor or miR. Gene sets defined by conserved sequence only and those not meeting the criteria NES ≥ 1.6, p ≤ 0.01, and FDR ≤ 0.25 were omitted.
Table 3.
Gene sets enriched in offspring BNST following paternal stress

Members of c3 gene sets share a binding motif for designated transcription factor or miRNA. c3 gene sets sharing the same name represent distinct binding sites for the transcription factor or miR given. Members of c5 gene sets share common ontolgy and do not necessarily represent coregulated genes. Gene sets defined by conserved sequence only and those not meeting the criteria NES ≥ 1.6, p ≤ 0.01, and FDR ≤ 0.25 were omitted. HAT, Histone acetyltransferase.
Analysis of sperm miR environment
To examine the role of paternal stress exposure in regulation of sperm miR environment, we assayed changes in sperm miR expression following breeding of stressed and control sires. Hierarchical clustering analysis using Pearson's correlation as a metric successfully segregated pubertal and adult stress samples from control samples, as visualized in a heat map (Fig. 4A). Expression of the following miRs was significantly increased in both pubertal stress and adult stress samples: miR-193-5p (F(2,9) = 7.3, p = 0.01), miR-204 (F(2,9) = 5.7, p = 0.02), miR-29c (F(2,8) = 5.3, p = 0.03), miR-30a (F(2,9) = 17.2, p = 0.0008), miR-30c (F(2,8) = 16.6, p = 0.001), miR-32 (F(2,6) = 5.3, p = 0.046), miR-375 (F(2,8) = 13.5, p = 0.003), miR-532–3p (F(2,7) = 5.4, p = 0.04), and miR-698 (F(2,8) = 6.3, p = 0.02) (Fig. 4B). Genes predicted to be targets of at least three of the nine significantly increased miRs are shown in Table 4.
Figure 4.

Paternal stress experienced throughout puberty or in adulthood produced robust changes in sperm miR content. A, Hierarchical clustering of sperm miR expression isolated a single clade containing all pubertal and adult stress males, and no controls (bolded red line). n = 4 per group. B, Analyses of significant differences in expression of specific miRs revealed nine that were significantly increased in both paternal stress groups. Data are presented as mean ± SEM. *p < 0.05, different from controls; #p < 0.05, different from pubertal stress.
Table 4.
Common predicted gene targets of the nine identified sperm miRNAs
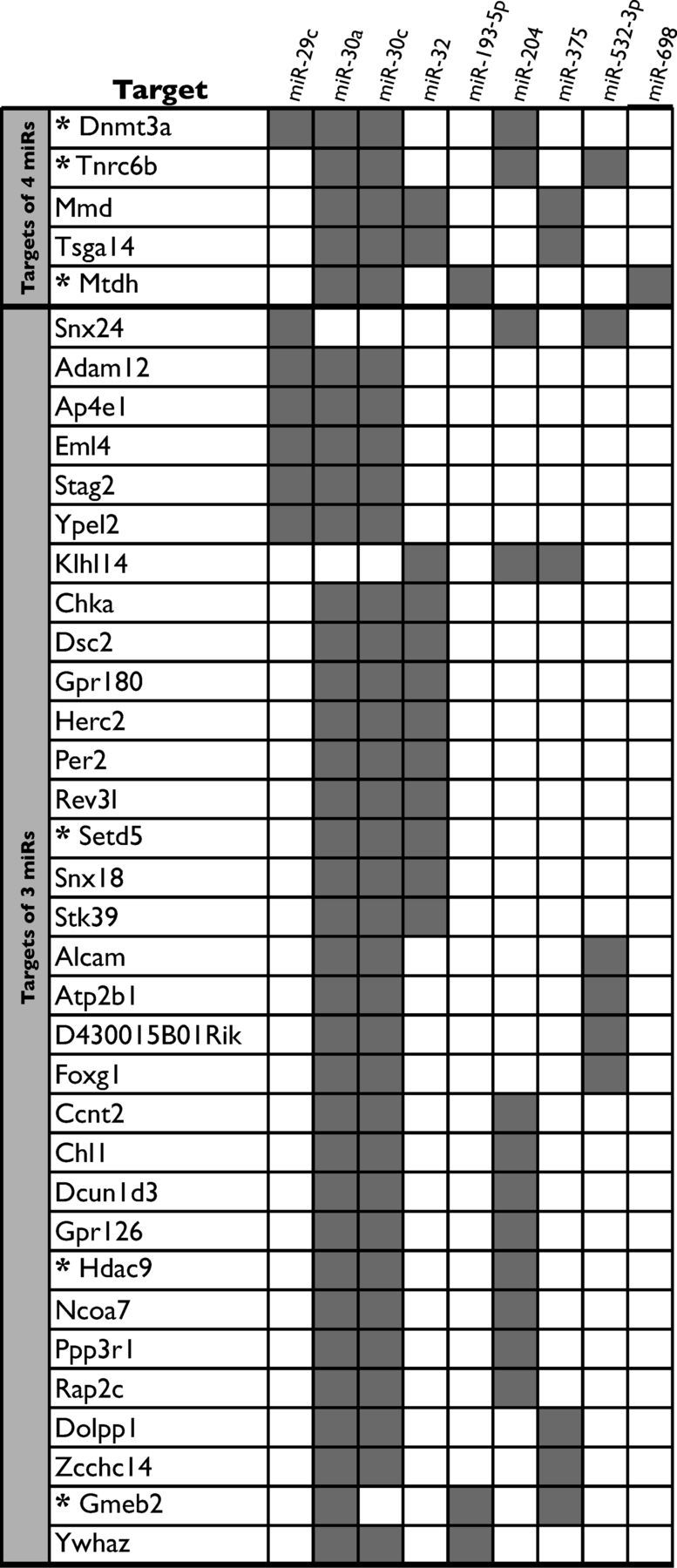
Gray boxes indicate that the given gene was a predicted miR target in miRanda, miRDB, miRwalk, and TargetScan alorigthms. Asterisks denote a predicted function in chromatin regulation, DNA methylation, or miR processing.
Discussion
Dysregulation of stress neurocircuitry is a common underlying feature in neuropsychiatric disease (Arborelius et al., 1999; Moghaddam, 2002; Nestler et al., 2002; Walker et al., 2008; Corbett et al., 2009). Adverse parental life experiences likely contribute to offspring HPA stress axis dysregulation through germ cell epigenetic modifications (Bale et al., 2010; Franklin et al., 2012). Previous studies have demonstrated that transgenerational metabolic, endocrine, and stress phenotypes can transmit through the male lineage following an initial maternal exposure, findings that emphasize the dynamic reprogramming of male germ cells (Jimenez-Chillaron et al., 2009; Franklin et al., 2010, 2011; Guerrero-Bosagna et al., 2010; Dunn and Bale, 2011; Morgan and Bale, 2011). Furthermore, identification of offspring physiological and behavioral changes following direct paternal perturbations has highlighted sperm epigenetic marks as a potential mechanism of transmission (Carone et al., 2010; Ng et al., 2010; Dietz et al., 2011; Vassoler et al., 2013). Therefore, in our study, male mice received 6 weeks of stress before breeding, and offspring outcomes were evaluated. As epidemiological studies suggest that the greatest effects on offspring and grand-offspring health occur during developmental windows of increased germ cell vulnerability, we exposed males to stress either throughout puberty or only in adulthood (Kaati et al., 2002, 2007; Pembrey et al., 2006). To identify potential epigenetic marks in sperm associated with the transmission of offspring phenotype, sperm miR content was assessed.
In our HPA stress axis assessment, we found that offspring from both paternal stress groups displayed significantly blunted corticosterone responses to acute restraint. While maternal stress has been shown to elicit sex-specific responses in stress-related outcomes, paternal stress surprisingly affected stress reactivity similarly in both sexes, suggesting distinct modes of maternal versus paternal transmission (Mueller and Bale, 2007, 2008; Franklin et al., 2010, 2011; Kapoor and Matthews, 2011; Morgan and Bale, 2011). It was also unexpected that HPA stress axis dysregulation occurred regardless of the timing of paternal stress exposure. Though epidemiological studies of Swedish famine years highlight preadolescence as a period of heightened germ cell susceptibility to malnutrition, our data illustrate that germ cell reprogramming by stress exposure occurs in adulthood (Kaati et al., 2002, 2007). Relative germ cell vulnerability throughout the lifespan may depend on the type of perturbation encountered, with severe caloric restriction and chronic stress eliciting distinct mechanisms.
The specificity of HPA stress axis reprogramming by paternal stress was remarkable, as is highlighted by the lack of significant differences in any of the assessed behavioral measures. These outcomes support divergent functioning and regulation of physiological and behavioral stress reactivity. Stress pathway dysregulation, either increased or decreased reactivity, reflects an organism's inability to respond appropriately to a changing environment, and thus may characterize a disease state or a predisposition toward one. Alternatively, a reduced physiological stress response may reflect an adaptive response programmed by the paternal lineage as a protective measure, ensuring greater offspring fitness in what is expected to be a more stressful environment. Interestingly, rodents models have previously demonstrated offspring HPA axis dysregulation following maternal or paternal exposures to diverse challenges including alcohol, lead, chronic stress, or social defeat, suggesting that the HPA axis is especially sensitive to environmental manipulations—the proverbial canary in the coal mine—thus warranting greater mechanistic examination (Coe et al., 2003; Weaver et al., 2004; Virgolini et al., 2006; Hellemans et al., 2008; Kapoor and Matthews, 2008; Mueller and Bale, 2008; Dietz et al., 2011; Harris and Seckl, 2011; Morgan and Bale, 2011).
To assess the neural circuitry underlying the blunted HPA in paternal stress offspring, we first used a pharmacological approach of an acute administration of the SSRI, citalopram, to stimulate the HPA in the absence of external stress. The serotonin system is a potent modulator of HPA stress axis reactivity, and has been reported previously to be disrupted in offspring following maternal stress (Heisler et al., 2007; Mueller and Bale, 2008; Franklin et al., 2011; Huang et al., 2012). However, we found that citalopram evoked similar corticosterone production and recovery levels in control and paternally stressed offspring, suggesting that the blunted response detected following a restraint stress did not likely involve dysregulation of serotonergic projections to the PVN. Given intact serotonergic regulation following paternal stress, principal components of the HPA axis, the pituitary and adrenal glands, were also examined in paternally stressed offspring for altered gene expression that may explain stress pathway dysregulation. We found no expression differences between groups in the pituitary gland (CRF receptor-1, the CRF receptor on pituitary corticotropes, and proopiomelanocortin, the precursor to ACTH) or in the adrenal gland (melanocortin receptor-2, the ACTH receptor in the adrenal cortex, and 11βHSD-1, the rate limiting enzyme in corticosterone production), suggesting that the reprogramming of the offspring HPA axis by paternal stress exposure lies centrally.
By microarray analyses, we examined broad changes in gene expression in key stress regulatory brain regions, the PVN and BNST. We hypothesized that paternal stress would more likely affect transcriptional patterns, rather than single candidate genes, as a complex cascade of programming events separates an initial paternal perturbation from its eventual effects in the adult offspring brain. Gene set enrichment analyses provided a characterization of these patterns by analyzing expression of a priori defined gene sets based on known biological associations, e.g., shared biological pathway, conserved cis- regulatory element, or common gene ontology category (Mootha et al., 2003; Subramanian et al., 2005). The enrichment of gene sets related to HPA stress axis regulation provided intriguing potential mechanisms that may underlie shifts in offspring stress responsivity. Most notably, enriched PVN expression of glucocorticoid receptor responsive genes could confer tighter control of CRF neurons in the PVN in paternal stressed offspring. Additionally, the majority of enriched gene sets we found in the PVN and BNST were those where member genes shared transcription factor or miR regulation. These broad differences in gene expression suggest potential involvement of broad chromatin changes and epigenetic programming. In particular, enriched BNST expression of gene sets associated with the activity of CREB, histone acetyltransferases such as CREB-binding protein, and six members of the miR-154 family, which are transcribed from a maternally imprinted locus, support the hypothesis that epigenetic mechanisms are likely involved in reprogramming of the offspring HPA axis circuitry (Seitz et al., 2003, 2004).
Paternal models of transgenerational epigenetics offer the advantage of examining germ cell transmission in the absence of confounding maternal factors, which can make deciphering the timing and identification of parental epigenetic transmission difficult. Among the limited set of epigenetic marks carried in sperm, miRs are poised to be products of a dynamic paternal environment and to uniquely contribute to postfertilization gene expression. Therefore, we assessed the sperm miR content from paternal controls and stress groups. Hierarchical clustering of over 250 miR transcripts segregated all stressed sires' miR expression profiles into a single clade, supporting recent evidence that sperm miRs are amenable to environmental perturbations (Li et al., 2012; Marczylo et al., 2012). Furthermore, the finding that sperm miR content exhibited similar changes in both stress groups was consistent with the effects of paternal stress exposure on offspring stress responsivity, and therefore supports a continuous germ cell vulnerability to epigenetic reprogramming across the lifespan.
In examination of significant changes in specific sperm miRs, paternal stress both throughout puberty and in adulthood significantly increased the expression of nine (miR-193-5p, miR-204, miR-29c, miR-30a, miR-30c, miR-32, miR-375, miR-532–3p, and miR-698). As miRs are normally present at low basal levels in sperm, the observed variability in control miR expression and the directional increase in miR levels with stress were not surprising. Importantly, miRs have been shown to regulate gene expression after fertilization, and may impact offspring development by acting on oocyte stores of maternal mRNA (Giraldez et al., 2006; Rassoulzadegan et al., 2006; Grandjean et al., 2009). Assessment of the top predicted mRNA targets of the nine significantly increased miRs revealed DNMT3a (DNA methyltansferase 3a), a critical regulator of de novo DNA methylation important for imprinted genes, and two proteins involved in miR processing, trinucleotide repeat containing 6b (Tnrc6b) and metadherin (Mtdh) (Kaneda et al., 2004; Lazzaretti et al., 2009; Yoo et al., 2011). These genes, targeted by four of the nine upregulated miRs, suggest that paternal stress may affect offspring HPA axis by intervening in the epigenetic regulation of oocyte development. Alternatively, sperm miRs may have altered epigenetic programming earlier, during spermatogenesis (Amanai et al., 2006).
In summary, our findings demonstrate that male exposure to stress, either throughout puberty or in adulthood, reprograms paternal germ cells and results in transmission of an offspring HPA stress axis dysregulation phenotype. Whether such diminished stress reactivity would be detrimental or beneficial to offspring likely depends on the environment into which they were born, as well as genetic background factors. However, the finding that mild stress experience can produce such long-term changes in male germ cells provides an important and novel mechanism contributing to neuropsychiatric disease risk.
Footnotes
This work was supported by NIH Grants MH087597, MH091258, and MH099910. A.B.R. was supported by NIH Systems and Integrative Biology Training Grant T32-GM07517. We thank J. Fluharty for technical support, C. Howerton for statistical consultation, and K. Morrison for editorial assistance.
The authors declare no competing financial interests.
References
- Amanai M, Brahmajosyula M, Perry AC. A restricted role for sperm-borne microRNAs in mammalian fertilization. Biol Reprod. 2006;75:877–884. doi: 10.1095/biolreprod.106.056499. [DOI] [PubMed] [Google Scholar]
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2011;93:23–58. doi: 10.1016/j.pneurobio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, Bock C, Li C, Gu H, Zamore PD, Meissner A, Weng Z, Hofmann HA, Friedman N, Rando OJ. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne FA. Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol. 2008;29:386–397. doi: 10.1016/j.yfrne.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe CL, Kramer M, Czéh B, Gould E, Reeves AJ, Kirschbaum C, Fuchs E. Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile rhesus monkeys. Biol Psychiatry. 2003;54:1025–1034. doi: 10.1016/S0006-3223(03)00698-X. [DOI] [PubMed] [Google Scholar]
- Corbett BA, Schupp CW, Levine S, Mendoza S. Comparing cortisol, stress, and sensory sensitivity in children with autism. Autism Res. 2009;2:39–49. doi: 10.1002/aur.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curley JP, Mashoodh R, Champagne FA. Epigenetics and the origins of paternal effects. Horm Behav. 2011;59:306–314. doi: 10.1016/j.yhbeh.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz DM, Laplant Q, Watts EL, Hodes GE, Russo SJ, Feng J, Oosting RS, Vialou V, Nestler EJ. Paternal transmission of stress-induced pathologies. Biol Psychiatry. 2011;70:408–414. doi: 10.1016/j.biopsych.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 2011;152:2228–2236. doi: 10.1210/en.2010-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweep H, Sticht C, Pandey P, Gretz N. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Franklin TB, Russig H, Weiss IC, Gräff J, Linder N, Michalon A, Vizi S, Mansuy IM. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68:408–415. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- Franklin TB, Linder N, Russig H, Thöny B, Mansuy IM. Influence of early stress on social abilities and serotonergic functions across generations in mice. PLoS ONE. 2011;6:e21842. doi: 10.1371/journal.pone.0021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin TB, Saab BJ, Mansuy IM. Neural mechanisms of stress resilience and vulnerability. Neuron. 2012;75:747–761. doi: 10.1016/j.neuron.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Dulawa SC. Assessment of murine startle reactivity, prepulse inhibition, and habituation. Curr Protoc Neurosci Chapter. 2003;8 doi: 10.1002/0471142301.ns0817s24. Unit8.17. [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–79. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- Godmann M, Lambrot R, Kimmins S. The dynamic epigenetic program in male germ cells: its role in spermatogenesis, testis cancer, and its response to the environment. Microsc Res Tech. 2009;72:603–619. doi: 10.1002/jemt.20715. [DOI] [PubMed] [Google Scholar]
- Goel N, Bale TL. Sex differences in the serotonergic influence on the hypothalamic-pituitary-adrenal stress axis. Endocrinology. 2010;151:1784–1794. doi: 10.1210/en.2009-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel N, Plyler KS, Daniels D, Bale TL. Androgenic influence on serotonergic activation of the HPA stress axis. Endocrinology. 2011;152:2001–2010. doi: 10.1210/en.2010-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean V, Gounon P, Wagner N, Martin L, Wagner KD, Bernex F, Cuzin F, Rassoulzadegan M. The miR-124-Sox9 paramutation: RNA-mediated epigenetic control of embryonic and adult growth. Development. 2009;136:3647–3655. doi: 10.1242/dev.041061. [DOI] [PubMed] [Google Scholar]
- Guerrero-Bosagna C, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol Cell Endocrinol. 2012;354:3–8. doi: 10.1016/j.mce.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Bosagna C, Settles M, Lucker B, Skinner MK. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE. 2010;5:e13100. doi: 10.1371/journal.pone.0013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 2011;59:279–289. doi: 10.1016/j.yhbeh.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Pronchuk N, Nonogaki K, Zhou L, Raber J, Tung L, Yeo GS, O'Rahilly S, Colmers WF, Elmquist JK, Tecott LH. Serotonin activates the hypothalamic-pituitary-adrenal axis via serotonin 2C receptor stimulation. J Neurosci. 2007;27:6956–6964. doi: 10.1523/JNEUROSCI.2584-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans KGC, Verma P, Yoon E, Yu W, Weinberg J. Prenatal alcohol exposure increases vulnerability to stress and anxiety-like disorders in adulthood. Ann N Y Acad Sci. 2008;1144:154–175. doi: 10.1196/annals.1418.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Xu H, Li H, Yang H, Chen Y, Shi X. Pre-gestational stress reduces the ratio of 5-HIAA to 5-HT and the expression of 5-HT1A receptor and serotonin transporter in the brain of foetal rat. BMC Neurosci. 2012;13:22. doi: 10.1186/1471-2202-13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman CM, Sandin S, Levine SZ, Lichtenstein P, Reichenberg A. Advancing paternal age and risk of autism: new evidence from a population-based study and a meta-analysis of epidemiological studies. Mol Psychiatry. 2011;16:1203–1212. doi: 10.1038/mp.2010.121. [DOI] [PubMed] [Google Scholar]
- Jimenez-Chillaron JC, Isganaitis E, Charalambous M, Gesta S, Pentinat-Pelegrin T, Faucette RR, Otis JP, Chow A, Diaz R, Ferguson-Smith A, Patti ME. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes. 2009;58:460–468. doi: 10.2337/db08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents“ and grandparents” slow growth period. Eur J Hum Genet. 2002;10:682–688. doi: 10.1038/sj.ejhg.5200859. [DOI] [PubMed] [Google Scholar]
- Kaati G, Bygren LO, Pembrey M, Sjöström M. Transgenerational response to nutrition, early life circumstances and longevity. Eur J Hum Genet. 2007;15:784–790. doi: 10.1038/sj.ejhg.5201832. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Matthews SG. Prenatal stress modifies behavior and hypothalamic-pituitary-adrenal function in female guinea pig offspring: effects of timing of prenatal stress and stage of reproductive cycle. Endocrinology. 2008;149:6406–6415. doi: 10.1210/en.2008-0347. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Matthews SG. Testosterone is involved in mediating the effects of prenatal stress in male guinea pig offspring. J Physiol. 2011;589:755–766. doi: 10.1113/jphysiol.2010.200543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neurosci Biobehav Rev. 2008;32:1519–1532. doi: 10.1016/j.neubiorev.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaretti D, Tournier I, Izaurralde E. The C-terminal domains of human TNRC6A, TNRC6B, and TNRC6C silence bound transcripts independently of argonaute proteins. RNA. 2009;15:1059–1066. doi: 10.1261/rna.1606309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li M, Liu Y, Song G, Liu N. A microarray for microRNA profiling in spermatozoa from adult men living in an environmentally polluted site. Bull Environ Contam Toxicol. 2012;89:1111–1114. doi: 10.1007/s00128-012-0827-0. [DOI] [PubMed] [Google Scholar]
- Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH. Smoking induces differential miRNA expression in human spermatozoa: a potential transgenerational epigenetic concern? Epigenetics. 2012;7:432–439. doi: 10.4161/epi.19794. [DOI] [PubMed] [Google Scholar]
- Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol Metab. 2002;13:373–380. doi: 10.1016/S1043-2760(02)00690-2. [DOI] [PubMed] [Google Scholar]
- McEuen JG, Semsar KA, Lim MA, Bale TL. Influence of sex and corticotropin-releasing factor pathways as determinants in serotonin sensitivity. Endocrinology. 2009;150:3709–3716. doi: 10.1210/en.2008-1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry. 2002;51:775–787. doi: 10.1016/S0006-3223(01)01362-2. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Morgan CP, Bale TL. Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci. 2011;31:11748–11755. doi: 10.1523/JNEUROSCI.1887-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. 2006;88:605–614. doi: 10.1016/j.physbeh.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol Behav. 2007;91:55–65. doi: 10.1016/j.physbeh.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/S0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature. 2010;467:963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- Oakberg EF. Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am J Anat. 1956;99:507–516. doi: 10.1002/aja.1000990307. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjöström M, Golding J, ALSPAC Study Team Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet. 2006;14:159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- Rando OJ. Daddy issues: paternal effects on phenotype. Cell. 2012;151:702–708. doi: 10.1016/j.cell.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- Rosenfield PJ, Kleinhaus K, Opler M, Perrin M, Learned N, Goetz R, Stanford A, Messinger J, Harkavy-Friedman J, Malaspina D. Later paternal age and sex differences in schizophrenia symptoms. Schizophr Res. 2010;116:191–195. doi: 10.1016/j.schres.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, Ferguson-Smith AC, Cavaille J. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet. 2003;34:261–262. doi: 10.1038/ng1171. [DOI] [PubMed] [Google Scholar]
- Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaille J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MK, Guerrero-Bosagna C. Environmental signals and transgenerational epigenetics. Epigenomics. 2009;1:111–117. doi: 10.2217/epi.09.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassoler FM, White SL, Schmidt HD, Sadri-Vakili G, Pierce RC. Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci. 2013;16:42–47. doi: 10.1038/nn.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgolini MB, Bauter MR, Weston DD, Cory-Slechta DA. Permanent alterations in stress responsivity in female offspring subjected to combined maternal lead exposure and/or stress. Neurotoxicology. 2006;27:11–21. doi: 10.1016/j.neuro.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Walker E, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol. 2008;4:189–216. doi: 10.1146/annurev.clinpsy.4.022007.141248. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia S, Pannell L, Fisher PB, Sarkar D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology. 2011;53:1538–1548. doi: 10.1002/hep.24216. [DOI] [PMC free article] [PubMed] [Google Scholar]