

Abstract
Obesity is associated with ovulatory disorders, decreased rates of conception, infertility, early pregnancy loss and congenital abnormalities. Poor oocyte quality and reduced IVF success have also been reported in obese women. Recent attempts to understand the mechanism by which these defects occur have focused on mitochondria, essential organelles that are critical for oocyte maturation and subsequent embryo development. The oocyte relies on maternally supplied mitochondria until the resumption of mitochondrial replication in the peri-implantation period. Here we review current literature addressing the roles of mitochondria in oocyte function and how mitochondrial dysfunction can lead to fertility problems. The relationship between mitochondrial dysfunction and oocyte function is evaluated by examining the following examples of environmental exposures: tobacco smoke, aging, caloric restriction and hyperglycemia. Finally, we present new data from a mouse model of obesity that has demonstrated that oocyte mitochondria play a key role in obesity-associated reproductive disorders.
Keywords: obesity, oocyte, mitochondria, infertility
Obesity and infertility
The World Health Organization estimates that >1.4 billion people are overweight globally and that worldwide obesity has doubled since 1980 (WHO, 2012). As the obesity epidemic continues, the associated medical co-morbidities, including those affecting reproduction, will increase as well. The reproductive disorders associated with obesity include ovulatory disorders (Douchi et al., 2002; Castillo-Martinez et al., 2003), decreased rates of conception (Bolumar et al., 2000), infertility (Rich-Edwards et al., 1994), early pregnancy loss (Wang et al., 2002), congenital abnormalities (Practice Committee of American Society for Reproductive, 2008) and reduced assisted reproductive technology (ART) success (Dokras et al., 2006; Tamer Erel and Senturk, 2009; Depalo et al., 2011; Luke et al., 2011). A large analysis of 45 000 ART cycles concluded that increasing obesity correlated with decreasing likelihood of achieving pregnancy. However, this effect was only observed when autologous oocytes were used; if donor oocytes from a normal weight woman were used, the effect was mitigated (Luke et al., 2011). These data strongly indicate that an obese environment is detrimental to oocyte quality.
In this review, we will describe the effects of environmental factors on oocyte function and fertility, focusing on the critical role of mitochondria. As evidence that mitochondrial dysfunction in the oocyte can lead to subfertility, we will examine murine models of aging, caloric restriction and hyperglycemia. Lastly, we will focus on murine models for human diet-induced obesity. These models, especially that of a high-fat diet (HFD), have clearly demonstrated the detrimental effect of excess nutrition on oocyte quality and have allowed researchers to begin to address the mechanisms by which these defects occur. We will use published literature to argue that mitochondria are essential to the mechanisms underlying obesity-related infertility.
Mitochondria: overview of structure and function
The predominant function of mitochondria is to generate energy, in the form of adenosine triphosphate (ATP), for the cell through the process of oxidative phosphorylation. Given their key role in regulating cellular metabolism, mitochondria also play roles in cell signaling, cellular differentiation and regulation of cell growth. Mitochondria are composed of an outer membrane, an intermembrane space and an inner membrane that is highly folded into cristae to increase its surface area. Within the inner membrane is the mitochondrial matrix, which houses several copies of the mitochondrial genome (mtDNA) and is the site of the tricarboxylic acid pathway (TCA). This pathway, together with the electron transport chain in the inner membrane, is responsible for ATP production.
The mammalian mitochondrial genome is a circular double-stranded DNA of 16 569 base pairs encoding 37 genes. Included in this group are genes encoding 13 proteins of the oxidative phosphorylation machinery (Ramalho-Santos et al., 2009; Kogo et al., 2011). These include the core complexes for cellular respiration: two subunits of ATP synthase, cytochrome c oxidase, cytochrome b and seven subunits of complex I also known as NADH dehydrogenase. The remaining subunits are encoded by the nuclear genome of the cell. Replication of mtDNA relies on several proteins, including transcription factor A, mitochondria (TFAM) (Ekstrand et al., 2004; Kaufman et al., 2007) and the mtDNA-specific polymerase, POLG (Bratic et al., 2009). Because oxidative phosphorylation produces reactive oxygen species (ROS) which can be damaging to the cell, mtDNA transcription and translation, as well as mitochondrial activity, are tightly regulated for maintenance of optimal cellular metabolism (Fontanesi et al., 2006; Mick et al., 2010).
Mitochondria: involvement in oocyte function
Recent studies investigating developmental competence of oocytes, defined as the ability to produce a healthy, viable embryo, have revealed that developmental deficits, including chromosomal segregation disorders (Van Blerkom, 2011), maturation failure and arrested cell division (Van Blerkom, 2004), are often linked to defects in the mitochondria-dependent bioenergetic capacity of oocytes. Although their functions are the same, the mitochondria of oocytes are structurally different from those of somatic cells. In periovulatory oocytes, the mitochondria appear oval or spheroid with dense matrix and few cristae (Motta et al., 2000). During the post-fertilization and post-implantation period, the mitochondria in somatic cells transition to an elongated dumbbell shape with a lighter matrix and numerous transversally oriented cristae (Dvorak et al., 1982). The decrease in matrix density and increase in cristae number occur coincidently with increased glucose utilization and oxygen consumption and are thought to be reflective of an increase in oxidative phosphorylation that occurs during the blastocyst stage and post-implantation.
Because the oocyte depends on its mitochondria for ATP production and maintenance of cellular homeostasis (Duchen, 2000), the number and activity of mitochondria are regulated to ensure proper oocyte maturation, fertilization and subsequent embryo development (Reynier et al., 2001; Cummins, 2002). In mammals, mitochondrial DNA copy number increases over 30-fold during the stages of oocyte maturation (Bentov et al., 2010), whereas a primary oocyte (20 μm) has 500 copies of mtDNA, an MII oocyte (80 μm) can have 150 000–700 000 copies of mtDNA (Chen et al., 1995; Reynier et al., 2001; Santos et al., 2006; Cao et al., 2007). This large range in mtDNA copy number suggests high variability between individual human oocytes. In mouse oocytes, the mtDNA copy number ranges from a few thousand to over 350 000 depending on the stage of development, with the most mature oocytes containing the largest number (Ge et al., 2012; Mahrous et al., 2012). In mice, oocytes with low mtDNA copy number are more likely to be compromised than oocytes with higher mtDNA copy number (Ge et al., 2012). Similarly, human oocytes that are successfully fertilized in vitro contain more copies of mtDNA than do oocytes that fail to fertilize (Stojkovic et al., 2001; Santos et al., 2006). These findings suggest that mtDNA copy number is associated with oocyte quality and subsequent fertility. Thus, it is conceivable that potential stressors adversely affect the replication of mtDNA, leading to dysfunction or dysregulation of key mtDNA gene products that control oxidative phosphorylation, and thereby impair maturational and developmental potential of oocytes.
In addition to copy number, mitochondrial activity is regulated during oocyte maturation. For example, mitochondria undergo a significant increase in membrane potential (Van Blerkom and Davis, 2007) during oocyte maturation, which correlates with a concomitant increase in oxidative phosphorylation (Motta et al., 2000). The converse has also been found: the absence of an increase in membrane potential is associated with decreased developmental potential of oocytes (Acton et al., 2004). Similarly, ATP levels increase during oocyte maturation (Nagano et al., 2006; Duran et al., 2011) and oocytes with higher ATP levels have greater fertilization rates and embryo development than those with lower ATP levels (Stojkovic et al., 2001; Nagano et al., 2006).
Oocyte competence is directly related to the oocyte's ability to supply mitochondria to its daughter cells. Because mtDNA replication and mitochondrial amplification do not occur until the later stages of preimplantation development, the mitochondrial content of the oocyte must be sufficient to allow adequate numbers of organelles and genomes to be distributed to each cell of the embryo (Ebert et al., 1988). Interruption of this amplification process leads to insufficient copies, resulting in mtDNA depletion by embryonic days 7.5–8.5 and embryo lethality (Luoma et al., 2004; Wai et al., 2010). Mice deficient in either TFAM or POLG, both of which are required for mtDNA replication, are embryonic lethal; loss-of-function mutations or partial deletions in either of these genes result in reduced fertility and/or premature ovarian insufficiency (Luoma et al., 2004; Wai et al., 2010). The distribution of the mitochondria per embryonic cell is equal during the undifferentiated cleavage stage divisions; however, upon differentiation of the cells in the late morula to the blastocyst stage, the distribution of mitochondrial numbers begins to diverge. For instance, studies in mouse and hamster have shown that trophectoderm cells inherit more mitochondria than do the cells that make up the inner cell mass (ICM) (Barnett et al., 1996; Van Blerkom, 2008). The trophectoderm cells become the placenta and extraembryonic tissues, which have high-energy demands and require large numbers of mitochondria. In contrast, the ICM has lower demands for ATP production before implantation and thus requires fewer, less active mitochondria (Hewitson and Leese, 1993; Houghton, 2006).
Mitochondria: impact of environmental influences on oocyte function
The mammalian oocyte grows and matures within a follicular microenviroment that consists of multiple cell layers. Cooperative signaling between theca cells, granulosa cells, cumulus cells and the oocyte must occur to achieve optimal oocyte competence (Eppig, 2001). For example, the metabolism- and appetite-regulating hormone leptin and its receptor play a key role in cumulus cell-dependent mechanisms regulating oocyte maturation and developmental competence (Paula-Lopes et al., 2007). Additionally, upon leaving the primordial resting pool and during the follicular growth phase, the oocyte is particularly sensitive to changes in the follicular environment, especially those stressors induced by nutrient changes (reviewed in Ramalho-Santos et al., 2009). Environmental influences such as exposure to toxins or alterations in nutrition can affect the oocyte and subsequently, reproductive function. Over the past 10 years, numerous studies have revealed that these reproductive consequences of adverse environmental exposures are in large part mediated by effects on mitochondrial function.
Mitochondria and tobacco smoke
Tobacco smoke has been shown to cause an aberration in mitochondrial structure and function in alveolar macrophages of the lung; those that are directly exposed to smoke have more mutations in mtDNA than do those without smoke exposure (Ballinger et al., 1996; Van Blerkom, 2011). In the ovary, tobacco smoke has similarly been found to affect mitochondrial function. For example, antral follicles from the ovaries of smokers have higher levels of ROS, byproducts of mitochondrial dysfunction, than do ovaries of non-smokers, suggesting a potential mechanism for mtDNA damage and subsequent oocyte dysfunction caused by tobacco smoke (Paszkowski et al., 2002; Van Blerkom, 2011).
Investigations into the mechanisms by which tobacco smoke affects ovarian function have focused on a primary component of tobacco smoke, polycyclic aromatic hydrocarbons (PAHs) (Mattison et al., 1989; Sussman et al., 1999; Matikainen et al., 2001). PAHs bind to and activate the aromatic hydrocarbon receptor (AHR), which acts as a transcription factor to increase oocyte expression of Bax, a pro-apoptotic member of the Bcl-2 family of mitochondrial proteins, leading to apoptosis (Oltvai et al., 1993; Matikainen et al., 2001) (Fig. 1). In mice, exposure to PAHs leads to an accelerated loss of primordial follicles and premature exhaustion of the ovarian pool (Mattison et al., 1983; Jurisicova et al., 2007), but co-treatment with the AHR antagonist resveratrol rescues immature follicles from cell death (Jurisicova et al., 2007). Furthermore, inactivation of either Bax or AHR prevents ovarian damage (Matikainen et al., 2001), suggesting that AHR-driven transcription of the mitochondrial protein Bax, a cell-death signaling protein, is responsible for environmental toxicant-induced ovarian failure. Similarly, the addition of recombinant BCL-XL, an anti-apoptotic protein, can prevent early embryo arrest by improving mitochondrial bioenergetics, preventing the accumulation of ROS and enhancing preimplantation embryo development (Liu et al., 2011).
Figure 1.

The impact of mitochondria on oocyte function and fertility: example of the effect of smoking and aging on fertility. PAHs are pathogenic chemicals found in tobacco smoke. PAHs bind to and activate the AHR. This action leads to the expression of the pro-apoptotic Bcl-2 mitochondrial protein, Bax, in oocytes, which subsequently leads to apoptosis. PAHs cause an accelerated loss of primordial follicles, leading to premature exhaustion of the ovarian pool. The addition of the AHR antagonist resveratrol rescues immature follicles from death. Inactivation of either the transcription factor Bax or AHR prevents ovarian damage. Bax deficiency leads to oocytes whose function is sustained into very old age, suggesting the importance of this mitochondrial protein on ovarian function and fertility in relation to aging.
Mitochondria and the aging oocyte
Women of advanced maternal age are known to have increased meiotic spindle abnormalities and aneuploidy in oocytes, leading to infertility, miscarriage and chromosomal anomalies (Baird et al., 2005); these defects have been linked to mitochondrial dysfunction (Brenner et al., 1998). The mitochondria in oocytes of older women are more often swollen and have disrupted cristae than those of younger women (Muller-Hocker et al., 1996). Likewise, Kushnir et al. (2012) demonstrated that older mice had a prolonged time to conception, and their oocytes exhibited morphologic changes suggestive of altered mitochondrial biogenesis. Additionally, oocytes from old mice had 2.7-fold less mtDNA than those of younger mice, suggesting decreased fertilization potential. Iwata et al. (2011) has similarly demonstrated in cows that increasing maternal age correlated with an increase in rates of abnormal fertilization and a decrease in mtDNA copy number.
The mechanism by which mitochondria are affected by aging appears to be similar to the way in which they are affected by tobacco smoke and PAHs (Fig. 1). Evidence of this comes from Perez et al. (2007), who showed that mice without the pro-apoptotic Bax gene were able to sustain ovarian function into advanced age, extend their potential for fertility and minimize the health problems associated with menopause. In fact, Bax deficiency led to oocytes whose function was sustained into very old age, providing strong support for the importance of this mitochondrial protein in ovarian function and fertility in relation to aging.
Mitochondria and caloric restriction
Many age-related health complications are altered by caloric restriction, which is defined as a decrease in daily caloric intake by 40% (Masoro, 2005; Mair and Dillin, 2008; Selesniemi et al., 2011). Calorically restricted mice do not exhibit aging-related increases in oocyte aneuploidy, chromosomal misalignment on the metaphase plate or meiotic spindle abnormalities (Selesniemi et al., 2011). Caloric restriction alters the metabolic changes and oxidative stress associated with aging; these alterations are thought to be regulated by mitochondria (Sohal et al., 1994; Barja, 2002; Selesniemi et al., 2011). Evidence for this conclusion comes from the finding that the aging-related alterations of mitochondrial distribution and impaired ATP production in oocytes are not seen in the oocytes of calorically restricted mice (Selesniemi et al., 2011). Investigations into the mechanisms by which caloric restriction exerts its effects have focused on peroxisome proliferator-activated receptor coactivator 1α (PGC-1α), a transcription regulator that is responsive to nutritional cues. For example, PGC-1α is associated with modulating the expression of genes involved in mitochondrial respiration in response to nutritional cues (Lin et al., 2005; Rodgers et al., 2008; Selesniemi et al., 2011). Selesniemi et al. (2011) found that, like caloric restriction, loss of PGC-1α prevented maternal aging-associated oocyte aneuploidy and spindle defects in mice. These pathways illustrate the potential for alteration of oocyte function with mitochondrial dysfunction in response to nutritional cues.
Mitochondria and hyperglycemia
Diabetes and hyperglycemia occurring before conception and/or during pregnancy are associated with poor reproductive outcomes in women (reviewed in Amaral et al., 2008). To investigate the mechanisms involved, three different approaches have been used to generate mouse models of diabetes. The Akita mouse strain is hyperglycemic and hypoinsulinemic owing to a mutation in the insulin 2 gene. Models of hyperglycemia and insulin resistance can also be achieved chemically either by injecting mice with streptozotocin, which is toxic to the pancreatic beta cells, or by daily injections of insulin to induce a hyperglycemic (blood glucose levels of >14 mM, whereas normal is ∼5 mM) and insulin-resistant state. Oocytes from these mice have abnormal spindles and misaligned chromosomes (Wang et al., 2009), which can result in increased aneuploidy in ovulated oocytes from diabetic mice, consistent with the known pattern of increased rates of spontaneous miscarriage associated with maternal diabetes.
Examinations of mitochondria in the oocytes of diabetic mice have revealed several physical defects. First, there are alterations in the mitochondrial ultrastructure (Ratchford et al., 2007; Wang et al., 2009): narrowed inter-membrane space, rupture of the outer membrane and mitochondrial swelling, all of which are known markers for mitochondrial-dependent apoptosis (Senoo-Matsuda et al., 2005). Secondly, maternal diabetes disrupts mitochondrial redistribution during oocyte maturation, with increased perinuclear distribution and mitochondrial clustering relative to controls, suggesting an insufficient translocation of mitochondria (Wang et al., 2009). Thirdly, oocytes from diabetic mice have significantly higher mtDNA copy number than oocytes from healthy controls (312 000 versus 228 000) (Wang et al., 2009).
This increase in mtDNA copy number, however, does not translate to an increase in mitochondrial function in the diabetic model. Rather, mitochondrial function is reduced in oocytes from diabetic mice. The average ATP level in oocytes obtained from diabetic mice is significantly lower than in controls (Wang et al., 2009).
Additionally, the AMP to ATP ratio, a measure of cellular energy stores, is higher in oocytes obtained from diabetic oocytes than in oocytes from controls (Ratchford et al., 2007). The higher AMP/ATP ratio suggests increased AMP-activated protein kinase (AMPK) activity, which would lead to the conservation of ATP and up-regulation of alternate pathways for energy production within the oocyte, including fatty acid oxidation and amino acid catabolism. Diabetic mice, however, have dysfunctional AMPK, which may be the cause of delayed meiotic maturation rate or progression to germinal vesicle breakdown (Ratchford et al., 2007). Similarly, reduced levels of the TCA pathway metabolites citrate, aspartate and malate were noted in oocytes from diabetic mice (Wang et al., 2009). In addition to metabolic defects, oocytes from diabetic mice have increased concentrations of ROS and decreased concentrations of glutathione, suggesting that they have higher oxidative stress than do controls. Together, these data provide strong evidence that hyperinsulinemia and the resulting hyperglycemia lead to mitochondrial dysfunction in oocytes and contribute to poor oocyte quality in the diabetic mouse.
Mitochondria: impact of an HFD on oocyte function
Mouse models for human diet-induced obesity have demonstrated the potential detrimental effect of excess nutrition on oocyte quality. We have previously shown that mice fed a high fat, obesogenic diet (HFD) have oocytes with delayed maturation and decreased developmental competence (Jungheim et al., 2010). We propose that these problems with oocyte function that ultimately lead to problems with fertility are a direct result of mitochondrial dysfunction. As we will discuss next, an HFD leads to abnormalities of mitochondrial morphology, mitochondrial distribution within the oocyte, metabolism and spindle formation within the oocyte (Fig. 2).
Figure 2.
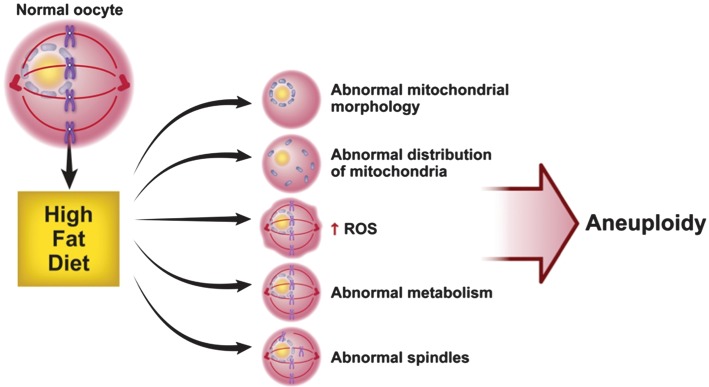
Maternal HFD leads to changes in the mitochondria of oocytes. Oocytes of mice on the HFD differ in several ways from those of control mice: (i) these oocytes have mitochondria with fewer cristae, increased cristae disarray, decreased electron density of the mitochondrial matrix, increased swelling and a greater number of vacuoles. (ii) Diet-induced obesity leads to an aberration in the pattern of mitochondrial distribution: mitochondria from obese females are distributed in a clumping, clustering pattern. (iii) The HFD leads to an increase in mitochondrial potential and ROS, suggestive of mitochondrial damage. (iv) Oocytes from mice fed an HFD have lower citrate levels than mice fed the control diet but normal, but ATP levels are normal; this suggests abnormal metabolism related to the HFD. (v) Oocytes from mice on HFD have fragmented spindles with ectopic organizing centers with chromosomes that are misplaced from the metaphase plate. These oocyte spindle defects and chromosome misalignments can result in high levels of aneuploidy.
HFD leads to abnormal mitochondrial morphology
Mitochondria from oocytes of mice on the HFD have fewer cristae, increased cristae disarray, decreased electron density of the mitochondrial matrix, increased swelling and a greater number of vacuoles than oocyte mitochondria from mice on the control diet have (Luzzo et al., 2012) (Fig. 3). Moreover, Igosheva et al. (2010) demonstrated that maternal diet-induced obesity in a murine model leads to an aberration in the pattern of mitochondrial distribution within the oocyte: instead of being uniformly distributed throughout the ooplasm with a perinuclear concentration as in controls, mitochondria from obese females are distributed in a clumping pattern without organization (Fig. 2). Perhaps not surprisingly, these defects are reminiscent of those observed in mouse models of diabetes, in which oocytes are exposed to a hyperglycemic state.
Figure 3.

Oocyte mitochondrial ultrastructure. Compared with the mitochondria from oocytes of mice on a control diet (A), the mitochondria from oocytes of mice from the HFD group (B) have fewer cristae, increased cristae disarray, decreased electron density of the mitochondrial matrix, increased swelling and a greater number of vacuoles. Reproduced with permission from Luzzo et al. (2012).
HFD leads to an increase in mitochondrial potential and ROS
To examine the possibility that an obesogenic state impairs oocyte mitochondrial function, Igosheva et al. (2010) used a low toxicity potentiometric fluorescent dye to examine oocyte mitochondria in mice on an HFD. They found that maternal diet-induced obesity leads to a dramatic increase in the inner mitochondrial membrane potential in oocytes. In contrast, Wu et al. (2010)used the dye JC-1 and found that membrane potential of oocyte mitochondria from mice on an HFD was lower than that of oocyte mitochondria from mice fed a control diet. This discrepancy may be explained by the fact that JC-1 is sensitive to factors other than inner mitochondrial membrane potential (Reers et al., 1991). Other differences between the two reports include the use of a different diet and strain of mice.
To assess the redox state of the oocytes, Igosheva et al. evaluated NADPH levels and found that, compared with oocytes from mice on a control diet, those from mice fed the HFD were more oxidized, suggesting that they were in a state of oxidative stress. The increased oxidation of flavoproteins in oocytes from obese females further supported the idea that obesity leads to oocyte oxidative stress. Additionally, the rate of ROS production was 2.1-fold higher in oocytes and zygotes from obese females than from those of lean mice. Together, these findings suggest a model in which excess nutrition leads to increased mitochondrial activity with concurrent increased oxidative stress (Fig. 2), which ultimately results in mitochondrial dysfunction and deleterious effects on oocyte function.
HFD leads to increased mitochondrial biogenesis and fission as a compensatory mechanism for stress-induced damage
Igosheva et al. also reported that mtDNA copy number is significantly higher in oocytes from obese mice than in those from lean controls (Igosheva et al., 2010). Similarly, we have previously shown that mitochondrial DNA copy numbers are significantly higher in oocytes from HFD mice than from mice on the control diet (Luzzo et al., 2012) similar to what was observed in oocytes of diabetic mice. A possible explanation for these findings is that mtDNA replication is a compensatory mechanism in response to oxidative-stress-induced mitochondrial damage. Evidence to support this hypothesis was revealed when we measured mitochondrial metabolism capacity in single oocytes and found that oocytes from mice fed an HFD had significantly lower citrate levels than mice fed the control diet (Luzzo et al., 2012). However, we observed no change in ATP levels. Thus, this change in citrate, a biochemical marker of the TCA cycle, suggests mitochondrial stress without significant compromise of overall metabolism. We also found that oocytes from mice on the HFD had increased expression of PGC-1α and Drp-1, markers for mitochondrial biogenesis and fission, respectively (Luzzo et al., 2012). Igosheva et al. similarly found that expression of nuclear genes encoding mtDNA transcription factors essential to mtDNA biogenesis, mtTFAM and NRF1, were also elevated in oocytes from obese females (Igosheva et al., 2010). These findings suggest that oxidative stress from an HFD model of obesity induces mitochondrial damage that leads to a compensatory response of increased mitochondrial biogenesis and mtDNA replication.
HFD leads to spindle defects and chromosome misalignment
Oocytes from control mice exhibit typical barrel-shaped spindles and chromosomes organized on the metaphase plate (Fig. 4). In contrast, oocytes from mice on HFD have fragmented spindles with ectopic organizing centers and chromosomes that are misplaced from the metaphase plate (Fig. 4). Spindle defects and chromosome alignments are significantly higher in HFD mice (48% spindle and 36% chromosomal abnormalities) than in controls (13 and 17%, respectively). These findings are analogous to those of a recent clinical IVF study in which the investigators reported that morbidly obese women had a significantly higher prevalence of disarrayed meiotic spindles with non-aligned chromosomes than did normal-weight women (28.6 versus 8.6%) (Machtinger et al., 2012). Although a few studies have reported no increased incidence of aneuploid miscarriages in obese women (Ebert et al., 1988; Luoma et al., 2004), it is possible that oocyte abnormalities lead to failed fertilization or loss before implantation. We propose that oocyte spindle defects and chromosome misalignments seen in the HFD model lead to high levels of aneuploidy (Fig. 2), which can result in a high frequency of embryos that are abnormal and fail to reach the blastocyst stage, resulting in greater rates of spontaneous miscarriage and subsequent subfertility.
Figure 4.
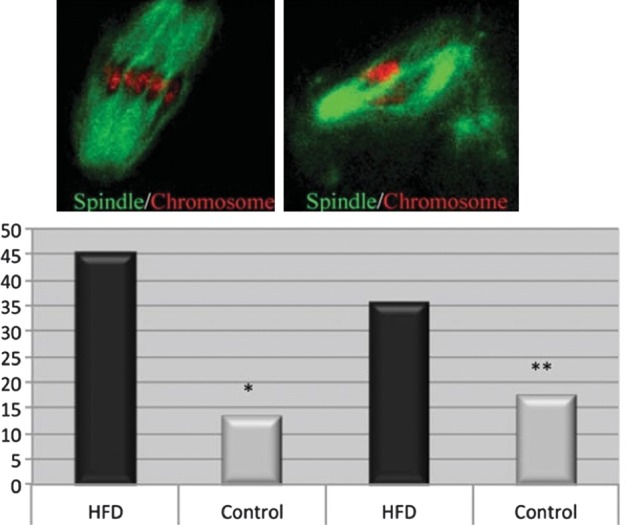
Oocyte aneuploidy and spindle formation/chromosome alignment. (top left) The MII oocytes from control mice display a typical barrel-shaped spindle and organized chromosomes on the metaphase plate. Oocytes from HFD mice (top right) have abnormal fragmented spindles and clustered chromosomes. (lower panel) Quantitative analysis demonstrates that both spindle defects and chromosome misalignment are higher in obese mice than in control mice. Reproduced with permission from Luzzo et al. (2012).
Genetic mouse models of obesity
Some attempts to use genetic models of obesity to examine the effects of excess nutrition on oocyte quality and fertility have led to the conclusion, which we argue is erroneous, that obesity does not affect oocyte quality. Two such models are db/db mice, which are subfertile and carry a mutation in the leptin receptor (Chen et al., 1996), and ob/ob mice, which are infertile and carry a mutation in the leptin gene (Zhang et al., 1994). A study published over 50 years ago argued that ob/ob mice were capable of becoming pregnant (Smithberg and Runner, 1957), but the success rate was extremely low—only 6% of ob/ob females delivered live pups after ovulation induction. Further complicating the conclusion of the paper is that no wild-type controls were used; it is known that the background strain generally produces over 7 pups per litter without ovulation induction. Thus, even though a few pregnancies were achieved, the phenotype was far from normal. In hindsight, this suggests that, in addition to a hypothalamic/pituitary problem (which was thought to be the only explanation for the obesity), the lack of leptin also affect oocyted quality, number or both. Around the same time that this study was published, another abstract was published in which the authors transplanted four ovaries from 3-month-old ob/ob mice into four lean control mice that had undergone ovariectomy (Hummel, 1957). Three months after the transplant, the control mice produced sired several litters and a total of 63 pups. The authors concluded that the obesity had no gonadal effects. The difficulty with this argument is that the recipient animals had normal leptin levels; thus compromised oocyte quality was reversed when ovaries were transplanted from an obese to a non-obese environment.
The final study to suggest that obesity does not affect oocyte quality (Chehab et al., 1996) exposed ob/ob mice to four different treatment regimens: two ob/ob groups differed by the length of leptin treatment, the third ob/ob group was treated with vehicle and the fourth ob/ob group, was subjected to a 65% caloric restriction diet. Leptin treatment for 6 months reversed the infertility, whereas severe food restriction and 50% weight loss in the first 40 days did not change fertility. The authors thus concluded that the reproductive defect was solely due to leptin deficiency in the brain and that obesity had nothing to do with the reproductive phenotype. At the time this conclusion was made, the hypothalamic effects of leptin and its receptor were not known; however, it is now clear that leptin and its receptor are expressed in cumulus cells and the oocyte and that they have a clear function in oocyte quality. The fact that caloric restriction did not rescue fertility can most likely be explained by the fact that the stress induced by such extreme caloric restriction can prevent ovulation or pregnancy. In addition, leptin treatment led to significant weight loss in the ob/ob mice. Therefore, the current interpretation of this study is that treatment with leptin not only normalizes leptin signaling in the oocyte and cumulus cells but also normalizes body weight and thus restores fertility.
Future directions
The negative impact of obesity on reproduction is well known, but the mechanisms responsible for developmental failure remain unclear. The HFD murine model for obesity has identified altered mitochondrial activity as one of the mechanisms of obesity-associated reproductive failure. Further studies are needed to evaluate obesity-related alterations in mitochondrial gene and protein expression that inappropriately regulate mitochondrial energy metabolism. We propose that further analysis of transcription factors such as Bax, similar to the work done on the effect of caloric restriction and tobacco smoke on oocyte function, will help elucidate how increased caloric exposure leads to mitochondrial dysfunction and, ultimately, oocyte dysfunction. We hope that with further identification of pathways involved in metabolic regulation, we will identify markers for improvement in oocyte quality and function.
Authors' roles
K.H.M. and N.M.G. conceptualized and organized the article together. N.M.G. did the research and drafting of the majority of the manuscript. K.H.M. critically revised the manuscript.
Funding
This work was supported by a National Institute of Health and Human Development grant (NIH), R01HD040390-07 (K.H.M.) and an American Diabetes Association Research Grant (K.H.M.).
Conflict of interest
Dr Moley is on the Scientific Advisory Board of Ovascience.
References
- Acton BM, Jurisicova A, Jurisica I, Casper RF. Alterations in mitochondrial membrane potential during preimplantation stages of mouse and human embryo development. Mol Hum Reprod. 2004;10:23–32. doi: 10.1093/molehr/gah004. [DOI] [PubMed] [Google Scholar]
- Amaral S, Oliveira PJ, Ramalho-Santos J. Diabetes and the impairment of reproductive function: possible role of mitochondria and reactive oxygen species. Curr Diabetes Rev. 2008;4:46–54. doi: 10.2174/157339908783502398. [DOI] [PubMed] [Google Scholar]
- Baird DT, Collins J, Egozcue J, Evers LH, Gianaroli L, Leridon H, Sunde A, Templeton A, Van Steirteghem A, Cohen J, et al. Fertility and ageing. Hum Reprod Update. 2005;11:261–276. doi: 10.1093/humupd/dmi006. [DOI] [PubMed] [Google Scholar]
- Ballinger SW, Bouder TG, Davis GS, Judice SA, Nicklas JA, Albertini RJ. Mitochondrial genome damage associated with cigarette smoking. Cancer Res. 1996;56:5692–5697. [PubMed] [Google Scholar]
- Barja G. Endogenous oxidative stress: relationship to aging, longevity and caloric restriction. Ageing Res Rev. 2002;1:397–411. doi: 10.1016/s1568-1637(02)00008-9. [DOI] [PubMed] [Google Scholar]
- Barnett DK, Kimura J, Bavister BD. Translocation of active mitochondria during hamster preimplantation embryo development studied by confocal laser scanning microscopy. Dev Dyn. 1996;205:64–72. doi: 10.1002/(SICI)1097-0177(199601)205:1<64::AID-AJA6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Bentov Y, Esfandiari N, Burstein E, Casper RF. The use of mitochondrial nutrients to improve the outcome of infertility treatment in older patients. Fertil Steril. 2010;93:272–275. doi: 10.1016/j.fertnstert.2009.07.988. [DOI] [PubMed] [Google Scholar]
- Bolumar F, Olsen J, Rebagliato M, Saez-Lloret I, Bisanti L. Body mass index and delayed conception: a European Multicenter Study on Infertility and Subfecundity. Am J Epidemiol. 2000;151:1072–1079. doi: 10.1093/oxfordjournals.aje.a010150. [DOI] [PubMed] [Google Scholar]
- Bratic I, Hench J, Henriksson J, Antebi A, Burglin TR, Trifunovic A. Mitochondrial DNA level, but not active replicase, is essential for Caenorhabditis elegans development. Nucleic Acids Res. 2009;37:1817–1828. doi: 10.1093/nar/gkp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner CA, Wolny YM, Barritt JA, Matt DW, Munne S, Cohen J. Mitochondrial DNA deletion in human oocytes and embryos. Mol Hum Reprod. 1998;4:887–892. doi: 10.1093/molehr/4.9.887. [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J, Yonekawa H. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet. 2007;39:386–390. doi: 10.1038/ng1970. [DOI] [PubMed] [Google Scholar]
- Castillo-Martinez L, Lopez-Alvarenga JC, Villa AR, Gonzalez-Barranco J. Menstrual cycle length disorders in 18- to 40-y-old obese women. Nutrition. 2003;19:317–320. doi: 10.1016/s0899-9007(02)00998-x. [DOI] [PubMed] [Google Scholar]
- Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- Chen X, Prosser R, Simonetti S, Sadlock J, Jagiello G, Schon EA. Rearranged mitochondrial genomes are present in human oocytes. Am J Hum Genet. 1995;57:239–247. [PMC free article] [PubMed] [Google Scholar]
- Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- Cummins JM. The role of maternal mitochondria during oogenesis, fertilization and embryogenesis. Reprod Biomed Online. 2002;4:176–182. doi: 10.1016/s1472-6483(10)61937-2. [DOI] [PubMed] [Google Scholar]
- Depalo R, Garruti G, Totaro I, Panzarino M, Vacca MP, Giorgino F, Selvaggi LE. Oocyte morphological abnormalities in overweight women undergoing in vitro fertilization cycles. Gynecol Endocrinol. 2011;27:880–884. doi: 10.3109/09513590.2011.569600. [DOI] [PubMed] [Google Scholar]
- Dokras A, Baredziak L, Blaine J, Syrop C, VanVoorhis BJ, Sparks A. Obstetric outcomes after in vitro fertilization in obese and morbidly obese women. Obstet Gynecol. 2006;108:61–69. doi: 10.1097/01.AOG.0000219768.08249.b6. [DOI] [PubMed] [Google Scholar]
- Douchi T, Kuwahata R, Yamamoto S, Oki T, Yamasaki H, Nagata Y. Relationship of upper body obesity to menstrual disorders. Acta Obstet Gynecol Scand. 2002;81:147–150. doi: 10.1034/j.1600-0412.2002.810210.x. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran HE, Simsek-Duran F, Oehninger SC, Jones HW, Jr, Castora FJ. The association of reproductive senescence with mitochondrial quantity, function, and DNA integrity in human oocytes at different stages of maturation. Fertil Steril. 2011;96:384–388. doi: 10.1016/j.fertnstert.2011.05.056. [DOI] [PubMed] [Google Scholar]
- Dvorak M, Tesarik J, Pilka L, Travnik P. Fine structure of human two-cell ova fertilized and cleaved in vitro. Fertil Steril. 1982;37:661–667. doi: 10.1016/s0015-0282(16)46279-5. [DOI] [PubMed] [Google Scholar]
- Ebert KM, Liem H, Hecht NB. Mitochondrial DNA in the mouse preimplantation embryo. J Reprod Fertil. 1988;82:145–149. doi: 10.1530/jrf.0.0820145. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Eppig JJ. Oocyte control of ovarian follicular development and function in mammals. Reproduction. 2001;122:829–838. doi: 10.1530/rep.0.1220829. [DOI] [PubMed] [Google Scholar]
- Fontanesi F, Soto IC, Horn D, Barrientos A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am J Physiol Cell Physiol. 2006;291:C1129–C1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- Ge H, Tollner TL, Hu Z, Dai M, Li X, Guan H, Shan D, Zhang X, Lv J, Huang C, et al. The importance of mitochondrial metabolic activity and mitochondrial DNA replication during oocyte maturation in vitro on oocyte quality and subsequent embryo developmental competence. Mol Reprod Dev. 2012;79:392–401. doi: 10.1002/mrd.22042. [DOI] [PubMed] [Google Scholar]
- Hewitson LC, Leese HJ. Energy metabolism of the trophectoderm and inner cell mass of the mouse blastocyst. J Exp Zool. 1993;267:337–343. doi: 10.1002/jez.1402670310. [DOI] [PubMed] [Google Scholar]
- Houghton FD. Energy metabolism of the inner cell mass and trophectoderm of the mouse blastocyst. Differentiation. 2006;74:11–18. doi: 10.1111/j.1432-0436.2006.00052.x. [DOI] [PubMed] [Google Scholar]
- Hummel KP. Transplantation of ovaries in the obese mouse. Anat Rec. 1957;128:569. [Google Scholar]
- Igosheva N, Abramov AY, Poston L, Eckert JJ, Fleming TP, Duchen MR, McConnell J. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One. 2010;5:e10074. doi: 10.1371/journal.pone.0010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata H, Goto H, Tanaka H, Sakaguchi Y, Kimura K, Kuwayama T, Monji Y. Effect of maternal age on mitochondrial DNA copy number, ATP content and IVF outcome of bovine oocytes. Reprod Fertil Dev. 2011;23:424–432. doi: 10.1071/RD10133. [DOI] [PubMed] [Google Scholar]
- Jungheim ES, Schoeller EL, Marquard KL, Louden ED, Schaffer JE, Moley KH. Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinology. 2010;151:4039–4046. doi: 10.1210/en.2010-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurisicova A, Taniuchi A, Li H, Shang Y, Antenos M, Detmar J, Xu J, Matikainen T, Benito Hernandez A, Nunez G, et al. Maternal exposure to polycyclic aromatic hydrocarbons diminishes murine ovarian reserve via induction of Harakiri. J Clin Invest. 2007;117:3971–3978. doi: 10.1172/JCI28493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogo N, Tazaki A, Kashino Y, Morichika K, Orii H, Mochii M, Watanabe K. Germ-line mitochondria exhibit suppressed respiratory activity to support their accurate transmission to the next generation. Dev Biol. 2011;349:462–469. doi: 10.1016/j.ydbio.2010.11.021. [DOI] [PubMed] [Google Scholar]
- Kushnir VA, Ludaway T, Russ RB, Fields EJ, Koczor C, Lewis W. Reproductive aging is associated with decreased mitochondrial abundance and altered structure in murine oocytes. J Assist Reprod Genet. 2012;29:637–642. doi: 10.1007/s10815-012-9771-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Liu X, Fernandes R, Gertsenstein M, Perumalsamy A, Lai I, Chi M, Moley KH, Greenblatt E, Jurisica I, Casper RF, et al. Automated microinjection of recombinant BCL-X into mouse zygotes enhances embryo development. PLoS One. 2011;6:e21687. doi: 10.1371/journal.pone.0021687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke B, Brown MB, Stern JE, Missmer SA, Fujimoto VY, Leach R. Female obesity adversely affects assisted reproductive technology (ART) pregnancy and live birth rates. Hum Reprod. 2011;26:245–252. doi: 10.1093/humrep/deq306. [DOI] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- Luzzo KM, Wang Q, Purcell SH, Chi M, Jimenez PT, Grindler N, Schedl T, Moley KH. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7:e49217. doi: 10.1371/journal.pone.0049217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machtinger R, Combelles CM, Missmer SA, Correia KF, Fox JH, Racowsky C. The association between severe obesity and characteristics of failed fertilized oocytes. Hum Reprod. 2012;27:3198–3207. doi: 10.1093/humrep/des308. [DOI] [PubMed] [Google Scholar]
- Mahrous E, Yang Q, Clarke HJ. Regulation of mitochondrial DNA accumulation during oocyte growth and meiotic maturation in the mouse. Reproduction. 2012;144:177–185. doi: 10.1530/REP-12-0113. [DOI] [PubMed] [Google Scholar]
- Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126:913–922. doi: 10.1016/j.mad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, et al. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat Genet. 2001;28:355–360. doi: 10.1038/ng575. [DOI] [PubMed] [Google Scholar]
- Mattison DR, Shiromizu K, Nightingale MS. Oocyte destruction by polycyclic aromatic hydrocarbons. Am J Ind Med. 1983;4:191–202. [PubMed] [Google Scholar]
- Mattison DR, Singh H, Takizawa K, Thomford PJ. Ovarian toxicity of benzo(a)pyrene and metabolites in mice. Reprod Toxicol. 1989;3:115–125. doi: 10.1016/0890-6238(89)90045-2. [DOI] [PubMed] [Google Scholar]
- Mick DU, Vukotic M, Piechura H, Meyer HE, Warscheid B, Deckers M, Rehling P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J Cell Biol. 2010;191:141–154. doi: 10.1083/jcb.201007026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta PM, Nottola SA, Makabe S, Heyn R. Mitochondrial morphology in human fetal and adult female germ cells. Hum Reprod. 2000;15(Suppl. 2):129–147. doi: 10.1093/humrep/15.suppl_2.129. [DOI] [PubMed] [Google Scholar]
- Muller-Hocker J, Schafer S, Weis S, Munscher C, Strowitzki T. Morphological-cytochemical and molecular genetic analyses of mitochondria in isolated human oocytes in the reproductive age. Mol Hum Reprod. 1996;2:951–958. doi: 10.1093/molehr/2.12.951. [DOI] [PubMed] [Google Scholar]
- Nagano M, Katagiri S, Takahashi Y. Relationship between bovine oocyte morphology and in vitro developmental potential. Zygote. 2006;14:53–61. doi: 10.1017/S0967199406003510. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Paszkowski T, Clarke RN, Hornstein MD. Smoking induces oxidative stress inside the Graafian follicle. Hum Reprod. 2002;17:921–925. doi: 10.1093/humrep/17.4.921. [DOI] [PubMed] [Google Scholar]
- Paula-Lopes FF, Boelhauve M, Habermann FA, Sinowatz F, Wolf E. Leptin promotes meiotic progression and developmental capacity of bovine oocytes via cumulus cell-independent and -dependent mechanisms. Biol Reprod. 2007;76:532–541. doi: 10.1095/biolreprod.106.054551. [DOI] [PubMed] [Google Scholar]
- Perez GI, Jurisicova A, Wise L, Lipina T, Kanisek M, Bechard A, Takai Y, Hunt P, Roder J, Grynpas M, et al. Absence of the proapoptotic Bax protein extends fertility and alleviates age-related health complications in female mice. Proc Natl Acad Sci USA. 2007;104:5229–5234. doi: 10.1073/pnas.0608557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Practice Committee of American Society for Reproductive M. Obesity and reproduction: an educational bulletin. Fertil Steril. 2008;90:S21–S29. doi: 10.1016/j.fertnstert.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos J, Varum S, Amaral S, Mota PC, Sousa AP, Amaral A. Mitochondrial functionality in reproduction: from gonads and gametes to embryos and embryonic stem cells. Hum Reprod Update. 2009;15:553–572. doi: 10.1093/humupd/dmp016. [DOI] [PubMed] [Google Scholar]
- Ratchford AM, Chang AS, Chi MM, Sheridan R, Moley KH. Maternal diabetes adversely affects AMP-activated protein kinase activity and cellular metabolism in murine oocytes. Am J Physiol Endocrinol Metab. 2007;293:E1198–E1206. doi: 10.1152/ajpendo.00097.2007. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- Reynier P, May-Panloup P, Chretien MF, Morgan CJ, Jean M, Savagner F, Barriere P, Malthiery Y. Mitochondrial DNA content affects the fertilizability of human oocytes. Mol Hum Reprod. 2001;7:425–429. doi: 10.1093/molehr/7.5.425. [DOI] [PubMed] [Google Scholar]
- Rich-Edwards JW, Goldman MB, Willett WC, Hunter DJ, Stampfer MJ, Colditz GA, Manson JE. Adolescent body mass index and infertility caused by ovulatory disorder. Am J Obstet Gynecol. 1994;171:171–177. doi: 10.1016/0002-9378(94)90465-0. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos TA, El Shourbagy S, St John JC. Mitochondrial content reflects oocyte variability and fertilization outcome. Fertil Steril. 2006;85:584–591. doi: 10.1016/j.fertnstert.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Selesniemi K, Lee HJ, Muhlhauser A, Tilly JL. Prevention of maternal aging-associated oocyte aneuploidy and meiotic spindle defects in mice by dietary and genetic strategies. Proc Natl Acad Sci USA. 2011;108:12319–12324. doi: 10.1073/pnas.1018793108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Igaki T, Miura M. Bax-like protein Drob-1 protects neurons from expanded polyglutamine-induced toxicity in Drosophila. EMBO J. 2005;24:2700–2713. doi: 10.1038/sj.emboj.7600721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithberg M, Runner MN. Pregnancy induced in genetically sterile mice. J Heredity. 1957;48:97–100. [Google Scholar]
- Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Stojkovic M, Machado SA, Stojkovic P, Zakhartchenko V, Hutzler P, Goncalves PB, Wolf E. Mitochondrial distribution and adenosine triphosphate content of bovine oocytes before and after in vitro maturation: correlation with morphological criteria and developmental capacity after in vitro fertilization and culture. Biol Reprod. 2001;64:904–909. doi: 10.1095/biolreprod64.3.904. [DOI] [PubMed] [Google Scholar]
- Sussman NB, Mazumdar S, Mattison DR. Modeling adverse environmental impacts on the reproductive system. J Womens Health. 1999;8:217–226. doi: 10.1089/jwh.1999.8.217. [DOI] [PubMed] [Google Scholar]
- Tamer Erel C, Senturk LM. The impact of body mass index on assisted reproduction. Curr Opin Obstet Gynecol. 2009;21:228–235. doi: 10.1097/GCO.0b013e32832aee96. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J. Mitochondria in human oogenesis and preimplantation embryogenesis: engines of metabolism, ionic regulation and developmental competence. Reproduction. 2004;128:269–280. doi: 10.1530/rep.1.00240. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J. Mitochondria as regulatory forces in oocytes, preimplantation embryos and stem cells. Reprod Biomed Online. 2008;16:553–569. doi: 10.1016/s1472-6483(10)60463-4. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion. 2011;11:797–813. doi: 10.1016/j.mito.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J, Davis P. Mitochondrial signaling and fertilization. Mol Hum Reprod. 2007;13:759–770. doi: 10.1093/molehr/gam068. [DOI] [PubMed] [Google Scholar]
- Wai T, Ao A, Zhang X, Cyr D, Dufort D, Shoubridge EA. The role of mitochondrial DNA copy number in mammalian fertility. Biol Reproduct. 2010;83:52–62. doi: 10.1095/biolreprod.109.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JX, Davies MJ, Norman RJ. Obesity increases the risk of spontaneous abortion during infertility treatment. Obes Res. 2002;10:551–554. doi: 10.1038/oby.2002.74. [DOI] [PubMed] [Google Scholar]
- Wang Q, Ratchford AM, Chi MM, Schoeller E, Frolova A, Schedl T, Moley KH. Maternal diabetes causes mitochondrial dysfunction and meiotic defects in murine oocytes. Mol Endocrinol. 2009;23:1603–1612. doi: 10.1210/me.2009-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. World Health Organization. Obesity and overweight: fact sheet N311. 2012.
- Wu LL, Dunning KR, Yang X, Russell DL, Lane M, Norman RJ, Robker RL. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology. 2010;151:5438–5445. doi: 10.1210/en.2010-0551. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]