

Abstract
Approximately 20%–30% of metastatic breast cancers show increased expression of the human epidermal growth factor receptor-2 (HER2) tyrosine kinase. Two HER2-specific therapies are currently approved for clinical treatment of patients with HER2-overexpressing metastatic breast cancer. Trastuzumab is a monoclonal antibody against HER2 and is approved for first-line treatment of HER2-positive metastatic breast cancer. Lapatinib is a small molecule dual inhibitor of epidermal growth factor receptor and HER2 tyrosine kinases, and is approved for trastuzumab-refractory disease. Although trastuzumab is a highly effective therapy for patients with HER2-overexpressing metastatic breast cancer, a significant number of patients in the initial clinical trials of trastuzumab monotherapy showed resistance to trastuzumab-based therapy. Further, among those who did respond, the initial trials indicated that the median time to progression was less than 1 year. Similarly, lapatinib is effective in a subset of trastuzumab-refractory cases, but the majority of patients display resistance. This review discusses the multiple molecular mechanisms of resistance that have been proposed in the literature. In addition, novel agents that are being tested for efficacy against HER2-positive breast cancer, including the antibodies pertuzumab and trastuzumab-DM1 and the immunotoxin affitoxin, are reviewed. The introduction of trastuzumab has revolutionized the clinical care of patients with HER2-positive metastatic breast cancer and has resulted in dramatic reductions in recurrences of early-stage HER2-positive breast cancer. The development and implementation of gene- and protein-based assays that measure potential molecular predictors of trastuzumab resistance will allow individualization of HER2-targeted therapeutic approaches, and may ultimately improve treatment of HER2-positive breast cancer.
Keywords: ErbB2, Herceptin, trastuzumab, drug resistance, neratinib, lapatinib, affitoxin
Introduction
Breast cancer continues to be the most common malignancy diagnosed in women within the United States, with approximately 226,000 new cases and almost 40,000 deaths estimated annually.1 Breast cancers have been grouped into four distinct molecular subtypes based upon gene expression profiling: luminal A, luminal B, basal-like, and human epidermal growth factor receptor 2 (HER2) positive.2,3 The HER2-positive subgroup comprises approximately 20%–30% of all metastatic breast cancers, generally due to amplification of the HER2 gene.2
HER2/ErbB2 is a receptor tyrosine kinase member of the epidermal growth factor receptor (EGFR) family, which also consists of HER3/ErbB3 and HER4/ErbB4. HER2 is the only member for which a specific ligand has not been identified. HER3 is the only member that lacks intrinsic kinase activity.4 Kinase activity is activated upon receptor dimerization, either with the same receptor type (homodimerization) or with another family member (heterodimerization). HER2 is the preferred heterodimerization partner, as HER2 heterodimers have increased ligand binding affinity and increased catalytic activity relative to other heterodimer complexes.5 Kinase activity results in autophosphorylation and subsequent downstream signaling through the phosphatidylinositol-3 kinase (PI3K)/Akt and Ras/mitogen-activated protein kinase (MAPK) cascades. Presence of homodimers or heterodimers appears to determine which downstream signaling pathways are activated, as HER2 homodimers have been shown to preferentially induce MAPK, while heterodimers activate MAPK and PI3K signaling.6 Activation of downstream PI3K signaling is thought to underlie much of the oncogenic activity of HER2, and, because of multiple PI3K recognition sites in the HER3 cytoplasmic tail, the most potent dimer pair is HER2/HER3.7–9
When HER2 is overexpressed, it is constitutively dimerized and active. Constitutive HER2 signaling leads to increased proliferation, angiogenesis, and reduced apoptosis. Overexpression of HER2 has been associated with highly aggressive disease, increased metastatic potential, and reduced survival relative to other breast cancer subtypes.10,11 The discovery that abnormal signaling from HER2 is associated with poor prognosis in breast cancer led to the search for therapeutic approaches (Figure 1) that specifically target this oncogene addiction.
Figure 1.
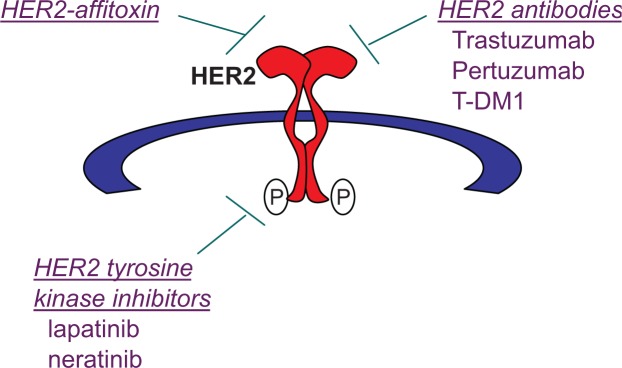
Therapeutic strategies to target human epidermal growth factor receptor 2 (HER2): (1) HER2-targeted immunotoxins that combine a HER2-targeted component with a toxic component, such as HER2-affitoxin; (2) antibodies that directly target extracellular epitopes of HER2 include trastuzumab, pertuzumab, and trastuzumab-DM1 (T-DM1) antibody-drug conjugate; and (3) small molecule tyrosine kinase inhibitors (TKIs) of HER2 include the dual epidermal growth factor receptor/HER2 TKI lapatinib and the pan-ErbB TKI neratinib.
Current treatments for HER2-positive breast cancer
Trastuzumab
Initial efforts to therapeutically target the HER2 protein led to the development of 4D5, a mouse monoclonal antibody12 that targets the extracellular domain of HER2.13 Monoclonal antibody 4D5 specifically inhibited growth of HER2- overexpressing breast cancer cell lines and prevented HER2-transformed NIH 3T3 cells from forming colonies in soft agar.13 Further, 4D5 suppressed phosphorylation of HER2 in HER2-overexpressing breast cancer cell lines and partially downregulated expression of total HER2.14 Combined treatment with 4D5 and cytotoxic chemotherapeutic drugs resulted in synergistic tumor regression and improved tumor-free survival in mouse models of human HER2-over-expressing breast tumor xenografts.15,16 Because monoclonal antibody (mAb) 4D5 is of mouse origin, the human immune system recognizes it as foreign and produces neutralizing antibodies against 4D5. Thus, a “humanized” antibody containing only the antigen-binding loops from mAb 4D5 and human variable region framework residues plus immuno-globulin G1 constant domains was constructed.17 The most effective recombinant humanized anti-HER2 antibody that was generated is now called trastuzumab (Herceptin®; Genen-tech, San Francisco, CA).
Trastuzumab is less antigenic than mAb 4D5, and it binds to HER2 with an affinity three times greater than that of 4D5.17 Preclinical studies showed significant regression of human HER2-overexpressing tumor xenografts in mice treated with trastuzumab as a single agent or in combination with a number of cytotoxic agents, including paclitaxel, cisplatin, and doxorubicin.18–20 The anticancer activity of trastuzumab is believed to be due to a number of mechanisms.21–26 Cell culture experiments suggested that trastuzumab may cause downregu-lation of cell surface HER2,27,28 which may contribute to the reduced downstream PI3K/Akt and MAPK signaling observed with trastuzumab treatment. Studies have also suggested that trastuzumab may selectively inhibit HER2-HER3 heterodi-merization,28,29 which would also reduce HER3-associated PI3K signaling. The extracellular domain of HER2 can be proteolytically cleaved, resulting in a membrane-bound truncated 95 kDa HER2 protein that has constitutively active tyrosine kinase activity (called p95 HER2). Some studies have shown that trastuzumab can prevent this cleavage of the HER2 extracellular domain.28,30 The cytostatic growth inhibitory activity of trastuzumab is due in part to increased expression of the cyclin-dependent kinase (CDK) inhibitor p27 Kip1 and subsequent inhibition of CDK2 activity.31,32 Trastuzumab has also been shown to reduce angiogenesis, resulting in reduced volume, diameter, and permeability of blood vessels.33 This normalization of the vasculature may be one reason that trastuzumab improves response to chemotherapeutic agents through more efficient delivery of the agents to the tumor. Finally, trastuzumab-bound cells are recognized by immune cells, resulting in activation of an antibody-dependent cellular cytotoxicity (ADCC) response.34,35 In fact, trastuzumab failed to induce tumor regression in mice lacking Fc-gamma receptors, which are normally present on immune natural killer cells and macrophages,35 suggesting that an intact immune system may improve the probability of achieving an optimal response to trastuzumab.
In 1998 the US Food and Drug Administration approved trastuzumab for the treatment of metastatic HER2-overex-pressing breast cancer. The initial clinical trials of trastuzumab as a single agent in HER2-overexpressing metastatic breast cancer demonstrated response rates ranging from 12% to 34% for a median duration of 9 months.36–38 Phase III trials of combination trastuzumab plus paclitaxel or docetaxel demonstrated increased response rates, time to disease progression, and overall survival versus single-agent chemotherapy.39–41 In patients with metastatic breast cancer showing HER2 amplification and no history of previous chemotherapy for metastatic disease, the median time to progression in response to single-agent chemotherapy was 4.9 months, compared with 7.4 months in patients receiving trastuzumab plus chemotherapy.41 The results of these trials indicate that trastuzumab is a highly effective therapy for patients with HER2-overexpressing metastatic breast cancer. However, a significant number of patients showed partial or complete resistance to trastuzumab-based therapy. Further, among those who did respond, the median time to acquire partial or complete resistance and show disease progression was less than 1 year. Multiple mechanisms contributing to trastuzumab resistance have been proposed and are discussed later in this review.
Lapatinib
Lapatinib (Tykerb®; GlaxoSmithKline, Research Triangle Park, NC) is a small molecule reversible inhibitor of the EGFR and HER2 tyrosine kinase domains. Early preclinical studies established lapatinib (GW572016) as an inhibitor of baseline and ligand-stimulated EGFR and HER2 signaling, resulting in reduced downstream PI3K and MAPK signaling in vitro and in vivo in models of HER2-overexpressing breast cancer.42,43 Lapatinib induced apoptosis of HER2-positive breast cancer cells as a single agent,43 sensitized HER2-positive breast cancer cells to radiation,44 and restored tamoxifen sensitivity to tamoxifen-resistant breast cancer models.45 The knockdown of EGFR using small interfering RNA did not affect lapatinib sensitivity of HER2-overexpressing breast cancer cell lines, whereas HER2 small interfering RNA caused cells to become more resistant to lapatinib.46 Thus, although lapatinib is an inhibitor of both the EGFR and HER2 kinase domain, its activity appears to be due primarily to HER2 inhibition.
Lapatinib was approved in 2007 in combination with capecitabine for use against HER2-overexpressing breast cancers that show disease progression on trastuzumab. Lapatinib is also approved as first-line therapy in combination with letrozole for hormone receptor–positive, HER2-positive metastatic breast cancer. Combination lapatinib plus chemotherapy achieved an overall response rate of 22% and a clinical benefit rate of 27%, with a median time to progression of 8.4 months.47 As a single agent, lapatinib showed clinical benefit rates ranging from 12.4% to 25% in populations pretreated with trastuzumab.48,49 Thus, lapatinib shows benefit in a subset of trastuzumab-refractory breast cancers, although the majority of trastuzumab-resistant disease shows partial resistance to lapatinib.
Another potential benefit of lapatinib may be in the setting of metastatic disease to the brain. HER2-positive breast cancers have an increased tendency to metastasize to the brain, possibly because of increased survival rates in patients since the introduction of trastuzumab, as well as the inability of a large antibody (trastuzumab) to cross the blood-brain barrier. In contrast, the small molecular size of lapatinib may facilitate its use as a treatment for HER2-positive breast cancer that has metastasized to the brain. Indeed, two phase II trials in patients with brain metastases showed reductions in the volume of brain lesions.50,51 The 2-year overall survival rate was higher in patients who responded to lapatinib than in those with stable or progressive disease (66% versus 44%, respectively). Thus, although lapatinib is an effective agent for treating HER2-positive metastatic breast cancer, novel approaches to improve response rates to lapatinib are warranted.
Mechanisms of resistance to current HER2-targeted treatments
Multiple molecular mechanisms have been proposed to contribute to trastuzumab resistance (Figure 2). At the receptor level, mutations in the HER2 kinase domain have been associated with reduced response to trastuzumab in non-small cell lung cancer,52,53 but this has not been reported in breast cancer. In addition, cell culture studies suggested that resistance is not due to changes in HER2 gene amplification, nor to reduced expression of the HER2 protein on the cell surface.54 However, recent clinical studies suggest that metastatic lesions from primary HER2-positive breast cancers do not always maintain HER2 amplification.55,56 In one study, up to one-third of patients with significant residual disease remaining after neoadjuvant trastuzumab-based therapy showed loss of HER2 amplification in association with poor survival rates.55 Another study showed that 43 of 182 patients (24%) with HER2-positive primary tumors had HER2-negative metastatic tumors.56 Another plausible mechanism by which tumors may escape trastuzumab anticancer activity is epitope masking, meaning that the drug (trastuzumab) cannot recognize or physically interact with the antigen (HER2). Whether epitope masking is a clinically relevant mechanism remains unclear. However, preclinical studies suggest membrane proteins that interact with HER2 may impede accessibility of the epitope by trastuzumab. For example, MUC4 is a transmembrane glycoprotein that is overexpressed in some breast tumors. MUC4 interacts with and activates HER2,57 impeding binding of trastuzumab to HER2. Exposure of cells to anti-MUC4 antibodies further reduced trastuzumab-HER2 interactions, suggesting that the mechanism of MUC4-mediated resistance is steric hindrance of the HER2-trastuzumab interaction. Partial masking of HER2 by MUC4 has been shown in a HER2-positive breast cancer cell line that has primary resistance to trastuzumab (JIMT1).58 Other HER2-directed antibodies that bind to epitopes different than trastuzumab showed higher effi-ciencies of binding, suggesting that HER2 is only partially masked. In addition, the knockdown of MUC4 increased the binding of trastuzumab to JIMT1 cells. Thus, the role of cell surface proteins such as MUC4 in limiting accessibility of HER2 by trastuzumab should be examined in vivo.
Figure 2.

Mechanisms of resistance to trastuzumab: (1) epitope masking, such that a cell surface molecule (MUC4) hides the epitope to which trastuzumab binds, preventing recognition and binding of human epidermal growth factor receptor 2 (HER2) by trastuzumab; (2) receptor cross talk, where cell surface receptor kinase such as insulin-like growth factor 1 receptor (IGF-IR) and Met stimulate activation of HER2 signaling; (3) increased expression and secretion of ligands that activate HER2 signaling, including growth differentiation factor 15 (GDF15), transforming growth factor (TGF) alpha, and heregulin (HRG); (4) increased phosphatidylinositol-3 kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling; (5) reduced expression of G1 arrest mediator p27Kip1 (a cyclin-dependent kinase inhibitor), and increased expression of apoptotic inhibitors B-cell lymphoma 2 (Bcl-2), myeloid cell leukemia sequence 1 (Mcl-1), and survivin.
Compensatory signaling mechanisms that sustain HER2 activity have been documented to abrogate trastuzumab sensitivity.21 Mechanisms include heterodimerization and cross talk with insulin-like growth factor 1 receptor (IGF-IR),59,60 where IGF-I stimulation caused HER2 phos-phorylation and IGF-IR inhibition reduced HER2 signaling in resistant cells.60 Importantly, antibody blockade or kinase inhibition of IGF-IR restored trastuzumab sensitivity in models of acquired trastuzumab resistance,60,61 suggesting that IGF-IR is a potential therapeutic target in trastuzumab-refractory HER2-overexpressing breast cancers. Increased expression of the ErbB ligands heregulin and transforming growth factor alpha62 has also been reported in models of trastuzumab-resistant HER2-positive breast cancer, leading to increased phosphorylation of HER2 in HER2-HER3 or HER2-EGFR heterodimers. In addition, the Met receptor tyrosine kinase has been shown to increase in trastuzumab-treated cells.63 Stimulation of Met with its ligand hepatocyte growth factor reduced response to trastuzumab in vitro, whereas knockdown or pharmacological inhibition of Met improved response to trastuzumab.63
Another potential mediator of cross talk is the transforming growth factor beta–related cytokine growth differentiation factor 15 (GDF15), which induced phosphorylation of HER2 in HER2-overexpressing breast cancer cells64–66 GDF15-mediated phosphorylation of HER2 has been shown to reduce trastuzumab sensitivity, whereas the HER2 tyrosine kinase inhibitor lapatinib abrogated GDF15-mediated Akt and extracellular signal-regulated kinase 1/2 phosphorylation and blocked GDF15-mediated trastuzumab resistance.64 HER2-overexpressing breast cancer cells with primary or acquired resistance to trastuzumab expressed increased levels of GDF15. Further, GDF15 knockdown using len-tiviral GDF15 short hairpin RNA increased trastuzumab sensitivity in resistant cells, supporting GDF15-mediated activation of HER2 as a novel mechanism of trastuzumab resistance. Interestingly, some mechanisms that confer trastuzumab resistance do not lead to lapatinib resistance, as trastuzumab-resistant cells with increased IGF-I signaling67 and GDF15 overexpression64 maintain sensitivity to lapatinib. Thus, it is important to understand which markers of trastuzumab resistance may predict for lapatinib sensitivity or resistance.
In addition to increased growth factor receptor signaling, expression levels and activity of endogenous cell cycle regulatory proteins affects response to trastuzumab and lapatinib. Reduced endogenous expression,54 genetic knockdown,68 or cytoplasmic localization of p27 Kip169 has been associated with increased CDK2 activity and proliferation and reduced growth inhibition in response to trastuzumab in vitro. Indeed, strategies that restore sensitivity to trastuzumab also result in increased expression or nuclear localization of p27 Kip1,54,59,63,70–72 suggesting that p27 Kip1 downregulation may be a predictor of resistance to trastuzumab and increased p27 Kip1 activity may improve response.
Increased expression of anti-apoptotic proteins has also been associated with resistance to HER2-targeted therapies. B-cell lymphoma 2 (Bcl-2) has been reported to upregulate in cells with acquired trastuzumab resistance.73 Resistant cells showed increased sensitivity to the BH3 mimetic ABT-737. Further, ABT-737 improved trastuzumab-mediated growth inhibition in resistant cells. Current data support further study of the Bcl-2 inhibitor ABT-737 in trastuzumab-resistant breast cancer. Other Bcl-2 antagonists have also been studied in the context of HER2-overexpressing breast cancer. Obatoclax (GX15-070), which inhibits Bcl-2, Bcl– extra large, and myeloid cell leukemia sequence 1 (Mcl-1), increased lapatinib-mediated autophagic cell death independent of endogenous EGFR or HER2 level in breast cancer cells.74 Similarly, synergistic cell death was induced by lapatinib combined with the Bcl-2 inhibitor HA14–1 or obatoclax in MCF-7, MCF-7 HER2 stable transfectant, or MCF-7 tamoxifen-resistant cells.75 A number of published studies support modulation of apoptotic regulators including Bcl-2 family members as a potential strategy for treating HER2-overexpressing breast cancer. Stable transfection of wild-type or mutant HER2 into breast cancer cells has been shown to increase Bcl-2 expression,76–79 supporting Bcl-2 as a potential target in HER2-dependent cancers. In addition, genetic knockdown of other anti-apoptotic proteins (Mcl-1 and survivin) has been shown to induce apoptosis in HER2-overexpressing breast cancer cell lines that were resistant to trastuzumab and lapatinib.80 The CDK inhibitor flavopiridol has been shown to suppress Mcl-1 expression.81 Combination flavopiridol plus lapatinib or trastuzumab resulted in synergistic cell death in HER2-overexpressing breast cancer cells.81–84 In addition, overexpression of Mcl-1 or knockdown of Bax or Bak abrogated the apoptotic effects of combination flavopiridol plus lapatinib,82 indicating that regulation of Bcl-2 family members is critical to achieving response to this drug combination.
One well-accepted molecular mechanism leading to trastuzumab resistance is increased PI3K signaling, which can occur subsequent to loss of the PTEN phosphatase gene or can be due to hyper-activating mutations in the PIK3CA catalytic subunit of PI3K.85,86 Esteva et al87 showed that phos-phorylation of Akt or the mammalian target of rapamycin (mTOR) substrate p70S6 K were not independently associated with trastuzumab resistance, but when considered together, phosphorylated Akt (p-Akt), p-p70S6K, and loss of PTEN were strongly associated with lower response to trastuzumab. Pharmacological inhibition of PI3K has been extensively studied in preclinical models as a strategy to improve response to trastuzumab.88,89 Clinical studies with mTOR inhibitor everolimus in trastuzumab-refractory disease showed very promising results. Response rates greater than 40% and disease control rates greater than 70% were achieved in metastatic HER2-overexpressing breast cancers resistant to trastuzumab and chemotherapy when treated with trastuzumab, paclitaxel, and everolimus.90 In addition, the combination of everolimus and trastuzumab achieved partial responses in 15% and persistent stable disease in 19% of patients, resulting in a clinical benefit rate of 34%.91
In contrast to trastuzumab resistance, where PI3K signaling clearly plays a role, the contribution of increased PI3K activation to lapatinib resistance is more controversial. A genome-wide loss-of-function short hairpin RNA screen performed to identify mediators of lapatinib resistance showed that loss of PTEN or PIK3CA mutations also contributed to lapatinib resistance. Further, treatment with a dual inhibitor of PI3K/mTOR inhibited colony formation and proliferation of lapatinib-resistant cells harboring genetic defects in PI3K signaling.92 In contrast, O’Brien et al93 suggested that lapatinib resistance was not associated with loss of PTEN or PIK3CA mutations and that lapatinib could block the hyperactive PI3K signaling associated with trastuzumab resistance. Wang et al94 examined 57 primary tumor samples from lapatinib-treated patients with HER2-overexpressing breast cancer heavily pretreated with chemotherapy and trastuzumab. Patients with loss of PTEN or hyperactivating mutations in PIK3CA had a significantly lower clinical benefit rate and a significantly lower overall response rate than those patients whose tumors did not show PI3K pathway activation (clinical benefit rate: 36.4% versus 68.6%, respectively; overall response rate: 9.1% versus 31.4%, respectively).
Gayle et al95 recently showed that a lower response to lapatinib was associated with an inability of lapatinib to block PI3K/mTOR signaling. Transfection of constitutively active Akt into lapatinib-sensitive cells abrogated response to lapatinib, while kinase-dead Akt improved lapatinib sensitivity, suggesting that inhibition of Akt phosphorylation is critical to achieving response to lapatinib. Further, inhibition of mTOR using rapamycin or ridaforolimus reduced p-Akt levels and increased response to lapatinib in cells that showed poor response to single-agent lapatinib, including those transfected with constitutively active Akt. Single-agent mTOR inhibition was associated with feedback signaling activating of Akt and extracellular signal-regulated kinase 1/2, which was overcome by co-treatment with lapatinib. Combination mTOR inhibition plus lapatinib resulted in synergistic growth inhibition of HER2-overexpressing trastuzumab-resistant breast cancer cells and xenografts. Our data95 indicated that p-Akt is a critical downstream target of lapatinib, whose inhibition must be intact in order to achieve optimal response to lapatinib. In cases where lapatinib alone does not effectively block PI3K/mTOR, our data95 support strategies that combine lapatinib with mTOR inhibition in the context of primary trastuzumab-resistant HER2-overexpressing breast cancer.
Emerging new HER2-directed therapies
Pertuzumab
Pertuzumab (Omnitarg®; Genentech) is a humanized anti-HER2 monoclonal antibody that binds to an extracellular epitope distinct from that to which trastuzumab binds. Pertuzumab disrupts HER2-EGFR and HER2-HER3 heterodimers. Combination trastuzumab plus pertuzumab showed enhanced tumor regression compared with either agent alone in xenograft models.96 Potential mechanisms of synergy between trastuzumab and pertuzumab include increased ADCC and reduced HER2 extracellular domain cleavage. Phase II testing of pertuzumab plus trastuzumab showed an objective response rate of 24.2% and clinical benefit rate of 50% in patients who had progressed on prior trastuzumab therapy.97 In contrast, another trial98 that tested pertuzumab monotherapy versus trastuzumab plus pertu-zumab showed disease progression in all patients treated with single-agent pertuzumab. The objective response rate and clinical benefit rate for the pertuzumab group was 3.4% and 10.3%, compared with 17.6% and 41.2% when tras-tuzumab was added. Progression-free survival (PFS) was 17.4 and 7.1 weeks in the pertuzumab and the combination groups, respectively. Another phase II trial reported patho-logic complete response rates of 45.8% and 29% in patients treated with trastuzumab, pertuzumab, and docetaxel versus trastuzumab plus docetaxel, respectively.99 A recent phase III clinical trial showed that the combination of trastuzumab plus pertuzumab and chemotherapy increased PFS and improved overall survival when compared with trastuzumab plus chemotherapy.100 The median PFS was 18.5 months in the combination arm versus 12.4 months in the control arm. Thus, combination trastuzumab plus pertuzumab appears to be an effective therapeutic strategy despite previous progression on trastuzumab.
Trastuzumab-DM1
Trastuzumab-DM1 (T-DM1) is an antibody-drug conjugate of trastuzumab and emtansine (DM1), a highly potent micro-tubule polymerization inhibitor.101 T-DM1 maintains the HER2-targeting function of trastuzumab, allowing selective delivery of DM1 to HER2-positive cells. T-DM1 is currently in phase III development for HER2-positive breast cancer. In preclinical models of trastuzumab and lapatinib resistance, T-DM1 maintained the anticancer molecular effects of trastuzumab, including inhibition of PI3K signaling, inhibition of HER2 extracellular domain cleavage, and ADCC.101–103 A phase I dose escalation trial of T-DM1 in trastuzumab-refractory HER2-positive metastatic breast cancer indicated a clinical benefit rate of 73% in 15 patients receiving the maximum tolerated dose every 3 weeks.104 Two large multicenter phase II trials of T-DM1 in patients with HER2-positive metastatic breast cancer previously treated with chemotherapy and trastuzumab or lapatinib showed overall response rates of 25.9%–34.5%, with PFS ranging from 4.6 to 6.9 months.105,106 An ongoing randomized phase III trial (EMILIA) will compare efficacy and toxicity of T-DM1 versus capecitabine plus lapatinib in patients with HER2-positive metastatic breast cancer who have previously received trastuzumab-based therapy. Importantly, patients who have been heavily pretreated with HER2-targeted therapies including T-DM1 may still derive clinical benefit from ongoing anti-HER2 regimens that incorporate trastuzumab or lapatinib.107
Neratinib
Neratinib (HKI-272; Pfizer Inc, New York, NY) is an irreversible pan-ErbB small molecule receptor tyrosine kinase inhibitor. It covalently binds to the cysteine residue in the adenosine triphosphate–binding pocket of the kinase domain of EGFR, HER2, and HER4, blocking autophos-phorylation and subsequent phosphorylation of downstream signaling molecules. Initial biochemical assays demonstrated that neratinib reduced kinase activity of the HER2 cytoplasmic domain with a half maximal inhibitory concentration (IC50) of 59 nM, and EGFR kinase activity with an IC50 of 92 nM.108 In cell culture, neratinib inhibited proliferation of HER2-overexpressing SKBr3 and BT474 cells with a very low-dose IC50 of 2–3 nM,108 which is lower than the previously observed IC50 of 25–32 nM for the reversible EGFR/HER2 tyrosine kinase inhibitor (TKI) lapatinib in these same cell lines.109 Similarly, autophosphorylation and phosphorylation of downstream molecules was reduced in BT474 cells with neratinib 2–5 nM, in contrast to trastuzumab 30 μg/mL, which, despite blocking proliferation, did not completely suppress signaling activity. Tumor regression was reported in xenograft models of HER2-overexpressing BT474 and SKOv3 cancer in association with reduced phos-phorylation of HER2.108
Neratinib has been compared with the approved TKI lapatinib with respect to mechanisms of action and biological efficacy in preclinical models. Neratinib mediated G1 arrest in HER2-overexpressing cells in association with induction of p27 Kip1 and reduced expression of cyclin D1,108 similar to what has been observed for lapatinib.109 In addition, both neratinib and lapatinib suppressed ligand-stimulated HER2-HER3 dimerization in MCF7 breast cancer cells, which express normal levels of HER2, in contrast to other reversible and irreversible HER2 inhibitors. Regarding preformed (non-ligand-stimulated) receptor dimerization, neratinib showed more rapid disruption (50% inhibition within 30 minutes) of HER2-HER3 dimers than lapatinib (50% of dimers were disrupted within 90 minutes). In addition, in contrast to other HER2 TKIs, lapatinib and neratinib reduced ligand-stimulated downregulation of total HER2 expression on the cell membrane, resulting in increased trastuzumab-mediated antibody-dependent cellular cytotoxic activity. To more closely examine the mechanisms behind neratinib- and lapatinib-mediated disruption of receptor dimerization, crystal structures of EGFR bound to various TKIs were obtained.110 These structures showed that neratinib, an irreversible TKI, and lapatinib, a reversible TKI, bind EGFR in the inactive (closed) kinase conformation, compared with reversible EGFR TKIs gefitinib and erlotinib, which can bind EGFR in the active (open) or inactive conformation. Lapatinib and neratinib are unique in that they each have an extra aromatic group in comparison with the other EGFR TKIs; this reduces the flexibility of the kinase to revert to the open position. Locking receptors in this inactive conformation prevents stable interactions between ErbB proteins. With respect to potential biomarkers or predictors of response, neratinib and lapatinib appear to impact common pathways that converge on PI3K signaling,111 and thus PI3K activity may serve as an indicator of potential response to these TKIs.
A phase II trial of neratinib in patients with HER2-overexpressing metastatic breast cancer showed PFS rates of 59% and 78% in patients refractory to trastuzumab or trastu-zumab naïve, respectively.112 The median PFS was 22.3 and 39.6 weeks and the objective response rates were 24% and 56% for refractory and trastuzumab-naïve disease, respectively. The major adverse event requiring dose reduction was diarrhea, with no high-grade cardiotoxicity observed.112,113 An earlier trial also reported antitumor action for neratinib in patients whose breast cancers were refractory to trastuzumab, taxanes, or anthracyclines and had HER2 levels of 2+ or 3+, as measured by immunohistochemistry.113 Thus, neratinib appears to show clinical benefit in patients with metastatic HER2-overexpressing breast cancer, including those who have progressed on trastuzumab-based treatment, and it may be a promising candidate for cancers that show poor response to lapatinib.
HER2-affitoxin
Immunotoxins are hybrid proteins composed of a targeting moiety such as an antibody and a toxic component derived from a plant (such as ricin) or bacteria (such as exotoxin A). HER2-affitoxin is a HER2-targeted immunotoxin that combines a HER2-specific affibody molecule with a truncated version of Pseudomonas exotoxin A called PE38KDEL.114 Affibody molecules are small proteins of approximately 7 kDa in size, making them 20 times smaller than an antibody. The amino acid sequence of an affibody molecule is based on a portion of the Staphylococcus aureus protein A. Production of affitoxin was performed by Zielinski et al114,115 by subcloning the PE38 toxin sequence into a HER2-affibody expression construct, transforming bacteria with the construct, and inducing expression of affitoxin by stimulating with isopro-pyl-beta-D-thiogalactopyranoside. Nickel-affinity (HisTrap HP; GE Healthcare, Hertfordshire, UK) and anion exchange columns were used to purify affitoxin from cell lysates.
HER2-affitoxin showed anticancer effects in models of HER2-overexpressing breast cancer.114 MCF7 breast cancer cells that were stably transfected with a HER2 expression plasmid (MCF7/HER2) showed higher binding of the HER2-affitoxin molecule relative to MCF7 parental cells. In addition, MCF7/HER2 cells were 30 times more sensitive to affitoxin than MCF7 cells, with cytotoxic activity achieved at picoMolar doses. Similarly, BT474 breast cancer cells, which have endogenous overexpression of HER2, showed stronger affinity for HER2-affitoxin and were 80 times more sensitive than MDA468 cells, which do not overexpress HER2. HER2-affitoxin binds an epitope of HER2 distinct from that which trastuzumab binds, suggesting that this novel therapy may be effective in patients whose cancers have become refractory to trastuzumab-based treatment.
HER2-affitoxin caused tumor regression in xenograft models of HER2-overexpressing breast cancer.115 HER2-targeted affitoxin (1.5 mg/kg), a control non-HER2-targeted affitoxin, or saline was delivered by intravenous injection every 3 days for a total of 24 days to athymic mice with BT474 xenografts. In contrast to the controls, HER2-affitoxin caused rapid tumor shrinkage of approximately 60% of the initial size within the first 3 days of treatment. At the end of the experiment, tumors in the HER2-affitoxin group were approximately 5% of the initial volume. Importantly, tumor regrowth was not observed within 76 days following the last day of treatment, suggesting long-term anticancer benefit from HER2-affitoxin. All animals tolerated treatment well, with slight (10%) body weight loss.
Pharmacokinetics data indicated that the half-life of HER2-affitoxin in the bloodstream of mice was almost 9 minutes with an initial concentration in plasma of almost 7 μg/mL. Dose-limiting toxicity studies indicated that mice could be treated with 0.25 or 0.5 mg/kg of HER2-affitoxin. Liver enzyme tests on mice treated with HER2-affitoxin showed that the average alanine aminotransferase plasma level was three times higher than in control animals but that the aspartate aminotransferase ratio was less than 2.115 No significant differences in alkaline phosphatase or bilirubin levels were observed.
In addition to the tumor efficacy studies using BT474 xenograft models, the HER2-overexpressing ovarian cancer cell line SKOv3 was also tested for response to HER2-affitoxin. Three doses of HER2-affitoxin at 0.25 mg/kg every 2 days were delivered by intravenous injection. Significant tumor regression was observed; however, in contrast to BT474 xenografts, regrowth of SKOv3 tumors occurred within 1 month of the last treatment. SKOv3 was also used as a model of metastatic HER2-positive cancer to determine if HER2-affitoxin reduces dissemination of cancer cells from the primary tumor. Luciferase-expressing SKOv3 cells were injected intraperitoneally, and tumor growth was examined by bioluminescence imaging.115 HER2-affitoxin significantly delayed tumor progression versus control saline treatment but failed to completely eradicate the tumors. Since SKOv3 is a highly invasive and aggressive cell line resistant to trastuzumab, these initial preclinical results suggest that HER2-affitoxin is a promising new therapeutic approach for HER2-overexpressing cancers that are nonresponsive to currently available HER2-targeted agents.
Conclusion and future perspectives
The introduction of trastuzumab has revolutionized the clinical care of patients with HER2-positive metastatic breast cancer and has resulted in dramatic reductions in recurrences of early-stage HER2-positive breast cancer. Trastuzumab-refractory disease may respond to new chemotherapy-trastuzumab combinations or to reintroduction of trastuzumab at a later time. Further, treating with a combination of multiple HER2-targeted agents may be the most effective strategy, as each HER2-directed drug appears to possess some nonoverlapping mechanisms of action, increasing the likelihood of synergy between different HER2-targeted therapies. Ultimately, identification of predictors of response will be critical for rational patient selection and for limiting nonspecific toxicities in those who are unlikely to benefit. One strategy is to develop a gene signature of trastuzumab resistance, including PIK3CA mutations that activate PI3K. Serum-based markers would be especially useful, as they can be measured by noninvasive, easily standardized assays. Such assays would measure secreted proteins such as ErbB ligands and GDF15 as predictors of trastuzumab resistance. The development and implementation of these gene- and protein-based assays that measure potential molecular predictors of resistance will allow individualization of HER2-targeted therapeutic approaches and may ultimately improve the treatment of HER2-positive breast cancer.
Acknowledgments
The author wishes to acknowledge funding support from the National Cancer Institute (K01CA118174 and 3K01CA-18174-5S1), The Mary Kay Foundation, the Georgia Cancer Coalition Distinguished Cancer Scholar Award, and P30 CA138292 (Winship Cancer Institute).
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 3.Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL., 3rd Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A. 1994;91(17):8132–8136. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16(7):1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh R, Narasanna A, Wang SE, et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011;71(5):1871–1882. doi: 10.1158/0008-5472.CAN-10-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100(15):8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HH, Sierke SL, Koland JG. Epidermal growth factor-dependent association of phosphatidylinositol 3-kinase with the ErbB3 gene product. J Biol Chem. 1994;269(40):24747–24755. [PubMed] [Google Scholar]
- 9.Soltoff SP, Carraway KL, 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14(6):3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eccles SA. The role of c-erbB-2/HER2/neu in breast cancer progression and metastasis. J Mammary Gland Biol Neoplasia. 2001;6(4):393–406. doi: 10.1023/a:1014730829872. [DOI] [PubMed] [Google Scholar]
- 11.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 12.Mabuchi S, Kawase C, Altomare DA, et al. mTOR is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clin Cancer Res. 2009;15(17):5404–5413. doi: 10.1158/1078-0432.CCR-09-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol. 1989;9(3):1165–1172. doi: 10.1128/mcb.9.3.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar R, Shepard HM, Mendelsohn J. Regulation of phosphorylation of the c-erbB-2/HER2 gene product by a monoclonal antibody and serum growth factor(s) in human mammary carcinoma cells. Mol Cell Biol. 1991;11(2):979–986. doi: 10.1128/mcb.11.2.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pietras RJ, Fendly BM, Chazin VR, Pegram MD, Howell SB, Slamon DJ. Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene. 1994;9(7):1829–1838. [PubMed] [Google Scholar]
- 16.Pietras RJ, Pegram MD, Finn RS, Maneval DA, Slamon DJ. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene. 1998;17(17):2235–2249. doi: 10.1038/sj.onc.1202132. [DOI] [PubMed] [Google Scholar]
- 17.Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the anti-tumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58(13):2825–2831. [PubMed] [Google Scholar]
- 19.Pegram MD, Slamon DJ. Combination therapy with trastuzumab (Herceptin) and cisplatin for chemoresistant metastatic breast cancer: evidence for receptor-enhanced chemosensitivity. Semin Oncol. 1999;26(4 Suppl 12):89–95. [PubMed] [Google Scholar]
- 20.Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, Fox JA. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin) Semin Oncol. 1999;26(4 Suppl 12):60–70. [PubMed] [Google Scholar]
- 21.Nahta R. Pharmacological strategies to overcome HER2 crosstalk and trastuzumab resistance. Curr Med Chem. 2012;19(7):1065–1075. doi: 10.2174/092986712799320691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastu-zumab resistance. Breast Cancer Res. 2006;8(6):215. doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nahta R, Esteva FJ. Herceptin: mechanisms of action and resistance. Cancer Lett. 2006;232(2):123–138. doi: 10.1016/j.canlet.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 24.Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26(25):3637–3643. doi: 10.1038/sj.onc.1210379. [DOI] [PubMed] [Google Scholar]
- 25.Nahta R, Shabaya S, Ozbay T, Rowe DL. Personalizing HER2-targeted therapy in metastatic breast cancer beyond HER2 status: what we have learned from clinical specimens. Curr Pharmacogenomics Person Med. 2009;7(4):263–274. doi: 10.2174/187569209790112337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 27.Cuello M, Ettenberg SA, Clark AS, et al. Down-regulation of the ErbB-2 receptor by trastuzumab (Herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress ErbB-2. Cancer Res. 2001;61(12):4892–4900. [PubMed] [Google Scholar]
- 28.Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263–275. doi: 10.1586/era.10.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 30.Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (Herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61(12):4744–4749. [PubMed] [Google Scholar]
- 31.Lane HA, Motoyama AB, Beuvink I, Hynes NE. Modulation of p27/CDK2 complex formation through 4D5-mediated inhibition of HER2 receptor signaling. Ann Oncol. 2001;12(Suppl 1):S21–S22. doi: 10.1093/annonc/12.suppl_1.s21. [DOI] [PubMed] [Google Scholar]
- 32.Le XF, Claret FX, Lammayot A, et al. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem. 2003;278(26):23441–23450. doi: 10.1074/jbc.M300848200. [DOI] [PubMed] [Google Scholar]
- 33.Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416(6878):279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 34.Arnould L, Gelly M, Penault-Llorca F, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–267. doi: 10.1038/sj.bjc.6602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 36.Baselga J, Tripathy D, Mendelsohn J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14(3):737–744. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 37.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 38.Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastu-zumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 39.Esteva FJ, Valero V, Booser D, et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(7):1800–1808. doi: 10.1200/JCO.2002.07.058. [DOI] [PubMed] [Google Scholar]
- 40.Seidman AD, Fornier MN, Esteva FJ, et al. Weekly trastuzumab and paclitaxel therapy for metastatic breast cancer with analysis of efficacy by HER2 immunophenotype and gene amplification. J Clin Oncol. 2001;19(10):2587–2595. doi: 10.1200/JCO.2001.19.10.2587. [DOI] [PubMed] [Google Scholar]
- 41.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 42.Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- 43.Xia W, Mullin RJ, Keith BR, et al. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/ErbB2 and downstream ERK1/2 and AKT pathways. Oncogene. 2002;21(41):6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 44.Zhou H, Kim YS, Peletier A, McCall W, Earp HS, Sartor CI. Effects of the EGFR/HER2 kinase inhibitor GW572016 on EGFR- and HER2-overexpressing breast cancer cell line proliferation, radiosensitization, and resistance. Int J Radiat Oncol Biol Phys. 2004;58(2):344–352. doi: 10.1016/j.ijrobp.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 45.Chu I, Blackwell K, Chen S, Slingerland J. The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res. 2005;65(1):18–25. [PubMed] [Google Scholar]
- 46.Zhang D, Pal A, Bornmann WG, et al. Activity of lapatinib is independent of EGFR expression level in HER2-overexpressing breast cancer cells. Mol Cancer Ther. 2008;7(7):1846–1850. doi: 10.1158/1535-7163.MCT-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecit-abine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 48.Blackwell KL, Burstein HJ, Storniolo AM, et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28(7):1124–1130. doi: 10.1200/JCO.2008.21.4437. [DOI] [PubMed] [Google Scholar]
- 49.Toi M, Iwata H, Fujiwara Y, et al. Lapatinib monotherapy in patients with relapsed, advanced, or metastatic breast cancer: efficacy, safety, and bio-marker results from Japanese patients phase II studies. Br J Cancer. 2009;101(10):1676–1682. doi: 10.1038/sj.bjc.6605343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin NU, Carey LA, Liu MC, et al. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2008;26(12):1993–1999. doi: 10.1200/JCO.2007.12.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin NU, Diéras V, Paul D, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15(4):1452–1459. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 52.Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med. 2006;354(24):2619–2621. doi: 10.1056/NEJMc060020. [DOI] [PubMed] [Google Scholar]
- 53.Wang SE, Narasanna A, Perez-Torres M, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10(1):25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 54.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64(11):3981–3986. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 55.Mittendorf EA, Wu Y, Scaltriti M, et al. Loss of HER2 amplification following trastuzumab-based neoadjuvant systemic therapy and survival outcomes. Clin Cancer Res. 2009;15(23):7381–7388. doi: 10.1158/1078-0432.CCR-09-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niikura N, Liu J, Hayashi N, et al. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J Clin Oncol. 2012;30(6):593–599. doi: 10.1200/JCO.2010.33.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Price-Schiavi SA, Jepson S, Li P, et al. Rat MUC4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for Herceptin resistance. Int J Cancer. 2002;99(6):783–791. doi: 10.1002/ijc.10410. [DOI] [PubMed] [Google Scholar]
- 58.Nagy P, Friedländer E, Tanner M, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a Herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65(2):473–482. [PubMed] [Google Scholar]
- 59.Huang X, Gao L, Wang S, et al. Heterotrimerization of the growth factor receptors ErbB2, ErbB3, and insulin-like growth factor-I receptor in breast cancer cells resistant to Herceptin. Cancer Res. 2010;70(3):1204–1214. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- 60.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 61.Rowe DL, Ozbay T, Bender LM, Nahta R. Nordihydroguaiaretic acid, a cytotoxic insulin-like growth factor-I receptor/HER2 inhibitor in trastuzumab-resistant breast cancer. Mol Cancer Ther. 2008;7(7):1900–1908. doi: 10.1158/1535-7163.MCT-08-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ritter CA, Perez-Torres M, Rinehart C, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13(16):4909–4919. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 63.Shattuck DL, Miller JK, Carraway KL, 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of HER2-overexpressing breast cancer cells. Cancer Res. 2008;68(5):1471–1477. doi: 10.1158/0008-5472.CAN-07-5962. [DOI] [PubMed] [Google Scholar]
- 64.Joshi JP, Brown NE, Griner SE, Nahta R. Growth differentiation factor 15 (GDF15)-mediated HER2 phosphorylation reduces trastuzumab sensitivity of HER2-overexpressing breast cancer cells. Biochem Pharmacol. 2011;82(9):1090–1099. doi: 10.1016/j.bcp.2011.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim KK, Lee JJ, Yang Y, You KH, Lee JH. Macrophage inhibitory cytokine-1 activates AKT and ERK-1/2 via the transactivation of ErbB2 in human breast and gastric cancer cells. Carcinogenesis. 2008;29(4):704–712. doi: 10.1093/carcin/bgn031. [DOI] [PubMed] [Google Scholar]
- 66.Park YJ, Lee H, Lee JH. Macrophage inhibitory cytokine-1 transacti-vates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep. 2010;43(2):91–96. doi: 10.5483/bmbrep.2010.43.2.091. [DOI] [PubMed] [Google Scholar]
- 67.Nahta R, Yuan LX, Du Y, Esteva FJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6(2):667–674. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- 68.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62(14):4132–4141. [PubMed] [Google Scholar]
- 69.Kute T, Lack CM, Willingham M, et al. Development of Herceptin resistance in breast cancer cells. Cytometry A. 2004;57(2):86–93. doi: 10.1002/cyto.a.10095. [DOI] [PubMed] [Google Scholar]
- 70.Cardoso F, Durbecq V, Laes JF, et al. Bortezomib (PS-341, Velcade) increases the efficacy of trastuzumab (Herceptin) in HER-2-positive breast cancer cells in a synergistic manner. Mol Cancer Ther. 2006;5(12):3042–3051. doi: 10.1158/1535-7163.MCT-06-0104. [DOI] [PubMed] [Google Scholar]
- 71.Esparís-Ogando A, Ocaña A, Rodríguez-Barrueco R, Ferreira L, Borges J, Pandiella A. Synergic antitumoral effect of an IGF-IR inhibitor and trastuzumab on HER2-overexpressing breast cancer cells. Ann Oncol. 2008;19(11):1860–1869. doi: 10.1093/annonc/mdn406. [DOI] [PubMed] [Google Scholar]
- 72.Tseng PH, Wang YC, Weng SC, et al. Overcoming trastuzumab resistance in HER2-overexpressing breast cancer cells by using a novel celecoxib-derived phosphoinositide-dependent kinase-1 inhibitor. Mol Pharmacol. 2006;70(5):1534–1541. doi: 10.1124/mol.106.023911. [DOI] [PubMed] [Google Scholar]
- 73.Crawford A, Nahta R. Targeting Bcl-2 in Herceptin-resistant breast cancer cell lines. Curr Pharmacogenomics Person Med. 2011;9(3):184–190. doi: 10.2174/187569211796957584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martin AP, Mitchell C, Rahmani M, Nephew KP, Grant S, Dent P. Inhibition of Mcl-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol Ther. 2009;8(21):2084–2096. doi: 10.4161/cbt.8.21.9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Witters LM, Witkoski A, Planas-Silva MD, Berger M, Viallet J, Lipton A. Synergistic inhibition of breast cancer cell lines with a dual inhibitor of EGFR-HER-2/neu and a Bcl-2 inhibitor. Oncol Rep. 2007;17(2):465–469. [PubMed] [Google Scholar]
- 76.Cittelly DM, Das PM, Salvo VA, Fonseca JP, Burow ME, Jones FE. Oncogenic HER2∆16 suppresses miR-15a/16 and deregulates Bcl-2 to promote endocrine resistance of breast tumors. Carcinogenesis. 2010;31(12):2049–2057. doi: 10.1093/carcin/bgq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB. Overex-pression of HER2 modulates Bcl-2, Bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res. 1996;2(7):1215–1219. [PubMed] [Google Scholar]
- 78.Mitra D, Brumlik MJ, Okamgba SU, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–2162. doi: 10.1158/1535-7163.MCT-09-0295. [DOI] [PubMed] [Google Scholar]
- 79.Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008;8:129. doi: 10.1186/1471-2407-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valabrega G, Capellero S, Cavalloni G, et al. HER2-positive breast cancer cells resistant to trastuzumab and lapatinib lose reliance upon HER2 and are sensitive to the multitargeted kinase inhibitor sorafenib. Breast Cancer Res Treat. 2011;130(1):29–40. doi: 10.1007/s10549-010-1281-5. [DOI] [PubMed] [Google Scholar]
- 81.Gojo I, Zhang B, Fenton RG. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin Cancer Res. 2002;8(11):3527–3538. [PubMed] [Google Scholar]
- 82.Mitchell C, Yacoub A, Hossein H, et al. Inhibition of Mcl-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther. 2010;10(9):903–917. doi: 10.4161/cbt.10.9.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nahta R, Iglehart JD, Kempkes B, Schmidt EV. Rate-limiting effects of cyclin D1 in transformation by ErbB2 predicts synergy between Herceptin and flavopiridol. Cancer Res. 2002;62(8):2267–2271. [PubMed] [Google Scholar]
- 84.Nahta R, Trent S, Yang C, Schmidt EV. Epidermal growth factor receptor expression is a candidate target of the synergistic combination of trastuzumab and flavopiridol in breast cancer. Cancer Res. 2003;63(13):3626–3631. [PubMed] [Google Scholar]
- 85.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 86.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 87.Esteva FJ, Guo H, Zhang S, et al. PTEN, PIK3CA, p-AKT, and p-p70S6 K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177(4):1647–1656. doi: 10.2353/ajpath.2010.090885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu CH, Wyszomierski SL, Tseng LM, et al. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res. 2007;13(19):5883–5888. doi: 10.1158/1078-0432.CCR-06-2837. [DOI] [PubMed] [Google Scholar]
- 89.Ozbay T, Durden DL, Liu T, O’Regan RM, Nahta R. In vitro evaluation of pan-PI3-kinase inhibitor SF1126 in trastuzumab-sensitive and trastuzumab-resistant HER2-over-expressing breast cancer cells. Cancer Chemother Pharmacol. 2010;65(4):697–706. doi: 10.1007/s00280-009-1075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andre F, Campone M, O’Regan R, et al. Phase I study of everoli-mus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol. 2010;28(34):5110–5115. doi: 10.1200/JCO.2009.27.8549. [DOI] [PubMed] [Google Scholar]
- 91.Morrow PK, Wulf GM, Ensor J, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011;29(23):3126–3132. doi: 10.1200/JCO.2010.32.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eichhorn PJ, Gili M, Scaltriti M, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–9230. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Brien NA, Browne BC, Chow L, et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9(6):1489–1502. doi: 10.1158/1535-7163.MCT-09-1171. [DOI] [PubMed] [Google Scholar]
- 94.Wang L, Zhang Q, Zhang J, et al. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer. 2011;11:248. doi: 10.1186/1471-2407-11-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gayle SS, Arnold SL, O’Regan RM, Nahta R. Pharmacologic inhibition of mTOR improves lapatinib sensitivity in HER2-overexpressing breast cancer cells with primary trastuzumab resistance. Anticancer Agents Med Chem. 2012;12(2):151–162. doi: 10.2174/187152012799015002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009;69(24):9330–9336. doi: 10.1158/0008-5472.CAN-08-4597. [DOI] [PubMed] [Google Scholar]
- 97.Baselga J, Swain SM. CLEOPATRA: a phase III evaluation of pertu-zumab and trastuzumab for HER2-positive metastatic breast cancer. Clin Breast Cancer. 2010;10(6):489–491. doi: 10.3816/CBC.2010.n.065. [DOI] [PubMed] [Google Scholar]
- 98.Cortés J, Fumoleau P, Bianchi GV, et al. Pertuzumab monotherapy after trastuzumab-based treatment and subsequent reintroduction of trastuzumab: activity and tolerability in patients with advanced human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2012 Mar 5; doi: 10.1200/JCO.2011.37.4207. Epub. [DOI] [PubMed] [Google Scholar]
- 99.Gianni L, Pienkowski T, Im YH, et al. Efficacy and safety of neoad-juvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32. doi: 10.1016/S1470-2045(11)70336-9. [DOI] [PubMed] [Google Scholar]
- 100.Baselga J, Cortés J, Kim SB, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17(20):6437–6447. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 102.Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–356. doi: 10.1007/s10549-010-1090-x. [DOI] [PubMed] [Google Scholar]
- 103.Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 104.Krop IE, Beeram M, Modi S, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28(16):2698–2704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 105.Burris HA, 3rd, Rugo HS, Vukelja SJ, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29(4):398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 106.Krop IE, LoRusso P, Miller K, et al. A phase 2 study of the HER2 antibody-drug conjugate trastuzumab-DM1 (T-DM1) in patients (pts) with HER2-positive metastatic breast cancer (MBC) previously treated with trastuzumab, lapatinib, and chemotherapy; 35th European Society for Medical Oncology Conference; October 8–12, 2010; Milan, Italy. abstract 2770. [Google Scholar]
- 107.Olson EM, Lin NU, DiPiro PJ, et al. Responses to subsequent anti-HER2 therapy after treatment with trastuzumab-DM1 in women with HER2-positive metastatic breast cancer. Ann Oncol. 2012;23(1):93–97. doi: 10.1093/annonc/mdr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64(11):3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 109.Hegde PS, Rusnak D, Bertiaux M, et al. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther. 2007;6(5):1629–1640. doi: 10.1158/1535-7163.MCT-05-0399. [DOI] [PubMed] [Google Scholar]
- 110.Sánchez-Martín M, Pandiella A. Differential action of small molecule HER kinase inhibitors on receptor heterodimerization: therapeutic implications. Int J Cancer. 2011 doi: 10.1002/ijc.26358. Epub August 8, 2011. [DOI] [PubMed] [Google Scholar]
- 111.Seyhan AA, Varadarajan U, Choe S, et al. A genome-wide RNAi screen identifies novel targets of neratinib sensitivity leading to neratinib and paclitaxel combination drug treatments. Mol Biosyst. 2011;7(6):1974–1989. doi: 10.1039/c0mb00294a. [DOI] [PubMed] [Google Scholar]
- 112.Burstein HJ, Sun Y, Dirix LY, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28(8):1301–1307. doi: 10.1200/JCO.2009.25.8707. [DOI] [PubMed] [Google Scholar]
- 113.Wong KK, Fracasso PM, Bukowski RM, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15(7):2552–2558. doi: 10.1158/1078-0432.CCR-08-1978. [DOI] [PubMed] [Google Scholar]
- 114.Zielinski R, Lyakhov I, Jacobs A, et al. Affitoxin: a novel recombinant, HER2-specific, anticancer agent for targeted therapy of HER2-positive tumors. J Immunother. 2009;32(8):817–825. doi: 10.1097/CJI.0b013e3181ad4d5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zielinski R, Lyakhov I, Hassan M, et al. HER2-affitoxin: a potent therapeutic agent for the treatment of HER2-overexpressing tumors. Clin Cancer Res. 2011;17(15):5071–5081. doi: 10.1158/1078-0432.CCR-10-2887. [DOI] [PMC free article] [PubMed] [Google Scholar]