

Abstract
Inactivation of tumor suppressor genes via homozygous deletion is a prototypic event in the cancer genome, yet such deletions often encompass neighboring genes. We hypothesized that homozygous deletions in such passenger genes can expose cancer-specific therapeutic vulnerabilities in the case where the collaterally deleted gene is a member of a functionally redundant family of genes exercising an essential function. The glycolytic gene Enolase 1 (ENO1) in the 1p36 locus is deleted in Glioblastoma (GBM), which is tolerated by expression of ENO2. We demonstrate that shRNA-mediated extinction of ENO2 selectively inhibits growth, survival, and tumorigenic potential of ENO1-deleted GBM cells and that the enolase inhibitor phosphonoacetohydroxamate (PhAH) is selectively toxic to ENO1-deleted GBM cells relative to ENO1-intact GBM cells or normal astrocytes. The principle of collateral vulnerability should be applicable to other passenger deleted genes encoding functionally-redundant essential activities and provide an effective treatment strategy for cancers harboring such genomic events.
Large scale analysis of the cancer genome has provided an unprecedentedly detailed picture of the genetic anatomy of cancer1 which has been, and continues to serve as a blueprint for the development of molecular targeted therapies. Targeted therapies directed against amplified or mutant-activated key driver oncoproteins have provided encouraging clinical progress2, whereas exploiting loss-of-function mutations or gene deletions has received considerably less attention and has thus far been less successful. Previous therapeutic work in the area of loss-of-function mutations and deletions has focused specifically on tumor suppressor genes via strategies that include synthetic lethal approaches. One striking example of a synthetic lethal interaction is the response of BRCA1-mutant cancers to PARP inhibitors, although this interaction appears to be dependent on genetic context3,4, and can be bypassed in late stage tumors3,4,5. Most other synthetic lethal interactors of inactivated tumor suppressors appear to be less robust in eliciting cancer cell death5, perhaps because such genes typically do not perform fundamental housekeeping functions.
Cancer genomes are characterized by numerous copy number amplifications and deletions, which target driver oncogenes and tumor suppressor genes, respectively. Often, these genomic alterations are large regional events, affecting many other genes in addition to the intended target(s). The fact that such broad genomic alterations are not negatively selected against in cancer cells implies that, on their own, the copy number alterations of these neighboring passengers must not carry severely detrimental biological consequences. That said, it is conceivable that these passenger genomic events can create unintended (collateral) vulnerabilities unique to those cells; such as when a passenger being co-deleted is a member of a redundant multi-gene family serving an essential housekeeping function. A large body of genetic interaction studies in invertebrates as well as mice indicates that many essential cellular housekeeping functions are carried out by multiple homologous genes that encode overlapping functions; this redundancy enables cell viability upon loss of one homologue but causes lethality upon loss of multiple homologues6–10 (Supplementary Fig. 1). In this conceptual framework, we hypothesized that the homozygous deletion of redundant essential housekeeping genes could create cancer-specific vulnerabilities (Supplementary Fig. 1a) whereby pharmacological inactivation of the second, non-deleted homologue would result in complete loss of activity in tumor cells carrying the deletion, without compromising the health of normal cells, in which both genes are intact and expressed (Supplementary Fig. 1b).
ENO1 is an essential redundant housekeeping gene homozygoulsy deleted in glioblastoma
By examining the Cancer Genome Atlas (TCGA) GBM data set for homozygous deletions targeting genes involved in essential cell activities1, we identified various such candidates, including the ENO1 gene, which resides at the 1p36 tumor suppressor locus (See Table 1 for a summary and Supplementary Table 1 for more detailed methodological support). Enolase, which is encoded by three homologous genes, is an essential enzyme that catalyzes the second to last step of glycolysis, converting 2-phosphoglyceric acid into phosphoenolpyruvate11. In mammals, enolase activity is encoded by three genes: ENO1, which is ubiquitously expressed12,13; ENO2, which is expressed exclusively in neural tissues12,14; and ENO3, which is expressed in muscle tissues15 (Supplementary Table 2). ENO1 is the major enolase isoform in GBM, accounting for 75–90% of cellular enolase activity12. Given the critical importance of glycolysis for energy generation and anabolic processes in normal and especially tumor cells16, GBM tumors homozygous null for ENO1 would be predicted to be highly sensitive to inhibition of enolase 2, whereas normal neural tissues should not be affected because of the functional redundancy of enolase 1 (Fig. 1a,b). Correspondingly, ENO2 knockout mice are viable and fertile, suggesting that pharmacological inhibition of enolase 2 is likely to be well tolerated at the organism level (Supplementary Table 2). Moreover, Saccharomyces cerevisiae, which possesses several enolase homologues, shows weak phenotypes with single mutants and incurs cell lethality only when all homologues are deleted8–10; whereas, Caenorhabditis elegans and Drosophila possess only one gene encoding enolase activity, and its deletion is lethal17,18.
Table 1. Collaterally homozygously deleted essential-redundant housekeeping genes in GBM.
Table 1 shows an evidence-based filtered list of genes homozygously deleted in GBM which are likely to execute an essential housekeeping function and have redundant (and potentially druggable) homologues. A more detail methods description is provided in Supplementary Table S1.
| HOMOZYGOUSLY DELETED GENE |
CHROMOSOMAL LOCUS |
Proximal Tumor suppressor gene |
TARGET HOMOLOGUE |
Pathway |
|---|---|---|---|---|
| ENO1 | 1p36.2 | Various | ENO2 | Glycolysis and Gluconeogenesis |
| H6PD | 1p36.2 | Various | G6PD | Pentose Phosphate Shunt |
| KIF1B | 1p36.2 | Various | KIF1A/C | Chromosomal Seggregation |
| NMNAT1 | 1p36.2 | Various | NMNAT2/3 | NAD+ Biosynthesis |
| UBE4B | 1p36.2 | Various | UBE4A | Polyubiquitin dependent degradation |
| ACO1 | 9p21.1 | INK/ARF | ACO2/ACO3 | Regulation of Iron Metabolism/Citric acid cycle |
| KLHL9 | 9p22 | INK/ARF | KLHL13 | Chromosomal segregation |
| PANK1 | 10q23.31 | PTEN | PANK3 | Acetyl-CoA Biosynthesis |
| KIF20B | 10q23.31 | PTEN | KIF20A | Chromosomal segregation/cytokinesis |
Figure 1. Homozygous deletions in ENO1 sensitize tumors to molecular targeting of ENO2.
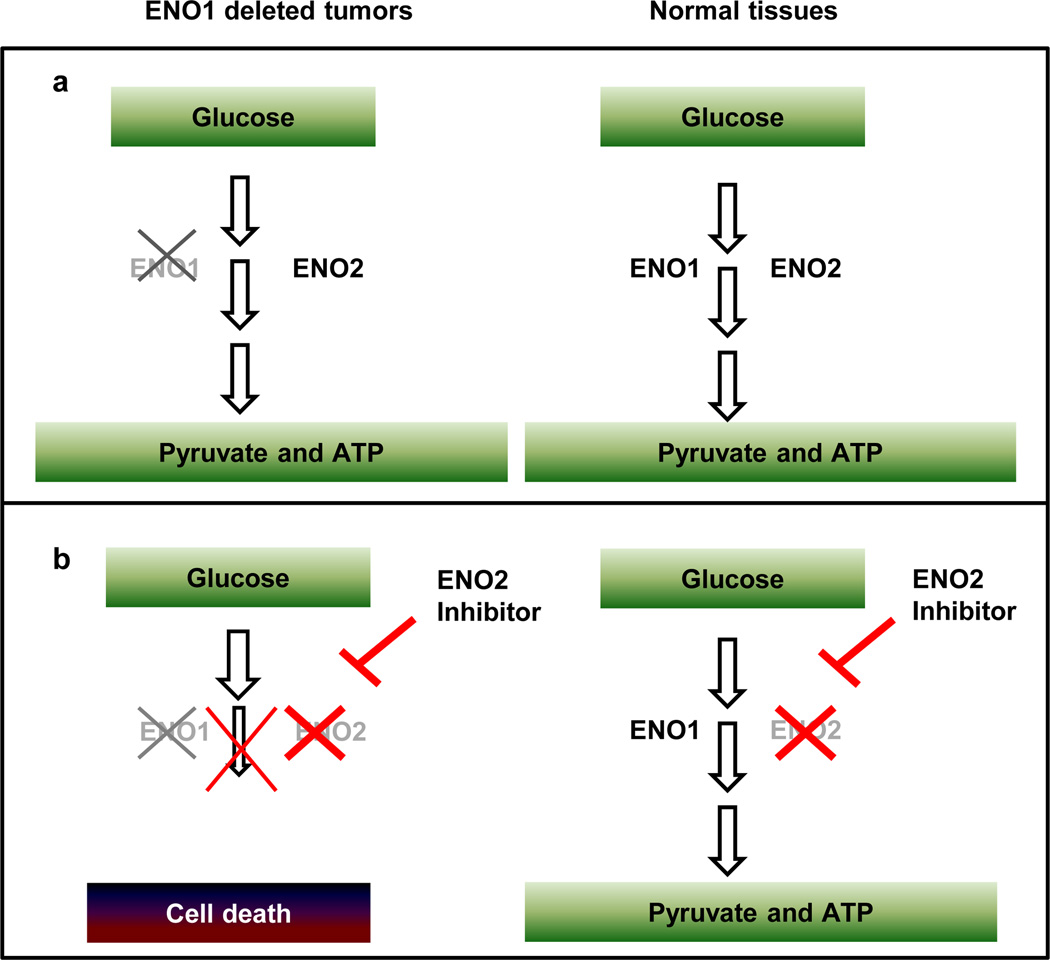
a, ENO1 is homozygously deleted in glioblastomas as part of the 1p36 locus. Loss of ENO1 is tolerable to the tumor because ENO2 is still expressed. b, A specific inhibitor of ENO2 should completely eliminate enolase activity in ENO1 null tumor cells (hence blocking glycolysis and ATP synthesis) but leave genomically intact normal tissues unaffected because enolase activity is still present because ENO1 is still expressed.
The 1p36 locus, which contains several candidate tumor suppressor genes, including CHD5 and CAMTA119,20, sustains frequent deletion in GBM (Fig. 2a). The 1p36 locus is homozygously deleted in 1–5% of GBMs1,21,22 (as well as oligodendrogliomas23 and large-cell neuroendocrine lung tumors24) and ENO1 is often included in the deletion. By examining the TCGA copy number aberrations (single nucleotide polymorphism [SNP] and array comparative genomic hybridization [aCGH] data)1 and expression profiles, we identified 5/359 GBM samples with homozygous deletion of ENO1 and associated near-complete absence of its expression (Fig. 2b and Supplementary Fig. 2). We identified two GBM cell lines, D423-MG22 and Gli5625, with homozygous deletions at the 1p36 locus spanning ENO1. A third GBM cell line, D502-MG22, also incurs homozygous deletion of many genes in this locus yet leaves ENO1 intact and thus serves as an excellent control (Fig. 2c). Western blot analysis confirmed the loss of enolase 1 and the retention of enolase 2 protein in D423-MG and Gli56, whereas both proteins were present in D502-MG and in all other glioma and normal glial cell lines tested (Fig. 2d).
Figure 2. Homozygous deletion of the 1p36 locus in GBM results in loss of ENO1 expression in primary tumors and cell lines.
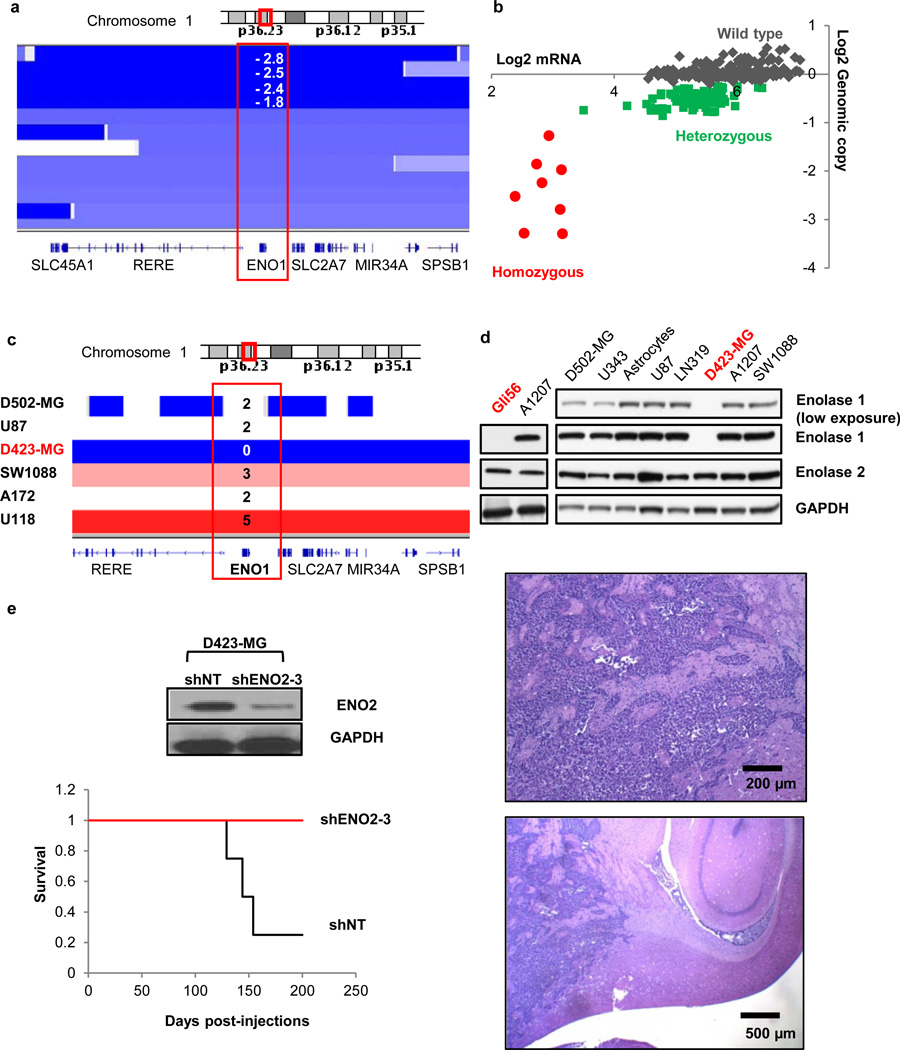
a, TCGA Affymetrix aCGH data show four primary GBMs with log2 copy number < −1, indicating homozygous deletion of the 1p36 locus. b, DNA copy number correlates with mRNA expression; expression is highest in tumors with n = 2 copies (WT) and lowest in tumors with n = 0 copies (null) of ENO1. c, The D423-MG cell line was identified as homozygously deleted by SNP arrays from the Wellcome Trust Sanger Institute data set. d, The complete absence of enolase 1 protein in D423-MG and Gli56 cells was confirmed by western blotting. e, shRNA knock-down of ENO2 in D423-MG ENO1 null cells ablated intracranial tumorigenesis in vivo (n=4 mice per group).
ENO2 knockdown inhibits growth of ENO1-deleted cells and not wild type cells
We used the D502-MG (ENO1 wild-type [WT]) and D423-MG (ENO1-null) cell lines to assess the impact of shRNA-mediated knockdown of ENO2 in an ENO1 WT or null context. Two independent ENO2 shRNAs (pLKO.1 vector) resulted in robust protein reduction and led to a profound inhibition of cell growth only in the context of ENO1 genomic deletion (Supplementary Fig. 3). We obtained the same result using an additional, independent ENO2 shRNA (pGIPZ vector) (Supplementary Fig. 4a,b). Furthermore, shRNA ablation of ENO2 in ENO1-null cells also resulted in decreased soft agar colony formation and blocked the in vivo tumorigenic potential of intracranially injected cells (Figure 2e and Supplementary Fig. 4c). Finally, the selective toxicity of ENO2 ablation to ENO1-null cells was demonstrated in an isogenic context using the doxycycline-inducible TRIPZ vector. When we used this doxycycline-inducible system in ENO1 WT cell lines (U87, A1207, LN319), two independent shRNAs reduced enolase 2 protein levels by >70% (Fig. 3a) with no impact on enolase 1 levels (data not shown). This ablation of ENO2 resulted in a profound inhibition of cell proliferation only in the ENO1-null D423-MG cell line (Fig. 3b). Furthermore, enforced expression of hairpin-resistant ENO2 cDNA fully reversed the deleterious effects of the shENO2 hairpin (Supplementary Fig. 5), showing that the inhibitory effect of the hairpin was indeed specific to diminished ENO2 expression and was not an off-target effect. Finally, when ENO1 was ectopically re-expressed in D423-MG (ENO1 null) cell lines at levels similar to those observed in ENO1-WT GBM lines, the deleterious effect of shRNA ablation of ENO2 was completely abrogated (Supplementary Fig. 6).
Figure 3. shRNA ablation of ENO2 affects ENO1-null but not ENO1-WT GBM cells.
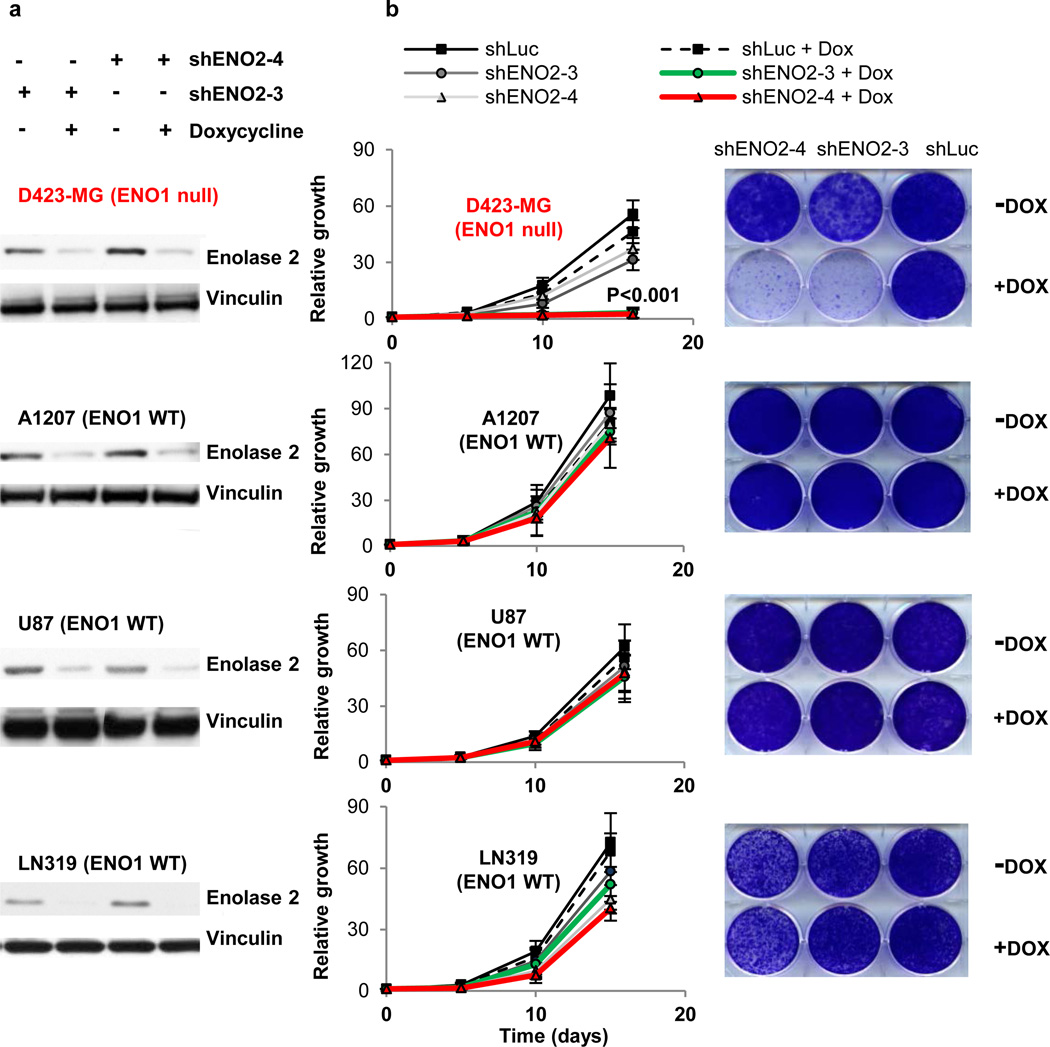
a, shRNA ablation by two independent doxycycline (Dox)-inducible TRIPZ hairpins against ENO2 (shENO2-3, shENO2-4) resulted in >70% reduction in enolase 2 protein levels in both ENO1 WT (A1207, U87, LN319) and ENO1-null (D423-MG) cell lines. b, Ablation of ENO2 dramatically inhibited growth of ENO1-null but not ENO1 WT cells, whereas non-targeting shRNA against luciferase (shLuc) had no effect in any cell line (n=3 biological replicates, S.E.M, t-test). Representative plates at the last time point of growth for cells infected with shLuc, shENO2-3, or shENO2-4, with or without Dox induction, are shown alongside growth curves for each cell line.
Pharmacologic inhibition of enolase is selectively toxic for ENO1-deleted glioma cells
Next, we assessed the impact of pharmacological inhibition of enolase activity in ENO1 WT and null cells. Previous studies have focused on the pharmacological inhibition of enolase, in particular for antiparasitic purposes26,27 and many compounds have been characterized, most of which act as reaction-intermediate analogues (Supplementary Table 3). The most potent enolase inhibitor is PhAH27, which is thought to act as a transition-state analogue with an inhibitory constant of 15 pM on yeast enolase. Although PhAH has not been tested on human enolases, previous work demonstrated inhibitory effects on enolases from distantly related organisms27,28, suggesting its potential use over a large phylogenetic distance. We find that PhAH was indeed capable of potent inhibition of enolase in vitro in native lysates of human GBM cell lines, with an IC50 of around 20 nM (Fig. 4b, data not shown). We used PhAH in concentrations ranging from 0.625 µM to 50 µM and observed marked toxicity in ENO1-null cells (Fig. 4a,c and Supplementary Fig. 7) and minimal impact on the ENO1-WT controls, which show at least 10-times greater enolase activity relative to the ENO1-null cells (because ENO1 accounts for 90% of total cellular enolase activity12, Fig. 4b). Although the IC50 of PhAH is similar for ENO1 and ENO2 in vitro (data not shown) the greater toxicity of the inhibitor to ENO1 null cells (Gli56, D423-MG) derives from the fact that in these cells enolase activity is already 90% lower as compared to ENO1-WT cell lines and consequently, a much lower dose is required to decrease total enolase activity below toxicity threshold. Further data indicate a direct relationship between the levels of enolase activity and the sensitivity to PhAH across different cell lines and in the same cell line with different levels of enforced enolase expression. First, U343 and D502-MG cells, which have intermediate levels of enolase activity (and enolase 1 protein expression, Fig. 2d) compared with the other cell lines, have intermediate levels of sensitivity to PhAH (Fig. 4), which in the case of U343 can be rescued by ectopic overexpression of ENO1 or ENO2 (data not shown). A systematic titration of PhAH in D423-MG cell lines with varying levels of enforced ENO1 or ENO2 expression, shows a direct relationship between the level of enolase expression/activity and the ensuing resistance to PhAH (Supplementary Fig. 7). PhAH toxicity was also abrogated in Gli56 ENO1-null cells by ectopic expression of physiological levels of ENO1 or overexpression of ENO2 (Supplementary Fig. 7). Regarding the mechanism of toxicity, cell cycle and apoptosis analysis demonstrated that PhAH treatment for 48 h induced a marked decrease of S-phase followed by a marked increase of apoptosis in D423-MG but not in ENO1 WT U373 cells (Supplementary Table 4). This effect was completely rescued by ENO2 overexpression (data not shown). The fact that this growth inhibition and subsequent apoptosis is due to energy crisis is substantiated by a strong induction of phosphorylated AMPK (Thr172)29, which was observed in D423-MG but not ENO1 WT cell lines (data not shown). It is tempting to speculate that this energy stress response exerts a protective effect and thus concomitant addition of an AMPK inhibitor with PhAH could result in further toxicity. Finally, it is worth noting that ENO1-null cells do not show any greater sensitivity to other molecular targeted therapies, such as a combination of receptor tyrosine kinase inhibitors30 (lapatinib, sorafenib, and PHA665752) (Supplementary Fig. 8) and rapamycin (data not shown) compared to ENO1 WT cells. These data indicate that D423-MG cells are not broadly susceptible to other anticancer agents and that PhAH selectively targets ENO1-null GBM cells.
Figure 4. Extreme sensitivity of ENO1-null cells to the pan-enolase inhibitor PhAH.

a, D423-MG and Gli56 ENO1-null lines are highly sensitive to PhAH toxicity while ENO1 WT cell lines and normal astrocytes are not. b, The sensitivity of GBM lines to PhAH treatment correlated with their overall enolase activity. Pre-incubation of the lysates with 1 µM PhAH inhibited enolase enzymatic activity by >95% (average, n = 2 technical replicates). c, PhAH minimally affected the growth of ENO1 WT GBM cells and normal astrocytes except at concentrations higher than 50 µM. Low concentrations of PhAH stall the growth of ENO1-null cells, while ENO1 heterozygous cells (D502-MG and U343) showed intermediate sensitivity (n = 4 biological replicates, S.E.M.).
Discussion
In this study, we sought to determine the impact of collateral deletion of genes in tumor suppressor loci that belong to redundant gene families playing cell essential roles and to assess whether extinction of remaining gene family members would create cancer-specific vulnerabilities. We provide genetic and pharmacological evidence that enolase 2 inhibition is lethal in cells with 1p36 homozygous deletion with collateral loss of ENO1, whereas ENO1-intact cells can rely on enolase 1 to undergo glycolysis and support survival. These findings are in agreement with genetic data from invertebrates8–10. Given that several homozygously deleted housekeeping genes can occur in the same deletion on 1p36 (e.g., H6PD, Supplementary Table 1), it may be possible to further increase the effectiveness and cancer-cell-specific killing by combining the inhibition of ENO2 with that of another homologue of a simultaneously deleted housekeeping gene.
Attempts to therapeutically exploit general metabolic differences between normal and cancer cells, such as glucose addiction (the Warburg effect) and glutamine or serine addiction16,31,32, remain areas of active preclinical investigation and clinical development. The approach described here is distinguished from these attempts in that it does not rely on any general aspect of cancer cell metabolism, but rather rests on genetically determined metabolic differences between normal and cancerous tissue to generate cancer-cell-specific vulnerabilities. Indeed, we propose that collateral vulnerability may be extended to other passenger homozygously deleted housekeeping genes in loci sustaining frequent deletion, such as 9p21 (CDKN2A) and 10q23 (PTEN), which contain members of functionally redundant housekeeping gene families (Table 1, Supplementary Table 1 and Supplementary Fig. 9). The strongest, pharmacologically targetable candidates in this list are the homologues of the dual-function energy metabolism/iron regulator aconitase 1 (ACO1; 9p21) and the coenzyme A biosynthetic enzyme, pantothenate kinase 1 (PANK1; 10q23). Importantly, many of the compounds targeting the homologues of these passenger genes represent novel molecular entities with respect to cancer treatment.
By one estimate, 11% of all protein coding genes in the human genome are deleted in human cancers33. Thus, given the large number of homozygous deletions across many different cancer types spanning many hundreds of genes33–36, the paradigm described here for GBM should be applicable to the development of personalized treatments for many additional cancer types.
METHODS
Cell culture
The cell lines D423-MG (1p36 homozygously deleted, including ENO1) and D502-MG (1p36 homozygously deleted, excluding ENO1) were kindly provided by D. Bigner22. (D423 and D502 are referred to as H423 and H502 in ref.22 but as D423-MG and D502-MG in the Wellcome Trust Sanger Institute database, the nomenclature we adopt here (www.sanger.ac.uk). Gli56 was obtained from David N. Louis as described in ref.25. The deletion in D423-MG spans the CAMTA1, VAMP3, PER3, UTS2, TNFRSF9, PARK7, ERRFI1, SLC45A1, RERE, ENO1, CA6, SLC2A5, GPR157, MIR34A, H6PD, SPSB1, and SLC25A33 genes while the deletion in Gli56 spans the UTS2, TNFRSF9, PARK7, ERRFI1, SLC45A1, RERE, ENO1, CA6, SLC2A5, GPR157, MIR34A, H6PD, SPSB1, SLC25A33, TMEM201, C1orf200, PIK3CD, CLSTN1, CTNNBIP1, LZIC, NMNAT1, RBP7 and UBE4B loci. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 20% fetal bovine serum (FBS). For comparison, the cell lines U87, LN319, SW1088, U343, U373, and A1207 were grown under the same conditions. Normal human astrocytes were obtained from ScienCell.
shRNA knockdown of ENO2 expression
We screened 22 hairpins targeting ENO2 and found 4 independent ones that reduced protein levels by <50%. Two of these hairpins were in the pLKO.1 vector (shENO2-1 and shENO2-2), and the remaining two were in the Expression Arrest GIPZ (shENO2-3) and TRIPZ (shENO2-4) shRNAmir vectors (Open Biosystems). The ENO2 shRNA sequences are as follows:
shENO2-1: 5’-CAAGGGAGTCATCAAGGACAA-3’; NM_001975
shENO2-2: 5’-CGCCTGGCTAATAAGGCTTTA-3’; NM_001975
shENO2-3: 5’-CGGCCTTCAACGTGATCAA-3’; NM_001975
shENO2-4: 5-’GGGACTGAGAACAAATCCA-3’. NM_001975
The hairpin in the GIPZ vector was cloned into the TRIPZ vector using a protocol provided by the manufacturer. The TRIPZ vector is a doxycycline-inducible system with a red fluorescent protein reporter that is expressed only upon doxycycline induction. Recombinant lentiviral particles were produced by transient transfection of 293T cells following a standard protocol. Briefly, 72 µg of the shRNA plasmid, 54 µg of delta 8.9 plasmid, and 18 µg of VSVG plasmid were transfected using FuGene (Roche) into 293T cells plated in 245 mm2 dishes. Viral supernatant was collected 72 h after transfection, concentrated by centrifugation at 23,000 rpm, and resuspended in cell growth medium. For transduction, viral solutions were added to cell culture medium containing 4 µg/mL polybrene; 48 h after infection, cells were selected using 2 µg/mL puromycin and tested for ENO2 knockdown by western blotting.
Proliferation assays and anchorage-independent growth
Cell growth of shRNA- or PhAH-treated cell lines was assayed either through crystal violet staining or using the Promega CellTiter-Glo proliferation kit (Roche) or alternatively, in vivo, by measuring confluence with the IncuCyte (Essen BioScience). Growth curves using the IncuCyte were generated by imaging every 2 hours with quadruplicate replicates. For crystal violet assays, 104 cells were seeded in a 6-well plate for each time point. At the indicated time point, cells were fixed with 10% formalin and stained with crystal violet solution for 1 h. Dye extraction was performed using 10% acetic acid solution, and absorbance was read at 590 nm. CellTiter-Glo experiments were performed according to the manufacturer’s instructions; 103 cells/well were plated in a 96-well plate for each time point, and luminescence readings were taken every 24 h. All experiments were performed in triplicate. Soft agar (anchorage-independent) growth was monitored in 6-well plates seeded with 104 cells of the indicated genotype. The medium contained DMEM with 10% FBS; the top agar contained 0.4% low melting agarose, while the bottom agar contained 1% low melting agarose. Growth was monitored by fluorescence (GFP) and after 28 days colonies were stained with iodonitrotetrazolium chloride (Sigma-Aldrich) and counted.
Orthotopic brain tumor formation
The in vivo tumorigenic potential of D423-MG cells transduced with non-targeting hairpin or shENO2-3 delivered through pGIPZ was determined as previously described37. SCID mice (Charles River) under deep anesthesia were placed into a stereotactic apparatus equipped with a z axis (Stoelting). Then, 3 × 105 cells were injected intracranially into the right caudate nucleus 3 mm below the surface of the brain, using a 10-µl Hamilton syringe. The animals were followed daily for development of neurological deficits. All mice experiments were performed with the approval of the Harvard Cancer Center and Dana-Farber Cancer Institute Institutional Animal Care and Use Committee.
Enolase activity assay
Enolase activity was measured via NADH oxidation in a pyruvate kinase–lactate dehydrogenase coupled assay as previously described12. Briefly, cells were lysed in 20 mM Tris HCl, 1 mM EDTA, and 1 mM β-mercaptoethanol (pH 7.4) and homogenized using a Polytron homogenizer three times for a period of 10 s followed by sonication. Enolase activity was recorded by measuring oxidation of NADH either spectrophotometrically by absorbance at 340 nm or fluorescently by excitation at 340 nm and emission at 460 nm.
Western blotting
After two washes with phosphate-buffered saline (PBS), cells were incubated in RIPA buffer for 15 min with gentle shaking. Lysates were then collected, sonicated, and centrifuged at 14,000 rpm for 10 min at 4 °C. SDS-PAGE and western blotting were performed as described previously37. The following antibodies were used: enolase 1, CST#3810; enolase 2, #9536; and GAPDH CST# 3683; phosphor-AMPK Thr172 CST# 2535 from Cell Signaling Technologies (Danvers, MA) and vinculin from Sigma-Aldrich (St. Louis, MO).
Inhibitor studies
PhAH lithium salt was custom synthesized by TCRS (Bristol, PA), following the protocol of Anderson et al28. Structure and purity were verified by NMR. PhAH was dissolved in PBS at 50 mM stock and stored frozen at −80 °C until use. Given the instability of the compound, the medium was replaced every 5 days and fresh inhibitor added with fresh medium. Rapamycin, sorafenib, lapatinib, and PHA665752 were obtained from LC Laboratories (Woburn, MA) and Tocris Bioscience (Bristol, UK), respectively.
Ectopic expression of ENO1, ENO2 and shRNA-resistant ENO2
Rescue of the phenotypic effects of knocking down ENO2 in the cell line D423-MG was performed by overexpressing an shRNA-resistant form of ENO2. Briefly, 6 silent mutations were introduced into the ENO2 coding region targeted by shENO2-4, using the QuikChange site-directed mutagenesis kit (Stratagene). The shRNA-resistant ENO2 coding region was cloned into the pHAGE-CMV lentiviral vector (a generous gift of D.N. Kotton) and overexpressed in the D423-MG cell line carrying shENO2-4, in the presence or absence of doxycycline. As a control, the same cell line was infected with a lentiviral vector carrying the green fluorescent protein (GFP) gene. For the ectopic re-expression of ENO1 or ENO2, sequenced verified cDNA clones were gateway cloned into the pHAGE-CMV lentiviral vector and lentivirally transduced into glioma cell lines as described above.
Cell cycle analysis
The D423-MG and U373 cell lines were treated for 48 h in the presence or absence of PhAH (25 µM) and fixed in 75% ethanol at −20 °C overnight. The following day, the cells were washed with cold PBS, treated with 100 µg of RNase A (Qiagen), and stained with 50 µg of propidium iodide (Roche). Flow cytometric acquisition was performed using a three-color FACScan flow cytometer and CellQuest software (Becton Dickinson). For each sample, 104 events were gated. Data analysis was performed using ModFit LT (Verity Software House).
Annexin V/7-AAD assay for apoptosis
The D423-MG and U373 cell lines were treated for 96 h in the presence or absence of PhAH (25 µM). For Annexin V/7-AAD assay cells were stained with Annexin V-PE and 7-AAD, and evaluated for apoptosis by flow cytometry according to the manufacturer's protocol (Biovision). The apoptotic cells were determined using a Becton-Dickinson FACScan cytometer. Both early apoptotic (annexin V-positive, 7-AAD-negative) and late apoptotic (annexin V-positive and 7-AAD-positive) cells were included in cell death determinations.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Keith Ligon, Cecile Maire, David N. Louis, James Kim, Gayatry Mohapatra for sharing bioinformatics data from their tumor neurosphere banks. We also thank D. Bigner for sharing the D423-MG and D502-MG cell lines and David N. Louis and James Kim for sharing the Gli56 cell line. We thank Gerry Chu and David Jakubosky for assistance with necropsy and histopathological analysis. F.L.M. was supported by a training grant from the National Institutes of Health (NIH T32-CA009361) and a fellowship from the American Cancer Society (115992-PF-08-261-01-TBE). S.C. was supported by a Dana-Farber Cancer Institute/Harvard Cancer Center Myeloma SPORE career development grant. E.A. was supported by the Howard Hughes Medical Institute Medical Research Fellowship # 57006984. V.M. was supported by a Harvard PRISE fellowship. F.L.M. thanks Jessica Mohr for assistance with preparation of the figures. We also thank Karen Muller for assistance with manuscript editing. We thank all members of the DePinho and Chin labs for fruitful suggestions and discussions. This work is supported by the NIH (P01CA95616 to C.B., L.C. and R.A.D.) and by the Ben and Catherine Ivy Foundation to R.A.D. and L.C.
Footnotes
AUTHOR CONTRIBUTIONS
F.L.M. and R.A.D. generated the original hypothesis. F.L.M. performed all bioinformatics work, including scanning the TCGA data set (with initial assistance from J.H.) and identifying candidates for collateral lethality, with the exception of KLHL9, which was identified by E.F. E.A. obtained the D423-MG cell line and designed and carried out the pLKO and pGIPZ shRNA experiments. S.C. designed and performed all shRNA experiments with the inducible vectors and rescue experiments. F.L.M. and E.A. identified PhAH, F.L.M. procured the compound, and F.L.M. and S.C. performed all inhibitor treatment experiments. L.N. generated shRNA-resistant constructs of ENO2. S.C., D.O., and E.F. performed cell cycle and apoptosis assays. R.N., V.M., D.E., P.D. and J.L. performed cell culture, crystal violet staining, western blotting, and associated experiments and assisted in the preparation of figures. C.B. provided extensive unpublished genomic data and reagents from his primary brain tumor and neurosphere bank for Table S1. E.A., M.L., B.H., and G.G. performed tumor cell injections. D.H., E.S., L.K., Y.A.W. and L.C. provided critical intellectual contributions throughout the project. F.L.M., E.A., S.C., Y.A.W., L.C., and R.A.D. wrote the paper.
CITED REFERENCES
- 1.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–4817. doi: 10.1182/blood-2008-07-077958. 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 3.Guha M. PARP inhibitors stumble in breast cancer. Nat Biotechnol. 2011;29:373–374. doi: 10.1038/nbt0511-373. 10.1038/nbt0511-373. [DOI] [PubMed] [Google Scholar]
- 4.De Soto JA, Deng CX. PARP-1 inhibitors: are they the long-sought genetically specific drugs for BRCA1/2-associated breast cancers? Int J Med Sci. 2006;3:117–123. doi: 10.7150/ijms.3.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weidle UH, Maisel D, Eick D. Synthetic lethality-based targets for discovery of new cancer therapeutics. Cancer Genomics Proteomics. 2011;8:159–171. [PubMed] [Google Scholar]
- 6.Vavouri T, Semple JI, Lehner B. Widespread conservation of genetic redundancy during a billion years of eukaryotic evolution. Trends Genet. 2008;24:485–488. doi: 10.1016/j.tig.2008.08.005. 10.1016/j.tig.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Brookfield JF. Genetic redundancy. Adv Genet. 1997;36:137–155. doi: 10.1016/s0065-2660(08)60308-9. [DOI] [PubMed] [Google Scholar]
- 8.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLuna A, et al. Exposing the fitness contribution of duplicated genes. Nat Genet. 2008;40:676–681. doi: 10.1038/ng.123. 10.1038/ng.123. [DOI] [PubMed] [Google Scholar]
- 10.Deutscher D, Meilijson I, Kupiec M, Ruppin E. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat Genet. 2006;38:993–998. doi: 10.1038/ng1856. 10.1038/ng1856. [DOI] [PubMed] [Google Scholar]
- 11.Poyner RR, Reed GH. Structure of the bis divalent cation complex with phosphonoacetohydroxamate at the active site of enolase. Biochemistry. 1992;31:7166–7173. doi: 10.1021/bi00146a020. [DOI] [PubMed] [Google Scholar]
- 12.Joseph J, Cruz-Sanchez FF, Carreras J. Enolase activity and isoenzyme distribution in human brain regions and tumors. J Neurochem. 1996;66:2484–2490. doi: 10.1046/j.1471-4159.1996.66062484.x. [DOI] [PubMed] [Google Scholar]
- 13.Stefanini M. Chronic hemolytic anemia associated with erythrocyte enolase deficiency exacerbated by ingestion of nitrofurantoin. Am J Clin Pathol. 1972;58:408–414. doi: 10.1093/ajcp/58.5.408. [DOI] [PubMed] [Google Scholar]
- 14.Kobayakawa K, et al. Innate versus learned odour processing in the mouse olfactory bulb. Nature. 2007;450:503–508. doi: 10.1038/nature06281. 10.1038/nature06281. [DOI] [PubMed] [Google Scholar]
- 15.Comi GP, et al. Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol. 2001;50:202–207. doi: 10.1002/ana.1095. [DOI] [PubMed] [Google Scholar]
- 16.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buszczak M, et al. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics. 2007;175:1505–1531. doi: 10.1534/genetics.106.065961. 10.1534/genetics.106.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonnichsen B, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434:462–469. doi: 10.1038/nature03353. 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 19.Henrich KO, et al. CAMTA1, a 1p36 tumor suppressor candidate, inhibits growth and activates differentiation programs in neuroblastoma cells. Cancer Res. 2011;71:3142–3151. doi: 10.1158/0008-5472.CAN-10-3014. 10.1158/0008-5472.CAN-10-3014. [DOI] [PubMed] [Google Scholar]
- 20.Bagchi A, Mills AA. The quest for the 1p36 tumor suppressor. Cancer Res. 2008;68:2551–2556. doi: 10.1158/0008-5472.CAN-07-2095. 10.1158/0008-5472.CAN-07-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin D, et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res. 2009;7:665–677. doi: 10.1158/1541-7786.MCR-08-0270. 10.1158/1541-7786.MCR-08-0270. [DOI] [PubMed] [Google Scholar]
- 22.Duncan CG, et al. Integrated genomic analyses identify ERRFI1 and TACC3 as glioblastoma-targeted genes. Oncotarget. 2010;1:265–277. doi: 10.18632/oncotarget.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotliarov Y, et al. High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances. Cancer Res. 2006;66:9428–9436. doi: 10.1158/0008-5472.CAN-06-1691. 10.1158/0008-5472.CAN-06-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng WX, et al. Array-based comparative genomic hybridization analysis of high-grade neuroendocrine tumors of the lung. Cancer Sci. 2007;96:661–667. doi: 10.1111/j.1349-7006.2005.00092.x. 10.1126/science.1142946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller W, et al. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene. 2005;26:583–593. doi: 10.1038/sj.onc.1209805. 10.1111/j.1349-7006.2005.00092.x. [DOI] [PubMed] [Google Scholar]
- 26.Guha-Chowdhury N, Clark AG, Sissons CH. Inhibition of purified enolases from oral bacteria by fluoride. Oral Microbiol Immunol. 1997;12:91–97. doi: 10.1111/j.1399-302x.1997.tb00623.x. [DOI] [PubMed] [Google Scholar]
- 27.de ASNMV. Structural flexibility in Trypanosoma brucei enolase revealed by X-ray crystallography and molecular dynamics. FEBS J. 2007;274:5077–5089. doi: 10.1111/j.1742-4658.2007.06027.x. 10.1111/j.1742-4658.2007.06027.x. [DOI] [PubMed] [Google Scholar]
- 28.Anderson VE, Weiss PM, Cleland WW. Reaction intermediate analogues for enolase. Biochemistry. 1984;23:2779–2786. doi: 10.1021/bi00307a038. [DOI] [PubMed] [Google Scholar]
- 29.Jing M, Ismail-Beigi F. Critical role of 5'-AMP-activated protein kinase in the stimulation of glucose transport in response to inhibition of oxidative phosphorylation. Am J Physiol Cell Physiol. 2007;292:C477–C487. doi: 10.1152/ajpcell.00196.2006. 10.1152/ajpcell.00196.2006. [DOI] [PubMed] [Google Scholar]
- 30.Stommel JM, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. 10.1038/sj.onc.1209805. [DOI] [PubMed] [Google Scholar]
- 31.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. nature10350 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raj L, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. nature10167 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox C, et al. A survey of homozygous deletions in human cancer genomes. Proc Natl Acad Sci U S A. 2005;102:4542–4547. doi: 10.1073/pnas.0408593102. 10.1073/pnas.0408593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tonon G, et al. High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A. 2005;102:9625–9630. doi: 10.1073/pnas.0504126102. 10.1073/pnas.0504126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.