

Summary
De novo mutation plays an important role in Autism Spectrum Disorders (ASDs). Notably, pathogenic copy number variants (CNVs) are characterized by high mutation rates. We hypothesize that hypermutability is a property of ASD genes, and may also include nucleotide-substitution hotspots. We investigated global patterns of germline mutation by whole genome sequencing of monozygotic twins concordant for ASD and their parents. Mutation rates varied widely throughout the genome (by 100-fold) and could be explained by intrinsic characteristics of DNA sequence and chromatin structure. Dense clusters of mutations within individual genomes were attributable to compound mutation or gene conversion. Hypermutability was a characteristic of genes involved in ASD and other diseases. In addition, genes impacted by mutations in this study were associated with ASD in independent exome-sequencing datasets. Our findings suggest that regional hypermutation is a significant factor shaping patterns of genetic variation and disease risk in humans.
Introduction
Spontaneous germline mutation plays an important role in human disease. For severe neurodevelopmental disorders such as Autism Spectrum Disorders (ASDs), highly-penetrant alleles are under strong negative selection (Uher, 2009). Such alleles segregate in the population over few generations or can frequently be observed as de novo mutations (DNMs) in affected individuals. Thus, in order to understand this aspect of the genetics of ASD and other human diseases, we must understand the mutational processes that give rise to human genetic diversity and the intrinsic and extrinsic forces that shape patterns of variation in the genome.
Mutation is a random process. However, the probability of mutation at a given site is not uniform throughout the genome. Regional mutation rates are subject to a variety of intrinsic characteristics (Ellegren et al., 2003) and extrinsic factors such as parental age (Crow, 2000). This is particularly evident for structural variation (SV). Rates of structural mutation can vary between 10−4 and 10−6 (Lupski, 2007), and there are numerous examples of hotspots for structural mutation where recurrent mutations are mediated by non-allelic homologous recombination (NAHR) between tandem segmental duplications (Lupski, 1998; Malhotra and Sebat, 2012).
Regional rates of nucleotide substitution are also variable (Ellegren et al., 2003); however the factors that influence regional mutability are not well understood. In contrast to the SV hotspots described above which are predominantly driven by meiotic recombination, rates of nucleotide substitution occur by a variety of mechanisms and the mutation rate is influenced to a much greater extent by mitotic mechanisms (Crow, 2000). Comparisons of genomes from the human and chimpanzee have found evidence that regional mutability is influenced by G+C content (Chimpanzee Sequencing and Analysis Consortium, 2005; Coulondre et al., 1978), recombination rate (Hardison et al., 2003; Hellmann et al., 2005; Lercher and Hurst, 2002) and chromosome banding patterns (Chimpanzee Sequencing and Analysis Consortium, 2005). These studies indicate that regional mutation rates are influenced by various properties of the genome and that no single factor can explain the observed patterns of genetic diversity and divergence in humans. However previous studies do not represent a complete and unbiased view of germline mutation. The full extent of variation in mutation rates genome-wide remains unclear (Francino and Ochman, 1999; Nelis et al., 1996; Webster et al., 2003), and the relevance of hypermutability to common diseases such as ASD is not known.
We have investigated global and regional rates of nucleotide substitution by direct detection of germline mutations in monozygotic (MZ) twins concordant for ASD and their parents. We show that the distribution of mutations in the genome is non-random. Wide variation in regional mutation rates can be explained by intrinsic characteristics of the genome. Furthermore we find significant evidence that genes impacted by de novo mutations in twins are associated with autism in independent cohorts.
Results
Detection of Germline Mutations by Whole Genome Sequencing in Monozygotic Twins
We applied a whole genome sequencing (WGS) strategy to characterizing patterns of germline mutation (Supplemental Figure 1). Central to our approach was the selection of a MZ-twin family sample and the development of a custom machine-learning based method for DNM calling.
Cell line-derived genomic DNAs from ten MZ twin pairs concordant for ASD and their parents were obtained from the NIMH genetics initiative biorepository (http://www.nimhgenetics.org). Concordant MZ twins afford significant advantages to this study. Disease risk can be more directly attributed to risk factors in the germline. In addition, having complete genome sequences on identical twins allows us to readily distinguish germline mutation from somatic and cell line mutations. To improve our power to account for paternal age effects on mutation rate, half of the twin pairs were selected to have younger fathers (16–29 y/o) and half were selected to have older fathers (38–41 y/o).
Deep (40X) WGS was performed at BGI using the Illumina HiSeq platform. Raw sequence files were processed at UCSD with a WGS pipeline consisting of automated tools for alignment and variant calling (see methods).
DNM detection was performed using a machine-learning based tool as described in methods. Using an internal “gold standard” set generated by exhaustive validation of putative DNM calls in one quad family (family 74–0352, see methods), we trained a Random Forest classifier (forestDNM) that discriminates validated de novo mutations (DNMs) from invalidated putative DNMs, based on combinations of the associated quality metrics. When presented with new sites and quality metrics, the trained classifier could discriminate true DNMs from false positives with high sensitivity and low false discovery rate (FDR). Based on misclassification error on the internal test set, we estimated that sensitivity was 91% (67 of 74 recovered) and FDR was 11.8% (9 false positives out of 76 called positives). Software specifications and validation studies are described in the supplemental material.
We applied forestDNM to the detection of DNMs in quads, and adapted forestDNM further to mutation detection of DNMs in trios. Mutations that were shared by monozygotic twins constitute germline mutations. Mutations that were not shared by MZ twins constitute somatic or cell line mutations (Koren et al, in press).
A total of 668 putative germline DNMs were detected and subject to comprehensive validation studies by Sanger sequencing and Sequenom genotyping. Validation results on mother, father and offspring were obtained for 652 sites and incomplete data was obtained for 16 sites (Supplementary Table 1). De novo mutations were confirmed for 565 sites (87%), and 87 DNM calls were invalidated, of which 34 (6%) were false positive variant calls and 53 (9%) were true-positive inherited SNPs falsely called as negative in one parent. Thus we confirm the high accuracy of forestDNM on new data. After excluding invalidated DNMs, subsequent analyses were performed on the remaining set of 581 DNM calls.
Base composition of DNMs detected in this study was similar to the base composition of segregating SNPs and DNMs reported in previous studies (Conrad et al., 2011; Lynch, 2010) (see Extended Experimental Methods). In addition, DNM calls were similar in depth and quality to variant calls for segregating SNPs (Supplemental Table 1). Phred-like quality scores and alternate-allele counts were slightly higher on average for validated DNM calls as compared to randomly sampled heterozygous SNPs (by one additional alternate allele read on average). This subtle skewing toward higher quality SNP calls had no effect on the overall genomic distribution of variants (Extended Experimental Methods).
Variation in Genome-Wide Rates of Germline Mutation
A total of 581 germline DNMs were detected in 10 MZ-twin pairs. A mean of 58 DNMs per offspring suggests that the average genome-wide mutation rate in humans is 1×10−8 per generation. Our estimate is lower than theoretical estimates by a factor of two (Haldane, 1935; Kondrashov and Crow, 1993), but consistent with empirical estimates from other whole genome sequencing studies (Conrad et al., 2011), Total mutation burden varied between 42 and 75 DNMs per offspring, consistent with previous observations of mutation rate variation (Conrad et al., 2011). Paternal age accounted for a substantial proportion of the variability in mutation burden in offspring. (P = 0.004, R2 = 0.44), see Figure 1, while maternal age was not significant. To account for any unforeseen deviations from the assumptions of the Poisson regression model, we also applied a permutation-based test: the one-sided P value for the effect of paternal age on mutation rate was 0.0226. These results allow us to quantify the accumulation of nucleotide substitutions in spermatogonial cells, which occurs at an average rate of 1 new mutation per year. Parent of origin was determined for 131 DNMs, of which 97 (74%) originated from the father (Supplemental Table 1).
Figure 1. Paternal age effect explains 44% of variation in genome-wide mutation rates.

Data points represent the total number of autosomal DNMs detected in offspring. See also Supplemental Figure 1 and Supplemental Table 1.
Global Patterns of Germline Mutation
Germline de novo mutations (DNMs) displayed a remarkably non-random positioning in the genome (P = 4.4 × 10−5, KS test). Compared to a random mutation model (uniform probability across the assembled genome, see methods for details), there was an overrepresentation of DNM pairs spaced more closely than the expectation (Fig. 2). The effect is significant when considering only the distribution within the individual (intra-individual DNM spacing) and when considering only the distribution across individuals (inter-individual DNM spacing).
Figure 2. Non-random distribution of DNMs in the genome.

Quantile-quantile plots of the observed distribution of inter-DNM distances within and between individuals and the expected distribution based on a random mutation model. Differences are statistically significant at α=0.05 by the KS test. See also Supplemental Figure 4 and Supplemental Table 2.
The observed distribution of DNMs reflects the underlying patterns of mutation and does not reflect non-uniformity in our ascertainment or validation of DNMs, as mentioned previously (see Extended Experimental Methods). As we describe in the following sections, the distribution of mutations can be explained by intrinsic characteristics of the genome.
Mutation Clusters
As evident from Figure 2, within individual genomes we observed striking enrichment of very closely-spaced DNMs (inter-DNM distance < 100 kb). Where parent of origin information was known for three loci (chr16:1823255–1823256, chr3:90077648– 90077664, chr8:3872643–3892698) closely-spaced pairs of DNMs had a single parent of origin. These results are consistent with very closely-spaced mutations arising as part of a single mutation event (Wang et al., 2007) within a narrow region of a chromosome. Defining a “cluster” as 2 or more DNMs located within a 100 kb span, a total of ten mutation clusters were identified on 8 chromosomes (see Supplemental Table 2), suggesting that mutation clusters occur at a rate of approximately 1 per generation. One mutation cluster in subject 75-0355 is evident in the 8p23 region illustrated in Figure 5. The observation of ten dense clusters of DNMs was statistically significant by permutation test (P<0.001).
Figure 5. Landscape of mutability in the genome.

(A) The 1 kb average mutability index (MI) across a 20 Mb genomic region of chromosome 8p21–23 indicates the existence of extended regions of hypermutability. “Hotspots” (red), “warm spots” (orange) as well as “cold spots” were defined by segmenting the MI scores using a 5 state HMM (see Supplemental Table 7). Predicted mutation rates (y-axis) were computed by multiplying the arithmetic mean MI by the baseline mutation rate of 10−8, then transforming to the log10 scale. The genome- and exome-wide distributions of MI are depicted in Supplemental Figure 7. The locations of DNMs are also shown and include a dense cluster of DNMs from individual 74–0355 (red, DNMs < 100 kb apart marked by asterisk). (B) The lower panel displays segmentation results for a second genomic region at 15q11–13. This region is notable for having a high rate of recurrent structural mutation. In the same region, the predicted rate of nucleotide substitutions is highly elevated.
Multiple mutational mechanisms could explain these findings, including compound mutation (Schrider et al., 2011) or gene conversion (Chen et al., 2007). Non-allelic gene conversion, which requires the presence of a paralogous sequence variant elsewhere in the genome could be ruled out for a majority (8/10) clusters (see Extended Experimental Methods). Clusters could instead be explained by compound mutation or by de novo nucleotide substitutions that occur during allelic gene conversion events (Hurles, 2002; Rattray et al., 2002). In all cases, we could rule out the possibility that mutation clusters are a spurious observation due to the mismapping of reads containing a paralogous sequence variant (see Extended Experimental Methods).
Determining Intrinsic Properties of the Genome that Influence Mutability
Regional mutation rates are subject to a combination of influences. In order to investigate the effect of intrinsic properties of the genome, we used logistic regression to discriminate DNMs from random genome background sites, based on the characteristics of the genome at these sites (and not relative or absolute genomic positions of DNMs). See Extended Experimental Methods for details on the genomic features used. Numerous features were found to influence site mutability. The most significant features were DNase hypersensitivity, GC content, nucleosome occupancy, recombination rate, simple repeats and the trinucleotide sequence surrounding the site (Fig. 3). We note that the UCSC Dennis and MEC nucleosome occupancy tracks (Gupta et al., 2008) use scores opposite in direction to indicate nucleosome occupancy. For both tracks, nucleosome occupancy was associated with suppressed mutation.
Figure 3. Individual associations of genome features with de novo mutation.
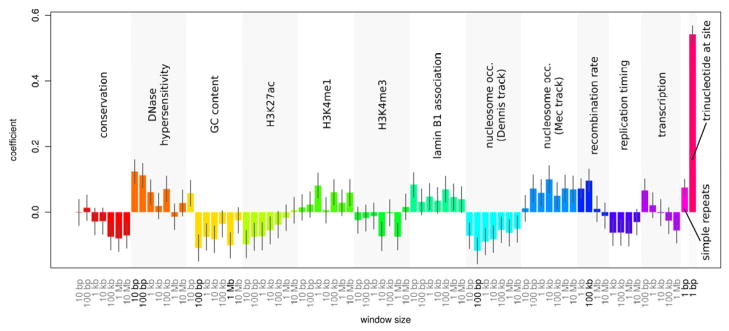
Predisposition to de novo mutation is influenced by sequence and chromatin characteristics. A variety of quantitative genome data were tested for associations with de novo mutation sites, including conservation, DNase hypersensitivity, GC content, histone marks, lamin B1 association, nucleosome occupancy, recombination rate, replication timing, transcription in human embryonic stem cells, simple repeats at the site of DNM, and the particular trinucleotide sequence centered at the site of DNM. The data were tested for association at different scales (i.e. window sizes at which the genome data were averaged), indicated on the x-axis. The strength and direction of association between the features and DNM are indicated by logistic regression coefficients (y-axis), which are shown with their standard errors. Significant associations (FDR < 0.10) are indicated in bold type. A summary of the relationship between these features and the principal components used in the predictive model is provided in Supplemental Figure 6. A detailed legend of the feature names and their descriptions is provided in Supplemental Table 6, and further details relating to the origin and construction of the features can be found in methods.
In addition to testing marginal associations, we investigated whether interactions between these genome features were predictive of mutation rate. This was done by performing two-way ANOVA for all possible combinations of features. After correcting for multiple testing, no two-way interactions were significant.
We next sought to construct a predictive model that could estimate nucleotide level mutation rates, based on information contained in all the features. This was accomplished by performing principal components analysis (PCA) on the features, and then using the principal components (PCs) as predictors in a regularized logistic regression model (again using “observed DNM” and “genome background” as the class labels). The output of the model is a measure of mutability that we call the mutability index (MI). MI is an estimate of relative mutation rate at single-nucleotide resolution (see methods for details). Throughout, we use the term “mutability” to refer to the mutability index and we use the term “mutation rate” to refer to the observed rate of DNMs for a given site or region.
Wide Variation in Site-Specific and Regional Mutation Rates
MI was highly predictive of site-specific (1bp resolution) mutation rates. (Fig 4), and could explain ~90% of the variability in mutation rates at sites across and genome. As expected, mutability was greatest for CpG dinucleotides. However, our model was highly predictive of mutation rate independent of this phenomenon. CpG sites and non-CpG sites varied widely in their mutability (10 fold and 100-fold respectively) and the range of CpG mutability overlapped considerably with the range for non-CpG sites (Supplemental Fig. 2).
Figure 4. Intrinsic characteristics of the genome explain variation in observed mutation rates.

Mutability index at the site level (1 bp) is highly predictive of the mutation rate in (A) ASD genomes in this study, (B) Control genomes in Conrad et al., 2011, and in ASD cases (C) and controls (D) of previous exome studies (combined data from O’Roak et al. 2011 and 2012, Iossifov et al., 2012, Sanders et al., 2012, and Neale et al., 2012). Mutability index explains a majority of the variability in site specific mutation rates, and the degree of mutation rate variation was similar in cases and controls. CpG sites and non-CpG sites varied widely in their mutability and the range of CpG mutability overlapped considerably with the range for non-CpG sites (Supplemental Fig. 2). Mutability index was also predictive of regional mutation rates (Supplemental Fig. 3).
The validity of MI was confirmed in independent datasets of de novo mutation. MI was highly correlated with mutation rate variation in a genome wide dataset from two trios (Conrad et al., 2011) (Fig. 4B) and exome datasets from 4 independent trio-based studies of autism (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2011; O’Roak et al., 2012; Sanders et al., 2012) (Fig 4C–D). In all datasets examined, observed mutation rates varied consistently by greater than two orders of magnitude ranging from 10−8.5 to 10−6.5. Mutability index explained ~90% of variation of the observed mutation rates in these studies. We conclude that our statistical model of mutability can explain a majority of the variance in site-specific mutation rates. Having confirmed the validity and accuracy of the MI, we apply this measure to the analysis of regional mutability.
We examined the landscape of mutability throughout the genome. Mean MI (in non-overlapping 1Kb windows) revealed broad genomic regions of hypermutability, generally tens to hundreds of kilobases in size. These included “hotspots” with highly elevated mutability (≥ 7 fold) and “warm spots” with moderately increased mutability (2–3 fold) (Fig. 5).
Genomic regions of hypermutability were then defined by segmenting the MI scores using a 5-state hidden Markov model (HMM). Parameters of the HMM were derived through numerical optimization by fitting a 5 component Gaussian mixture to the overall distribution of mean MI of 1kb windows of the genome. The sequence of hidden states along each chromosome was calculated using the Viterbi algorithm. Altogether approximately 9% and 0.02% of the genome was defined as “warm” and “hot” respectively, with 54%, 37%, and 0.5% being segmented as “baseline”, “cool”, and “cold”.
Hypermutability of these genomic regions was confirmed in this study and in 5 independent mutation datasets. The observed rate of DNMs in genomic segments was positively correlated with the mean MI of segments (Supplementary Fig. 3A–B). Likewise exonic mutation rates were highly correlated with the mean MI of exons (Supplementary Fig. 3C–D).
These results confirm that the landscape of mutability, like other characteristics of the genome, is highly non-random, in part explaining the distribution of DNMs that we originally observed. When we sampled a null model such that representation of sites was consistent with predicted mutability, the observed inter-DNM distances were closer to this null distribution (Supplemental Fig. 4).
Considering that our statistical model was developed based on a dataset of genomes largely derived from individuals diagnosed with ASD, we examined whether the mutation rate variation we observe is related to autism. We compared the predictive value of MI in cases and controls in the exome datasets. MI was equivalently predictive of mutation rate in healthy individuals (R2 = 0.91, slope = 0.78) and in ASD cases (R2 = 0.90, slope = 0.79) (Fig. 4C–D). Likewise, we compared the total burden of DNMs in hypermutable genes (average exonic log10 MI > 0.5), in cases and controls and observed no association with ASD (OR=0.73, P=0.227). Therefore the mutation rate variation that we observe cannot be attributable only to a subset of disease-associated mutations that are present in autism genomes.
A U-shaped Relationship between Genome Mutability and Evolutionary Conservation
We examined the relationship between regional rates of mutation, evolutionary change and genetic diversity in the human genome. Our results confirm that some hotspots have undergone rapid evolutionary change, consistent with previous studies (Chimpanzee Sequencing and Analysis Consortium, 2005). However patterns of germline mutation, particularly within the exome reveal many highly-mutable regions that change little over evolutionary time, an observation that challenges the common definition of the “evolutionary hotspot”.
A plot of mutability, divergence and diversity reveals a distinctly U-shaped relationship between mutability and sequence conservation (Fig. 6A). In regions that are less conserved (the left arm of the “U”), there is clear a correlation of hypermutability, hyperdivergence and hyperdiversity, consistent with such regions undergoing rapid evolutionary change. Surprisingly, in highly-conserved regions (the right arm of the “U”) the opposite trend is evident: hypermutability is correlated with highly-conserved sequence and low genetic diversity.
Figure 6. U-shaped relationship of mutability and evolutionary conservation.
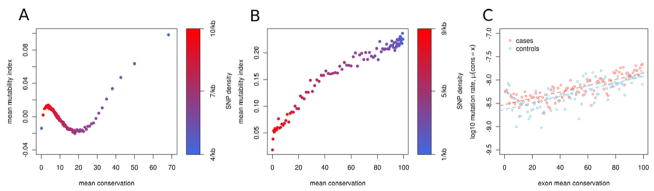
(A) Throughout the genome, we observe a correlation of hypermutability, hyperdivergence and hyperdiversity, consistent with previous studies. By contrast, in highly conserved regions the opposite trend is evident. MI and conservation were averaged in 1kb windows genome-wide. Windows were then binned according to percentiles of conservation. (B) Specifically within exons, there is a strong positive correlation of mutability and evolutionary conservation (also binned by percentiles of conservation). (C) The positive correlation between mutation rate and average exon conservation was confirmed by data from exome studies. Note that the positive relationship exists for both cases and controls. Under the null hypothesis, in which exons are hit with probability proportional to their length, this relationship is not observed.
We performed a similar analysis of exons and confirmed a strong positive correlation between MI and conservation (Fig. 6B). However the right arm of the “U” (Fig. 6A) could not be entirely explained by exonic sequences. We repeated the analysis in Figure 6A after excluding all exons, and the same U-shaped relationship was observed (data not shown). Lastly, we confirmed a positive correlation of conservation and mutation rate in protein-coding exons in exome datasets of ASD (Fig. 6C). Notably, the positive correlation between mutation rate and conservation was similar in cases and in controls. Therefore, this trend does not appear to reflect patterns that are unique to mutations that are detected in subjects with ASD. The above results suggest that mutability in the genome is, to some extent, coupled with functionality.
There are multiple genomic features that vary with evolutionary conservation in a similar fashion, most notably GC content. However, this feature alone does not explain patterns of mutability (see Figure 3). Importantly, genotype quality of SNPs and DNMs was not correlated with conservation (data not shown); hence, these observations do not appear to be an artifact of variable ascertainment.
Hypermutability is Common among Disease Genes
Genes that are subject to high mutation rates and strong purifying selection could be of particular importance to human disease. Mutability was significantly elevated for essential genes derived from the Online gEne Essentiality (OGEE) database (Chen et al., 2012) and human disease genes derived from the Online Mendelian Inheritance in Man (OMIM) database and varied by the modes of inheritance (Fig. 7A–B). Mutability was highest for essential genes and genes associated with dominant disorders. Mutability was elevated to a lesser extent for genes involved in recessive or polygenic traits.
Figure 7. Disease genes are characterized by high mutability.

Disease genes are more mutable than non-disease genes (A) within genes and (B) within exons. In both cases, mutability is highest for genes involved in dominant disorders and mutability is increased to a lesser extent for genes involved recessive and polygenic traits. Mutability is significantly elevated for genes preferentially expressed in the brain (C- D) as well as genes involved in ASD (see methods for details). An asterisk indicates a significant difference compared to the respective background set (at α=0.01 by a two-sided t-test). See also Supplemental Table 3 and Supplemental Table 4 for the mean mutability index of exons and genes, respectively.
Of relevance to our disease of interest, mutability of brain-expressed genes was significantly higher on average. In addition, mutability was elevated in a literature-based set of genes that have been implicated in ASD and in a set of genes that are associated with “syndromic” forms of autism (Fig 7C–D). Examples of hypermutable ASD-associated genes include NRXN1, AUTS2, GABRB3, SHANK2 and KCNMA1, which have one or more exons that rank among the top 20% most highly mutable in the exome (Supplemental Table 3 and Supplemental Table 4). Another particularly striking example of a disease-associated hotspot (see Figure 5B) is the 15q11–13 region. This region is well known for having an elevated structural mutation rate due to its local segmental duplication architecture, where recurrent duplications are associated with ASD and deletions are associated with Prader Willi/Angelman syndrome (Ledbetter et al., 1981). As we observe here, mutability of the DNA sequence within 15q11–13 is also predicted to be high independent of the local duplication architecture, suggesting that rates of nucleotide substitution are also elevated in this region.
Exonic Mutations in MZ Twins are Significantly Associated with ASD
DNMs detected in our MZ twin samples impacted a total of 34 genes (Supplemental Table 1), including 29 protein-coding genes and 5 non-coding RNAs. We hypothesize that genetic risk in our patient population is explained in part by de novo mutations in some of the above genes. We investigated the frequency of DNMs in protein coding genes in larger exome datasets on 962 cases and 590 controls from recent studies of ASD (1,035 mutations in 969 genes in cases, 564 mutations in 536 genes in controls) (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2011; O’Roak et al., 2012; Sanders et al., 2012).
In our set of 29 genes, 0 exonic hits were reported in controls, consistent with the low probability of observing an overlapping gene by chance. By contrast, seven de novo coding mutations of five genes were detected in cases, and two genes (KIRREL3 and GPR98) were hit twice. This constitutes a significant genetic overlap between genes mutated in concordant MZ twins and sporadic-ASD cases for total number of hits (P = 0.006), number of double hits (P=0.005), and number of genes (P = 0.04). See Extended Experimental Methods for details on the calculation of these empirical P values.
Discussion
Our model of intrinsic mutability, based on the unbiased ascertainment of germline mutations in families, reveals wide variation in mutation rates throughout the genome. The results of our study provide a global view of this landscape of mutability and its corresponding influence on genetic diversity and susceptibility to disease in humans. We show that hypermutability is a characteristic of disease genes, including genes that have been implicated in autism.
Mutability is explained by multiple influences acting in combination. For instance a specific di- or tri-nucleotide motif may have an elevated mutation rate. However, mutability of the site can be further modulated by other factors, including factors acting on larger scales such as nucleosome occupancy (~102 bp), recombination rate (~104 bp) and replication timing (~106 bp). Mutation rate variation in somatic cells (Koren et al, in press) and cancer (Schuster-Bockler and Lehner, 2012) is also influenced by aspects of chromatin structure, consistent with partially-overlapping mutational mechanisms acting in germ cells and somatic cells.
The patterns of mutability that we have uncovered provide new insights into the relationship between mutation, genetic diversity and disease that were not evident from studies of segregating genetic variation (Ellegren et al., 2003). We demonstrate that genome mutability and evolutionary conservation have a U-shaped relationship. Paradoxically, some of the mostly highly mutable sequences in the genome are in fact highly conserved.
The correlation of hypermutability and high evolutionary conservation is surprising and could not have been predicted from previous studies based on segregating variation in humans. We consider three possible theories to explain this finding. The first is the hypothesis that regional hypermutability itself is a trait that could be selected for under certain conditions, for instance where greater genetic diversity at a specific locus provides a fitness advantage. This hypothesis is reminiscent of the classic concept of “adaptive mutation” (Delbrück and Bailey, 1946; Rosenberg, 2001), a process by which genome wide mutation rates in bacteria increase in response to selective pressure. The second is the hypothesis that certain functional and highly-conserved elements originated from ancient mutation hotspots, and have since been subject to intense purifying selection. The third is the hypothesis that conserved hotspots could be explained simply by the fact that some DNA repair mechanisms are coupled with gene regulation (van Attikum and Gasser, 2005) or transcription (Svejstrup, 2002). Thus, the most highly transcribed regions in a given tissue could be the most susceptible to mutation. Further studies are needed to determine the underlying mutational and evolutionary mechanisms, but these findings have a significant implication regardless: patterns of mutation in the human genome appear to favor genetic changes that influence biological function.
Hypermutability in the genome has implications for human disease. Mutability is highest for essential genes and genes involved in dominant disorders. To a lesser extent mutability is elevated for genes primarily involved in recessive disorders or polygenic traits (polygenes). Likewise, hypermutable loci are likely to be important in neurodevelopmental disorders. Mutability was significantly elevated for a large set of genes that are preferentially expressed in the brain and genes that have been implicated in ASD. Our results are consistent with a prominent role for recurrent de novo mutations in autism and in other traits that have a contribution from dominant-acting alleles. We view these results, and the previous observation that mutation occurs at higher rates in highly-conserved elements, as possibly two sides of the same coin. Presumably, the selective pressures that constrain evolutionary divergence and nucleotide diversity in mutational hotspots are acting upon disease phenotypes such as ASD.
The genome-wide rate of mutation in individuals with ASD was not high. The average mutation rate in the genomes of patients in this study was 1×10−8. While the present study was under review, three studies were published using whole genome sequencing to estimate the human mutation rate. These studies yielded estimates in the same range as ours (0.89–2.3 ×10−8) (Campbell et al., 2012; Kong et al., 2012; Sun et al., 2012). Also, one study documented the occurrence of compound mutations and gene conversion events (Campbell et al., 2012). A second study also documented a paternal age effect on germline mutation rates (Kong et al., 2012). Collectively, these studies suggest that the true mutation rate in humans is lower than previous theoretical estimates (Haldane, 1935; Kondrashov and Crow, 1993; Nachman and Crowell, 2000), possibly by as much as a factor of two. This knowledge has led some to consider a recalibration of the time scales of human evolution and the divergence of human populations. (Scally and Durbin, 2012). These results also suggests that, after accounting for any effects due to paternal age, genome-wide rate of mutation in most individuals with autism is not significantly elevated.
The mutation rate variation that we observed in this study reflects patterns of mutation in a sample of subjects with ASD. This fact raises the possibility that disease mutations in our dataset could have an influence on the overall distribution of DNMs and estimates of site-specific mutability. When we compared mutation rates in exomes of cases and controls, we did not find evidence that mutation rate variation differs between affected and unaffected individuals. However, due to a paucity of available genome-wide data on controls, we are not able to compare regional mutation rates of intronic and intergenic regions in cases and controls. Thus, we cannot rule out the possibility that the distribution of DNMs in individuals with ASD might tend to exhibit a higher level of clustering around disease genes.
The set of genes impacted by de novo mutations in concordant MZ twins demonstrated a significant association with autism in independent samples, a result that was equally surprising and tantalizing. Given that the majority of exonic DNMs in autism cohorts are likely to be unrelated to disease (Neale et al., 2012; Sanders et al., 2012), we anticipated the same to be true for DNMs in our MZ-twin pairs. To the contrary, a set of independent exome sequencing studies (962 cases and 590 controls) detected 7 exonic mutations in 5 genes exclusively in cases, a result that is unlikely to occur by chance. This result suggests that exonic mutations in our MZ twin sample may be enriched in causal variants as compared to DNMs in the more typical sporadic/simplex cases.
These results do not provide conclusive evidence implicating individual genes in autism. However mutations detected in our concordant twin sample and in independent studies highlight some intriguing candidates. These include GPR98 and KIRREL3, where three de novo point mutations of each have been detected exclusively in cases and a balanced translocation disrupting KIRREL3 has been reported in a recent study (Talkowski et al., 2011). In addition, the TCF4 gene is a strong candidate given the documented involvement of this gene in Pitt Hopkins syndrome (Amiel et al., 2007; Zweier et al., 2007) and Intellectual disability (Hamdan et al., 2012; Need et al., 2012), and the observation of multiple de novo mutations of TCF4 in ASD (this study and in O’Roak et al. 2012).
As exemplified by early success from the preceding waves of CNV (Sebat et al., 2007) and exome-sequencing (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012) studies in ASD, new technologies for detection of mutations in the genome hold promise for understanding the genetic basis of this disorder. WGS provides another major boost to our ability to ascertain point mutations and CNVs (Michaelson and Sebat, 2012). This considerable improvement in mutation discovery comes at a relatively modest increase in sequencing cost. As the field continues its rapid transition toward incorporating comprehensive data on genomic variation and de novo mutations into genetic studies, we anticipate progress toward a deeper understanding on the underlying mechanisms of genome evolution and disease.
Experimental Procedures
Whole Genome Sequencing
Genomic DNAs from ten identical twin pairs and their parents were obtained from the NIMH genetics initiative biorepository (http://www.nimhgenetics.org). All DNA samples were derived from EBV-immortalized lymphoblastoid cell lines. A list of the 40 samples is provided in the Extended Experimental Methods. Deep (40X) whole genome sequencing was performed at BGI using the Illumina HiSeq Platform (500 bp library, 90 bp reads). Prior to sequencing, samples were randomized to minimize batch effects. Genomes were aligned to hg18 with BWA (Li and Durbin, 2009), and all subsequent analyses were performed with hg18 as the reference unless otherwise stated.
Alignment and Variant Calling
Alignment and variant (SNP) calls were generated on quad families using our WGS analysis pipeline implemented on the Triton compute cluster at UCSD (http://tritonresource.sdsc.edu/). Short reads were mapped to hg18 reference genome by BWA version 0.59 with the following parameters: “aln -o 1 -e 63 -i 15 -I -l 31 -k 2 -t 6”. Subsequent processing was carried out using SAMtools version 0.18, GATK version 1.2–52 (DePristo et al., 2011), and Picard tools version 1.52, which consisted of following steps: merging and sorting of the BAM files, indel realignment, fixing mate pairs, removal of duplicate reads, base quality score recalibration for each individual. Variant calls for each family were made (in “trio” mode) by running the unified genotyper for all four family members.
DNM Detection
Based on experience from our earlier CNV-based studies of de novo mutation (Malhotra et al., 2011; Nord et al., 2011; Sebat et al., 2007), an unfiltered set of putative DNMs is highly enriched for errors. In order to accurately distinguish true de novo variants from errors, we employed a custom machine learning pipeline we call forestDNM. A detailed description of the development and validation of this software is provided in Extended Experimental Methods. Briefly, a Random Forest (RF) classifier was trained based using quality metrics (see Supplemental Table 5) on an initial set of positive and negative training examples obtained by comprehensive validation of unfiltered putative DNMs from a single family (family 74–0352). See Supplemental Figure 5 for a depiction of the predictive importance of each quality metric. The trained classifier had an estimated sensitivity of 91% (67 of 74 recovered) and an estimated specificity of 11.8% (9 false positives out of 76 called positives. We used this trained RF classifier to predict the validation status of the putative DNMs in all families. In total, we predicted 668 DNMs in the 10 families.
Experimental Validation
Putative DNMs were validated by genotyping offspring using two independent validation methods. Sanger sequencing and Sequenom MassArray genotyping technologies (see Extended Experimental Methods). Parental genotypes were obtained using the Sequenom platform and additionally by Sanger sequencing if an informative Sequenom assay could not be designed. A total of 565/668 putative DNMs sites were validated and 87 sites were invalidated (34 as false heterozygous calls in the twins, 53 as inherited variants), corresponding to an overall observed FDR of 13%. In all subsequent analyses, we combine sites with complete validation data (565) with sites with incomplete validation data (16) for a total of 581 DNMs. Given the demonstrated low FDR of the classifier, we only expect 2 of the 16 incompletely validated sites to be false positives, so their inclusion is justifiable.
Parental Age Effect
We used Poisson regression to test the relationship between paternal and maternal age and DNM burden (Fig. 1 and Supplemental Table 1). In a fit using both paternal and maternal age as covariates, paternal age was significant (P=0.01) but maternal age was not (P=0.6). We thus discarded maternal age and fit a model using only paternal age, which had a significant effect (P=0.0039) and a slope of approximately 1 DNM/year.
Analysis of the Effect of DNA and Chromatin Features on de novo Mutation
We investigated whether quantitative genomic features (see Extended Experimental Methods for details on training data and features) had individual associations to DNM mutation by fitting logistic regression models (classes: observed DNM or genome background site), using each of the genome features as covariates. The coefficients and their standard errors are shown in Figure 3, and those features with significant (FDR < 0.10) associations have been noted in bold-faced type. Positive coefficients indicate a positive association between the value of the feature and DNM as the predicted class, whereas negative coefficients indicate a negative association.
Modeling Intrinsic Mutability of the Genome
With an unbiased set of germline mutations as training data, we used regularized logistic regression to predict mutability of sites based on intrinsic characteristics of the genome (see Extended Experimental Methods for details). We assembled quantitative genome features (conservation, transcription, GC content, simple repeat entropy, replication timing, recombination rate, DNase hypersensitivity, histone marks, nucleosome occupancy, lamin B1 association) and summarized them at several scales by taking the mean value in windows of 10 bp, 100 bp, 1 kb, 10 kb, 100 kb, 1 Mb, 10 Mb. The bulk of these data were derived from UCSC Genome Browser tracks (http://genome.ucsc.edu), and their provenance is outlined in detail in Supplemental Table 6. In addition to these features, we included a numerical variable that indicated predisposition to DNM, based on the trinucleotide sequence centered at the site (see Extended Experimental Methods for details).
Using these genomic features directly in the model would be problematic because they are highly correlated, with large-scale variation in GC content being one major source of the correlations. In order to mare fully exploit the information carried in the features, we performed principal components analysis (PCA), to produce 78 decorrelated features (i.e. the principal components or PCs).
PCs represent the unique signals in the data, and were used in place of the genome features as predictors in the model. Using these, we fit the model to the training data and defined a linear relationship between the class membership probability and the logarithm of the fold DNM excess at that probability. The relationship between genomic features, the PCs, and the model coefficients is shown in Supplemental Figure 6. We define the “mutability index” (MI) as the log10 of the fold-excess of training set DNMs observed for a given predicted class probability. This fold excess is an estimate of relative mutation rate. Using the model, we determined MI for every position in the genome. The genome and exome-wide distribution of MI at the single nucleotide level is given in Supplemental Figure 7.
To define regional patterns of mutability, we segmented the genome-wide map of mutability with a 5-state (cold, cool, baseline, warm, hot) hidden Markov model (HMM), see Supplemental Table 7. In all analyses involving exons, the exon boundaries were used to define regions, and the mean MI over the exon was used as the representative mutability.
Genomic Distribution of DNMs
We computed two types of inter-DNM distance, considering first the nearest neighboring DNM within an offspring (i.e. a twin pair) and then the nearest neighboring DNM in another unrelated offspring. We call these the within-individual inter-DNM distance and the between-individuals inter-DNM distance, respectively. We then computed null distributions by sampling random positions from the genome (excluding assembly gaps) while maintaining the number and family-wise allocation per chromosome of DNMs, then calculating both inter-DNM distances as described. Using the KS test we found that both observed distributions were significantly enriched (at α=0.05) for smaller inter-DNM distances compared to the simulated null distributions (Fig. 2), suggesting that observed DNMs are spaced more closely than expected by chance.
In light of our exploration of genome wide mutability, we hypothesized that if the null distributions were sampled such that a site’s probability of inclusion in the sample were proportional to its MI, the deviation of the observed inter-DNM distance distribution from the expectation would be attenuated. This was indeed the case for both inter-DNM distance measures (Supplemental Fig. 4), as shown by the difference in P values where both uniform sampling and weighted (i.e. by MI) sampling were used to construct the null distributions.
Correlation of Mutability Index with Mutation rate
Site-level analysis
The genome was binned with respect to nucleotide-resolution MI in increments of 0.1 on the log10 scale, and both the proportion of the genome scored within that bin, as well as the diploid mutation rate of sites scored within that bin, were calculated. We used sites from this work, (Conrad et al., 2011) and (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2011; O’Roak et al., 2012; Sanders et al., 2012) which were lifted over from hg19 to hg18 coordinates, in the calculation of these mutation rates (Fig. 4).
Regional analysis
We examined whether the trend of increasing mutation rate with increasing MI also held when looking at a regional scale. For whole genome sequencing studies (this study and (Conrad et al., 2011)), we used the previously described HMM segments, with their mean MI as the representation of regional mutability. We then binned such that each bin contained an equivalent number of DNMs (10%), and then calculated the diploid mutation rate (Supplemental Fig. 3A–B). For exome studies (Supplemental Fig. 3C–F) we used the mean MI of exons as the measure of regional mutability, again binned the exons such that each bin contained 10% of DNMs from the respective study, and finally calculated the diploid mutation rate. Linear regression models were fit for each study independently, and all studies showed a positive correlation between MI and mutation rate (all slopes were significant at α=0.01 except (Conrad et al., 2011) which had the fewest DNMs).
Conservation, Segregating Variation, and Mutability
We investigated the relationship between MI, segregating variation, and evolutionary conservation (Fig. 6) by first binning regions (genomic HMM segments and exons) according to the percentiles of their mean conservation values (yielding 100 bins). We then calculated the bin’s mean MI, mean conservation, and SNP density. SNPs were compiled from the families in this study, and the total number of observed SNPs was counted per bin, rather than the number of polymorphic sites (this places more emphasis on common variation).
Mutability and the Genetic Mode of Disease Genes
We compared trends in mutability when classifying genes according to the genetic basis of their related disease phenotype (Fig. 7A–B). For polygenic disease traits, we consulted the NHGRI GWAS catalog (Hindorff et al., 2009) and selected the most commonly studied diseases: diabetes (types I and II), coronary heart disease, Crohn’s disease, ulcerative colitis, multiple sclerosis, and rheumatoid arthritis. We selected genes that had a SNP (i.e. within its boundaries) referenced in the GWAS catalog and classified them as “polygene” (296 genes). For the recessive and dominant categories, we downloaded the OMIM database (http://omim.org) and extracted genes that were connected to diseases with “recessive” and “dominant” in the title, respectively (122 and 86 genes). Essential genes were extracted from the OGEE database (Chen et al., 2012) for a total of 1394 genes. Together, these sets of genes comprised our “disease genes”, and all remaining genes were considered as the background set. We computed gene and exon mean MI for all genes, and compared trends in mutability by performing a t-test on each category, with the “background” set of genes as the reference group. To show the trends of enriched mutability in each category, we calculated a bootstrapped group mean. This was accomplished by bootstrap sampling equal numbers (100 and 1000 for genes and exons, respectively) from the category under consideration (background, polygene, recessive, dominant, or essential) and computing the mean of each sample. This was performed 1000 times for each group.
Mutability of Brain and Autism Genes
An approach similar to that described above was used for investigating the trend of mutability in brain and autism genes, compared to the background set of all other genes (Fig. 7C–D). A list of genes preferentially expressed in the brain was assembled (totaling 2577) according to the approach used in (Raychaudhuri et al., 2010). We also assembled two sets of autism genes. The first was an inclusive set of ASD genes based on the strength of their connection to autism in the literature. This was accomplished by using NCBI mappings between Entrez gene IDs and PubMed IDs together with Fisher’s exact test to find genes significantly associated with autism publications. We thresholded the list at FDR < 0.01, resulting in 93 literature-supported genes that have been implicated in ASD. The second was a partially-overlapping set which included only “syndromic-ASD” genes (CACNA1C, CNTNAP2, FMR1, MECP2, NLGN3, NLGN4X, PTEN, SHANK3, TSC1, TSC2 and UBE3A) and “ASD-related” genes (AGTR2, ARX, ATRX, CDKL5, FOXP2, HOXA1, NF1, SLC6A8) from a previous study (Sakai et al., 2011). We added to this list 3 additional syndromic ASD genes, including KCNMA1 (Laumonnier et al., 2006), AUTS2 (Huang et al., 2010), SHANK2 (Berkel et al., 2010). Again we used t-tests and bootstrapped means to compare the distributions of brain and autism-implicated genes against the background set of all other genes.
Supplementary Material
Acknowledgments
This manuscript is dedicated in honor of James F. Crow, a pioneer in human genetics. This study was supported by grants to JS from the National Institutes of Health (MH076431 and HG005725) and the Simons Foundation Autism Research Initiative (SFARI 178088). LMI was supported by NIH RO1 HD065288 and NIH RO1 MH091350. We wish to thank Joe Gleeson for helpful comments on the manuscript. Special thanks to the San Diego Supercomputing Center (SDSC) for providing computing resources. Samples used in this study were provided by the NIMH genetics initiative (http://www.nimhgenetics.org). Acknowledgments for autism biomaterials are provided in supplemental material.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, Plouin P, Carter NP, Lyonnet S, Munnich A, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–993. doi: 10.1086/515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L, Vives L, O’Roak BJ, Sudmant PH, Shendure J, et al. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet. 2012 doi: 10.1038/ng.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Chuzhanova N, Ferec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 2007;8:762–775. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- Chen WH, Minguez P, Lercher MJ, Bork P. OGEE: an online gene essentiality database. Nucleic Acids Res. 2012;40:D901–906. doi: 10.1093/nar/gkr986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, Depristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- Delbrück M, Bailey WT. Induced Mutations in Bacterial Viruses. Cold Spring Harb Symp Quant Biol. 1946;11:33–37. [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Smith NG, Webster MT. Mutation rate variation in the mammalian genome. Curr Opin Genet Dev. 2003;13:562–568. doi: 10.1016/j.gde.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Francino MP, Ochman H. Isochores result from mutation not selection. Nature. 1999;400:30–31. doi: 10.1038/21804. [DOI] [PubMed] [Google Scholar]
- Gupta S, Dennis J, Thurman RE, Kingston R, Stamatoyannopoulos JA, Noble WS. Predicting human nucleosome occupancy from primary sequence. PLoS Comput Biol. 2008;4:e1000134. doi: 10.1371/journal.pcbi.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. The rate of spontaneous mutation of a human gene. J Genet. 1935:317–326. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Patry L, Dionne-Laporte A, Spiegelman D, Dobrzeniecka S, Rouleau GA, Michaud JL. Parent-child exome sequencing identifiesa de novo truncating mutation in TCF4 in non-syndromic intellectual disability. Clin Genet. 2012 doi: 10.1111/j.1399-0004.2012.01890.x. [DOI] [PubMed] [Google Scholar]
- Hardison RC, Roskin KM, Yang S, Diekhans M, Kent WJ, Weber R, Elnitski L, Li J, O’Connor M, Kolbe D, et al. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 2003;13:13–26. doi: 10.1101/gr.844103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE. Why do human diversity levels vary at a megabase scale? Genome Res. 2005;15:1222–1231. doi: 10.1101/gr.3461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XL, Zou YS, Maher TA, Newton S, Milunsky JM. A de novo balanced translocation breakpoint truncating the autism susceptibility candidate 2 (AUTS2) gene in a patient with autism. Am J Med Genet A. 2010;152A:2112–2114. doi: 10.1002/ajmg.a.33497. [DOI] [PubMed] [Google Scholar]
- Hurles M. Are 100,000 “SNPs” useless? Science. 2002;298:1509. doi: 10.1126/science.298.5598.1509a. author reply 1509. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS, Crow JF. A molecular approach to estimating the human deleterious mutation rate. Hum Mutat. 1993;2:229–234. doi: 10.1002/humu.1380020312. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Roger S, Guerin P, Molinari F, M’Rad R, Cahard D, Belhadj A, Halayem M, Persico AM, Elia M, et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am J Psychiatry. 2006;163:1622–1629. doi: 10.1176/ajp.2006.163.9.1622. [DOI] [PubMed] [Google Scholar]
- Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–329. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002;18:337–340. doi: 10.1016/s0168-9525(02)02669-0. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, McCarthy S, Michaelson JJ, Vacic V, Burdick KE, Yoon S, Cichon S, Corvin A, Gary S, Gershon ES, et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Sebat J. forestSV: structural variant discovery through statistical learning. Nat Methods. 2012;9:819–821. doi: 10.1038/nmeth.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, Meisler MH, Goldstein DB. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49:353–361. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelis E, Van Broeckhoven C, De Jonghe P, Lofgren A, Vandenberghe A, Latour P, Le Guern E, Brice A, Mostacciuolo ML, Schiavon F, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4:25–33. doi: 10.1159/000472166. [DOI] [PubMed] [Google Scholar]
- Nord AS, Roeb W, Dickel DE, Walsh T, Kusenda M, O’Connor KL, Malhotra D, McCarthy SE, Stray SM, Taylor SM, et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet. 2011;19:727–731. doi: 10.1038/ejhg.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, McGill CB, Strathern JN. The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics. 2002;162:1063–1077. doi: 10.1093/genetics/162.3.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Korn JM, McCarroll SA, Altshuler D, Sklar P, Purcell S, Daly MJ. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 2010:6. doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM. Evolving responsively: adaptive mutation. Nat Rev Genet. 2001;2:504–515. doi: 10.1038/35080556. [DOI] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY. Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med. 2011;3:86ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet. 2012;13:745–753. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
- Schrider DR, Hourmozdi JN, Hahn MW. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol. 2011;21:1051–1054. doi: 10.1016/j.cub.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012 doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JX, Helgason A, Masson G, Ebenesersdottir SS, Li H, Mallick S, Gnerre S, Patterson N, Kong A, Reich D, et al. A direct characterization of human mutation based on microsatellites. Nat Genet. 2012;44:1161–1165. doi: 10.1038/ng.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svejstrup JQ. Mechanisms of transcription-coupled DNA repair. Nat Rev Mol Cell Biol. 2002;3:21–29. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, Kirby A, Liu S, Muddukrishna B, Ohsumi TK, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet. 2011;88:469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uher R. The role of genetic variation in the causation of mental illness: an evolution-informed framework. Mol Psychiatry. 2009;14:1072–1082. doi: 10.1038/mp.2009.85. [DOI] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol. 2005;6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- Wang J, Gonzalez KD, Scaringe WA, Tsai K, Liu N, Gu D, Li W, Hill KA, Sommer SS. Evidence for mutation showers. Proc Natl Acad Sci U S A. 2007;104:8403–8408. doi: 10.1073/pnas.0610902104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MT, Smith NG, Ellegren H. Compositional evolution of noncoding DNA in the human and chimpanzee genomes. Mol Biol Evol. 2003;20:278–286. doi: 10.1093/molbev/msg037. [DOI] [PubMed] [Google Scholar]
- Zweier C, Peippo MM, Hoyer J, Sousa S, Bottani A, Clayton-Smith J, Reardon W, Saraiva J, Cabral A, Gohring I, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) Am J Hum Genet. 2007;80:994–1001. doi: 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.