

Abstract
Aims
To determine the extent and time-course of hepatic and intestinal cytochrome P450 3A (CYP3A) inactivation due to the mechanism-based inhibitor clarithromycin.
Methods
Intestinal and hepatic CYP3A inhibition was examined in 12 healthy volunteers following the administration of single and multiple doses of oral clarithromycin (500 mg). Intestinal biopsies were obtained under intravenous midazolam sedation at baseline and after the first dose, on days 2–4, and on days 6–8 of the clarithromycin treatment. The formation of 1′-hydroxymidazolam in biopsy tissue and the serum 1′-hydroxymidazolam:midazolam ratio were indicators of intestinal and hepatic CYP3A activity, respectively.
Results
Intestinal CYP3A activity decreased by 64 % (p=0.0029) following the first dose of clarithromycin, but hepatic CYP3A activity did not significantly decrease. Repeated dosing of clarithromycin caused a significant decrease in hepatic CYP3A activity (p=0.005), while intestinal activity showed little further decline. The CYP3A5 or CYP3A4*1B genotype were unable to account for inter-individual variability in CYP3A activity.
Conclusions
Following the administration of clarithromycin, the onset of hepatic CYP3A inactivation is delayed compared to that of intestinal CYP3A. The time-course of drug–drug interactions due to clarithromycin will vary with the relative contribution of intestinal and hepatic CYP3A to the clearance and bioavailability of a victim substrate.
Keywords: Clarithromycin, CYP3A, Intestine, Mechanism-based inactivation, Midazolam
The macrolide antibiotic clarithromycin is a potent mechanism-based inhibitor of both hepatic and intestinal cytochrome P450 3A (CYP3A) activity [1], thereby reducing the clearance of other CYP3A substrates, including cyclosporine, midazolam, omeprazole, and tacrolimus [2–4]. Consequently, clarithromycin may cause significant drug–drug interactions that lead to severe toxicity when co-administered with CYP3A substrates that have a narrow therapeutic index. Clarithromycin inhibits the CYP3A enzyme via the formation of a metabolic-intermediate complex (MIC) in human liver microsomes in vitro [5]. Such MICs are formed irreversibly under physiological conditions and may explain the potency of many clinically important CYP3A inhibitors [6]. In contrast to CYP3A inactivation by grapefruit juice, formation of the clarithromycin MIC complex does not result in rapid protein degradation [7]. The in vivo inhibitory effect of a mechanism-based inactivator is generally thought to be more prominent after multiple-dose administration and to last longer than that of a reversible inhibitor because new protein must be synthesized to restore activity [8]. In one study, the administration of clarithromycin for 7 days reduced small intestine CYP3A activity by 74 %, but tissue inhibitor concentrations were too low to cause significant competitive inhibition, consistent with the hypothesis that clarithromycin likely causes irreversible inhibition of intestinal CYP3A activity [9].
Recently, we proposed a semi-physiological pharmacokinetic model that describes the pharmacokinetic interaction between clarithromycin and midazolam [10]. This model incorporates the hepatic and intestinal metabolism of clarithromycin and midazolam and mechanism-based inactivation of intestinal and hepatic CYP3A by clarithromycin. The model accurately predicted the extent of the interaction at steady-state dosing, based on the fold-change in the intravenous (IV) and oral area under the time–concentration curve (AUC) of midazolam. In addition, the recovery of intestinal and hepatic CYP3A was consistent with the observed fold-change in the oral and IV midazolam AUC at 1, 36, 72, and 144 h after discontinuation of clarithromycin dosing (500 mg every 12 h for 7 days). This interaction model indicates that a single dose of clarithromycin produces near-maximal CYP3A inactivation in the intestine but that hepatic CYP3A requires several doses before maximum inactivation occurs. However, no data on the onset of CYP3A inactivation by clarithromycin were available to validate this finding. Thus, this study was designed to elucidate the timing and extent of CYP3A inhibition in the liver and intestine during treatment with clarithromycin, using midazolam as a probe substrate. The effect of the CYP3A5 genotype on hepatic and intestinal CYP3A activity before and after inhibition was also investigated.
Materials and methods
Subjects
The study was performed in 12 healthy subjects (6 men, 6 women), aged ≥18 years, who had consumed no prescription or over-the-counter medications for at least 2 weeks before the study. Individuals with an intolerance to macrolide antibiotics or benzodiazepines or a significant medical history of smoking or alcohol intake were excluded from the study. Subjects were excluded from participating in the study if the blood tests revealed a low platelet cell count (<100,000/mL) or a high international normalized ratio (>1.5). For at least 2 weeks prior to the start of the study until its conclusion, volunteers were restricted from consuming any food or beverage containing grapefruit or grapefruit juice, vegetables from the mustard green family (kale, broccoli, watercress, collard greens, kohlrabi, Brussels sprouts, mustard, etc) and charbroiled meats. Additionally, food or beverage items containing xanthenes (e.g. coffee, tea, chocolate, colas) were not consumed by the subjects 48 h prior to beginning of the baseline phase and throughout the study. The Clarian and Indiana University Purdue University Indianapolis (IUPUI) Institutional Review Board approved this study. All subjects provided written informed consent.
Study design
Each volunteer underwent four endoscopies prior to and during a weekly course of oral clarithromycin 500 mg twice daily (Biaxin XL®; Abbot Laboratories, Abbot Park, IL). The first (baseline) endoscopy was performed within 1 week (day −7 to day −1) prior to the subject starting clarithromycin treatment. Three additional endoscopies were performed 12 h after the first dose and on days 2–4 and 6–8 of clarithromycin treatment (Fig. 1). Following an overnight fast, individuals were dosed with IV midazolam (Roche Pharmaceuticals, Nutley, NJ) to achieve effective conscious sedation (Table 1) for the esophagogastroduodenoscopy (EGD). Mucosal biopsy specimens were obtained from the second portion of the duodenum and were immediately snap frozen in liquid nitrogen and stored at −80 °C until analysis. During each endoscopy visit, blood samples were obtained prior to and 1, 2, and 3 h after midazolam administration. Compliance with clarithromycin dosing and diet, alcohol, and drug restrictions was assessed by pill count and subject interviews.
Fig. 1.
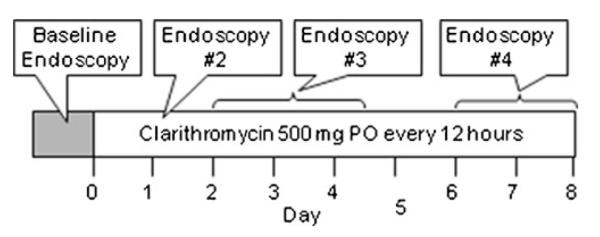
Study design. Volunteers received clarithromycin 500 mg orally (PO) every 12 h for 8 days. Duodenal biopsies were obtained under sedation with intravenous midazolam, prior to and after the first dose of clarithromycin and on days 2–4 and 6–8 of treatment
Table 1.
Volunteer demographic data, doses of intravenous midazolam and the number of 500 mg oral clarithromycin tablets received prior to each endoscopy
| Subject | Sex | Race | CYP3A5 genotype | Age (years) | Midazolam dose (mg) |
Consecutive clarithromycin tablets |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E1 | E2 | E3 | E4 | E1 | E2 | E3 | E4 | |||||
| 1 | F | White | *3/*3 | 24 | 4 | 6 | 6 | 8 | 0 | 1 | 6 | 12 |
| 2 | M | White | *3/*3 | 35 | 2 | 6 | 7 | 8 | 0 | 1 | 4 | 12 |
| 3 | M | Black | *3/*6 | 26 | 5.5 | 8 | 7 | 10 | 0 | 1 | 2 | 12 |
| 4 | M | Black | *1/*1 | 32 | 6 | 6 | 4 | 6 | 0 | 1 | 5 | 13 |
| 5 | F | White | *3/*3 | 25 | 7 | 8 | 10 | 10 | 0 | 1 | 5 | 13 |
| 6 | F | White | *3/*3 | 22 | 8 | 10 | 10 | 10 | 0 | 1 | 2 | 12 |
| 7 | F | White | *3/*3 | 25 | 10 | 8 | 10 | 10 | 0 | 1 | 2 | 12 |
| 8 | M | Black | *1/*1 | 48 | 7 | 6 | 6 | 10 | 0 | 1 | 2 | 12 |
| 9 | M | White | *3/*3 | 30 | 10 | 9 | 8 | 8 | 0 | 1 | 2 | 12 |
| 10 | M | White | *3/*3 | 25 | 8 | 10 | 10 | 10 | 0 | 1 | 2 | 10 |
| 11 | F | White | *1/*3 | 24 | 8 | 8 | 10 | 10 | 0 | 1 | 2 | 10 |
| 12 | F | Black | *1/*3 | 37 | 10 | 10 | 10 | 10 | 0 | 3 | 5 | 15 |
CYP3A5, Cytochrome P450 3A5; E1, E2, E3, E4, first, second, third, fourth endoscopy, respectively; F, female; M, male
Duodenal homogenates
Small bowel biopsy specimens (approximately 20 μg wet tissue weight) were homogenized as described previously [9]. Duodenal biopsy specimens were homogenized by a hand-held glass homogenizer in 1 mL ice-cold buffer consisting of 50 mmol/L Tris(hydroxymethyl) aminomethane, 2 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L benzamidine, 50 μg aprotinin, and 20 % glycerol (pH 7.4). The resultant homogenate was immediately snap frozen in liquid nitrogen and stored at −80 °C until assayed. Homogenate protein concentrations were determined by the method of Lowry et al. [11] using bovine serum albumin as the standard.
Immunoblotting
An aliquot (15 μg) of small-bowel homogenate was mixed with 5× nonreducing sample buffer (Pierce, Rockford, IL) and heated to 95 °C for 5 min. The samples were electrophoresed in 9 % polyacrylamide/0.1 % sodium dodecyl sulfate gels using a Bio-Rad Protean II Unit (Bio-Rad, Hercules, CA) and then transferred onto a Hybond-P hydrophobic polyvinylidene difluoride membrane (Amersham Pharmacia Biotech, Piscataway, NJ) with 25 mmol/LTris and 192 mmol/L glycine buffer at 100 V for 1.5 h. The blots were first blocked for 1 h in Tris-buffered saline containing 5 % nonfat dry milk, and the membrane was then incubated overnight with a 1:500 dilution of polyclonal antibody to CYP3A4 (Gentest, Woburn, MA) or with villin antibody (Thermo Scientific, Rockford, IL) in Tris buffered saline. After extensive washing with Tris-buffered saline solution containing 0.1 % Tween 20, the membranes were probed with a horseradish peroxidase secondary antibody and developed with an enhanced chemifluorescence kit (GE Healthcare, Piscataway, NJ). The membrane was exposed to BioMax Light 2 film (Kodak, Rochester, NY), and optical densities of the bands on the film were converted to quantitative numbers using the Kodak Electrophoresis Documentation and Analysis System 290 (EDAS 290) and Kodak one-dimensional image analysis software (Kodak, Rochester, NY). The amount of CYP3A4 in the biopsy specimens was obtained by comparison with serial dilutions of recombinant human CYP3A4 (Gentest). The standards were run on the same gel as the biopsy samples. The concentrations of CYP3A4 in the enterocytes were expressed relative to the concentration of the enterocyte-specific protein villin in the same sample.
Midazolam hydroxylation activity
An aliquot of 0.5 mL of small-bowel homogenate was diluted with 0.5 mL of 100 mmol/L sodium phosphate buffer (pH 7.4) and incubated at 37 °C with 5 or 50 μmol/L midazolam. The reaction was initiated with the addition of 1 μmol NADPH and was quenched after 10 min with 1 mL of ethyl acetate/hexane (50:50, v/v) and then with 1 mL of 1 N sodium hydroxide/1-mol/L glycine (pH 11.3) buffer on ice; N-desmethyldiazepam was added to the mixture as an internal standard. The mixture was then extracted with an additional 3 mL of ethyl acetate/hexane (50:50, v/v). The organic phase was dried under vacuum, and samples were reconstituted in 100 μL of mobile phase and analyzed by liquid chromatography/mass spectrometry (LC/MS) as described below.
Midazolam and clarithromycin assays
Midazolam and 1′-hydroxymidazolam were quantified using desmethyldiazepam as the internal standard, as previously described in detail [12]. Compounds of interest were separated by use of a Luna C18 column (5 μm, 4.6×150–mm internal diameter; Phenomenex, Torrance, CA) equipped with a Brownlee RP-18 guard column (Perkin Elmer, Shelton, CT). The mobile phase [acetonitrile/20 mmol/L ammonium acetate, pH 7.4/methanol (40:40:20, v/v/v)] was delivered isocratically at 1 mL/min. Desmethyldiazepam, midazolam, and 1′-hydroxymidazolam were quantified by use of a mass spectrometer (Navigator; Finnigan, San Jose, CA) equipped with an APCI interface. The eluent was analyzed by atmospheric pressure chemical ionization (APCI) in the positive mode with a cone voltage of 25 V and source and probe temperatures of 200 °C and 550 °C, respectively. N-desmethyldiazepam, midazolam, and 1′-hydroxymidazolam were detected with selected ion monitoring at mass-to-charge ratios of 271, 326, and 342, respectively. The limit of quantification in serum was 0.25 ng/mL for midazolam and 1′-hydroxymidazolam; the respective corresponding coefficient of variation and relative error for midazolam and 1′-hydroxymidazolam at 1.4, 10, and 40 ng/mL were less than 7 and 10 %.
Serum clarithromycin, 14-hydroxyclarithromycin, and N-desmethylclarithromycin concentrations were determined using troleandomycin as the internal standard, as previously described in detail [13]. In brief, the compounds of interest were separated by passage through a C8 column (5 μm, 100 × 4.6–mm internal diameter; Brownlee, Perkin Elmer) and a mobile phase of 20 mmol/L ammonium acetate, pH 7/methanol (20:80, v/v). The mobile phase was delivered isocratically at 1 mL/min. Detection was achieved by use of a mass spectrometer with an APCI inlet (Navigator; Finnigan, San Jose, CA). The limits of quantitation were 2.5 ng/mL for clarithromycin and its metabolites. The respective corresponding coefficients of variation and relative error for clarithromycin and its metabolites at 10 ng/mL were less than 8 and 9 %.
CYP3A5 genotype
Genomic DNA was extracted from whole blood by use of the Qiagen Midi kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). Genotyping of CYP3A5*3, CYP3A5*6, CYP3A5*7, and CYP3A4*1B was carried out using a previously described method [14, 15]; the PCR cycling conditions were: 5 min at 95 °C; 35 cycles of 0.5 min at 94 °C, 0.5 min at 55 °C, and 1 min at 72 °C, followed by a final 10-min cycle at 72 °C. CYP3A5 high expressers were defined as having at least one CYP3A5*1 allele. CYP3A5 low expressers were defined as having two variant alleles, CYP3A5*3, *6, or *7, that result in reduced protein expression or the expression of proteins with greatly reduced activity.
Intestinal and hepatic CYP3A activities
Intestinal CYP3A activity was determined by the ex vivo formation of 1′-hydroxymidazolam in homogenized biopsy tissue. Vmax (maximum reaction rate) and Km (concentration of substrate that gives half-maximal activity) were obtained from the double-reciprocal plot of the data and the ex vivo CLint (intrinsic clearance) was calculated as Vmax/Km. The ratio of serum 1′-hydroxymidazolam to midazolam obtained 3 h after the midazolam dose was used as an in vivo indicator of hepatic CYP3A activity based on previous data [16].
Statistical analysis
Data are reported as mean and standard deviation (SD). Recruitment of 12 subjects provided 83 % power to detect a twofold change in formation rates of 1′-hydroxymidazolam, with 5 % type I error. Changes in hepatic and intestinal activity from baseline and the effect of CYP3A5 genotype were compared by analysis of variance (ANOVA) and the Wilcoxon test with the Bonferroni correction for multiple comparisons. All statistical analyses were performed using R statistical software [17].
Results
The 12 subjects completed the study without adverse effects. The subject demographics and the doses of clarithromycin and midazolam administered prior to each endoscopy are shown in Table 1. One subject (subject #12) received three doses of clarithromycin prior to the second endoscopy; thus only the data from 11 subjects was included for the 12-h time-point analysis. The third and fourth endoscopies were performed as per protocol except that one subject (subject #12) received a total of 15 doses of oral clarithromycin prior to the last endoscopy.
CYP3A4 and CYP3A5 genotype
Four individuals were CYP3A5 high expressers: two homozygous and two heterozygous (Table 1). CYP3A4*1B was expressed by five individuals, including the four CYP3A5 high expressers and subject 3 who carried the CYP3A5*6 allele.
Intestinal CYP3A activity and expression
A reduction in intestinal CYP3A activity was observed for all individuals after the first dose of clarithromycin (Fig. 2). With the exception of one subject, intestinal activity remained suppressed throughout the course of treatment. Mean intestinal CYP3A activity decreased by 64 % from 0.22 pmol min−1 mg−1 [95 % confidence interval (CI) 0.13–0.31] to 0.10 pmol min−1 mg−1 (95 % CI 0.05–0.14) following the first dose of clarithromycin (p=0.0029; Fig. 3). The mean intestinal CYP3A activity further decreased to 0.06 pmol min−1 mg−1 (95 % CI 0.03–0.08) and 0.08 pmol min−1 mg−1 (95 % CI 0.01–0.15) by the third and fourth endoscopies, respectively (Fig. 3). CYP3A4 protein from ten of the 12 volunteers was quantified by Western blot and normalized to villin expression. A large variability in expression was noted (Fig. 4), but there was no difference between the endoscopies in amount of CYP3A4 protein expressed (p=0.92).
Fig. 2.

Change in cytochrome P450 3A (CYP3A) activity following the administration of clarithromycin. Individual changes in intestinal (a) and hepatic (b) CYP3A activity based on the formation of 1′-hydroxymidazolam and the 3-h serum 1′-hydroxymidazolam:midazolam ratio, respectively. Open triangles CYP3A5 high expressers, closed triangles CYP3A5 low expressers
Fig. 3.
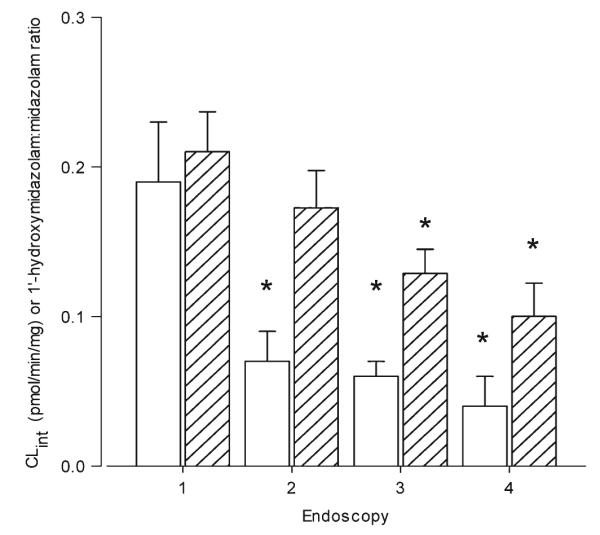
Intestinal and hepatic CYP3A activity at each endoscopy (1–4). Intestinal (white bars) CYP3A activity was determined by the formation of 1′-hydroxymidazolam in intestinal homogenates. Hepatic (bars with diagonal lines) activity was based on 3-h serum 1′-hydroxymidazolam:midazolam ratios. Data represent the mean ± standard error (SE). *p<0.05 compared to baseline endoscopy (i.e., first endoscopy (1)]
Fig. 4.
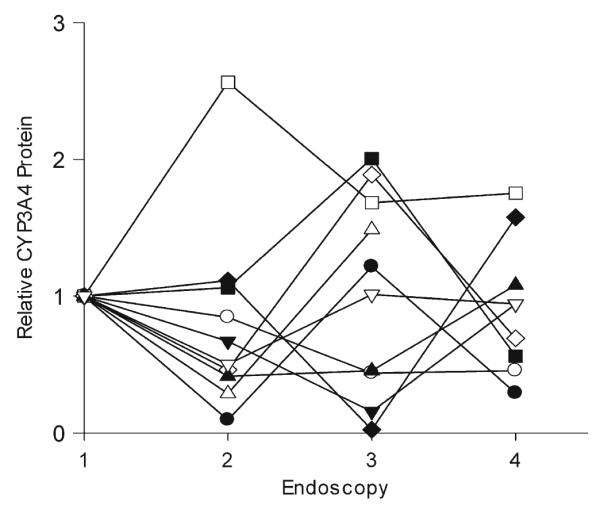
CYP3A4 protein expression in intestinal mucosa. Duodenal biopsy tissue was homogenized, and CYP3A4 protein expression was quantified by western blot analysis. The relative amount of CYP3A4, normalized for villin content, at each endoscopy (1–4) is shown for ten subjects (different symbols)
Hepatic CYP3A activity
Hepatic CYP3A activity, as assessed by the 3-h 1′-hydroxymidazolam:midazolam serum ratio, was less rapidly inhibited by clarithromycin than intestinal CYP3A activity. As shown in Fig. 2, there was a greater variability in hepatic CYP3A activity than in intestinal CYP3A activity following the first dose of clarithromycin, with three individuals showing increased clearance of midazolam. With the exception of one individual, hepatic activity was suppressed after 1 week of treatment with clarithromycin (Fig. 2). The mean hepatic 1′-hydroxymidazolam:midazolam ratio after the first dose of oral clarithromycin was not significantly different from baseline, i.e., 0.22 (95 % CI 0.17–0.27) and 0.19 (95 % CI 0.14–0.24), respectively (p = 0.92; Fig. 3). A significant decrease in hepatic CYP3A activity was seen with subsequent doses of clarithromycin. Hepatic CYP3A activity decreased by 42 (p=0.0088), and 48 % (p=0.07) from baseline by the third and fourth endoscopies, respectively (Fig. 3). When the three individuals for whom hepatic activity increased by >1.1-fold following the first dose of clarithromycin does were excluded, the decrease in hepatic activity between the first and second EGD remained non-significant following the first dose of clarithromycin (71±26 % of baseline; p=0.2)
Clarithromcyin concentration
Clarithromycin and 14-hydroxyclarithromycin were detected in all plasma samples, with mean clarithromycin concentrations of 13.3±9.4, 22.1±12.4, and 27.8±13.5 μM and mean 14-hydroxyclarithromcyin concentrations of 3.12±1.25, 4.47±2.27, and 4.18±1.52 μM at endoscopies 2, 3, and 4, respectively. The clarithromcyin plasma concentration was weakly but statistically associated with hepatic CYP3A activity (r2=0.13, p=0.03), but was not associated with intestinal CYP3A activity (p=0.44).
CYP3A5 genotype and inhibition of intestinal CYP3A activity
There was no significant (p=0.37) difference in the baseline intestinal CYP3A activity between CYP3A5 high expressers (n=4) and low expressers (n=8). When stratified by their CYP3A5 genotype, the decrease in intestinal CYP3A activity with the first dose of oral clarithromycin was not significant for CYP3A5 high expressers (p=0.55), but it was significant for CYP3A5 low expressers (p=0.046; Fig. 5). There was no significant difference between the high expressers and low expressers in terms of the extent of intestinal CYP3A inhibition at the time of the fourth endoscopy (p>0.99).
Fig. 5.

Relative change in CYP3A activity based on CYP3A5 genotype. The relative change in intestinal (a) and hepatic (b) CYP3A activity was assessed in CYP3A5 high expressers (n=4, white bars) and low expressers (n=8, black bars). Data are presented as the mean ± SE. *p=0.046 compared to baseline activity
CYP3A5 genotype and inhibition of hepatic CYP3A activity
There was no significant (p=0.81) difference in the baseline hepatic CYP3A activity between CYP3A5 high expressers and low expressers. When stratified by their CYP3A5 genotype, the decrease in the hepatic CYP3A activity with the first dose of oral clarithromycin was not significant for either the CYP3A5 low expressers or high expressers (Fig. 5). CYP3A5 low expressers showed a significant drop in hepatic CYP3A activity from the baseline by the third endoscopy (p=0.046), whereas hepatic activity did not significantly decrease by the third endoscopy in CYP3A5 high expressers (p=0.94). There was no significant difference between the high expressers and low expressers in terms of the extent to which hepatic CYP3A was inhibited at the time of the fourth endoscopy (p=0.68).
Discussion
The accurate prediction of drug–drug interactions involving mechanism-based inactivators requires a knowledge of drug-specific properties, such as the KI (inhibitor concentration for half-maximal inactivation) and kinact (maximal inactivation rate constant), as well as the enzyme-specific half-life. These properties, combined with the pharmacokinetic profile of the inactivator, determine the extent of the drug–drug interaction. By evaluating the change in 1′-hydroxymidazolam formation during treatment with clarithromycin, we demonstrated a differential effect of inactivation on intestinal and hepatic CYP3A. Twelve hours after a single dose of clarithromycin, the intestinal CYP3A activity was reduced by 64 %, indicating significant inactivation of CYP3A in the gut wall. However, several doses of clarithromycin were required before maximum inactivation of hepatic CYP3A was achieved.
Midazolam is a selective CYP3A probe both in vitro and in vivo [1, 18], and its disposition is not dependent on the activity of the p-glycoprotein efflux transporter (MDR1) [19, 20]. Midazolam is metabolized by CYP3A to form 1′-hydroxymidazolam and a minor metabolite, 4-hydroxymidazolam in vitro and in vivo in humans [18, 21]; less than 1 % of the midazolam dose is excreted unchanged in the urine following IV administration. In this study, midazolam was administered IV at a dose required for sedation (5–10 mg), and the ratio of 1′-hydroxymidazolam to midazolam concentration in plasma was used to assess hepatic CYP3A activity, as previously described [9, 16]. Midazolam is efficiently extracted by intestinal CYP3A, with up to 75 % of the first-pass loss of midazolam following oral administration occurring in the intestinal wall [1, 22]. Thus, we also used midazolam as an ex vivo measure of CYP3A activity in intestinal biopsy samples, as previously described [9].
A potential confounder to the in vitro assay measuring the formation of 1′-hydroxymidazolam may be residual metabolite concentrations in the enterocytes due to IV use of midazolam as a sedative. The in vitro rate of 1′-hydroxymidazolam formation in incubations with 5 μM midazolam was, on average, 0.39 pmol min−1 mg−1. Incubations lasted 10 min, resulting in the formation of 1,300 ng of 1′-hydroxmidazolam per milligram protein. The average plasma concentration of 1′-hydroxymidazolam observed 2–3 hours after midazolam administration was 0.14–0.16 ng/mL. Thus, any 1′-hydroxymidazolam present in the homogenate would be negligible and not impact the observed in vitro clearance of midazolam. Therefore, we do not expect residual in vivo metabolites of midazolam to have confounded the estimation of 1′-hydroxymidazolam formation rate. Additionally, we have shown previously [8] that the residual concentration of clarithromycin in the homogenates is minimal and lower than its competitive inhibition rate constant (KI) for CYP3A.
The quantification of hepatic CYP3A activity was obtained from the single-point plasma measurement of the ratio of 1′-hydroxymidazolam to midazolam at 3 h after IV midazolam dosing. The single point ratio of 1′-hydroxymidazolam:midazolam has been used extensively as a measure of CYP3A activity [23–29]. In particular, this ratio is strongly correlated with hepatic CYP3A4 content (r2=0.87) [30] and with midazolam clearance (r2=0.89) [31].
Inter-individual variability in CYP3A activity may occur due to the presence of CYP3A4 or CYP3A5 polymorphisms [32]. The rate of 1′-hydroxymidazolam formation is twofold greater for CYP3A5 than for CYP3A4 [18, 33], and differential inhibition kinetics of CYP3A4 and CYP3A5 have been reported [34–36]. The catalytic activity of CYP3A5 has been shown to be less susceptible than CYP3A4 to time-dependent inhibition by erythromycin and verapamil, and CYP3A5 has also been found to have a slower maximum rate of enzyme inactivation than CYP3A4 in vitro [37, 38]. Both CYP3A4 and CYP3A5 contribute to the total CYP3A activity in vivo and, therefore, the time-course and maximal inhibitory effect was postulated to be different in CYP3A5 high expressers compared to low expressers. However, the four CYP3A5 high expressers in our study did not differ from CYP3A5 low expressers in either baseline activity or extent of inactivation of intestinal or hepatic CYP3A activities after 7 days of treatment with clarithromycin. Statistically significant reductions were observed in intestinal activity in CYP3A5 low expressers—but not high expressers—after the first dose of clarithromycin. It should be noted, however, that this study was not powered to detect a difference between genotypes, and the linkage disequilibrium noted between CYP3A4*1B and CYP3A5*1 makes it impossible to distinguish between the two genotypes without a large sample size.
The results of this study are consistent with those previously evaluating the turn-over rate of CYP3A enzyme. Half-life estimates for CYP3A4 range from 20 to 85 h [39–42]. Greenblatt et al. estimated the turnover rate of intestinal CYP3A to be 23 h based on the time-course of recovery of intestinal CYP3A following single doses of grapefruit juice [39]. Okudaira et al. found that the AUC of oral midazolam increased by 2.32- and 3.38-fold following 2 and 4 days of erythromycin 200 mg four times daily. They estimated the turnover of CYP3A as 22.8 h [42]. As in our study, Okudaira et al. found a large inter-individual variability in the extent of CYP3A inhibition by erythromycin. However, they did not account for CYP3A5 genotype, nor were they able to separately examine intestinal and hepatic CYP3A activity.
Bartkowski et al. examined the effect of erythromycin on the clearance of IV alfentanil [43]. Similar to our findings, no effect was observed following the first dose of erythromycin (500 mg), but the systemic clearance of alfentanil was increased by 1.6-fold following a 7-day course of erythromycin [43]. Compared to erythromycin, clarithromycin is a more potent CYP3A mechanism-based inhibitor, with the in vitro KI estimated to be between 4 and 31 mg/L and with a kinact of 2.5–4.3 in human liver microsomes [5, 44]. In the present study, hepatic CYP3A exhibited a 50 % reduction in activity following 7 days of clarithromycin.
The results of this study are consistent with a model we have developed for the inactivation of intestinal and hepatic CYP3A by clarithromycin [10]. The semi-physiologically based pharmacokinetic model incorporates intestinal and hepatic inactivation of CYP3A by clarithromycin. This CYP3A inactivation directly reduces the clearance of both clarithromycin and midazolam, and the intestinal activity appears to be well-predicted by the model. However, the extent of inactivation of hepatic CYP3A is slightly overestimated compared to that observed in this study. As shown by this model (Fig. 6), intestinal CYP3A is exposed to a higher concentration of clarithromycin (Fig. 6b) than is hepatic CYP3A (Fig. 6a) following the first dose of inhibitor. We postulate that this high concentration of clarithromycin in the intestines results in a rapid inactivation of CYP3A (Fig. 6d), reaching a near maximal extent following a single dose. However, the presence of this high concentration of clarithromycin in the gut wall is short-lived, with no detectable clarithromycin present 12 h after dosing [9]. Thus, intestinal CYP3A activity begins to recover within the 12-h dosing interval. On the other hand, the concentration of clarithromycin in the liver following the initial dose is lower than that in the gastrointestinal tract. However, unlike the gastrointestinal tract where new enterocytes are produced, the inhibitor may accumulate in the hepatocytes upon subsequent dosing. Therefore, less CYP3A inactivation occurs following each dose of clarithromycin, but the enzyme does not have time to recover before the next dose (Fig. 6c). This results in an additional inactivation with each dose until a steady-state is reached.
Fig. 6.

Simulated effect of clarithromycin on hepatic and intestinal CYP3A activity. A semi-physiologically based pharmacokinetic model incorporating effects of clarithromycin inactivation in the intestine and liver was developed. Clarithromycin 500 mg was administered orally every 12 h. a, b Simulated hepatic and gut-wall concentrations of clarithromycin. c Predicted (line) and observed (box plot/point estimates) hepatic CYP3A activity. d Predicted (line) and observed (box plot/point estimates) gut-wall CYP3A activity
The timing of the inhibition of intestinal CYP3A is critical to understanding when a patient taking an oral concomitant CYP3A substrate is at risk of experiencing a drug–drug interaction. This timing will be dependent on not only the properties of the inhibitor, but also on those of the substrate drug. The bioavailability of CYP3A substrates with high gut-wall extraction (i.e., low gut-wall availability) will be increased following a single dose of clarithromycin. These drugs, such as simvastatin, atorvastatin, and buspirone, are also characterized by substantial increases in oral exposure following inhibition of gut-wall CYP3A by grapefruit juice [45–47]. However, CYP3A substrates with higher gut-wall availability, such as alprazolam, nifedipine and quinidine, are not dependent on gut-wall CYP3A, and the inhibitory effects of clarithromycin will mirror the hepatic inactivation of CYP3A and will be delayed, only appearing after multiple doses of inhibitor. Similarly, the absorption properties of the inhibitor may affect the time-course of intestinal inactivation. Clarithromycin was administered as a slow-release formulation (Biaxin XL®), which may have increased its contact time with gut-wall CYP3A, thereby delaying enzyme recovery. Gut-wall enzyme recovery may begin sooner following the administration of a mechanismbased inhibitor with a faster absorption rate and reduced intestinal residence time.
In conclusion, the onset of intestinal CYP3A inhibition occurs following the first dose of the mechanism-based CYP3A inhibitor clarithromycin. In contrast, reduction in hepatic CYP3A activity is delayed, and several doses of clarithromycin are required to reach the nadir. Although the inter-individual variability in CYP3A baseline activity and the rate of inactivation were both high, these findings could not be explained by CYP3A genotype.
Acknowledgments
This work was supported by NIH Grants T32GM08425 and GM067308.
Abbreviations
- CYP450
Cytochrome P450
- MIC
Metabolic-intermediate complex
Footnotes
Conflicts of interest None.
Contributor Information
Sara K. Quinney, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA
Srikar R. Malireddy, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA
Raj Vuppalanchi, Gastroenterology/Hepatology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA.
Mitchell A. Hamman, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA
Naga Chalasani, Gastroenterology/Hepatology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA.
J. Christopher Gorski, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA.
Stephen D. Hall, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA; Eli Lilly and Company, Lilly Corporate Center, Drop Code 0720, Indianapolis, IN 46285, USA
References
- 1.Gorski JC, Jones DR, Haehner-Daniels BD, Hamman MA, O’Mara EM, Jr, Hall SD. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin Pharmacol Ther. 1998;64(2):133–143. doi: 10.1016/S0009-9236(98)90146-1. [DOI] [PubMed] [Google Scholar]
- 2.Gustavson LE, Kaiser JF, Edmonds AL, Locke CS, DeBartolo ML, Schneck DW. Effect of omeprazole on concentrations of clarithromycin in plasma and gastric tissue at steady state. Antimicrob Agents Chemother. 1995;39(9):2078–2083. doi: 10.1128/aac.39.9.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabada B. Re-evaluation of the gastrointestinal effects of beta-lactam inhibitors of beta-lactamase. Rev Esp Quimioter. 1998;11(4):377–379. [PubMed] [Google Scholar]
- 4.Wolter K, Wagner K, Philipp T, Fritschka E. Interaction between FK 506 and clarithromycin in a renal transplant patient. Eur J Clin Pharmacol. 1994;47(2):207–208. doi: 10.1007/BF00194974. [DOI] [PubMed] [Google Scholar]
- 5.Mayhew BS, Jones DR, Hall SD. An in vitro model for predicting in vivo inhibition of cytochrome P450 3A4 by metabolic intermediate complex formation. Drug Metab Dispos. 2000;28(9):1031–1037. [PubMed] [Google Scholar]
- 6.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. doi:10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 7.Lin HL, Kenaan C, Hollenberg PF. Identification of the residue in human CYP3A4 that is covalently modified by bergamottin and the reactive intermediate that contributes to the grapefruit juice effect. Drug Metab Dispos. 2012;40(5):998–1006. doi: 10.1124/dmd.112.044560. doi:10.1124/dmd.112.044560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin JH, Lu AY. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet. 1998;35(5):361–390. doi: 10.2165/00003088-199835050-00003. [DOI] [PubMed] [Google Scholar]
- 9.Pinto AG, Wang YH, Chalasani N, Skaar T, Kolwankar D, Gorski JC, Liangpunsakul S, Hamman MA, Arefayene M, Hall SD. Inhibition of human intestinal wall metabolism by macrolide antibiotics: effect of clarithromycin on cytochrome P450 3A4/5 activity and expression. Clin Pharmacol Ther. 2005;77(3):178–188. doi: 10.1016/j.clpt.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Quinney SK, Zhang X, Lucksiri A, Gorski JC, Li L, Hall SD. Physiologically based pharmacokinetic model of mechanism-based inhibition of CYP3A by clarithromycin. Drug Metab Dispos. 2010;38(2):241–248. doi: 10.1124/dmd.109.028746. doi:10.1124/dmd.109.028746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 12.Belle DJ, Callaghan JT, Gorski JC, Maya JF, Mousa O, Wrighton SA, Hall SD. The effects of an oral contraceptive containing ethinyloestradiol and norgestrel on CYP3A activity. Br J Clin Pharmacol. 2002;53(1):67–74. doi: 10.1046/j.0306-5251.2001.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu SY, Wilson DS, Guay DR, Craft C. Clarithromycin pharmacokinetics in healthy young and elderly volunteers. J Clin Pharmacol. 1992;32(11):1045–1049. doi: 10.1002/j.1552-4604.1992.tb03809.x. [DOI] [PubMed] [Google Scholar]
- 14.Liu TC, Lin SF, Chen TP, Chang JG. Polymorphism analysis of CYP3A5 in myeloid leukemia. Oncol Rep. 2002;9(2):327–329. [PubMed] [Google Scholar]
- 15.van Schaik RH, van der Heiden IP, van den Anker JN, Lindemans J. CYP3A5 variant allele frequencies in Dutch Caucasians. Clin Chem. 2002;48(10):1668–1671. [PubMed] [Google Scholar]
- 16.Lin YS, Lockwood GF, Graham MA, Brian WR, Loi CM, Dobrinska MR, Shen DD, Watkins PB, Wilkinson GR, Kharasch ED, Thummel KE. In-vivo phenotyping for CYP3A by a single-point determination of midazolam plasma concentration. Pharmacogenetics. 2001;11(9):781–791. doi: 10.1097/00008571-200112000-00006. [DOI] [PubMed] [Google Scholar]
- 17.R Development Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna: 2011. [Google Scholar]
- 18.Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol. 1994;47(9):1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 19.Schmiedlin-Ren P, Thummel KE, Fisher JM, Paine MF, Lown KS, Watkins PB. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1 alpha,25-dihydroxyvitamin D3. Mol Pharmacol. 1997;51(5):741–754. doi: 10.1124/mol.51.5.741. [DOI] [PubMed] [Google Scholar]
- 20.Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, Roden MM, Belas F, Chaudhary AK, Roden DM, Wood AJ, Wilkinson GR. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res. 1999;16(3):408–414. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]
- 21.Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36(1):89–96. [PubMed] [Google Scholar]
- 22.Thummel KE, O’Shea D, Paine MF, Shen DD, Kunze KL, Perkins JD, Wilkinson GR. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59(5):491–502. doi: 10.1016/S0009-9236(96)90177-0. [DOI] [PubMed] [Google Scholar]
- 23.Perera MA, Thirumaran RK, Cox NJ, Hanauer S, Das S, Brimer-Cline C, Lamba V, Schuetz EG, Ratain MJ, Di Rienzo A. Prediction of CYP3A4 enzyme activity using haplotype tag SNPs in African Americans. Pharmacogenomics J. 2009;9(1):49–60. doi: 10.1038/tpj.2008.13. doi:10.1038/tpj.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krupka E, Venisse N, Lafay C, Gendre D, Diquet B, Bouquet S, Perault MC. Probe of CYP3A by a single-point blood measurement after oral administration of midazolam in healthy elderly volunteers. Eur J Clin Pharmacol. 2006;62(8):653–659. doi: 10.1007/s00228-006-0159-2. doi:10.1007/s00228-006-0159-2. [DOI] [PubMed] [Google Scholar]
- 25.Lepper ER, Baker SD, Permenter M, Ries N, van Schaik RH, Schenk PW, Price DK, Ahn D, Smith NF, Cusatis G, Ingersoll RG, Bates SE, Mathijssen RH, Verweij J, Figg WD, Sparreboom A. Effect of common CYP3A4 and CYP3A5 variants on the pharmacokinetics of the cytochrome P450 3A phenotyping probe midazolam in cancer patients. Clin Cancer Res. 2005;11(20):7398–7404. doi: 10.1158/1078-0432.CCR-05-0520. doi:10.1158/1078-0432.CCR-05-0520. [DOI] [PubMed] [Google Scholar]
- 26.Chaobal HN, Kharasch ED. Single-point sampling for assessment of constitutive, induced, and inhibited cytochrome P450 3A activity with alfentanil or midazolam. Clin Pharmacol Ther. 2005;78(5):529–539. doi: 10.1016/j.clpt.2005.08.004. doi:10.1016/j.clpt.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Fellay J, Marzolini C, Decosterd L, Golay KP, Baumann P, Buclin T, Telenti A, Eap CB. Variations of CYP3A activity induced by antiretroviral treatment in HIV-1 infected patients. Eur J Clin Pharmacol. 2005;60(12):865–873. doi: 10.1007/s00228-004-0855-8. doi:10.1007/s00228-004-0855-8. [DOI] [PubMed] [Google Scholar]
- 28.Gurley BJ, Gardner SF, Hubbard MA, Williams DK, Gentry WB, Khan IA, Shah A. In vivo effects of goldenseal, kava kava, black cohosh, and valerian on human cytochrome P450 1A2, 2D6, 2E1, and 3A4/5 phenotypes. Clin Pharmacol Ther. 2005;77(5):415–426. doi: 10.1016/j.clpt.2005.01.009. doi:10.1016/j.clpt.2005.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eap CB, Fellay J, Buclin T, Bleiber G, Golay KP, Brocard M, Baumann P, Telenti A. CYP3A activity measured by the midazolam test is not related to 3435 C>T polymorphism in the multiple drug resistance transporter gene. Pharmacogenetics. 2004;14(4):255–260. doi: 10.1097/00008571-200404000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Thummel KE, Shen DD, Podoll TD, Kunze KL, Trager WF, Bacchi CE, Marsh CL, McVicar JP, Barr DM, Perkins JD. Use of midazolam as a human cytochrome P450 3A probe: II. Characterization of inter- and intraindividual hepatic CYP3A variability after liver transplantation. J Pharmacol Exp Ther. 1994;271(1):557–566. [PubMed] [Google Scholar]
- 31.Carrillo JA, Ramos SI, Agundez JA, Martinez C, Benitez J. Analysis of midazolam and metabolites in plasma by high-performance liquid chromatography: probe of CYP3A. Ther Drug Monit. 1998;20(3):319–324. doi: 10.1097/00007691-199806000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54(10):1271–1294. doi: 10.1016/s0169-409x(02)00066-2. [DOI] [PubMed] [Google Scholar]
- 33.Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383–391. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 34.Gibbs MA, Thummel KE, Shen DD, Kunze KL. Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos. 1999;27(2):180–187. [PubMed] [Google Scholar]
- 35.Jones DR, Gorski JC, Hamman MA, Mayhew BS, Rider S, Hall SD. Diltiazem inhibition of cytochrome P-450 3A activity is due to metabolite intermediate complex formation. J Pharmacol Exp Ther. 1999;290(3):1116–1125. [PubMed] [Google Scholar]
- 36.Khan KK, He YQ, Domanski TL, Halpert JR. Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: an evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol Pharmacol. 2002;61(3):495–506. doi: 10.1124/mol.61.3.495. [DOI] [PubMed] [Google Scholar]
- 37.Huang W, Lin YS, McConn DJ, II, Calamia JC, Totah RA, Isoherranen N, Glodowski M, Thummel KE. Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab Dispos. 2004;32(12):1434–1445. doi: 10.1124/dmd.104.001313. doi:10.1124/dmd.104.001313. [DOI] [PubMed] [Google Scholar]
- 38.Wang YH, Jones DR, Hall SD. Differential mechanism-based inhibition of cyp3a4 and cyp3a5 by verapamil. Drug Metab Dispos. 2005;33(5):664–671. doi: 10.1124/dmd.104.001834. [DOI] [PubMed] [Google Scholar]
- 39.Greenblatt DJ, von Moltke LL, Harmatz JS, Chen G, Weemhoff JL, Jen C, Kelley CJ, LeDuc BW, Zinny MA. Time course of recovery of cytochrome p450 3A function after single doses of grapefruit juice. Clin Pharmacol Ther. 2003;74(2):121–129. doi: 10.1016/S0009-9236(03)00118-8. doi:10.1016/S0009-9236(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 40.Fromm MF, Kroemer HK, Eichelbaum M. Differential induction of prehepatic and hepatic metabolism of verapamil by rifampin. Hepatology. 1996;24(4):796–801. doi: 10.1002/hep.510240407. [DOI] [PubMed] [Google Scholar]
- 41.Hsu A, Granneman GR, Witt G, Locke C, Denissen J, Molla A, Valdes J, Smith J, Erdman K, Lyons N, Niu P, Decourt JP, Fourtillan JB, Girault J, Leonard JM. Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected subjects. Anti-microb Agents Chemother. 1997;41(5):898–905. doi: 10.1128/aac.41.5.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura K, Watanabe A, Okudaira N, Okazaki O, Sudo K. Effect of ion suppression on judgment of enzyme inhibition and avoidance of error by utilizing a stable isotope-labeled probe substrate: example of CYP3A4 inhibition with [13 C4,15N] labeled midazolam as a substrate. Drug Metab Pharmacokinet. 2007;22(2):113–118. doi: 10.2133/dmpk.22.113. [DOI] [PubMed] [Google Scholar]
- 43.Bartkowski RR, Goldberg ME, Larijani GE, Boerner T. Inhibition of alfentanil metabolism by erythromycin. Clin Pharmacol Ther. 1989;46(1):99–102. doi: 10.1038/clpt.1989.112. [DOI] [PubMed] [Google Scholar]
- 44.Ito K, Ogihara K, S-I K, Itoh T. Prediction of the in vivo interaction between midazolam and macrolides based on in vitro studies using human liver microsomes. Drug Metab Dispos. 2003;31(7):945–954. doi: 10.1124/dmd.31.7.945. doi:10.1124/dmd.31.7.945. [DOI] [PubMed] [Google Scholar]
- 45.Lilja JJ, Kivisto KT, Neuvonen PJ. Grapefruit juice-simvastatin interaction: effect on serum concentrations of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. Clin Pharmacol Ther. 1998;64(5):477–483. doi: 10.1016/S0009-9236(98)90130-8. doi:10.1016/s0009-9236(98)90130-8. [DOI] [PubMed] [Google Scholar]
- 46.Lilja JJ, Kivisto KT, Neuvonen PJ. Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin Pharmacol Ther. 1999;66(2):118–127. doi: 10.1053/cp.1999.v66.100453001. doi:10.1053/cp.1999.v66.100453001. [DOI] [PubMed] [Google Scholar]
- 47.Lilja JJ, Kivisto KT, Backman JT, Lamberg TS, Neuvonen PJ. Grapefruit juice substantially increases plasma concentrations of buspirone. Clin Pharmacol Ther. 1998;64(6):655–660. doi: 10.1016/S0009-9236(98)90056-X. doi:10.1016/S0009-9236(98)90056-X. [DOI] [PubMed] [Google Scholar]