

Bacteria and archaea have evolved adaptive immune defenses termed clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems that use short RNA to direct degradation of foreign nucleic acids. Here, we engineer the type II bacterial CRISPR system to function with custom guide RNA (gRNA) in human cells. For the endogenous AAVS1 locus, we obtained targeting rates of 10 to 25% in 293T cells, 13 to 8% in K562 cells, and 2 to 4% in induced pluripotent stem cells. We show that this process relies on CRISPR components; is sequence-specific; and, upon simultaneous introduction of multiple gRNAs, can effect multiplex editing of target loci. We also compute a genome-wide resource of ~190 K unique gRNAs targeting ~40.5% of human exons. Our results establish an RNA-guided editing tool for facile, robust, and multiplexable human genome engineering.
Bacterial and archaeal clustered regularly interspaced short palindromic repeats (CRISPR) systems rely on CRISPR RNAs (crRNAs) in complex with CRISPR-associated (Cas) proteins to direct degradation of complementary sequences present within invading viral and plasmid DNA (1-3). A recent in vitro reconstitution of the Streptococcus pyogenes type II CRISPR system demonstrated that crRNA fused to a normally trans-encoded tracrRNA is sufficient to direct Cas9 protein to sequence-specifically cleave target DNA sequences matching the crRNA (4). The fully defined nature of this two-component system suggested that it might function in the cells of eukaryotic organisms such as yeast, plants, and even mammals. By cleaving genomic sequences targeted by RNA sequences (4-6), such a system could greatly enhance the ease of genome engineering.
Here, we engineer the protein and RNA components of this bacterial type II CRISPR system in human cells. We began by synthesizing a human codon–optimized version of the Cas9 protein bearing a C-terminal SV40 nuclear localization signal and cloning it into a mammalian expression system (Fig. 1A and fig. S1A). To direct Cas9 to cleave sequences of interest, we expressed crRNA-tracrRNA fusion transcripts, hereafter referred to as guide RNAs (gRNAs), from the human U6 polymerase III promoter. Directly transcribing gRNAs allowed us to avoid reconstituting the RNA-processing machinery used by bacterial CRISPR systems (Fig. 1A and fig. S1B) (4, 7-9). Constrained only by U6 transcription initiating with G and the requirement for the PAM (protospacer-adjacent motif) sequence -NGG following the 20–base pair (bp) crRNA target, our highly versatile approach can, in principle, target any genomic site of the form GN20GG (fig. S1C; see supplementary text S1 for a detailed discussion).
Fig. 1. Genome editing in human cells using an engineered type II CRISPR system.
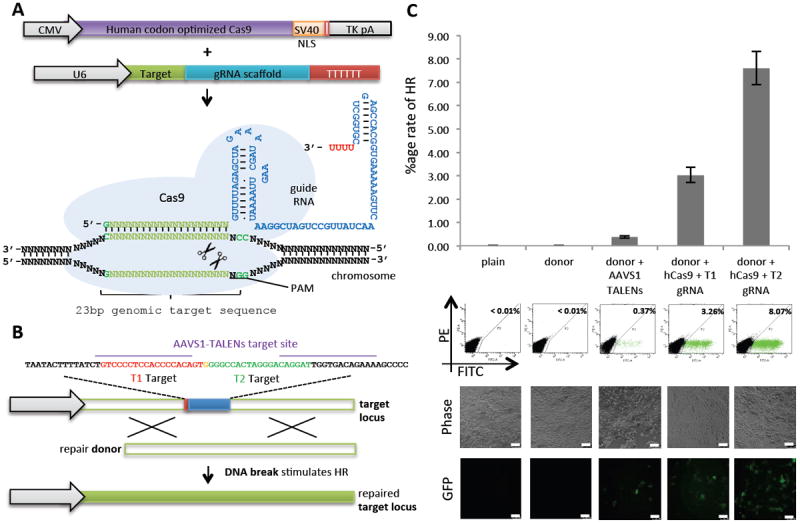
(A) RNA-guided gene targeting in human cells involves coexpression of the Cas9 protein bearing a C-terminal SV40 nuclear localization signal (NLS) with one or more gRNAs expressed from the human U6 polymerase III promoter. Cas9 unwinds the DNA duplex and cleaves both strands upon recognition of a target sequence by the gRNA, but only if the correct PAM is present at the 3′ end. Any genomic sequence of the form GN20GG can, in principle, be targeted. CMV, cytomegalovirus promoter; TK, thymidine kinase; pA, polyadenylation signal. (B) A genomically integrated GFP coding sequence is disrupted by the insertion of a stop codon and a 68-bp genomic fragment from the AAVS1 locus. Restoration of the GFP sequence by HR with an appropriate donor sequence results in GFP+ cells that can be quantified by FACS. T1 and T2 gRNAs target sequences within the AAVS1 fragment. Binding sites for the two halves of the TALEN are underlined. (C) Bar graph depicting HR efficiencies induced by T1, T2, and TALEN-mediated nuclease activity at the target locus, as measured by FACS. Representative FACS plots and microscopy images of the targeted cells are depicted below. (Scale bar, 100 μm.) Data are shown as means ± SEM (N = 3).
To test the functionality of our implementation for genome engineering, we developed a green fluorescent protein (GFP) reporter assay (Fig. 1B) in human embryonic kidney HEK 293T cells similar to one previously described (10). Specifically, we established a stable cell line bearing a genomically integrated GFP coding sequence disrupted by the insertion of a stop codon and a 68-bp genomic fragment from the AAVS1 locus that renders the expressed protein fragment nonfluorescent. Homologous recombination (HR) using an appropriate repair donor can restore the normal GFP sequence, which enabled us to quantify the resulting GFP+ cells by flow-activated cell sorting (FACS).
To test the efficiency of our system at stimulating HR, we constructed two gRNAs, T1 and T2, that target the intervening AAVS1 fragment (Fig. 1B) and compared their activity to that of a previously described TAL effector nuclease heterodimer (TALEN) targeting the same region (11). We observed successful HR events using all three targeting reagents, with gene correction rates using the T1 and T2 gRNAs approaching 3% and 8%, respectively (Fig. 1C). This RNA-mediated editing process was notably rapid, with the first detectable GFP+ cells appearing ~20 hours post transfection compared with ~40 hours for the AAVS1 TALENs. We observed HR only upon simultaneous introduction of the repair donor, Cas9 protein, and gRNA, which confirmed that all components are required for genome editing (fig. S2). Although we noted no apparent toxicity associated with Cas9/gRNA expression, work with zinc finger nucleases (ZFNs) and TALENs has shown that nicking only one strand further reduces toxicity. Accordingly, we also tested a Cas9D10A mutant that is known to function as a nickase in vitro, which yielded similar HR but lower nonhomologous end joining (NHEJ) rates (fig. S3) (4, 5). Consistent with (4), in which a related Cas9 protein is shown to cut both strands 3 bp upstream of the PAM, our NHEJ data confirmed that most deletions or insertions occurred at the 3′ end of the target sequence (fig. S3B). We also confirmed that mutating the target genomic site prevents the gRNA from effecting HR at that locus, which demonstrates that CRISPR-mediated genome editing is sequence-specific (fig. S4). Finally, we showed that two gRNAs targeting sites in the GFP gene, and also three additional gRNAs targeting fragments from homologous regions of the DNA methyl transferase 3a (DNMT3a) and DNMT3b genes could sequence-specifically induce significant HR in the engineered reporter cell lines (figs. S5 and S6). Together, these results confirm that RNA-guided genome targeting in human cells is simple to execute and induces robust HR across multiple target sites.
Having successfully targeted an integrated reporter, we next turned to modifying a native locus. We used the gRNAs described above to target the AAVS1 locus located in the PPP1R12C gene on chromosome 19, which is ubiquitously expressed across most tissues (Fig. 2A). We targeted 293Ts, human chronic myelogenous leukemia K562 cells, and PGP1 human induced pluripotent stem (iPS) cells (12) and analyzed the results by next-generation sequencing of the targeted locus. Consistent with our results for the GFP reporter assay, we observed high numbers of NHEJ events at the endogenous locus for all three cell types. The two gRNAs T1 and T2 achieved NHEJ rates of 10 and 25% in 293Ts, 13 and 38% in K562s, and 2 and 4% in PGP1-iPS cells, respectively (Fig. 2B). We observed no overt toxicity from the Cas9 and gRNA expression required to induce NHEJ in any of these cell types (fig. S7). As expected, NHEJ-mediated deletions for T1 and T2 were centered around the target site positions, which further validated the sequence-specificity of this targeting process (figs. S7 to S9). Simultaneous introduction of both T1 and T2 gRNAs resulted in high-efficiency deletion of the intervening 19-bp fragment (fig. S8), which demonstrated that multiplexed editing of genomic loci is feasible using this approach.
Fig. 2. RNA-guided genome editing of the native AAVS1 locus in multiple cell types.
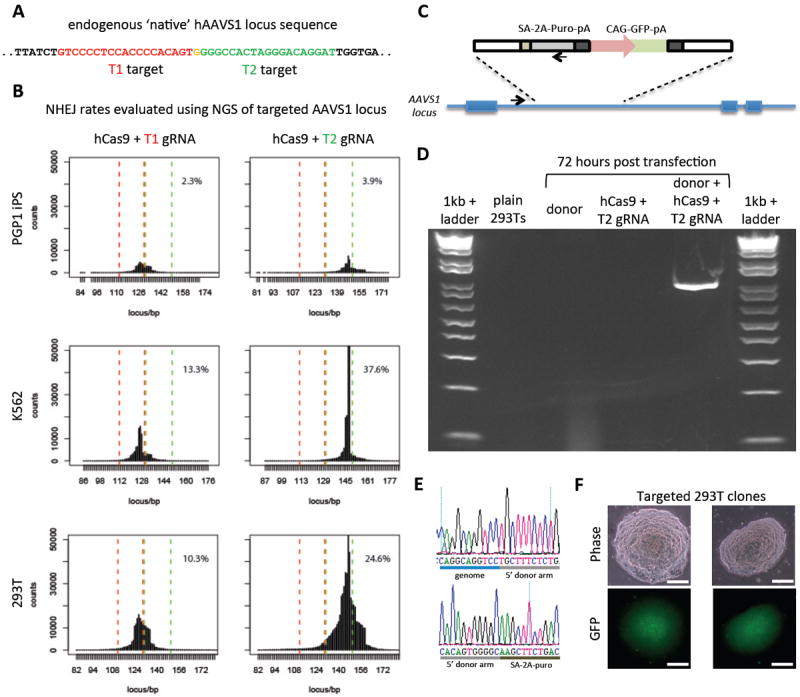
(A) T1 (red) and T2 (green) gRNAs target sequences in an intron of the PPP1R12C gene within the chromosome 19 AAVS1 locus. (B) Total count and location of deletions caused by NHEJ in 293Ts, K562s, and PGP1 iPS cells after expression of Cas9 and either T1 or T2 gRNAs as quantified by next-generation sequencing. Red and green dashed lines demarcate the boundaries of the T1 and T2 gRNA targeting sites. NHEJ frequencies for T1 and T2 gRNAs were 10% and 25% in 293T, 13% and 38% in K562, and 2% and 4% in PGP1 iPS cells, respectively. (C) DNA donor architecture for HR at the AAVS1 locus, and the locations of sequencing primers (arrows) for detecting successful targeted events, are depicted. (D) PCR assay 3 days after transfection demonstrates that only cells expressing the donor, Cas9 and T2 gRNA exhibit successful HR events. (E) Successful HR was confirmed by Sanger sequencing of the PCR amplicon, which showed that the expected DNA bases at both the genome-donor and donor-insert boundaries are present. (F) Successfully targeted clones of 293T cells were selected with puromycin for 2 weeks. Microscope images of two representative GFP+ clones is shown. (Scale bar, 100 μm.)
Last, we attempted to use HR to integrate either a double-stranded DNA donor construct (13) or an oligo donor into the native AAVS1 locus (Fig. 2C and fig. S10). We confirmed HR-mediated integration, using both approaches, by polymerase chain reaction (PCR) (Fig. 2D and fig. S10) and Sanger sequencing (Fig. 2E). We also readily derived 293T or iPS clones from the pool of modified cells using puromycin selection over 2 weeks (Fig. 2F and fig. S10). These results demonstrate that Cas9 is capable of efficiently integrating foreign DNA at endogenous loci in human cells.
Our versatile RNA-guided genome-editing system can be readily adapted to modify other genomic sites by simply modifying the sequence of our gRNA expression vector to match a compatible sequence in the locus of interest. To facilitate this process, we bioinformatically generated ~190,000 specific gRNA-targetable sequences targeting ~40.5% exons of genes in the human genome (refer to Methods and table S1). We incorporated these target sequences into a 200-bp format compatible with multiplex synthesis on DNA arrays (14) (fig. S11 and tables S2 and S3). This resource provides a ready genome-wide reference of potential target sites in the human genome and a methodology for multiplex gRNA synthesis.
Our results demonstrate the promise of CRISPR-mediated gene targeting for RNA-guided, robust, and multiplexable mammalian genome engineering. The ease of retargeting our system to modify genomic sequences greatly exceeds that of comparable ZFNs and TALENs, while offering similar or greater efficiencies (4). Existing studies of type II CRISPR specificity (4) suggest that target sites must perfectly match the PAM sequence NGG and the 8- to 12-base “seed sequence” at the 3′ end of the gRNA. The importance of the remaining 8 to 12 bases is less well understood and may depend on the binding strength of the matching gRNAs or on the inherent tolerance of Cas9 itself. Indeed, Cas9 will tolerate single mismatches at the 5′ end in bacteria and in vitro, which suggests that the 5′ G is not required. Moreover, it is likely that the target locus’s underlying chromatin structure and epigenetic state will also affect the efficiency of genome editing in eukaryotic cells (13), although we suspect that Cas9’s helicase activity may render it more robust to these factors, but this remains to be evaluated. Elucidating the frequency and underlying causes of off-target nuclease activity (15, 16) induced by CRISPR, ZFN (17, 18), and TALEN (19, 20) genome-engineering tools will be of utmost importance for safe genome modification and perhaps for gene therapy. Potential avenues for improving CRISPR specificity include evaluating Cas9 homologs identified through bioinformatics and directed evolution of these nucleases toward higher specificity. Similarly, the range of CRISPR-targetable sequences could be expanded through the use of homologs with different PAM requirements (9) or by directed evolution. Finally, inactivating one of the Cas9 nuclease domains increases the ratio of HR to NHEJ and may reduce toxicity (figs. S1A and fig. S3) (4, 5), whereas inactivating both domains may enable Cas9 to function as a retargetable DNA binding protein. As we explore these areas, we note that another parallel study (21) has independently confirmed the high efficiency of CRISPR-mediated gene targeting in mammalian cell lines. We expect that RNA-guided genome targeting will have broad implications for synthetic biology (22, 23), the direct and multiplexed perturbation of gene networks (13, 24), and targeted ex vivo (25-27) and in vivo gene therapy (28).
Supplementary Material
Materials and Methods
Supplementary Text
Figs. S1 to S11
Tables S1 to S3
References (29-46)
Acknowledgments
This work was supported by NIH grant P50 HG005550. We thank S. Kosuri for advice on the oligonucleotide pool designs and synthesis. G.M.C. and P.M. have filed a patent based on the findings of this study.
Footnotes
Supplementary Materials
References and Notes
- 1.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 2.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 3.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sapranauskas R, et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39:9275. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 8.Miyagishi M, Taira K. U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol. 2002;20:497. doi: 10.1038/nbt0502-497. [DOI] [PubMed] [Google Scholar]
- 9.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanjana NE, et al. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JH, et al. A robust approach to identifying tissue-specific gene expression regulatory variants using personalized human induced pluripotent stem cells. PLoS Genet. 2009;5:e1000718. doi: 10.1371/journal.pgen.1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosuri S, et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat Biotechnol. 2010;28:1295. doi: 10.1038/nbt.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King NM, Cohen-Haguenauer O. En route to ethical recommendations for gene transfer clinical trials. Mol Ther. 2008;16:432. doi: 10.1038/mt.2008.13. [DOI] [PubMed] [Google Scholar]
- 17.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rebar EJ, Pabo CO. Zinc finger phage: Affinity selection of fingers with new DNA-binding specificities. Science. 1994;263:671. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 19.Boch J, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 20.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:XXX. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalil AS, Collins JJ. Synthetic biology: Applications come of age. Nat Rev Genet. 2010;11:367. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purnick PE, Weiss R. The second wave of synthetic biology: From modules to systems. Nat Rev Mol Cell Biol. 2009;10:410. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
- 24.Zou J, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holt N, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 27.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 28.Li H, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makarova KS, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 31.Deveau H, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Ploeg JR. Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages. Microbiology. 2009;155:1966. doi: 10.1099/mic.0.027508-0. [DOI] [PubMed] [Google Scholar]
- 33.Rho M, Wu YW, Tang H, Doak TG, Ye Y. Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 2012;8:e1002441. doi: 10.1371/journal.pgen.1002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pride DT, et al. Analysis of streptococcal CRISPRs from human saliva reveals substantial sequence diversity within and between subjects over time. Genome Res. 2011;21:126. doi: 10.1101/gr.111732.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garneau JE, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 36.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrangou R, Horvath P. CRISPR: New horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3:143. doi: 10.1146/annurev-food-022811-101134. [DOI] [PubMed] [Google Scholar]
- 38.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dreszer TR, et al. The UCSC Genome Browser database: Extensions and updates 2011. Nucleic Acids Res. 2012;40(Database issue):D918. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karolchik D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lorenz R, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 45.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Q, Schlabach MR, Hannon GJ, Elledge SJ. Design of 240,000 orthogonal 25mer DNA barcode probes. Proc Natl Acad Sci U S A. 2009;106:2289. doi: 10.1073/pnas.0812506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Supplementary Text
Figs. S1 to S11
Tables S1 to S3
References (29-46)