

Abstract
The Acute Respiratory Distress Syndrome (ARDS) is a major public health problem and a leading source of morbidity in Intensive Care Units (ICUs). Lung tissue in patients with ARDS is characterized by inflammation, with exuberant neutrophil infiltration, activation and degranulation that is thought to initiate tissue injury through the release of proteases and oxygen radicals. Treatment of ARDS is supportive primarily because the underlying pathophysiology is poorly understood. This gap in knowledge must be addressed in order to identify urgently needed therapies. Recent research efforts in anti-inflammatory drug development have focused on identifying common control points in multiple signaling pathways. The protein kinase C (PKC) serine-threonine kinases are master regulators of proinflammatory signaling hubs, making them attractive therapeutic targets. Pharmacological inhibition of broad spectrum PKC activity and, more importantly, of specific PKC isoforms (as well as deletion of PKCs in mice) exerts protective effects in various experimental models of lung injury. Furthermore, PKC isoforms have been implicated in inflammatory processes that may be involved in the pathophysiologic changes that result in ARDS, including activation of innate immune and endothelial cells, neutrophil trafficking to the lung, regulation of alveolar epithelial barrier functions and control of neutrophil pro-inflammatory and pro-survival signaling. This review focuses on the mechanistic involvement of PKC isoforms in the pathogenesis of ARDS and highlights the potential of developing new therapeutic paradigms based on the selective inhibition (or activation) of specific PKC isoforms.
Keywords: inflammation, PAMPs, DAMPs, lung injury, neutrophils, cytokines, adhesion molecules, alveolar permeability
I. Introduction
The goals of this review are to i) describe the potential role played by activation of proinflammatory signaling pathways in the pathogenesis of ARDS with particular attention to cells of the innate immune system – alveolar macrophages and neutrophils - as well as the lung endothelium and alveolar epithelium; ii) systematically outline the role of protein kinase C (PKC) isoforms in the regulation of proinflammatory signaling relevant to ARDS and, most importantly, iii) summarize the mounting body of experimental evidence suggesting that selective modulation of PKC isoform activity may represent a previously unexplored pharmaceutical option in the management of ARDS.
Clinical Presentation, Definitions and Epidemiology of ARDS
ARDS is a term used to describe a highly lethal syndrome that is clinically characterized by acute onset, increasing hypoxemia with V/Q mismatch and heterogeneously distributed parenchymal abnormalities ranging from mild atelectasis to irreversible consolidation (1). Since ARDS was first described by Ashbaugh and colleagues in 1967 there have been several attempts to formally define ARDS and differentiate cases based on severity (2). In 1994, the American–European Consensus Conference (AECC) proposed standard definitions based on physiologic, radiologic and clinical abnormalities (3). In 2012, the AECC guidelines were revised by the ARDS Definition Task Force. The “Berlin Definition of ARDS” (4) defined the disorder as an “acute, diffuse inflammatory lung injury, leading to increased pulmonary vascular permeability, increased lung weight, and loss of aerated lung tissue. The clinical hallmarks are hypoxemia and bilateral radiographic opacities, associated with increased venous admixture, increased physiological dead space, and decreased lung compliance. The morphological hallmark of the acute phase is diffuse alveolar damage”. Definitions of ARDS are continually re-evaluated, as evidenced by a recent study demonstrating that post-mortem histopathological evidence of diffuse alveolar damage (DAD) was found in 69% of patients that experienced severe ARDS (according to the new Berlin definition) for > 72 hours, while only 12% of patients with mild ARDS displayed evidence of DAD(5). The new Berlin definition eliminates the term acute lung injury (ALI) because the pathophysiologic changes underlying both ALI and ARDS are the same (4). In the new formulation, ARDS is classified as mild (PaO2/FiO2 between 200 and 300 mmHg – formerly classified as Acute Lung Injury), moderate (PaO2/FiO2 between 100 and 200 mmHg), and severe (PaO2/FiO2 ≤ 100 mmHg) (6).
In the United States, approximately 200,000 patients are diagnosed with ARDS each year with a mortality rate of 25–40% (1, 7). The most recent data suggest that the age-adjusted incidence of ARDS has increased to 86 per 105 persons, leading to an annual requirement for 3.6 million hospital days (7). The incidence of ARDS was significantly higher for those between the ages of 75 through 84, estimated at 306 per 105 persons, a fact that may become more problematic as the proportion of the US population ≥ 65 years of age doubles from 35 million to 71 million people between 2010 and 2030 (8). Currently, there is no treatment to address the underlying pathophysiology of ARDS. Management is purely supportive and lung-protective ventilation strategies have proven beneficial with increased survival (9). While beneficial, this approach is designed to prevent secondary conditions and is not directed at the underlying pathologic defect, which remains poorly understood. The pressing need for therapy mandates exploration of the connection between proinflammatory signaling and the pathogenesis of lung injury.
Pathologically, ARDS involves damage to pulmonary endothelial cells, accumulation of extra-vascular lung water, neutrophil activation and influx into the alveolar space, and loss of epithelial integrity with sloughing of necrotic or apoptotic type I alveolar cells (10). Resident alveolar and fixed tissue macrophages are activated. Type II cells are characteristically preserved but impairment in function is reflected in depletion of surfactant and surfactant proteins. With progression, protein-rich fluid and debris from cellular disruption and death accumulates in alveoli and forms a dense, thick hyaline membrane that lines the denuded alveolar surface. These changes characterize the initial, “inflammatory” or “exudative” stage of the disorder, which lasts about seven days. The following 7–14 days, often referred to as the “proliferative” phase, results in substantial remodeling of the site of inflammation. After 14 or more days after the onset, progressive fibrosis typically consolidates the damaged lung parenchyma. It is important to note that some patients recover lung function and show no signs of permanent lung dysfunction. Whether the long term recovery and maintenance of lung function in these patients reflects regeneration of the damaged alveolar tissue, or other potential reparative mechanisms such as compensatory lung growth (11), is not currently known.
ARDS may result from direct lung injury, such as that caused by pneumonia, chest trauma, inhalation injury, near drowning, aspiration, reperfusion following cardiac arrest or emboli. Indirect inflammation, such as that caused by non-pulmonary infection, shock, hemorrhage, trauma, burns or extracorporeal circulation (including hemodialysis) also provokes the development of ARDS. Importantly, the mechanical ventilation required to manage ARDS can induce further lung injury (12). Common to all is activation of innate immunity in response to “danger signals” - pathogen-associated molecular patterns (PAMPs) in infection and damage associated molecular patterns (DAMPs) in response to injury and other inflammatory events (13, 14).
II. Pathogenesis of ARDS
The pathogenesis of ARDS involves a dysregulated inflammatory response within the lung, culminating in neutrophil-mediated lung tissue injury and the subsequent release of oxidants, proteases, leukotrienes and both pro- and anti-inflammatory mediators (Figure 1) (15). Activation of macrophages, microvascular endothelial cells, alveolar epithelial cells and recruited leukocytes, leads to production of cytokines (such as TNF-α, IL-6, IL-10 and IL-1β), chemokines (such as CXCL8 (IL-8)), increased expression of adhesion molecules (such as ICAM-1), and increased levels of reactive oxygen species (ROS) (16, 17). Activation of the bronchiolar and alveolar epithelium also results in increased epithelial permeability due to downregulation of junctional complex proteins (18, 19). This in combination with widespread epithelial injury (apoptosis and necrosis) leads to the leakage of protein-rich fluid, and recruitment of neutrophils into the bronchoalveolar space, as well as the release of DAMPs which further amplify the inflammatory response (Figure 1) (20). Decreased sodium pumping activity, decreased surfactant production and increased surfactant damage further hampers the resolution of edema and increases the work of breathing, respectively, both contributing to declining lung function and need for mechanical ventilation.
Figure 1.

Schematic illustrating some of the major events in the pathogenesis of ARDS. A: Top left panel: Normal alveolus with type I and type II epithelial cells, resident alveolar macrophage, capillary endothelial cells and inactivated circulating neutrophils. B: Top right panel: Activation of the innate immune system by indirect or direct lung injury (PAMPs/DAMPs) results in the release of proinflammatory cytokines and ROS by alveolar macrophages. Lung endothelial and epithelial activation further amplifies expression of cytokines and adhesion molecules that promote neutrophil adhesion and activation. C: Bottom right panel: The proinflammatory cascade drives robust neutrophil migration into a thickened, edematous interstitium and also into the alveolar space. Release of toxic mediators by recruited inflammatory cells drives cell death and lung tissue injury. Loss of epithelial barrier function results in flooding of the alveoli and hyaline membrane formation, thus impairing gas exchange.
Neutrophil migration into the lung is a hallmark of ARDS. The concentration of neutrophils in the bronchoalveolar lavage (BAL) fluid of ARDS patients correlates with disease severity and outcome (21, 22). In addition to these clinical correlations, neutrophil depletion prior to infection or injury significantly decreased the severity of lung injury in most animal models (23, 24). As a caveat, ARDS has been reported in neutropenic patients suggesting other mechanisms (25). Other cells such as alveolar macrophages also release toxic mediators that can damage lung tissue and may also contribute to the development of ARDS. Thus, a better understanding of the role played by each type of immune cell in ARDS is essential. The mechanisms that trigger neutrophil infiltration are poorly understood, but likely result from a severely proinflammatory phenotype in the lung. Proinflammatory cytokines and other alarmins (i.e. PAMPS and DAMPS) activate the nuclear transcription factors NFκB, AP-1, and C/EBP-β in alveolar macrophages (AM) as well as epithelial and endothelial cells (15, 26–30). These transcription factors regulate the expression of proinflammatory genes such as chemokines and adhesion molecules which promote neutrophil influx. Chemokines such as CXCL8 are elevated in BAL fluid (BALF) of patients with ARDS (17) and increased CXCL8 levels are associated with neutrophil infiltration (31, 32). Therefore, strategies that focus on modulating the early inflammatory response in the development of ARDS are needed in order to address the underlying pathophysiology.
III. Therapeutic Development in ARDS
While supportive measures have improved mortality rates in patients with ARDS, there is no definitive therapeutic. The pharmacological component of clinical management is based on pathophysiological observations, rather than treatments which selectively target underlying pathological mechanisms (33–35). In order to improve the considerable mortality associated with ARDS, therapeutic strategies that address the exaggerated inflammatory response are required. Potential therapeutic target sites include local control of the lung’s response to systemic (and local) inflammation, as well as direct modulation of neutrophil migration and activation. Immunoregulatory therapies focused on targeting individual mediators, including specific cytokines such as granulocyte macrophage colony stimulating factor (GM-CSF), have met with little success in the management of ARDS (36, 37). This is likely due to the fact that the inflammatory response is mediated by multiple overlapping and redundant signaling pathways in multiple cell types – suggesting the potential merits of targeting “signaling hubs” which influence multiple pathways. We and others have hypothesized that a more optimal strategy is to target molecules that are of central importance in multiple proinflammatory signaling pathways, thereby influencing the balance of pro- and anti-inflammatory signaling activity on a much broader scale. There are several control points appropriate for drug targeting and protein kinase inhibitors have become a major focus for the development of anti-inflammatory drugs (38–40). The family of PKC isoforms are key regulators of signaling in inflammatory diseases, making them attractive therapeutic targets for ARDS (41).
IV. The Protein Kinase C (PKC) Family
The PKC family is a highly conserved group of kinases that has evolved into a repertoire of at least 12 different isoforms in mammals (42). Some isoforms are ubiquitously expressed while others display higher degrees of tissue specificity. The overlap in expression of PKC isoforms is cell-type specific, allowing for a multiplicity of context-dependent functions. This contextual-dependency of PKC function often makes it difficult to determine the precise roles of PKC isoforms in normal and aberrant cellular processes. The development of selective activators and inhibitors has greatly improved our understanding of PKC isoform function in specific cells and tissues, and in various disease conditions (43, 44). In addition to providing significant insights into PKC isoform function, many of these compounds have provided a basis for the development of therapeutic strategies to treat diseases that involve hyperactive PKC signaling, such as inflammatory disorders (45).
Structure and Function of PKC Subfamilies
All PKC isoforms share a highly conserved carboxy-terminal kinase domain linked by a variable hinge region to the amino-terminal regulatory domain, which shows considerable divergence across isoforms (46). The regulatory domain contains a pseudosubstrate sequence that interacts with the substrate binding site of the kinase domain, resulting in conformational auto-inhibition of catalytic activity. Binding of the regulatory domain by allosteric effectors and/or secondary messengers disrupts binding of the pseudosubstrate sequence, freeing the kinase domain to interact with and phosphorylate substrates (Figure 2). PKC isoforms are classified based on structural arrangement, which controls mode of activation (Figure 2) (47). The classical PKC subfamily (cPKC: α, βI, βII and γ isoforms), is activated by calcium and the lipid second messenger diacylglycerol (DAG). The novel PKC subfamily (nPKC; δ, ε, θ and η isoforms) is activated by DAG, but not calcium. The atypical PKCs (aPKC; ι/λ, ζ isoforms) are not activated by calcium or DAG, but are sensitive to other lipid second messengers such as phosphatidylserine. DAG is produced by the cleavage of phophoinositide-4,5-bisphosphate in the cell membrane by phospholipase C (PLC) isoforms, to which cPKCs and nPKCs are reversibly recruited through zinc finger domains. PLC-γ and PLC-β-mediated production of DAG is typically initiated by receptor tyrosine kinases (RTKs) or G-protein coupled receptors (GPCRs) in activated membrane-associated signaling complexes (48).
Figure 2.
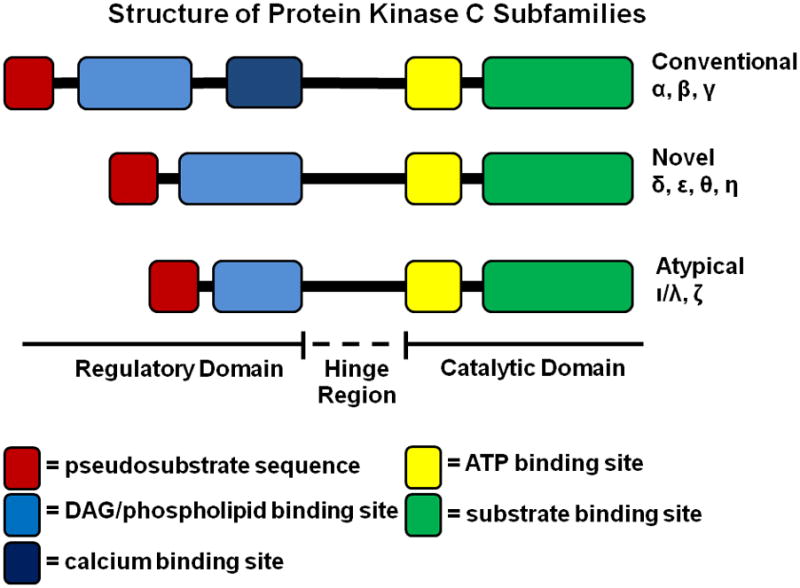
Schematic illustrating the structural arrangement of binding sites in the regulatory and catalytic domains of the conventional, novel and atypical PKC subfamilies. The catalytic subunit contains an ATP binding site and substrate binding domains which are largely conserved across isoforms. The regulatory domains are more divergent in terms of both structure and function. Only the classical isoforms contain a calcium binding activation site, while all the subfamilies contain binding sites for activation by DAG (classical and novel) or other phospholipid messengers (atypical). The regulatory domain in each subfamily contains a pseudosubstrate binding sequence that binds and inhibits kinase function in the absence of activating signals.
Priming and Activation of PKCs
In addition to the specific activators of each PKC subfamily, functional activity of PKC isoforms is also dependent on subcellular localization, which determines proximity to specific substrates and binding partners that facilitate PKC signal transduction (49, 50). The most important class of PKC binding partners are the isoform-specific receptors for activated C kinases (RACKs), which selectively and preferentially bind only the activated forms of their specific PKC (46, 51, 52). Binding to RACKs, and other scaffolding proteins such as the A kinase anchor proteins (53), brings PKCs into contact with their target substrates, making these protein-protein interactions the ultimate determinant of activated PKC function. Therefore, therapeutics that target isoform-specific protein-protein interaction domains in PKCs can provide potent selectivity.
Covalent modifications, specifically phosphorylation, are part of a maturation process that confers true functional activity of PKC isoforms. Even without pseudosubstrate binding mediated auto-inhibition, the unphosphorylated state of PKC isoforms prevent kinase activity in otherwise functionally competent structural conformations (54). At least 2 kinases are required for PKC “priming”, namely PDK-1 (55, 56) and the mTORC2 complex (57, 58). Thus, PKC regulation is multifaceted and dependent on: priming modifications (site-specific phosphorylation) required for activation, auto-inhibition exerted via pseudosubstrate binding of the catalytic domains, activation by calcium and lipid second messengers, and most importantly, subcellular localization, which is controlled by interactions with binding partners such as RACKs and other signaling complex scaffolds.
V. PKCs and Pathogenesis of ARDS
Using genetic knockout approaches and pharmacological inhibitors, PKCs have been implicated in a myriad of pathologies, including diabetes, fibrosis, oxidative stress, hypertension, thrombosis, cancer, cardiomyopathy, autoimmunity and other inflammatory disorders (59–61). Much of the data in the literature on the mechanisms of PKC isoform involvement in ARDS pathogenesis is inferred indirectly from studies in which pharmacological modulation of PKC activity (62–64), or the use of knockout mice for specific PKC isoforms (65, 66), is correlated with improvements in commonly employed experimental animal models of lung injury.
Animal Models of ARDS/Lung Injury
No consensus definition of ARDS in animal models existed prior to a report published by the American Thoracic Society’s Acute Lung Injury in Animal Models Study Group in 2011 (67). The definitions presented are a useful framework for investigators, but the scarcity of data that currently exists precludes establishment of absolute definitions of ARDS across the spectrum of available animal models. The main measurable features used to confirm ARDS in animal models are 1) Histological evidence of lung tissue injury – accumulation of neutrophils in the interstitium and alveoli, presence of hyaline membranes, proteinaceous debris in the alveoli and thickening of alveolar walls; 2) Disruption of the capillary-alveolar barrier – increased extravascular water, total (and high molecular weight) protein levels in BAL; 3) Lung inflammation – increased neutrophil counts in BAL, myeloperoxidase activity, cytokine levels; 4) Physiological dysfunction – hypoxemia, increased alveolar-arterial oxygen difference and reduced lung compliance. Animal models that capture these core pathophysiological features establish measurable outcomes for early testing of therapeutic paradigms. Furthermore, it is critical to evaluate putative anti-inflammatory therapeutics using models of lung injury induced by live bacteria (i.e. pneumonia or sepsis), in order to ensure that there are no adverse effects on host defense.
Broad Spectrum PKC Inhibition in Animal Models of ARDS/Lung Injury
With the limited availability of selective inhibitors of PKC isoforms, the vast majority of mechanistic studies on PKC signaling in ARDS involve the use of broad spectrum inhibitors. The clinical application of these compounds is not feasible or desirable due to the numerous unwanted effects in other pathways; nevertheless, the use of these compounds in experimental models has shed much light on the involvement of PKCs in ARDS pathogenesis. A mouse model of transfusion-related lung injury demonstrated increased levels of the macrophage inflammatory protein 1 (MIP-1) in plasma and BALF as a correlate for lung injury, and broad spectrum PKC inhibition in this model with the small molecules RO31-8425 (PKC-α, -β, -γ selectivity), RO31-8220 (broad spectrum PKC inhibitor, also an inhibitor of GSK-3 and an activator of JNK) and GF10 (broad spectrum inhibitor) resulted in decreased MIP-1 levels and protection from lung injury (68). The PKC inhibitor polymixin B (broad spectrum inhibitor which also binds endotoxin) attenuated lung injury and lowered levels of TNF-α, IL-1β, MCP-1 and IL-10 in a murine model of lung injury secondary to acute pancreatitis, suggesting that broad spectrum PKC inhibition dampened the overall inflammatory response in the lung (64). Similarly, systemic infusion of staurosporine (broad spectrum inhibitor which also inhibits PKA and PKG) and polymixin B both reduced pancreatic and pulmonary MPO levels in a rat model of acute pancreatitis (62).
There is also substantial evidence linking PKCs to the pathogenesis of ARDS through indirect modulation of PKC activity. In a sheep model of LPS-induced lung injury, recombinant human activated protein C (rhAPC) reduced extravascular water levels and improved oxygenation, along with other clinical improvements that correlated with reduced translocation of PKC-α and PKC-ε from the cytosolic fraction to the particulate fraction of lung tissue homogenates (69). Dapsone (DDS), a myeloperoxidase inhibitor, reduced PKC activation, preventing lung tissue damage in a murine model of paraquat-induced lung injury (70). Furthermore, inhibition of Neurokinin-1 receptor signaling with the small molecule SR140333 in a murine cecal ligation and puncture (CLP) model of polymicrobial sepsis exerted a protective effect against lung injury, primarily through reduced activation of PKC-α and the downstream transcription factors NFκB and activator protein 1 (AP-1) (71). Studies utilizing broad spectrum inhibitors fail to identify the involvement of specific PKC isoforms, however these studies established the framework for the concept of systematically breaking down the roles of individual PKC isoforms to establish more specific therapeutic targets.
VI. Proinflammatory Roles of PKCs in ARDS/Lung Injury
Activation of the Innate Immune System and Lung Inflammation: Role of PKCs
The importance of inflammatory signaling in lung injury has been recognized for over 30 years (72). In the lung, resident macrophages, neutrophils, dendritic cells, endothelial, and epithelial cells respond to pathogens or tissue injury by the detection of PAMPS and DAMPS (Figure 1). These mediators bind to pattern recognition receptors (PRRs) including Toll-like receptors (TLRs), C-type lectin receptors (CLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and purinergic receptors (73–76). Bacterial lipopolysaccharide (LPS) is the quintessential PAMP, which, along with other bacterial PAMPs such as flagellin and peptidoglycan bind to TLRs and activate proinflammatory signaling. Endothelial and epithelial injury and death results in the release of DAMPs, including intracellular proteins, such as high mobility group box-1 (HMGB1), heat shock proteins and fragmented DNA. These and other DAMPs, such as extracellular matrix (ECM) fragments liberated by protease digestion of lung tissue (77), bind to TLRs and the receptor for advanced glycation end products (RAGE), on resident inflammatory cells and recruited leukocytes, activating proinflammatory signaling.
PKCs are known mediators of inflammation and PKC-α, -δ, -ε, and -ζ are involved in multiple steps of TLR signaling pathways, implicating this family of kinases in PAMP and DAMP-driven pathogenesis in ARDS. The link between PKCs and PAMP-stimulated TLR signaling was established in studies demonstrating that pharmacological inhibition of PKCs attenuates LPS-induced cytokine production in macrophages and neutrophils (78, 79). Activation of downstream targets in TLR pathways, such as MAPKs, RhoA, TAK1 and NFκB, by PKCs promotes expression of proinflammatory genes (80). PKC-α is required for TLR2/1-mediated activation of MAPK, NFκB and AP-1, and the downstream expression of cytokines such as TNF-α, IL-6 and IL-10 (81). Inhibition and downregulation of PKC-δ in peritoneal macrophages and RAW264.7 cells reduced the activity of TIRAP, a TLR2/4 adaptor protein, thus depressing downstream activity of p38 MAPK and NFκB (82). These findings establish a direct link between PKC-δ and TLR4/TLR2 mediated proinflammatory signaling. Importantly, Puneet et al. (83) showed that sphingosine kinase-1 (SphK1) is upstream of PKC-δ activation required for NFκB-mediated inflammatory gene expression in macrophages, and that inhibition of SphK1 in vivo decreased cytokine production and mortality in a model of polymicrobial sepsis. Macrophages from PKC-ε−/− mice display decreased LPS-stimulated activation of NFκB and production of inflammatory mediators such as TNF-α, IL-1β, PGE2 and nitric oxide (NO) (84). Moreover, PKC-ε is activated by all TLRs that signal through the adaptor protein MyD88 (TLRs1-9; except TLR3) (85), and is known to be involved with TLR4 activation through the MyD88-independent TRAM-mediated pathway (86). PKC-ζ mediates phosphorylation-dependent degradation of IRAK in the TLR4 signaling complex, thereby functioning as part of a negative feedback loop to dampen receptor activation, highlighting the often times opposing roles of PKC isoforms in regulation of inflammatory signaling pathways (87).
PKCs and Activation of Alveolar Macrophages
Alveolar macrophages (AMs) are the sentinel cells of the innate immune system present within the distal airspaces, and are involved in both the initiation and resolution of inflammation. Activation of AMs by PAMPs/DAMPs and the subsequent release of proinflammatory cytokines, in an IRF- or NFκB-dependent manner, initiates the inflammatory response in the lung. These AM-derived cytokines induce expression of additional cytokines and chemokines in alveolar epithelial cells and tissue-resident macrophages in the interstitium that promote neutrophil recruitment (Figure 1) (88). Upregulation of adhesion molecules promotes migration of leukocytes to sites of inflammation; and microbicidal functions are elicited through the release of reactive oxygen species (ROS) and proteases, which if not appropriately regulated can contribute to lung injury.
Several PKC isoforms are critical regulators of AMs (89). PKC-ζ−/− mice display a reduction in LPS-induced inflammatory response in the lungs in vivo (90). Further, specific inhibition of PKC-ζ decreased the levels of LPS-induced proinflammatory cytokine production in wild type alveolar macrophages through attenuation of NFκB activation (90). PKC-α, and -δ were also identified as important regulators of LPS-induced cytokine production in AMs (91). PKC-β and RACK1 levels were indirectly correlated with maturation of cytokines and ROS production in rat alveolar macrophages (92). NOX-mediated ROS production is also linked to PKC-δ in AMs (93). Similarly, increased sepsis injury-induced mortality in Nrf-2 (an antioxidant response mediator) knockout mice was shown to involve increased PKC-dependent ROS production in AMs (94). Surfactant protein A (SPA) modulates activation of alveolar macrophages, enhancing phagocytosis of bacteria, through interactions of PKC-ζ and Rab7 (95). In contrast to the involvement of PKC-ζ in proinflammatory cytokine production by AMs, PKC-ζ is also implicated as an upstream mediator of SPA-induced anti-inflammatory activation by IκB-α in alveolar macrophages (96), highlighting the stimulus-specific and opposing functions of a single PKC isoform in a single cell type. In summary, PKC-α, -δ and -ζ have been implicated in multiple aspects of AM activation which are critical early events in ARDS pathogenesis suggesting that modulation of PKC activity in AMs may serve as an important point of control in ARDS.
PKCs and Endothelial Activation
Experimental animal models of lung injury have illustrated that endothelial injury, characterized by the formation of large intercellular gaps, necrosis, fragmentation and sloughing of the lung endothelium, is an early event in the development of lung injury (13). Endothelial cells express several PKC isoforms including α, β, δ, ε, η and ζ (97, 98). PKCs influence diverse cellular processes in endothelial cells, including cytoskeletal dynamics and expression of adhesion molecules and intercellular junctional complex proteins, all of which bear directly on leukocyte emigration and vascular permeability. There is a scarcity of data on PKC isoform regulation of proinflammatory signaling specifically in pulmonary microvascular endothelial cells; however, there is a significant body of work on the role of PKCs in vascular inflammation in other contexts. Selective inhibition of cPKCs with Go6976 attenuated TNF-α expression in pulmonary arterial endothelial cells (99). PKC-α and -ε have been implicated in endothelial cell contraction and intercellular gap formation associated with increased vascular permeability in the lung during ARDS (100). PKC-δ is required for PMA- and DAG-induced pulmonary microvascular hyperpermeability, through phosphorylation of the PKC substrate MARCKS (101). Important to the pathogenesis of ARDS, non-cardiogenic pulmonary edema is associated with increased translocation of PKCs to the plasma membrane in endothelial cells, and mediators of noncardiogenic edema such as TNF-α, thrombin and ROS all activate PKCs (102). PKC-ζ enhances thrombin-induced permeability (103), while PKC-α has been shown to mediate TNF-α and lysophosphatidylcholine-induced endothelial permeability in cell culture (104, 105). Similarly, PKC-β also is a positive regulator of the phorbol ester PMA-induced endothelial permeability in vitro (106). PKC regulation of ROS production in lung endothelial cells is poorly understood; however, PKC-α and -ζ are implicated in endothelial ROS production during cardiovascular inflammation (107).
An integral component of ARDS pathogenesis is the recruitment and activation of neutrophils through the production of chemokines/cytokines and expression of adhesion molecules such as intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in endothelial cells (108–110). Human lung microvascular endothelial cells infected with respiratory syncitial virus (RSV) in vitro upregulated ICAM-1 in a PKC- and PKA-dependent manner, potentially contributing to the increased influx of neutrophils observed in vivo (111). Using small molecule inhibitors and selective peptides, Minami et al. demonstrated that thrombin induced VCAM-1 expression in endothelial cells is dependent on PKC-δ (NFκB activation) and PKC-ζ (GATA activation) (112). Importantly, this study demonstrates that multiple PKC isoforms elicit overlapping redundant functions through independent signaling pathways. PKC-β positively regulates VCAM-1 and ICAM-1 expression in activated coronary endothelium (113). PKC-δ inhibition attenuated ICAM-1 expression in HUVECs, which corresponded to decreased binding of NFκB to the ICAM-1 promoter, placing this isoform in pathways activating NFκB-mediated expression of proinflammatory adhesion molecules (114). Therefore, PKC-δ is linked to pulmonary microvascular hyperpermeability, while -α, -β, -ε and -ζ have been shown to promote endothelial barrier dysfunction. It is important to note that these studies have been performed in endothelial cells from a myriad of tissue sources. Endothelial cells are heterogeneous and mechanisms regulating neutrophil transmigration are both tissue and stimulus dependent (115, 116), highlighting the need to determine the roles of specific PKC isoforms in pulmonary microvascular inflammation.
PKCs and Neutrophil Activation
Neutrophil priming, activation, extravasation and release of cytokines, ROS and proteases are hallmarks of ALI/ARDS, and represent potential points of control in the development of therapeutics. Human neutrophils are known to express PKC-α, PKC-β, PKC-δ, and PKC-ζ; while PKC-ι/λ and PKC-θ expression have also been reported (117–122). There is an absence of studies defining direct mechanistic involvement of specific PKCs in neutrophil-mediated events during ARDS pathogenesis, however much is known regarding the regulation of neutrophil functions by PKCs, especially the roles of PKC isoforms in regulation of oxygen radical production (118, 123, 124).
PKCs and Neutrophil Adhesion and Migration
A consequence of lung endothelial activation is upregulation of adhesion molecules and enhanced neutrophil adhesion. In combination with cytokines and chemokines, this adhesive interaction promotes neutrophil activation and facilitates migration into the lung. Commensurate with the roles of PKCs in endothelial activation described above, PKCs have well defined roles in neutrophil adhesion and migration. Both PKC-δ and PKC-ζ are critical regulators of neutrophil adhesion (125–127). In contrast, PKC-α inhibition had no effect on adhesion, indicating PKC selectivity (126). PKC-θ−/− mice demonstrated a functional role for this PKC isoform in integrin-mediated neutrophil adhesion during the process of extravasation at sites of inflammation. Impaired neutrophil rolling and arrest was observed in vivo in PKC-θ−/− mice and PKC-θ−/− neutrophils exhibited delayed cell spreading in vitro (128). Similarly, PKC-δ has been shown to promote neutrophil adhesion under flow conditions in vitro (125, 129, 130) and is selectively activated in human neutrophils following reperfusion injury in skeletal muscle in vivo (131).
Neutrophil recruitment to the lungs is significantly different than recruitment to other organs. In most organs, neutrophil accumulation is mediated through post-capillary venules, while in the lung, neutrophils move out of the pulmonary circulation across capillary endothelium and the lung architecture makes it an organ which is particularly susceptible to neutrophil accumulation. Neutrophils are physically sequestered in the lung microvasculature, where they are primed by cytokines and chemokines prior to entering to the tissue (Figure 1) (109, 132–135). Neutrophil priming in the lung microvasculature results in shape changes and cell “stiffening” based on cytoskeletal rearrangements, thus impeding passage of neutrophils through the pulmonary capillaries and leading to increased neutrophil pooling. A direct role for PKC isoforms involvement in priming and cytoskeletal-associated signaling in activated neutrophils has been established using the PKC inhibitors Go6976, a PKC-α/β inhibitor and the broad spectrum PKC inhibitor GF 109203X (selectively inhibits α, β, γ, δ and ε isoforms) (136, 137). Thus, PKC-δ, -θ and -ζ have clearly defined roles in neutrophil adhesion and emigration, while the PKC-α and -β isoforms have been implicated in the cytoskeletal rearrangements associated with the cell stiffening and shape changes during the priming of neutrophils entrapped in the pulmonary microvasculature.
PKCs in Neutrophil Pro-inflammatory and Anti-Apoptotic Signaling
Mature human neutrophils have a relatively short life span in the circulation and undergo spontaneous or constitutive apoptosis. This apoptotic process is thought to be a protective mechanism that minimizes the risk of host tissue damage caused by the release of neutrophil-derived toxic mediators. Neutrophils undergoing apoptosis have decreased oxygen radical production, degranulation, and phagocytosis in response to stimuli. During inflammatory diseases such as ARDS, neutrophil apoptosis is attenuated (138–140). The increased survival, and therefore accumulation, of neutrophils contributes to the pathophysiology of ARDS. Excessive release of neutrophil-derived toxic mediators in the lung results in alveolar tissue damage and respiratory dysfunction. Our group has shown that PKC-δ is involved in TNF-α-mediated suppression of neutrophil apoptosis. PKC-δ associates with the TNF-α receptor TNFR1 and is required for suppression of caspase-3 activity (141). TNF-α stimulation of human neutrophils causes association of PKC-δ and PI3K with the p60TNFR, and TNF-α-mediated anti-apoptotic signaling is blocked by PKC-δ inhibition, but not calcium-dependent cPKC isoforms (142). Further, PKC-δ is an important regulator in the assembly of the anti-apoptotic TNFR1 signaling complex in neutrophils (143). Other PKC isoforms have been identified in regulating cytokine and chemokine production in neutrophils (144). For example, PKC-α and -β mediate LPS- and peptidoglycan (PGN)-induced expression of TNF-α through TLR2/4 pathways in mouse neutrophils (145). Thus, PKC-δ mediates cytokine-induced anti-apoptotic signaling, and PKC-α and -β appear to be involved in cytokine/chemokine upregulation by neutrophils in response to proinflammatory signals.
The accumulation of activated neutrophils combined with impaired clearance results in an imbalance of oxidants to anti-oxidants, proteases to anti-proteases, and proinflammatory to anti-inflammatory cytokines – all of which contribute to lung tissue damage and amplification of the proinflammatory feedback loop through the release of DAMPs (28, 134). PKCs have been firmly placed in signaling pathways regulating ROS and protease production in neutrophils. PKC-α, -β, and -δ are implicated as regulators of the NADPH oxidase and superoxide anion (O2−) generation (117, 146, 147). Our group has shown that PKC-δ mediates TNF-α induced oxygen radical production by human neutrophils (123). The role for PKC-δ in regulating ROS production is stimulus-dependent and PKC-δ is not required for fMLP-induced O2− production in human neutrophils. PKC-α and -β were also shown to regulate O2− production, but not degranulation, in vitro (118, 124). Cells from PKC-δ−/− mice demonstrate limited inflammatory activities, including reduced degranulation and impaired TNF-α-induced O2− production (125). In addition to ROS production, PKC-δ also regulates cytokine-induced tertiary granule release in neutrophils (148). In summary, PKCs are central to the regulation of diverse neutrophil functions through multiple pathways in a stimulus-specific manner, establishing this family of kinases as prime targets for modulating inflammation.
PKCs and Epithelial Activation: Increased Permeability and Cytokine Expression
Activation of epithelial cells is a crucial aspect of ARDS pathogenesis (149). The loss of alveolar epithelial barrier function is critical to the development of the protein-rich edema in the distal airspaces, as evidenced by studies demonstrating that lung endothelial injury alone does not induce lung injury (150). Lung epithelial cells upregulate molecules such CXCL8, GM-CSF and ICAM-1 in response to proinflammatory stimuli. PKCs have been implicated in pathogen-mediated inflammatory signaling in alveolar epithelial cells (18). Lysophosphatidic acid (LPA)-induced production of CXCL8 in airway epithelial cells was attenuated by expression of a dominant negative PKC-δ (151). Similarly, PKC-δ inhibition decreased TNF-α- and PMA-induced expression of CXCL8 in airway epithelial cells, suggesting that PKC-δ is upstream of NFκB-mediated expression of cytokines and chemokines in lung epithelial cells stimulated by multiple ligands (152). Pharmacological inhibition and transient transfection with antisense PKC-δ nucleotides attenuated neutrophil adhesion to lung epithelium in vivo and downregulated expression of adhesion molecules in a lung epithelial cell line, implicating this isoform in neutrophil-epithelial adhesion (129).
High levels of CO2 occur with respiratory disease and hypercapnia decreases epithelial barrier integrity through disruption of Na/K-ATPase pumping function that clears fluid from the alveoli by establishing mini-osmotic gradients. PKC-ζ is required for the JNK-mediated downregulation of Na/K-ATPase that contributes to disruption of epithelial barrier integrity (153). PKC-ζ mediated Na/K-ATPase endocytosis in alveolar epithelial cells, leading to loss of function and impaired alveolar fluid clearance in a rabbit model of thrombin-induced pulmonary edema. In the presence of pulmonary edema, the alveolar epithelium becomes hypoxic, and PKCs are implicated in hypoxia-induced signaling. PKC-ζ inhibition attenuated hypoxia-induced loss of occludin protein at the plasma membrane of alveolar epithelial cells (19). LPS-induced alveolar type II cell injury leads to NAPDH oxidase-mediated expression of proinflammatory cytokines, a process which is PKC-ζ-dependent and promotes neutrophil influx, alveolar matrix degradation, and fibrotic tissue remodeling (154). Thus, PKC-ζ is an important signaling element involved in downregulation of junctional complex proteins and sodium/potassium pumps, thereby promoting leakage of fluid into the distal airspaces and impairing the resolution of edema, respectively. PKC-δ and -ζ appear to be involved in the upregulation of cytokines, chemokines and adhesion molecules in epithelial cells upon proinflammatory stimulation.
PKCs and Epithelial Apoptosis
The implications of widespread epithelial apoptosis in the pathogenesis of ARDs are twofold. First, death of alveolar type I and type II cells leads to formation of intercellular gaps and denuding of the alveolar surface, facilitating the leakage of protein-rich edema into the distal air spaces. Second, under conditions of widespread epithelial injury and impaired efferocytosis, necrosis of dying/dead epithelial cells leads to the release of intercellular contents which signal as DAMPs (20). PKCs have been implicated in epithelial apoptosis. Activation of PKC-α and PKC-δ were shown to play a permissive role upstream of p38 MAPK phosphorylation in experimental cadmium-induced alveolar type II and Clara cell apoptosis (155). Similarly, PKC-δ has been implicated in asbestos-related alveolar epithelial cell apoptosis (156). Therefore, targeting of PKC isoforms could potentially suppress epithelial apoptosis, preserve epithelial barrier integrity and sodium/potassium pump function, cytokine and chemokine production in alveolar epithelial cells; thereby targeting epithelial cells at multiple control points.
VII. Translational Studies: Selective Inhibition of PKC Isoforms in Animal Models of Lung Injury
The idea of targeting protein kinases in inflammatory disorders has gained considerable momentum in the last decade (38, 157, 158). We and others have hypothesized that selectively dampening inflammatory signaling, specifically protein kinase C isoform activation, will provide protection against lung injury through the following effects: i) decreased neutrophil influx, ii) preservation of microvascular barrier function, iii) preservation of alveolar epithelial barrier function, and iv) reduction proinflammatory cytokine/chemokine production (Figure 3, PKC-δ as a paradigm for selective inhibition strategies in the treatment of sepsis-induced lung injury). It will be essential to determine at which point in ARDS progression PKC inhibition is most effective. Anti-inflammatory therapeutics will likely be most effective when administered during the pre-acute and acute phase of ARDS. Recent improvements in early diagnosis of lung injury, prior to the need for mechanical ventilation, have significantly increased the feasibility of effectively using therapeutics targeting the early phases of lung injury (10).
Figure 3.

Targeting of PKC-δ in sepsis-induced lung injury. Selective inhibition of PKC-δ targets multiple control points in the development and early progression of ARDS as outlined in Figure 1. PKC-δ inhibition reduces activation of alveolar macrophages (decreased ROS production), endothelial cells (decreased NFκB activation, cytokine and adhesion molecule expression), and epithelial cells (decreased chemokine/cytokine production, increased survival). Importantly to the pathogenesis of ARDS, PKC-δ inhibition targets several key neutrophil functions including adhesion, migration, ROS production and prolonged (pathological) survival. Through inhibition of multiple arms of the proinflammatory cascade it is possible to protect against lung injury – and perhaps provide new tools for the clinical management of ARDS that address the underlying pathology.
Protection from Lung Injury in PKC KO mice
Studies demonstrating protection from experimentally induced lung injury in PKC-δ−/− (66), PKC-θ−/− (65) and PKC-ζ−/− (90) mice have implicated these isoforms in the pathogenesis of ARDS. PKC-θ−/− (65) and PKC-ζ−/− (90) mice exhibited protection from LPS-induced lung inflammation and injury. Similarly, LPS-induced lung injury was attenuated in PKC-δ−/− mice, and expression of a catalytically inactive PKC-δ in wild type (PKC-δ+/+) lung microvascular endothelial cells attenuated LPS-induced endothelial barrier dysfunction in vitro (66). In separate studies, PKC-δ−/− mice displayed decreased levels of asbestos-induced cytokine production and infiltration of neutrophils and macrophages in BAL and lung tissue (159). In summary, mice lacking specific PKC isoforms (δ, θ and ζ) demonstrated protection against lung injury, suggesting pharmacological inhibition of these isoforms as a potential therapeutic approach for ARDS. While mouse models provide important insight into the pathophysiology of lung injury, genetic differences do exist and may not accurately reflect human responses to inflammatory insults (160). Thus, it is essential to verify PKC isoform-specific involvement in larger animal models of ARDS and confirm observed biochemical effects in human cell culture systems.
Specific Inhibitors of PKC Isoforms: Applications in Experimental Lung Injury
The design of compounds with isoform selectivity is the core issue in development of therapeutics that target PKCs. Classical small compound inhibitors, which typically target the highly conserved catalytic domains, are known to exhibit broad inhibition of multiple kinases. The Mochly-Rosen group has developed peptide inhibitors that overcome this lack of selectivity by targeting the more variable regulatory regions, which typically regulate interactions with other proteins, therefore modulating subcellular localization and ultimate functional activity of PKCs (161). These peptides are coupled to a membrane-permeant TAT-peptide that allows safe and effective intracellular delivery into target cells in vitro and in vivo (162). These inhibitors have been tested extensively in experimental models of cardiovascular diseases, characterizing bio-distribution, pharmacokinetics and functional inhibition of PKC activity and translocation (162), as well as in phase I/II clinical trials for treatment of myocardial infarction, in which no toxicity was observed (163).
There is a considerably smaller, but growing, body of literature on the use of these compounds in experimentally induced lung injury. Acute pancreatitis often leads to ARDS with characteristic lung edema and extensive neutrophil influx into the lungs. PKC-δ mediates proinflammatory cytokine production in pancreatic acinar epithelial cells (164), which in turn amplifies the systemic inflammatory response leading to the collateral development of lung injury. PKC-δ inhibition with the selective TAT-peptide inhibitor attenuated lung edema and myeloperoxidase activity in a murine model of caerulein-induced pancreatitis (164), demonstrating a direct link between PKC-δ activity and neutrophil influx and activation in ARDS. The impairment of neutrophil influx in LPS-induced lung injury in PKC-θ−/− mice further suggests targeting of specific PKCs as a potential strategy for the treatment of ARDS (128). Kilpatrick and colleagues showed that intra-tracheal administration of the PKC-δ TAT-peptide inhibitor in a rat model of sepsis-induced lung injury significantly reduced pulmonary and circulating chemokine levels, limited inflammatory cell influx, attenuated capillary leak and the development of pulmonary edema and, overall, exerted a protective effect on the lung (63, 165). The novel PKC isoforms PKC-θ and PKC-δ have been shown to regulate key steps required for the development of lung injury in models of direct and indirect injury, respectively. Taken together, these findings further illustrate the cell- and stimulus-specific roles of PKCs in the pathogenesis of ARDS. Hence, with a deeper understanding of PKC isoform involvement in ARDS of various causes, targeted modulation of PKC-dependent proinflammatory signaling in a patient-specific manner could serve as a potent therapeutic approach in the management of ARDS.
VIII. Conclusions
In conclusion, we have summarized published observations that suggest PKC isoforms can serve as potential therapeutic targets for ARDS, specifically highlighting the roles of PKCs in neutrophil recruitment and emigration into the lung and the resultant neutrophil-mediated damage to endothelial and alveolar epithelial cells. Further mechanistic investigations will reveal the degree to which protection from lung injury by selective PKC inhibition is driven by local effects in the lung parenchymal cells versus dampening of the systemic inflammatory response. The balance between attenuating an exaggerated inflammatory response while preserving sufficient immune function to resolve the underlying insult will be crucial to the development of effective therapies. Translational studies should focus efforts on determining the optimal routes and methods of delivery to target specific cell types involved in the pathogenesis of ARDS, i.e. endotracheal installation (initial contact with alveolar macrophages and epithelium) vs. arterial infusion-based delivery (initial contact with endothelium and recruited leukocytes). It will also be critical to establish at which points in the progression of ARDS that potential therapies are most effective. Finally, due to the heterogeneity of PKC isoform expression and function in the multiple cell types involved in ARDS, combined therapies that target multiple PKC isoforms - based on mechanistic knowledge of which isoforms predominate over proinflammatory signals in these cell types - could potentially facilitate more effective therapeutic outcomes.
Acknowledgments
Grant Support: This work was supported by grants from the National Heart Lung and Blood Institute (R01HL111552) and the National Institute of General Medicine Sciences (R41GM103193) to L.E.K., the National Institute for Nursing Research (F31NR012100) to M.L., and the National Heart Lung and Blood Institute (5T32-HL007777) to M.J.M, National Institutes of Health, Bethesda, Maryland.
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Matthay MA, Zemans RL. The Acute Respiratory Distress Syndrome: Pathogenesis and Treatment. Annual Review of Pathology: Mechanisms of Disease. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashbaugh D, Bigelow D, Petty T, Levine B. Acute respiratory distress in adults. Lancet. 1967;12:319–323. doi: 10.1016/s0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- 3.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. American Journal of Respiratory and Critical Care Medicine. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 4.ARDS Definition Task Force. Ranieri V, Rubenfeld G, Thompson B, Ferguson N, Caldwell E, Fan E, Camporota L, Slutsky A. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 5.Thille AW, Esteban A, Fernandez-Segoviano P, Rodriguez JM, Aramburu JA, Penuelas O, Cortes-Puch I, Cardinal-Fernandez P, Lorente JA, Frutos-Vivar F. Comparison of the Berlin Definition for Acute Respiratory Distress Syndrome with Autopsy. American journal of respiratory and critical care medicine January 31, 2013. 2013 doi: 10.1164/rccm.201211-1981OC. [DOI] [PubMed] [Google Scholar]
- 6.Costa EL, Amato MB. The new definition for acute lung injury and acute respiratory distress syndrome: is there room for improvement? Curr Opin Crit Care. 2013;19:16–23. doi: 10.1097/MCC.0b013e32835c50b1. [DOI] [PubMed] [Google Scholar]
- 7.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and Outcomes of Acute Lung Injury. The New England journal of medicine. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. Public Health and Aging: Trends in Aging-United States and Worldwide. Morbidity and Mortality Weekly Report. 2003;52:101–106. [PubMed] [Google Scholar]
- 9.The Acute Respiratory Distress Syndrome Network. Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 10.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. The Journal of Clinical Investigation. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler JP, Loring SH, Patz S, Tsuda A, Yablonskiy DA, Mentzer SJ. Evidence for adult lung growth in humans. The New England journal of medicine. 2012;367:244–247. doi: 10.1056/NEJMoa1203983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oeckler RA, Hubmayr RD. Ventilator-associated lung injury: a search for better therapeutic targets. European Respiratory Journal. 2007;30:1216–1226. doi: 10.1183/09031936.00104907. [DOI] [PubMed] [Google Scholar]
- 13.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Seminars in respiratory and critical care medicine. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 14.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS. Characterization and Modulation of the Immunosuppressive Phase of Sepsis. Infect Immun. 78:1582–1592. doi: 10.1128/IAI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 16.Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14:523–535. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 17.Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 18.Evans SE, Kottom TJ, Pagano RE, Limper AH. Primary alveolar epithelial cell surface membrane microdomain function is required for Pneumocystis beta-glucan-induced inflammatory responses. Innate immunity. 2012;18:709–716. doi: 10.1177/1753425912436763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caraballo JC, Yshii C, Butti ML, Westphal W, Borcherding JA, Allamargot C, Comellas AP. Hypoxia increases transepithelial electrical conductance and reduces occludin at the plasma membrane in alveolar epithelial cells via PKC-zeta and PP2A pathway. American journal of physiology. Lung cellular and molecular physiology. 2011;300:L569–578. doi: 10.1152/ajplung.00109.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt EP, Tuder RM. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. Journal of cell death. 2010;2010:41–53. doi: 10.4137/JCD.S5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthay MA, Eschenbacher WL, Goetzl EJ. Elevated concentrations of leukotriene D4 in pulmonary edema fluid of patients with the adult respiratory distress syndrome. J Clin Immunol. 1984;4:479–483. doi: 10.1007/BF00916578. [DOI] [PubMed] [Google Scholar]
- 22.Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. American journal of respiratory and critical care medicine. 1994;150:113–122. doi: 10.1164/ajrccm.150.1.8025736. [DOI] [PubMed] [Google Scholar]
- 23.Till GO, Johnson KJ, Kunkel R, Ward PA. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. The Journal of clinical investigation. 1982;69:1126–1135. doi: 10.1172/JCI110548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. American journal of physiology. Lung cellular and molecular physiology. 2000;279:L1137–1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 25.Ognibene FP, Martin SE, Parker MM, Schlesinger T, Roach P, Burch C, Shelhamer JH, Parrillo JE. Adult respiratory distress syndrome in patients with severe neutropenia. The New England journal of medicine. 1986;315:547–551. doi: 10.1056/NEJM198608283150904. [DOI] [PubMed] [Google Scholar]
- 26.Park GY, Christman JW. Nuclear Factor Kappa B is a Promising Therapeutic Target in Inflammatory Lung Disease. Current Drug Targets. 2006;7:661–668. doi: 10.2174/138945006777435317. [DOI] [PubMed] [Google Scholar]
- 27.Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Medicine. 2004;30:1702–1714. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- 28.Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Molecular Medicine. 2010;16:69–82. doi: 10.2119/molmed.2009.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gea-Sorlí S, Guillamat R, Serrano-Mollar A, Closa D. Activation of lung macrophage subpopulations in experimental acute pancreatitis. The Journal of pathology. 2011;223:417–424. doi: 10.1002/path.2814. [DOI] [PubMed] [Google Scholar]
- 30.Christman JW, Sadikot RT, Blackwell TS. The role of nuclear factor-kappa B in pulmonary diseases. Chest. 2000;117:1482–1487. doi: 10.1378/chest.117.5.1482. [DOI] [PubMed] [Google Scholar]
- 31.Donnelly SC, Haslett C, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, Pollok AJ, Grant IS. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. The Lancet. 1993;341:643–647. doi: 10.1016/0140-6736(93)90416-e. [DOI] [PubMed] [Google Scholar]
- 32.Miller E, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis. 1992;146:427–432. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 33.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. The Lancet. 2007;369:1553–1564. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 34.The National Heart Lung Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Efficacy and Safety of Corticosteroids for Persistent Acute Respiratory Distress Syndrome. The New England journal of medicine. 2006;354:1671–1684. doi: 10.1056/NEJMoa051693. [DOI] [PubMed] [Google Scholar]
- 35.The Acute Respiratory Distress Syndrome N. Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. The New England journal of medicine. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 36.Bosma KJ, Lewis JF. Emerging therapies for treatment of acute lung injury and acute respiratory distress syndrome. Expert Opinion on Emerging Drugs. 2007;12:461–477. doi: 10.1517/14728214.12.3.461. [DOI] [PubMed] [Google Scholar]
- 37.Bosma KJ, Taneja R, Lewis JF. Pharmacotherapy for Prevention and Treatment of Acute Respiratory Distress Syndrome: Current and Experimental Approaches. Drugs. 2010;70:1255–1282. doi: 10.2165/10898570-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaestel M, Kotlyarov A, Kracht M. Targeting innate immunity protein kinase signalling in inflammation. Nature reviews. Drug discovery. 2009;8:480–499. doi: 10.1038/nrd2829. [DOI] [PubMed] [Google Scholar]
- 39.Philip C. Targeting protein kinases for the development of anti-inflammatory drugs. Current Opinion in Cell Biology. 2009;21:317–324. doi: 10.1016/j.ceb.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 40.Muller S, Knapp S. Targeting kinases for the treatment of inflammatory diseases. Expert Opinion on Drug Discovery. 2010;5:867–881. doi: 10.1517/17460441.2010.504203. [DOI] [PubMed] [Google Scholar]
- 41.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nature reviews. Drug discovery. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nature reviews. Molecular cell biology. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 43.Churchill EN, Qvit N, Mochly-Rosen D. Rationally designed peptide regulators of protein kinase C. Trends in Endocrinology & Metabolism. 2009;20:25–33. doi: 10.1016/j.tem.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kheifets V, Mochly-Rosen D. Insight into intra- and inter-molecular interactions of PKC: Design of specific modulators of kinase function. Pharmacological Research. 2007;55:467–476. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacological Research. 2007;55:523–536. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Steinberg SF. Structural Basis of Protein Kinase C Isoform Function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mellor H, Parker PJ. The extended protein kinase C superfamily. The Biochemical journal. 1998;332 ( Pt 2):281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diaz-Meco MT, Moscat J. The atypical PKCs in inflammation: NF-kappaB and beyond. Immunol Rev. 2012;246:154–167. doi: 10.1111/j.1600-065X.2012.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaken S, Parker PJ. Protein kinase C binding partners. BioEssays: news and reviews in molecular, cellular and developmental biology. 2000;22:245–254. doi: 10.1002/(SICI)1521-1878(200003)22:3<245::AID-BIES6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 50.Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- 51.Schechtman D, Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene. 2001;20:6339–6347. doi: 10.1038/sj.onc.1204778. [DOI] [PubMed] [Google Scholar]
- 52.Csukai M, Chen CH, De Matteis MA, Mochly-Rosen D. The coatomer protein beta′-COP, a selective binding protein (RACK) for protein kinase Cepsilon. The Journal of biological chemistry. 1997;272:29200–29206. doi: 10.1074/jbc.272.46.29200. [DOI] [PubMed] [Google Scholar]
- 53.Akakura S, Gelman IH. Pivotal Role of AKAP12 in the Regulation of Cellular Adhesion Dynamics: Control of Cytoskeletal Architecture, Cell Migration, and Mitogenic Signaling. Journal of signal transduction. 2012;2012:529179. doi: 10.1155/2012/529179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. Embo J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 56.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 57.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. Embo J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. Embo J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertram A, Ley K. Protein Kinase C Isoforms in Neutrophil Adhesion and Activation. Archivum Immunologiae et Therapiae Experimentalis. 59:79–87. doi: 10.1007/s00005-011-0112-7. [DOI] [PubMed] [Google Scholar]
- 60.Xu J, Yang G, Li T, Ming J, Liu L. Involvement of Cpi-17 and zipper-interacting protein kinase in the regulation of protein kinase C-alpha, protein kinase C-epsilon on vascular calcium sensitivity after hemorrhagic shock. Shock. 2010;33:49–55. doi: 10.1097/SHK.0b013e3181a76d77. [DOI] [PubMed] [Google Scholar]
- 61.Yang SL, Hsu C, Lue SI, Hsu HK, Yang J, Liu MS. Protein kinase C activity is increased in rat heart during the early hyperdynamic phase of sepsis. Shock. 1998;9:199–203. doi: 10.1097/00024382-199803000-00007. [DOI] [PubMed] [Google Scholar]
- 62.Shi C, Zhao X, Wang X, Zhao L, Andersson R. Potential effects of PKC or protease inhibitors on acute pancreatitis-induced tissue injury in rats. Vascul Pharmacol. 2007;46:406–411. doi: 10.1016/j.vph.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 63.Kilpatrick LE, Standage SW, Li H, Raj NR, Korchak HM, Wolfson MR, Deutschman CS. Protection against sepsis-induced lung injury by selective inhibition of protein kinase C-delta (delta-PKC) Journal of leukocyte biology. 2011;89:3–10. doi: 10.1189/jlb.0510281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao X, Shi C, Wang X, Andersson R. Protein kinase C modulates the pulmonary inflammatory response in acute pancreatitis. Respiratory physiology & neurobiology. 2006;152:16–26. doi: 10.1016/j.resp.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 65.Bertram A, Zhang H, von Vietinghoff S, de Pablo C, Haller H, Shushakova N, Ley K. Protein Kinase C-θ Is Required for Murine Neutrophil Recruitment and Adhesion Strengthening under Flow. The Journal of Immunology. 2012;188:4043–4051. doi: 10.4049/jimmunol.1101651. [DOI] [PubMed] [Google Scholar]
- 66.Chichger H, Grinnell KL, Casserly B, Chung CS, Braza J, Lomas-Neira J, Ayala A, Rounds S, Klinger JR, Harrington EO. Genetic disruption of protein kinase Cdelta reduces endotoxin-induced lung injury. American journal of physiology. Lung cellular and molecular physiology. 2012;303:L880–888. doi: 10.1152/ajplung.00169.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM on behalf of the Acute Lung Injury in Animals Study, G. An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhi L, Chi X, Gelderman MP, Vostal JG. Activation of platelet protein kinase C by ultraviolet light B mediates platelet transfusion-related acute lung injury in a two-event animal model. Transfusion 2012. 2012 Jul 31; doi: 10.1111/j.1537-2995.2012.03811.x. [DOI] [PubMed] [Google Scholar]
- 69.Waerhaug K, Kuklin VN, Kirov MY, Sovershaev MA, Langbakk B, Ingebretsen OC, Ytrehus K, Bjertnaes LJ. Recombinant human activated protein C attenuates endotoxin-induced lung injury in awake sheep. Crit Care. 2008;12:R104. doi: 10.1186/cc6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cho SC, Rhim JH, Choi HR, Son YH, Lee SJ, Song KY, Park SC. Protective effect of 4,4′-diaminodiphenylsulfone against paraquat-induced mouse lung injury. Exp Mol Med. 2011;43:525–537. doi: 10.3858/emm.2011.43.9.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hegde A, Koh YH, Moochhala SM, Bhatia M. Neurokinin-1 receptor antagonist treatment in polymicrobial sepsis: molecular insights. International journal of inflammation. 2010;2010:601098. doi: 10.4061/2010/601098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. The Lancet Saturday 12 August 1967. Critical care and resuscitation: journal of the Australasian Academy of Critical Care Medicine. 2005;7:60–61. [PubMed] [Google Scholar]
- 73.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. 2011;32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Williams MR, Azcutia VN, Newton G, Alcaide P, Luscinskas FW. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011;32:461–469. doi: 10.1016/j.it.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takeda K, Akira S. Toll-like receptors in innate immunity. International Immunology. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 76.Prince LR, Whyte MK, Sabroe I, Parker LC. The role of TLRs in neutrophil activation. Current Opinion in Pharmacology. 2011;11:397–403. doi: 10.1016/j.coph.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 77.Collins SL, Black KE, Chan-Li Y, Ahn YH, Cole PA, Powell JD, Horton MR. Hyaluronan fragments promote inflammation by down-regulating the anti-inflammatory A2a receptor. American journal of respiratory cell and molecular biology. 2011;45:675–683. doi: 10.1165/rcmb.2010-0387OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fronhofer V, Lennartz MR, Loegering DJ. Role of PKC isoforms in the Fc(gamma)R-mediated inhibition of LPS-stimulated IL-12 secretion by macrophages. Journal of Leukocyte Biology. 2006;79:408–415. doi: 10.1189/jlb.0805438. [DOI] [PubMed] [Google Scholar]
- 79.Zhou X, Yang W, Li J. Ca2+- and Protein Kinase C-dependent Signaling Pathway for Nuclear Factor-κB Activation, Inducible Nitric-oxide Synthase Expression, and Tumor Necrosis Factor-α Production in Lipopolysaccharide-stimulated Rat Peritoneal Macrophages. Journal of Biological Chemistry. 2006;281:31337–31347. doi: 10.1074/jbc.M602739200. [DOI] [PubMed] [Google Scholar]
- 80.Loegering DJ, Lennartz MR. Protein Kinase C and Toll-Like Receptor Signaling. Enzyme Research. 2011;2011:1–7. doi: 10.4061/2011/537821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langlet C, Springael C, Johnson J, Thomas S, Flamand V, Leitges M, Goldman M, Aksoy E, Willems F. PKC-alpha controls MYD88-dependent TLR/IL-1R signaling and cytokine production in mouse and human dendritic cells. Eur J Immunol. 2010;40:505–515. doi: 10.1002/eji.200939391. [DOI] [PubMed] [Google Scholar]
- 82.Kubo-Murai M, Hazeki K, Sukenobu N, Yoshikawa K, Nigorikawa K, Inoue K, Yamamoto T, Matsumoto M, Seya T, Inoue N, Hazeki O. Protein kinase Cdelta binds TIRAP/Mal to participate in TLR signaling. Mol Immunol. 2007;44:2257–2264. doi: 10.1016/j.molimm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 83.Puneet P, Yap CT, Wong L, Yulin L, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. SphK1 Regulates Proinflammatory Responses Associated with Endotoxin and Polymicrobial Sepsis. Science. 2010;328:1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]
- 84.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection. J Exp Med. 2001;194:1231–1242. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Faisal A, Saurin A, Gregory B, Foxwell B, Parker PJ. The scaffold MyD88 acts to couple protein kinase Cepsilon to Toll-like receptors. The Journal of biological chemistry. 2008;283:18591–18600. doi: 10.1074/jbc.M710330200. [DOI] [PubMed] [Google Scholar]
- 86.McGettrick AF, Brint EK, Palsson-McDermott EM, Rowe DC, Golenbock DT, Gay NJ, Fitzgerald KA, O’Neill LA. Trif-related adapter molecule is phosphorylated by PKC{epsilon} during Toll-like receptor 4 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9196–9201. doi: 10.1073/pnas.0600462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu J, Jacinto R, McCall C, Li L. Regulation of IL-1 receptor-associated kinases by lipopolysaccharide. J Immunol. 2002;168:3910–3914. doi: 10.4049/jimmunol.168.8.3910. [DOI] [PubMed] [Google Scholar]
- 88.Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Frontiers in immunology. 2011;2:65. doi: 10.3389/fimmu.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meldrum DR, Meng X, Sheridan BC, McIntyre RC, Jr, Harken AH, Banerjee A. Tissue-specific protein kinase C isoforms differentially mediate macrophage TNFalpha and IL-1beta production. Shock. 1998;9:256–260. doi: 10.1097/00024382-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 90.Yao H, Hwang JW, Moscat J, Diaz-Meco MT, Leitges M, Kishore N, Li X, Rahman I. Protein kinase C zeta mediates cigarette smoke/aldehyde- and lipopolysaccharide-induced lung inflammation and histone modifications. The Journal of biological chemistry. 2010;285:5405–5416. doi: 10.1074/jbc.M109.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martins JO, Ferracini M, Ravanelli N, Landgraf RG, Jancar S. Insulin inhibits LPS-induced signaling pathways in alveolar macrophages. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2008;21:297–304. doi: 10.1159/000129388. [DOI] [PubMed] [Google Scholar]
- 92.Corsini E, Viviani B, Lucchi L, Marinovich M, Racchi M, Galli CL. Ontogenesis of protein kinase C betaII and its anchoring protein RACK1 in the maturation of alveolar macrophage functional responses. Immunol Lett. 2001;76:89–93. doi: 10.1016/s0165-2478(00)00327-8. [DOI] [PubMed] [Google Scholar]
- 93.Bourdonnay E, Serezani CH, Aronoff DM, Peters-Golden M. Regulation of alveolar macrophage p40phox: hierarchy of activating kinases and their inhibition by PGE2. Journal of leukocyte biology. 2012;92:219–231. doi: 10.1189/jlb.1211590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kong X, Thimmulappa R, Kombairaju P, Biswal S. NADPH Oxidase-Dependent Reactive Oxygen Species Mediate Amplified TLR4 Signaling and Sepsis-Induced Mortality in Nrf2-Deficient Mice. The Journal of Immunology. 2010;185:569–577. doi: 10.4049/jimmunol.0902315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sender V, Moulakakis C, Stamme C. Pulmonary surfactant protein A enhances endolysosomal trafficking in alveolar macrophages through regulation of Rab7. J Immunol. 2011;186:2397–2411. doi: 10.4049/jimmunol.1002446. [DOI] [PubMed] [Google Scholar]
- 96.Moulakakis C, Adam S, Seitzer U, Schromm AB, Leitges M, Stamme C. Surfactant protein A activation of atypical protein kinase C zeta in IkappaB-alpha-dependent anti-inflammatory immune regulation. J Immunol. 2007;179:4480–4491. doi: 10.4049/jimmunol.179.7.4480. [DOI] [PubMed] [Google Scholar]
- 97.Rahman A, Bando M, Kefer J, Anwar KN, Malik AB. Protein Kinase C-Activated Oxidant Generation in Endothelial Cells Signals Intercellular Adhesion Molecule-1 Gene Transcription. Molecular Pharmacology. 1999;55:575–583. [PubMed] [Google Scholar]
- 98.Zimmerman GA, Albertine KH, Carveth HJ, Gill EA, Grissom CK, Hoidal JR, Imaizumi T, Maloney CG, McIntyre TM, Michael JR, Orme JF, Prescott SM, Topham MS. Endothelial activation in ARDS. Chest. 1999;116:18S–24S. doi: 10.1378/chest.116.suppl_1.18s. [DOI] [PubMed] [Google Scholar]
- 99.Tsai BM, Patel K, Wang M, Morrell ED, Crisostomo PR, Meldrum DR. Selective protein kinase C inhibition attenuates pulmonary artery cytokine expression without affecting hypoxic pulmonary vasoconstriction. Shock. 2007;27:36–39. doi: 10.1097/01.shk.0000235128.97610.b2. [DOI] [PubMed] [Google Scholar]
- 100.Siflinger-Birnboim A, Johnson A. Protein kinase C modulates pulmonary endothelial permeability: a paradigm for acute lung injury. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2003;284:L435–L451. doi: 10.1152/ajplung.00106.2002. [DOI] [PubMed] [Google Scholar]
- 101.Tinsley JH, Teasdale NR, Yuan SY. Involvement of PKCdelta and PKD in pulmonary microvascular endothelial cell hyperpermeability. American journal of physiology. Cell physiology. 2004;286:C105–111. doi: 10.1152/ajpcell.00340.2003. [DOI] [PubMed] [Google Scholar]
- 102.Daffara R, Botto L, Beretta E, Conforti E, Faini A, Palestini P, Miserocchi G. Endothelial cells as early sensors of pulmonary interstitial edema. J Appl Physiol. 2004;97:1575–1583. doi: 10.1152/japplphysiol.00236.2004. [DOI] [PubMed] [Google Scholar]
- 103.Li X, Hahn CN, Parsons M, Drew J, Vadas MA, Gamble JR. Role of protein kinase Czeta in thrombin-induced endothelial permeability changes: inhibition by angiopoietin-1. Blood. 2004;104:1716–1724. doi: 10.1182/blood-2003-11-3744. [DOI] [PubMed] [Google Scholar]
- 104.Ferro T, Neumann P, Gertzberg N, Clements R, Johnson A. Protein kinase C-alpha mediates endothelial barrier dysfunction induced by TNF-alpha. American journal of physiology. Lung cellular and molecular physiology. 2000;278:L1107–1117. doi: 10.1152/ajplung.2000.278.6.L1107. [DOI] [PubMed] [Google Scholar]
- 105.Huang F, Subbaiah PV, Holian O, Zhang J, Johnson A, Gertzberg N, Lum H. Lysophosphatidylcholine increases endothelial permeability: role of PKCalpha and RhoA cross talk. American journal of physiology. Lung cellular and molecular physiology. 2005;289:L176–185. doi: 10.1152/ajplung.00003.2005. [DOI] [PubMed] [Google Scholar]
- 106.Nagpala PG, Malik AB, Vuong PT, Lum H. Protein kinase C beta 1 overexpression augments phorbol ester-induced increase in endothelial permeability. J Cell Physiol. 1996;166:249–255. doi: 10.1002/(SICI)1097-4652(199602)166:2<249::AID-JCP2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 107.Churchill E, Budas G, Vallentin A, Koyanagi T, Mochly-Rosen D. PKC isozymes in chronic cardiac disease: possible therapeutic targets? Annu Rev Pharmacol Toxicol. 2008;48:569–599. doi: 10.1146/annurev.pharmtox.48.121806.154902. [DOI] [PubMed] [Google Scholar]
- 108.Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Current Opinion in Critical Care. 2008;14:22–30. doi: 10.1097/MCC.0b013e3282f269b9. [DOI] [PubMed] [Google Scholar]
- 109.Reutershan J, Ley K. Bench-to-bedside review: acute respiratory distress syndrome - how neutrophils migrate into the lung. Crit Care. 2004;8:453–461. doi: 10.1186/cc2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rahman A, Fazal F. Hug Tightly and Say Goodbye: Role of Endothelial ICAM-1 in Leukocyte Transmigration. Antioxidants & Redox Signaling. 2009;11:823–839. doi: 10.1089/ars.2008.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arnold R, Konig W. Respiratory syncytial virus infection of human lung endothelial cells enhances selectively intercellular adhesion molecule-1 expression. J Immunol. 2005;174:7359–7367. doi: 10.4049/jimmunol.174.11.7359. [DOI] [PubMed] [Google Scholar]
- 112.Minami T, Abid MR, Zhang J, King G, Kodama T, Aird WC. Thrombin Stimulation of Vascular Adhesion Molecule-1 in Endothelial Cells Is Mediated by Protein Kinase C (PKC)-δ-NF-κB and PKC-ζ-GATA Signaling Pathways. Journal of Biological Chemistry. 2003;278:6976–6984. doi: 10.1074/jbc.M208974200. [DOI] [PubMed] [Google Scholar]
- 113.Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114:681–687. doi: 10.1161/CIRCULATIONAHA.106.622514. [DOI] [PubMed] [Google Scholar]
- 114.Rahman A, Anwar KN, Uddin S, Xu N, Ye RD, Platanias LC, Malik AB. Protein kinase C-delta regulates thrombin-induced ICAM-1 gene expression in endothelial cells via activation of p38 mitogen-activated protein kinase. Molecular and cellular biology. 2001;21:5554–5565. doi: 10.1128/MCB.21.16.5554-5565.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aird WC. Phenotypic Heterogeneity of the Endothelium: I. Structure, Function, and Mechanisms. Circulation research. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 116.Hillyer P, Mordelet E, Flynn G, Male D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: discriminating the tissue-specific functions that affect leucocyte migration. Clinical & Experimental Immunology. 2003;134:431–441. doi: 10.1111/j.1365-2249.2003.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Majumdar S, Kane LH, Rossi MW, Volpp BD, Nauseef WM, Korchak HM. Protein kinase C isotypes and signal-transduction in human neutrophils: selective substrate specificity of calcium-dependent beta-PKC and novel calcium-independent nPKC. Biochim Biophys Acta. 1993;1176:276–286. doi: 10.1016/0167-4889(93)90056-u. [DOI] [PubMed] [Google Scholar]
- 118.Korchak HM, Rossi MW, Kilpatrick LE. Selective role for beta-protein kinase C in signaling for O-2 generation but not degranulation or adherence in differentiated HL60 cells. The Journal of biological chemistry. 1998;273:27292–27299. doi: 10.1074/jbc.273.42.27292. [DOI] [PubMed] [Google Scholar]
- 119.Korchak HM, Kilpatrick LE. Roles for beta II-protein kinase C and RACK1 in positive and negative signaling for superoxide anion generation in differentiated HL60 cells. The Journal of biological chemistry. 2001;276:8910–8917. doi: 10.1074/jbc.M008326200. [DOI] [PubMed] [Google Scholar]
- 120.Kent JD, Sergeant S, Burns DJ, McPhail LC. Identification and regulation of protein kinase C-delta in human neutrophils. J Immunol. 1996;157:4641–4647. [PubMed] [Google Scholar]
- 121.Sergeant S, McPhail LC. Opsonized zymosan stimulates the redistribution of protein kinase C isoforms in human neutrophils. J Immunol. 1997;159:2877–2885. [PubMed] [Google Scholar]
- 122.Dang PM, Hakim J, Perianin A. Immunochemical identification and translocation of protein kinase C zeta in human neutrophils. FEBS Letters. 1994;349:338–342. doi: 10.1016/0014-5793(94)00700-4. [DOI] [PubMed] [Google Scholar]
- 123.Kilpatrick LE, Sun S, Li H, Vary TC, Korchak HM. Regulation of TNF-induced oxygen radical production in human neutrophils: role of {delta}-PKC. Journal of leukocyte biology. 2010;87:153–164. doi: 10.1189/jlb.0408230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Korchak HM, Dorsey LB, Li H, Mackie D, Kilpatrick LE. Selective roles for alpha-PKC in positive signaling for O2− generation and calcium mobilization but not elastase release in differentiated HL60 cells. Biochim Biophys Acta. 2007;1773:440–449. doi: 10.1016/j.bbamcr.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 125.Chou WH, Choi DS, Zhang H, Mu D, McMahon T, Kharazia VN, Lowell CA, Ferriero DM, Messing RO. Neutrophil protein kinase Cdelta as a mediator of stroke-reperfusion injury. The Journal of clinical investigation. 2004;114:49–56. doi: 10.1172/JCI21655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Laudanna C, Mochly-Rosen D, Liron T, Constantin G, Butcher EC. Evidence of zeta protein kinase C involvement in polymorphonuclear neutrophil integrin-dependent adhesion and chemotaxis. The Journal of biological chemistry. 1998;273:30306–30315. doi: 10.1074/jbc.273.46.30306. [DOI] [PubMed] [Google Scholar]
- 127.Chakrabarti S, Patel KD. The atypical zeta (zeta) isoform of protein kinase C regulates CD11b/CD18 activation in human neutrophils. Microcirculation. 2008;15:555–567. doi: 10.1080/10739680801949732. [DOI] [PubMed] [Google Scholar]
- 128.Bertram A, Zhang H, von Vietinghoff S, de Pablo C, Haller H, Shushakova N, Ley K. Protein kinase C-theta is required for murine neutrophil recruitment and adhesion strengthening under flow. J Immunol. 2012;188:4043–4051. doi: 10.4049/jimmunol.1101651. [DOI] [PubMed] [Google Scholar]
- 129.Woo CH, Lim JH, Kim JH. VCAM-1 upregulation via PKCdelta-p38 kinase-linked cascade mediates the TNF-alpha-induced leukocyte adhesion and emigration in the lung airway epithelium. American journal of physiology. Lung cellular and molecular physiology. 2005;288:L307–316. doi: 10.1152/ajplung.00105.2004. [DOI] [PubMed] [Google Scholar]
- 130.Hendey B, Zhu CL, Greenstein S. Fas activation opposes PMA-stimulated changes in the localization of PKCdelta: a mechanism for reducing neutrophil adhesion to endothelial cells. Journal of leukocyte biology. 2002;71:863–870. [PubMed] [Google Scholar]
- 131.Huda R, Vergara LA, Solanki DR, Sherwood ER, Mathru M. Selective activation of protein kinase C delta in human neutrophils following ischemia reperfusion of skeletal muscle. Shock. 2004;21:500–504. doi: 10.1097/01.shk.0000124029.92586.5a. [DOI] [PubMed] [Google Scholar]
- 132.Moreland JG, Bailey G, Nauseef WM, Weiss JP. Organism-Specific Neutrophil-Endothelial Cell Interactions in Response to Escherichia coli, Streptococcus pneumoniae, and Staphylococcus aureus. J Immunol. 2004;172:426–432. doi: 10.4049/jimmunol.172.1.426. [DOI] [PubMed] [Google Scholar]
- 133.Burns AR, Smith CW, Walker DC. Unique Structural Features That Influence Neutrophil Emigration Into the Lung. Physiological Reviews. 2003;83:309–336. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 134.Perl M, Lomas-Neira J, Chung CS, Ayala A. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury-a unifying hypothesis? What we have learned from small interfering RNAs. Molecular Medicine. 2008;14:465–475. doi: 10.2119/2008-00011.Perl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Doerschuk CM. Mechanisms of Leukocyte Sequestration in Inflamed Lungs. Microcirculation. 2001;8:71–88. [PubMed] [Google Scholar]
- 136.Saiepour D, Sehlin J, Oldenborg PA. Insulin inhibits phagocytosis in normal human neutrophils via PKCalpha/beta-dependent priming of F-actin assembly. Inflamm Res. 2006;55:85–91. doi: 10.1007/s00011-005-0009-1. [DOI] [PubMed] [Google Scholar]
- 137.Wrann CD, Winter SW, Barkhausen T, Hildebrand F, Krettek C, Riedemann NC. Distinct involvement of p38-, ERK1/2 and PKC signaling pathways in C5a-mediated priming of oxidative burst in phagocytic cells. Cell Immunol. 2007;245:63–69. doi: 10.1016/j.cellimm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 138.Ertel W, Keel M, Infanger M, Ungethum U, Steckholzer U, Trentz O. Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up-regulation of protein-tyrosine phosphorylation. J Trauma. 1998;44:767–775. doi: 10.1097/00005373-199805000-00005. discussion 775-766. [DOI] [PubMed] [Google Scholar]
- 139.Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med. 2000;28:N105–113. doi: 10.1097/00003246-200004001-00013. [DOI] [PubMed] [Google Scholar]
- 140.Matute-Bello G, Liles WC, Radella F, 2nd, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. American journal of respiratory and critical care medicine. 1997;156:1969–1977. doi: 10.1164/ajrccm.156.6.96-12081. [DOI] [PubMed] [Google Scholar]
- 141.Kilpatrick LE, Sun S, Mackie D, Baik F, Li H, Korchak HM. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: role of {delta}-PKC and ERK1/2. J Leuk Biol. 2006;80:1512–1521. doi: 10.1189/jlb.0406284. [DOI] [PubMed] [Google Scholar]
- 142.Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-delta and PI 3-kinase in TNF-alpha-mediated antiapoptotic signaling in the human neutrophil. American journal of physiology. Cell physiology. 2002;283:C48–57. doi: 10.1152/ajpcell.00385.2001. [DOI] [PubMed] [Google Scholar]
- 143.Kilpatrick LE, Sun S, Korchak HM. Selective regulation by delta-PKC and PI 3-kinase in the assembly of the antiapoptotic TNFR-1 signaling complex in neutrophils. American journal of physiology. Cell physiology. 2004;287:C633–642. doi: 10.1152/ajpcell.00486.2003. [DOI] [PubMed] [Google Scholar]
- 144.Li J, D’Annibale-Tolhurst MA, Adler KB, Fang S, Yin Q, Birkenheuer AJ, Levy MG, Jones SL, Sung EJ, Hawkins EC, Yoder JA, Nordone SK. A MARCKS-Related Peptide Suppresses Cytokine mRNA and Protein Expression in LPS-Activated Canine Neutrophils. American journal of respiratory cell and molecular biology. 2012;48:314–321. doi: 10.1165/rcmb.2012-0278OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Asehnoune K, Strassheim D, Mitra S, Yeol Kim J, Abraham E. Involvement of PKCalpha/beta in TLR4 and TLR2 dependent activation of NF-kappaB. Cell Signal. 2005;17:385–394. doi: 10.1016/j.cellsig.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 146.Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41:7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 147.Brown GE, Stewart MQ, Liu H, Ha VL, Yaffe MB. A novel assay system implicates PtdIns(3,4)P(2), PtdIns(3)P, and PKC delta in intracellular production of reactive oxygen species by the NADPH oxidase. Molecular Cell. 2003;11:35–47. doi: 10.1016/s1097-2765(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 148.Chakrabarti S, Zee JM, Patel KD. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: novel pathways for tertiary granule release. Journal of leukocyte biology. 2006;79:214–222. doi: 10.1189/jlb.0605353. [DOI] [PubMed] [Google Scholar]
- 149.Manicone AM. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert review of clinical immunology. 2009;5:63–75. doi: 10.1586/177666X.5.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. The Journal of clinical investigation. 1991;88:864–875. doi: 10.1172/JCI115388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JG, Watkins T, He D, Saatian B, Natarajan V. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. The Journal of biological chemistry. 2004;279:41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 152.Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB. Regulation of airway epithelial cell NF-kappa B-dependent gene expression by protein kinase C delta. J Immunol. 2003;170:5681–5689. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- 153.Vadasz I, Dada LA, Briva A, Helenius IT, Sharabi K, Welch LC, Kelly AM, Grzesik BA, Budinger GR, Liu J, Seeger W, Beitel GJ, Gruenbaum Y, Sznajder JI. Evolutionary Conserved Role of c-Jun-N-Terminal Kinase in CO(2)-Induced Epithelial Dysfunction. PloS one. 2012;7:e46696. doi: 10.1371/journal.pone.0046696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leverence JT, Medhora M, Konduri GG, Sampath V. Lipopolysaccharide-induced cytokine expression in alveolar epithelial cells: role of PKCzeta-mediated p47phox phosphorylation. Chemico-biological interactions. 2011;189:72–81. doi: 10.1016/j.cbi.2010.09.026. [DOI] [PubMed] [Google Scholar]
- 155.Lag M, Refsnes M, Lilleaas EM, Holme JA, Becher R, Schwarze PE. Role of mitogen activated protein kinases and protein kinase C in cadmium-induced apoptosis of primary epithelial lung cells. Toxicology. 2005;211:253–264. doi: 10.1016/j.tox.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 156.Shukla A, Stern M, Lounsbury KM, Flanders T, Mossman BT. Asbestos-Induced Apoptosis Is Protein Kinase C{delta}-Dependent. Am J Respir Cell Mol Biol. 2003;29:198–205. doi: 10.1165/rcmb.2002-0248OC. [DOI] [PubMed] [Google Scholar]
- 157.Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Current Opinion in Cell Biology. 2009;21:317–324. doi: 10.1016/j.ceb.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 158.O’Neill LA. Targeting signal transduction as a strategy to treat inflammatory diseases. Nature reviews. Drug discovery. 2006;5:549–563. doi: 10.1038/nrd2070. [DOI] [PubMed] [Google Scholar]
- 159.Shukla A, Lounsbury KM, Barrett TF, Gell J, Rincon M, Butnor KJ, Taatjes DJ, Davis GS, Vacek P, Nakayama KI, Nakayama K, Steele C, Mossman BT. Asbestos-induced peribronchiolar cell proliferation and cytokine production are attenuated in lungs of protein kinase C-delta knockout mice. The American journal of pathology. 2007;170:140–151. doi: 10.2353/ajpath.2007.060381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG Inflammation and Host Response to Injury LSCRP. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Budas GR, Koyanagi T, Churchill EN, Mochly-Rosen D. Competitive inhibitors and allosteric activators of protein kinase C isoenzymes: a personal account and progress report on transferring academic discoveries to the clinic. Biochemical Society transactions. 2007;35:1021–1026. doi: 10.1042/BST0351021. [DOI] [PubMed] [Google Scholar]
- 162.Begley R, Liron T, Baryza J, Mochly-Rosen D. Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochemical and biophysical research communications. 2004;318:949–954. doi: 10.1016/j.bbrc.2004.04.121. [DOI] [PubMed] [Google Scholar]
- 163.Bode C, Costa M, Gibson CM, Granger C, Green C, Grimes K, Harrington R, Huber K, Kleiman N, Mochly-Rosen D, Roe M, Sadowski Z, Solomon S, Widimsky P DELTA MI Investigators. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. 2008;117:886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 164.Ramnath RD, Sun J, Bhatia M. PKC delta mediates pro-inflammatory responses in a mouse model of caerulein-induced acute pancreatitis. J Mol Med (Berl) 2010;88:1055–1063. doi: 10.1007/s00109-010-0647-9. [DOI] [PubMed] [Google Scholar]
- 165.Martins JO. Editorial: Can PKCδ be a novel therapeutic target? Journal of leukocyte biology. 2011;89:1–2. doi: 10.1189/jlb.0810452. [DOI] [PubMed] [Google Scholar]