

Abstract
During a single turnover of the hydroxylase component (MMOH) of soluble methane monooxygenase from Methylosinus trichosporium OB3b, several discrete intermediates are formed. The diiron cluster of MMOH is first reduced to the FeIIFeII state (Hred). O2 binds rapidly at a site away from the cluster to form the FeIIFeII intermediate O, which converts to an FeIIIFeIII-peroxo intermediate P and finally to the FeIVFeIV intermediate Q. Q binds and reacts with methane to yield methanol and water. The rate constants for these steps are increased by a regulatory protein, MMOB. Previously reported transient kinetic studies have suggested that an intermediate P* forms between O and P in which the g = 16 EPR signal characteristic of the reduced diiron cluster of Hred and O is lost. This was interpreted as signaling oxidation of the cluster, but low accumulation of P* prevented further characterization. In this study, three methods to directly detect and trap P* are applied together to allow its spectroscopic and kinetic characterization. First, the MMOB mutant His33Ala is used to specifically slow the decay of P* without affecting its formation rate, leading to its nearly quantitative accumulation. Second, spectra-kinetic data collection is used to provide a sensitive measure of the formation and decay rate constants of intermediates as well as their optical spectra. Finally, the substrate furan is included to react with Q and quench its strong chromophore. The optical spectrum of P* closely mimics those of Hred and O, but it is distinctly different from that of P. The reaction cycle rate constants allowed prediction of the times for maximal accumulation of the intermediates. Mössbauer spectra of rapid freeze quench samples at these times show that the intermediates are formed at almost exactly the predicted levels. The Mössbauer spectra show that the diiron cluster of P*, quite unexpectedly, is in the FeIIFeII state. Thus, the loss of the g = 16 EPR results from a change of the electronic structure of the FeIIFeII center rather than oxidation. The similarity of the optical and Mössbauer spectra of Hred, O, and P* suggest that only subtle changes occur in the electronic and physical structure of the diiron cluster as P* forms. Nevertheless, the changes that do occur are necessary for O2 to be activated for hydrocarbon oxidation.
The soluble form of methane monooxygenase (sMMO) found in many methanotrophs catalyzes the oxidation of methane to methanol as part of the metabolic pathway that allows these microorganisms to use methane as the sole carbon and energy source. 1 This demanding reaction (methane bond dissociation energy equals 105 kcal mole−1) 2 is catalyzed only by sMMO and particulate MMO (and to a lesser extent, pMMO-like ammonia monooxygenase in autotrophic ammonia-oxidizing bacteria). 3,4 sMMO is an enzyme system comprised of three protein components: i) a hydroxylase (MMOH) containing a diiron metal cluster in the active site, ii) a [2Fe-2S] cluster and FAD containing reductase (MMOR) that mediates electron transfer between NADH and the diiron cluster, and iii) a regulatory protein MMOB that is devoid of any cofactors. 5–7 The unique reactivity of sMMO has spurred research into its chemical mechanism and led to extensive efforts to mimic the chemistry using synthetic model compounds. 6–12 We have studied the mechanism of sMMO using the enzyme from the Type II methanotroph Methylosinus trichosporium OB3b (M. t. OB3b), while others have used the enzyme from the Type X methanotroph Methylococcus capsulatus Bath (M. c. Bath) with similar results.6–8,13
Single turnover transient kinetic studies in the presence of MMOB have been used to map the reaction cycle intermediates as shown in Scheme 1. 6,7,14–16 Table S1 summarizes the experimental evidence for the various intermediates of the reaction cycle. The resting diferric MMOH (Hox) can be reduced by two electrons from either MMOR or chemical reductants to form diferrous MMOH (Hred). 5 This form of the enzyme reacts with O2 to form intermediate O which has oxygen bound to the enzyme, but perhaps not to the diiron cluster. 17 Intermediate O decays to form intermediate P; the latter exhibits a weak optical band at 700 nm and Mössbauer parameters diagnostic of a diferric cluster with a peroxo moiety bound to the iron atoms. 18–20 The precise nature of the cluster in P is unknown, but comparison with model complexes and peroxo intermediates in related diiron enzymes suggests that it is a μ-1,2-peroxo dinuclear FeIII complex. 8 Intermediate P decays to form intermediate Q, which has been shown by Mössbauer and X-ray absorption studies to contain a unique bis-μ-oxo FeIV2 diamond core cluster. 14,20,21 Q exhibits a relatively intense optical spectrum with maxima at 330 and 430 nm (η430 = 7500 M−1 cm−1). This spectrum has allowed extensive studies of the mechanism of the reaction with methane and adventitious substrates.14,15,18,22–25 It is likely that the reaction occurs by hydrogen atom abstraction with a substantial tunneling component to form intermediate R.7,22,24,26–28 In contrast to intermediates O, P, and Q, intermediate R does not live long enough to be directly observed, but it is postulated to occur based on racemization during chiral 1H, 2H, 3H-ethane oxygenation and computational studies showing that a substrate radical is transiently formed. 26,29–31 Rebound of the cluster-bound hydroxyl group onto the substrate radical would yield the product complex, termed T, which dissociates to complete the cycle. 14
Scheme 1.

Reaction cycle intermediates of MMOH.
Our transient kinetic studies of the O to P conversion have indicated the presence of an intervening intermediate termed P*.19 Two experimental observations that led to this mechanistic proposal. The first evidence is that the rate constant of formation of P (10 s−1 at 4 °C, pH 7.0) is slower than the rate constant for the decay of O (26 s−1 at 4 °C, pH 7.0), indicating that there must be an intermediate in between. The second line of evidence arises from the observation that the decay rate constant of O is pH independent, in contrast to the pH dependent rate constant for the formation of P, showing that the reactions cannot be the same. To date, transient kinetic single turnover studies on MMOH from M. t. OB3b have failed to reveal any spectral features that can be attributed to P*. However, the fact that the g = 16 signal of diferrous cluster disappears as P* is formed was interpreted to indicate that either one or both cluster irons are oxidized due to formation of a metal-ligated superoxo or peroxo complex. 19
Recent studies using the M. c. Bath enzyme provide kinetic evidence for the presence of an intermediate before Hperoxo (equivalent to P) in the MMOH catalytic cycle. 16 A global fitting of the kinetic time traces at 420 nm and 720 nm of a single turnover reaction of reduced M. c. Bath MMOH with O2 in the presence of methane is consistent with an intermediate with an electronic absorption spectrum similar to that of Hperoxo, which is known to have an FeIIIFeIII cluster. Accordingly, it was reasoned that the intermediate (also named P*) and Hperoxo both have FeIIIFeIII clusters in similar electronic environments.
One particularly relevant aspect of the MMOH reaction cycle for the present study is the dramatic effect of MMOB on the rate of catalysis. 6,28,32 The overall rate constant for the conversion of intermediate Hred to P in the M. t. OB3b enzyme system is accelerated 1000 fold by the presence of MMOB. 17 For adventitious substrates, the regiospecificity of hydroxylation usually changes quite markedly when MMOB is added, and spectroscopic studies indicate that the diiron cluster environment of MMOH is altered in the MMOH-MMOB complex. 33–35 The residues that form the interface between MMOH and MMOB have been identified by spectroscopic studies. 36,37 A set of MMOB variants made by introducing site-specific mutations at these residues has been shown to alter the rate constants for interconversion between MMOH reaction cycle intermediates. 28,32 One of the variants, MMOB His33Ala (H33A), specifically decreases one or more of the rate constants for the steps in the conversion of Hred to P, potentially allowing the reactions in this sequence to be studied in detail.
Here, we show that MMOB H33A decreases the rate constant for the specific step of P formation from P* by about 30 fold without a significant decrease in the rate constant for O decay, resulting in nearly quantitative accumulation of P*. This permits the observation of the electronic absorption and Mössbauer spectra of P*. It is shown that P* has a different oxidation state than previously proposed, suggesting a new approach to formation of the peroxo complex in diiron cluster containing oxygenases.
EXPERIMENTAL PROCEDURES
Chemicals
3-(N-morpholino)-propanesulfonic acid, glycerol, ferrous ammonium sulfate, cysteine, urea, furan, sodium hydrosulfite and methyl viologen were purchased from Sigma-Aldrich.
Biological Materials
MMOH was purified from M. trichosporium OB3b with the following modifications to the published protocol. 38 All purification buffers contained 0.2 mM ferrous ammonium sulfate and 2.0 mM cysteine as a stabilizer for the active site iron of MMOH. These were added to the cold buffers at least 3 h before use and nitrogen purged for 1 h before and during use. The chromatography columns were also equilibrated with nitrogen-purged buffers before loading the iron/cysteine stabilizer containing buffers. The second high-resolution ion-exchange chromatography column was additionally scrubbed free of oxygen with 20 ml of 5.0 mM sodium hydrosulfite solution. The M. t. OB3b cell free extract was applied to the initial DEAE ion-exchange column in a batch-binding manner. The protein fractions containing MMOH were subsequently collected in sealed argon-purged glass vials. The MMOH containing protein fractions were pooled and concentrated. This concentrated eluent pool was desalted through a Sephadex G-25 column (22 × 2.8 cm) equilibrated in 25 mM MOPS, pH 6.8. The desalted protein pool was subsequently applied to a high-resolution Q-Sepharose column (12 × 2.8 cm) equilibrated in 25 mM MOPS, pH 6.8. MMOH is eluted with a 900 ml gradient from 0 M to 0.08 M NaCl in the same buffer at a linear flow rate of 23 cm/h. The MMOH containing protein fractions were collected in argon-purged sealed glass vials, pooled and concentrated via ultrafiltration. Glycerol was added to a final concentration of 5% (v/v) to the protein pool. Recombinantly expressed H33A MMOB mutant was purified according to the protocol previously described. 39
Transient Kinetic Experiments
Transient kinetic single turnover experiments were performed on an Applied Photophysics stopped-flow instrument (Model SX.18MV with SX Pro-Data upgrade). The sample preparation for MMOH involved making MMOH anaerobic under argon at 4 °C followed by a transfer into an anaerobic glove bag (Coy). The protein was reduced with a stoichiometric excess of sodium hydrosulfite and methyl viologen (10 % of the MMOH active site concentration) at room temperature for 20 minutes. The chemical reductants were separated from MMOH by passage through a Sephadex G-25 PD-10 desalting column (GE Healthcare) equilibrated in 100 mM MOPS buffer at the chosen pH point containing 0.2 mM ferrous ammonium sulfate and 2.0 mM cysteine. The iron/cysteine-containing buffer was incubated for 1.5 h at 4 °C before being made anaerobic. The reduced protein was loaded into one of the drive syringes on the stopped flow instrument using a Hamilton gas-tight syringe. The other drive syringe was loaded with a stoichiometric equivalent amount of H33A MMOB in oxygen-saturated buffer. If an sMMO substrate was utilized in the single turnover experiment, it is also added to this drive syringe. For both these experiments and the rapid freeze quench (RFQ) experiments described below, the results are not affected by pre-mixing MMOH and MMOB in one syringe or mixing them during the transient kinetic experiment. MMOH is much more soluble when mixed with MMOB so that pre-mixing facilitates sample handling when using very high enzyme concentrations in the RFQ experiments. On the other hand, reduced MMOH reacts much slower with O2 in the absence of MMOB, facilitating handling for lower concentration samples. The single-wavelength transient kinetic data was analyzed with the Pro-Data Viewer software from Applied Photophysics and fit to a summed exponential expression.15 Singular value decomposition of spectra-kinetic multiple-wavelength data was performed using the Pro-Kineticist global analysis software (Applied Photophysics). The protein concentration of MMOH used in the transient kinetic experiments is described in terms of reactive MMOH active sites. This description arises from the presence of two populations of MMOH active sites: i) a population that undergoes turnover with previously observed kinetic rates and forms catalytic intermediates, 14 ii) a population that undergoes a slow reaction with O2 and therefore does not accumulate catalytic intermediates during turnover. The presence of two populations of MMOH with only one population displaying intermediates in the single turnover catalytic cycle, is also observed in MMOH from M. c. Bath. 16 The reactive population of MMOH active sites comprises 40% of the total MMOH active sites in the MMOH protein used in most of the experiments described in this study, although preparations with as high as 60% have been obtained. The active fraction was determined from a comparison of the observed specific activity to the specific activity of a fully active preparation (1200 nmoles/min/mg).38 The active fraction can also be determined from the maximum observed yield of Q by optical detection and the known rate constants for the reaction cycle. The two methods give good agreement.
RFQ Mössbauer and EPR Experiments
For the preparation of rapid freeze-quench Mössbauer and EPR samples, MMOH was reduced with a stoichiometric amount of sodium hydrosulfite in the presence of methyl viologen (10 % of the MMOH active site concentration). A stoichiometric amount (per active site) of H33A MMOB was then added to reduced MMOH under anaerobic conditions and the mixture loaded in an RFQ syringe. The RFQ syringe was loaded on an Update Instrument Model 1019 RFQ apparatus. A low-temperature bath circulator (Neslab LT-50) maintained the reactants in the RFQ assembly at a temperature of 4 °C. The RFQ samples were produced by mixing the reduced MMOH enzyme with oxygen-saturated buffer and freezing the reaction at specified time points on counter-rotating aluminum wheels at liquid nitrogen temperature. 40 The frozen sample was then packed in RFQ Mössbauer cups or EPR tubes under liquid nitrogen. EPR spectra were collected using a Bruker Elexsys E-500 or Bruker ESP 300 spectrometer each equipped with a Bruker dual mode cavity and an Oxford ESR 910 liquid helium cryostat. Mössbauer spectroscopy was performed as previously described.40,41 Spectra were analyzed using the software WMOSS (SEE Co, Edina, MN, USA).
RESULTS
Single Turnover Reaction Using H33A MMOB in the Absence of Substrate Slows the P* to P Conversion Step
The photodiode-array traces for the single-turnover reaction of Hred in the presence of H33A MMOB but absence of substrate are shown in Figure 1. While the formation and decay of intermediate Q are readily seen in the 400–450 nm region, there is no evidence of the spectrum of intermediate P, normally seen in the 700 nm region. 15,16,18,28 Extraction of the rate constants for the reaction by multiple exponential fitting of the time course shown in the inset of Figure 1, indicates that the apparent formation rate constant of Q at 4 °C is decreased from 2.7 s−1 (observed with wild-type MMOB) to 0.38 s−1, similar to our previous findings. 28 When considered together, these results indicate that the use of MMOB H33A greatly slows formation of Q by slowing a step prior to the P to Q conversion.
Figure 1.

Photo-diode array spectra of a single-turnover reaction of MMOH with H33A MMOB (12 μM reactive MMOH active sites) in the absence of substrate, pH 7.0, 4 °C. The diferrous form of the enzyme Hred (black trace) oxidizes to form Q (red trace). Q subsequently decays to the resting diferric state Hox of the enzyme (blue trace). There is no optical evidence for the formation any other intermediate apart from Q in this reaction. Inset: Kinetic time course at 430 nm (red) extracted from the diode array data. The 2-exponential fit (black) shows that Q forms with a rate constant of 0.38 s−1.
The slow step in the O to P sequence can be determined by monitoring the disappearance of intermediate O using rapid freeze quench techniques and EPR spectroscopy. As shown in Figure 2, the rate constant of decay of the parallel mode g = 16 EPR signal of O is nearly unchanged when using H33A MMOB in place of WT MMOB (k = 28 s−1 and 26 s−1, respectively). Together the kinetic data show that it is the P* to P step which is greatly slowed by the mutation.
Figure 2.

Decay of the parallel mode g = 16 integer-spin signal of compound O with WT MMOB (▲) or H33A MMOB (●) at pH 7.0, 4 °C. A single exponential fit to the data yields rate constants of 26 s−1 or 28 s−1, respectively. The signal is normalized to that of a sample mixed with anaerobic buffer. EPR measurement conditions: T = 2.0 K, microwave power = 0.5 mW, microwave frequency = 9.405 GHz. The residual g =16 EPR intensity at 0.1 s arises from the slow-reacting fraction of reduced MMOH that reacts very slowly with oxygen. This represents approximately 45 % of the active sites in the particularly active MMOH sample utilized for EPR studies.
Single Turnover Reaction Using H33A MMOB in the Presence of a Substrate Facilitates Direct Detection of P*
The broad electronic absorption spectrum of Q with large extinction coefficients (Figure 1) masks the spectra of other intermediates with weaker optical absorbance. In order to circumvent this problem, we have added furan as a substrate to rapidly react with Q, and therefore quench its optical spectrum. 15 Since this substrate does not change the rate constants of the catalytic cycle prior to the decay of Q, 14,15the accumulation of intermediates preceding Q should not be altered. 14,15 We have also used methane instead of furan to quench Q with no change in the results. However, since methane is a gas, it limits the amount of O2 that can be dissolved in the buffer, thereby restricting the concentration range available for the kinetic experiments.
A search for the optical features of P* using the commonly employed diode array detector as in Figure 1 proved unsuccessful. This is due to the relative insensitivity of the diode array detector. As an alternative, a spectra-kinetic data accumulation method was used to observe the single turnover reaction of MMOH with H33A MMOB and furan. In this technique, a series of single wavelength time courses are monitored at 15 nm intervals between 325 nm and 685 nm. These traces are then used to reconstruct the electronic absorption spectra of O and the other intermediates in the catalytic cycle. While this requires much more enzyme than a single diode-array measurement, the input light intensity is greatly attenuated (avoiding potential photochemical bleaching), and much finer temporal resolution is obtained due to the shorter integration time, leading to substantially higher signal to noise ratios. Moreover, the higher dynamic range of the photomultiplier detector allows the 300–380 nm region to be accurately monitored at the protein concentrations used in the experiment. The latter was critical for the characterization of P*.
In the single turnover reaction of Hred with H33A MMOB and furan, we can observe the rapid formation of an intermediate species maximizing at 250 ms, which subsequently decays over 10 seconds to the resting diferric form of the enzyme, Hox (Figure 3). While the formation of this transient intermediate can be observed across the whole wavelength region scanned, the decay can be only seen at long wavelengths between 535 nm and 685 nm. The absence of this decay phase at shorter wavelengths occurs because the formation of diferric Hox has a comparable rate constant as the decay of the intermediate and because Hox possesses an electronic absorption band with a large extinction coefficient in the near-UV region (Figure 1), which masks the decay of the intermediate.
Figure 3.

Single wavelength kinetic trace at 625 nm for the single turnover reaction of Hred and stoichiometric (to total diiron cluster concentration) H33A MMOB (83 μM reactive MMOH active sites) and 6 mM furan with 0.9 mM O2 at pH 7.0 and 4 °C, indicates the rapid formation and decay of a transient species. The fit to the kinetic data (black trace) was obtained from a global fit to the multiple wavelength data (325 nm–685 nm range).
In order to obtain accurate rate constants and estimates for the relative accumulation of the intermediates, a global analysis was performed using the large data set accumulated over a range of wavelengths. This technique utilizes the entire data set and adds the additional constraint of accommodating the kinetic model at multiple wavelengths. The global fitting analysis of the spectra-kinetic data supports the following kinetic model for the single turnover reaction: O → P* → P → Hox. The rate constants determined for the steps in the reaction in the order shown are: k1 = 21.9 ± 0.5 s−1, k2 = 0.33 ± 0.03 s−1, k3 = 1.76 ± 0.05 s−1. The result of this fit is shown as an overlay of the time course at 625 nm in Figure 3. The formation rate constant of 21.9 s−1 for the first intermediate is similar to the decay rate constant of compound O, as measured by following the decay of its characteristic integer spin g = 16 EPR signal (Figure 2). 17 The decay rate constant for this intermediate (0.33 s−1) is also similar to the formation rate constant of 0.38 s−1 for Q (in actuality P) in the single turnover reaction monitored by diode array (Figure 1). 28 This confirms the identity of the observed intermediate as compound P*. It also implies that the observation of the single turnover reaction starts with compound O. The formation of O is not observed here as it likely occurs in the dead time of the stopped flow instrument and has spectral properties indistinguishable from those of Hred.
The global fit allows estimation of the time dependent accumulation of each intermediate as shown in Figure 4, top. P* is predicted to accumulate to approximately 90 % of the total active site concentration after 250 ms. Later, a small accumulation of intermediate P is also predicted as P* decays. The observed decay rate constant of P (1.7 s−1) is slightly smaller than is observed in a single-turnover reaction in the presence of wild type MMOB (2.7 s−1). 28 This is not unexpected because the functionally related MMOB H5A variant has been shown to decrease this rate constant to 1.71 s−1. 28 The global fit to the MMOH reaction does not require the accumulation of compound Q between the decay of P and the formation of Hox. This is expected because the presence of 6 mM furan will completely quench Q (kform = 1.7 s−1, kdecay ~ 72 s−1 as calculated from the second order rate constant of Q decay with furan). 14,28 Compound P* is a catalytically competent intermediate because its rate constant for decay exceeds the turnover number for furan as a substrate at 4 °C (kcat = 0.12 s−1). 14,28
Figure 4.

Speciation plots based on the global fitting analysis to the spectra-kinetic data of the single turnover reaction of MMOH in the presence of H33A MMOB. Top: in the presence of furan. Bottom: in the absence of a substrate. The top plot shows time courses for each species obtained directly from a global fit of the spectra-kinetic data from 325 nm–685 nm collected over a 10 s interval. For the simulation in the bottom panel, a numerical integration method using the rate constants determined here and the previously determined value for the rate constant for Q decay when using MMOB H33A in the absence of substrate was used to compute the time courses shown.28 The dashed vertical lines indicate the times at which rapid freeze quench samples were obtained for Mössbauer spectroscopy. In both top and bottom panels, the fractional accumulation refers to the reactive fraction of MMOH.
UV-Vis Spectrum of P*
Multicomponent analysis of the entire multi-wavelength data set from which the time course shown in Figure 3 was extracted allows the electronic absorption spectrum of P* to be approximated (Figure 5). It has a maximal absorption in the near-UV region (η325 = 12000 M−1 cm−1) which is very similar to that observed for O (and Hred) (η325 = 11100 M−1 cm−1). In fact, the entire UV-vis spectrum of P* is remarkably similar to, albeit distinguishable from, those of O and Hred (Figure 5, inset). This suggests that they are similar species and markedly different than the more strongly absorbing intermediate P.
Figure 5.

The pure component spectra of compound P* (red) as obtained by global fitting of spectra-kinetic data compared to those of compound O/Hred (black) and Hox (magenta). The pure component spectrum of P (blue) (from a Hred single turnover reaction using WT-MMOB) has been overlaid for a comparison with P*. Experimental conditions as in Figure 3. Inset: Expanded spectra showing the difference in absorbance for intermediates P*, O, and P the long wavelength region.
pH Dependence of P* Formation and Decay Steps
The pH dependence of the kinetic steps of formation and decay of compound P* has been studied in order to draw a comparison with the observation of pH dependence of O decay and P formation when using wild type MMOB. 19 The results clearly indicate that the formation of P* is pH independent while its decay is a proton dependent step (Figure 6). Based on the similarity with the pH dependence of the kinetic steps observed when using wild type MMOB, this data strongly corroborates the O → P* → P → Q → Hox kinetic model based on electronic absorption spectroscopy.
Figure 6.

pH dependence profile of the kinetic steps of P* formation and decay. The rate constants were obtained from a global fit of Hred single turnover reaction using H33A MMOB in the presence of furan at each pH point. The experimental error for the data in the right panel falls within the symbols used to indicate the data.
Mössbauer Characterization of Compound P* Reveals a Diferrous Center
Using 57Fe enriched Hred and H33A MMOB in the absence of a substrate, we have prepared three samples for Mössbauer characterization of P* (Figure 7A, B, and C). Of the three samples, the first is the fully reduced sMMO, while the latter two were rapid-freeze quenched at 320 ms and 3 s, respectively, after mixing with O2 at the times for which P* and Q are predicted to be near maximum levels (see Figure 4, bottom). Analyses of the Mössbauer spectra were complicated by the presence of a large fraction of slow-reacting diferrous MMOH (Hslred, ca. 60% in the Mössbauer samples, 45% in the EPR samples) that is always encountered for MMOH from both M. t. OB3b and M. c. Bath.18,20 In the absence of substrates, Hslred decays into an oxidized state Hslox with an overall rate constant of 0.023 s−1 at 4 °C, pH 7.17
Figure 7.

RFQ Mössbauer spectra from the time course of the reaction of Hred in the presence of stoichiometric H33A MMOB with a saturated solution of O2. Spectra were recorded at 4.2 K in zero magnetic field. (A) Spectrum of Hred. The black line is a representation of Hred drawn to represent 95% of the Fe in the sample. Contributions of the individual sites (47.5% each) are shown in red and blue. Red: ΔEQ = 3.22 mm/s, δ = 1.26 mm/s; Lorentzian lines with 0.35 mm/s FWHM. Blue: ΔEQ = 2.37 mm/s, δ = 1.35 mm/s; Voigt lines with −0.6 mm/s FWHM. The remainder of the sample (5%) is comprised of unidentified non-diferrous species. (B) Sample quenched at 320 ms. Purple line (86% of total Fe) represents all ferrous species in the sample. Magenta line shows contribution of P (3% of Fe); magenta arrow points at the high energy line of P. The sample also contains ~ 5% Hox (green line and green arrow) and ~5% unchanging non-diferrous species. (C) Sample quenched at 3 s. Black line is the sum of simulations for diferrous species (58%), intermediate Q (23%), intermediate P (magenta arrow, 7%) and Hox(green arrow, 7%). ~5% unchanging non-diferrous species are also present. (D) Spectrum obtained by subtracting the contributions of Hred from Figure 7C. The solid black line is the sum of the colored curves which correspond to Q (orange), P (ΔEQ = 1.55 mm/s, δ = 0.69 mm/s, magenta) and Hox (green). (E) Superposition of the raw data of the samples of (B, black hashed) and (C, cyan).
Measurements of MMOH samples exhibiting a range of activities have shown that the reactive and slow-reacting fractions exhibit indistinguishable Mössbauer spectra for both the diferric and diferrous states.42,43 Previous studies have established that the yield of product is >80% from a single turnover reaction in the presence of MMOB allowed to proceed until all of the diferrous MMOH has been oxidized independent of the fraction of slow-reactive MMOH present.17 We hypothesize that Hslred fails to form the proper complex with MMOB, thereby making O2 binding a rather slow and rate limiting step and masking intermediate formation. The diiron cluster itself is likely to remain in a uniform, active conformation.
The two iron sites of Hred are inequivalent and display distinct Mössbauer spectra.43 In the absence of an applied magnetic field each site yields a quadrupole doublet. The quadrupole splitting, ΔEQ(1), of site 1 is essentially the same for all molecules in the sample, i. e. the two lines of the doublet are sharp. ΔEQ(2), however, is substantially distributed about its mean value, resulting in the observation of broad absorption lines (such broadening is frequently observed for high-spin FeII complexes). We wondered whether these lines were truly broadened or whether they masked the presence of two doublets representing site 2 (as the latter case, for instance, might occur if a binding site for water is partially occupied). To address this point and to enhance the spectral resolution, we have removed the line width contribution of the 57Co Mössbauer source (~ 0.12 mm/s) using a Fourier transform procedure described by Dibar-Ure and Flinn and others.44,45 The resolution-enhanced spectra, shown and described in SI, suggest that the line positions of all doublets present have been identified and that site 2 produces one, albeit broadened, doublet rather than two. The sharp doublet of site 1 has quadrupole splitting ΔEQ = 3.22 mm/s and isomer shift δ = 1.26 mm/s (red line in Figure 7A). We have simulated the broadened doublet of site 2 (blue line in Figure 7A) by using Voigt line shapes, convoluting a Lorentzian of 0.15 mm/s full width into a Gaussian with full width 0.65 mm/s (this is accomplished by using a negative line width in WMOSS).
For this study, we were interested in modeling the shape of the high-energy lines of sites 1 and 2 such that a good value is obtained for the total amount of diferrous cluster in the sample (the spectral area to the right of the dashed line, which represents half of the ferrous absorption, is not contaminated by contributions from other species). The blue and red lines were obtained by simulating the ferrous high-energy feature, assuming a 1:1 site ratio (a 1:1 site ratio is strongly indicated by the 8.0 Tesla spectrum of Figure 13 of ref 43). We are not claiming that the black solid line in Figure 7A (sum of red and blue curves) is a unique representation for Hred (both reactive and slow-reacting), but the decomposition will serve well for the present purpose. The parameters used are quoted in the caption of Figure 7.
The black curve of Figure 7A suggests that ca. 95% of the iron in the sample prior to mixing with O2 is high-spin ferrous. The remaining 5% of the iron is probably originates from unreduced diferric species that remains constant in all samples. The high energy lines of this species would appear in the central part of the spectrum (see green arrows in Figure 7). In the following, all Fe percentages quoted refer to percentage of total Fe in a sample.
Figure 7B shows a Mössbauer spectrum of a sample freeze-quenched at 320 ms (P* sample). It can be seen that the spectra of Figures 7A and B are exceedingly alike. Fitting the rightmost feature as described above shows that ~ 86% of the iron of the P* sample is diferrous. Yet, nearly all of the g = 16 EPR feature belonging to the active fraction of MMOH, Hred and O, has disappeared at this time (Figure 2). If the disappearing fraction of the signal were to indicate oxidation of the cluster, we would expect to observe at most 55% of the diferrous cluster (the remaining Hslred) in the Mössbauer spectrum, suggesting that P* is diferrous (we will strengthen this argument shortly). The ~8 % Fe that disappeared from the ferrous pool accumulates in the spectral region where diferric intermediates P, Hox and Hslox absorb (Figure 7B arrows). We note that a P* sample of a second enzyme preparation produced a spectrum identical to that of Figure 7B.
Figure 7C shows a Mössbauer spectrum quenched 3 s after adding O2. The most conspicuous change between the spectra of the 320 ms and 3 s samples is a pronounced decline of the diferrous species. This change is readily appreciated by plotting (Figure 7E) the spectra of Figures 7B (hashed) and 7C (cyan) such that their spectral area represents the same amount of total iron. Analysis of the spectrum in Figure 7C shows that the ferrous pool now contains 58% of the total Fe. Hence between 320 ms and 3 s about 28 % of the iron in the sample is converted from the ferrous state into a higher oxidation state. By removing the remaining 58 % of the ferrous Fe from the spectrum of Figure 7C (this fraction is due to residual Hslred plus presumably some P*), we obtained the spectrum of Figure 7D, which shows the spectra of the species that result from the iron that vanished from the ferrous pool. The majority component in Figure 7D, representing 23 % of total Fe (57 % of the active iron), is a species (orange line) with ΔEQ = 0.53 mm/s and δ = 0.18 mm/s. Within the uncertainties (±0.02 mm/s for ΔEQ and ±0.01 mm/s for δ) these values agree with those previously reported for the FeIVFeIV intermediate Q.20,21 An additional ~7 % of the iron can be attributed to a doublet with parameters (ΔEQ = ~1.55 mm/s, δ = ~0.69 mm/s) indicative of the diferric peroxo intermediate P; its contribution is outlined by the magenta line in Figure 7D. Approximately 6 % of the total iron (over and above the original unreduced fraction) is diferric at 3 s (green curve).
As summarized in Table 1, the distribution of species revealed by the Mössbauer spectra correlates very well with the kinetics-derived speciation plot of Figure 4, bottom. At 3 s, the kinetics predict that the sample will be composed of 50 % Hslred, 10% P*, 4 % P, 22 % Q, 2 % Hox, 6 % Hslox and 5 % unidentified species that may include Hox that was never reduced. The observed fractions are 58 % diferrous (Hslred plus presumably P*), 7 % P, 23 % Q, 7 % diferric (Hox plus Hslox), and 5 % other unidentified non-diferrous species. The excellent agreement at this time point, particularly the correct prediction of the amount of Q observed, suggests that the speciation plot is accurate and can be used to predict the distribution of species at 320 ms. The 28 % loss of diferrous material observed between the 320 ms to 3 s time points is accounted for as a 6 % loss of Hslred and a 22 % loss of P*. P* is partially converted into P and then Q, while the small fraction of P present at 320 ms also flows into Q. Since the speciation plot and the Mössbauer spectra show that approximately the same amount of P is present at both time points, the net loss in diferrous P* (22%, Figure 7C) should approximately equal the net gain in Q (23%), as observed. The speciation plot predicts that P* declined by 67 % between 320 ms and 3 s, suggesting that 35% of the total iron was in the P* state when it had its maximum concentration near 0.3 s after reaction with O2. This value is also consistent with kinetic predictions and the observed Mössbauer total of 86 % diferrous cluster at 320 ms (predicted 55% Hslred plus 33% P*). Together these findings strongly support the assignment of P* as a diferrous species.
Table 1.
Predicted and observed distribution of intermediates in RFQ experiments
| t = 0 s | t = 320 ms | t = 3 s | |||
|---|---|---|---|---|---|
|
| |||||
| Prediction (At start) | Observation (Mössbauer) | Prediction (Kinetics) | Observation (Mössbauer) | Prediction (Kinetics) | Observation (Mössbauer) |
| 100 % Hred | 95 % diferrous | 55 % Hslred | 86 % diferrous | 50 % Hslred | 58 % diferrous |
| 33 % P* | 10 % P* | ||||
|
| |||||
| 3.4 % P | 3 % P | 4 % P | 7 % P | ||
| 3.6 % (Q + Hox) | 5 % Hox | 22 % Q | 23 % Q | ||
| 2 % Hox | 7 % Hox+ Hslox | ||||
| 6 % Hslox | |||||
|
| |||||
| 5 % unidentifieda | 5 % unidentitiedb | 5 % unidentifieda | 5 % unidentifiedb | 5 % unidentifieda | |
In all samples, approximately 5 % of the Mössbauer absorption could not be positively identified as it competes with the noise. This absorption is probably associated with diferric species (not a single one). It is certainly not originating from ferrous MMOH and may derive in part from unreduced enzyme.
Assumed from the Mössbauer spectrum of the t = 0 sample.
The line shape of the ferrous pool has small variations throughout the series of experiments, but these variations are so minor that we were not able to extract a unique spectrum for P* by examining difference spectra. Obviously, the Mössbauer spectrum of P* is very similar to that of Hred. As pointed out above, we explored the possibility that the distribution of the ΔEQ values at site 2 arises from the superposition of two or more distinct species (more than one form of Hred or Hred plus O or P*). However, the Fourier transform treated spectra shown in SI Figure S1 provide no evidence that site 2 contributes two different doublets which would indicate, across the molecular population, two ligand geometries for site 2.
As described above, the use of H33A MMOB shifts the kinetics such that P* can be trapped effectively in a 0.3–3 s time window. As a control, a sample was prepared at 0.3 s using WT MMOB. A 4.2 K, zero field Mössbauer spectrum is shown in Figure 8B. The spectrum of the H33A MMOB sample of Figure 7B is shown in Figure 8A for comparison. In spectrum 8B, ca. 15% of the iron belongs to intermediate P (magenta line) and 20 % is associated with intermediate Q (orange line), which is in excellent agreement with the expected amounts of these species based on the rate constants for intermediate conversion reported previously.15,19 We note that the amount of iron in P and Q represents essentially all iron belonging to the reactive MMOH fraction, and thus the formation of these species is significantly accelerated when using WT MMOB. The Mössbauer parameters of P are not readily obtained because the low-energy line of the doublet is invariably masked by the contribution of other species. Currently, our best estimate, obtained from analysis of the spectrum of Figure 8B is ΔEQ = 1.53 ± 0.06 mm/s and δ = 0.66 ± 0.03 mm/s. These values using WT MMOB agree well with ΔEQ =1.51 mm/s and δ = 0.66 mm/s reported for P (termed Hperoxo) for the M. c. Bath enzyme.18
Figure 8.

4.2 K Mössbauer spectra of rapid freeze quenched samples of the Hred single turnover reaction at 320 ms. The reaction for spectrum A uses H33A MMOB while the reaction for spectrum B uses WT MMOB. Here spectrum A is the same as that shown in Figure 7B. Spectrum B exhibits considerable accumulation of intermediates P (15% of Fe, magenta) and Q (20%, orange).
DISCUSSION
The combined use of H33A MMOB to provide a constriction in the flow of the reaction cycle, inclusion of substrate to eliminate the strong background chromophore from compound Q, and the use of sensitive spectra kinetic data collection has allowed the observation of the transient intermediate P* in the single turnover reaction of MMOH. Our early transient kinetic studies strongly suggested that a reaction cycle intermediate occurs after O and before P, and that it should accumulate to observable levels (maximum ~45 %).17,19,28 Nevertheless, direct detection has proven difficult. The current results show that this was the case because intermediates Hred, O, and P* have very similar optical spectra and also because the kinetics of intermediate interconversion dictate that a high background from the more strongly absorbing P and Q are always present when wild type MMOB is utilized in the reaction. Based on the observation that the decay of O results in the loss of the g = 16 EPR signal characteristic of the diferrous cluster, we originally proposed that P* would contain one or two FeIII ions in some sort of oxygen-bound cluster 17,19 The current study shows that this is not the case, suggesting that the critical step in the preparation of the cluster to bind and activate O2 occurs by a novel mechanism. This aspect of the MMOH reaction cycle is discussed here.
The Loss of the g = 16 EPR Signal in the Transformation from O to P* is Caused by a Structural Change of the Diferrous Cluster, not the Result of Oxidation
The view that any form of MMOH containing a diferrous cluster must exhibit a g = 16 EPR is too restrictive as is apparent from a consideration of its origin. As shown previously, the two high-spin ferrous sites of Hred are weakly coupled by ferromagnetic exchange, with J-values around −0.75 cm−1 in the
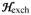 = J
S1•S2 convention (S1 = S2 = 2). Evaluated in the weak coupling scheme (see Figure 4 of ref 34) the geff = 16 resonance results from a particular combination of J and the zero-field splitting parameters D1, E1/D1, D2 and E2/D2 of the two sites. Spin concentration and line shapes of the geff = 16 signal are well described by choosing J = −0.75 cm−1, D1 = D2 = −5 ± 1 cm−1, E1/D1 = E2/D2 = 0.27 together with σE/D= 0.05 (σE/D describes a Gaussian distribution of E/D values, assumed to be the same for both sites). The assumption of equal D and E/D values for both sites is convenient but not crucial (see ref34) The g = 16 feature at X-band results from Δmi = 0 transitions between the members of a (quasi) doublet of m1,
m2 = ± 2 heritage which is split by Δ ≈ 9(E1/D1)4
D12/8J ≈ 0.2 cm−1. 34 The expression for Δ reveals that a minor structural change at the active site could change one of the relevant parameters such that Δ exceedes ≈ 0.3 cm−1, the energy of the microwave quantum at X-band, essentially abolishing the resonance. Given that J is already very small, a decline of J, for instance, by a mere 0.3 cm−1 would be sufficient to explain the disappearance of the EPR signal for P*. In principle, these changes could be probed at Q-band, but the interpretation of such experiments might be difficult as the signals from Hred and P* may overlap. Of course, in the transition from O to P*, the exchange coupling constant J may change sign to render an antiferromagnetically coupled system, with the consequence that the ground quasi doublet is EPR silent.
= J
S1•S2 convention (S1 = S2 = 2). Evaluated in the weak coupling scheme (see Figure 4 of ref 34) the geff = 16 resonance results from a particular combination of J and the zero-field splitting parameters D1, E1/D1, D2 and E2/D2 of the two sites. Spin concentration and line shapes of the geff = 16 signal are well described by choosing J = −0.75 cm−1, D1 = D2 = −5 ± 1 cm−1, E1/D1 = E2/D2 = 0.27 together with σE/D= 0.05 (σE/D describes a Gaussian distribution of E/D values, assumed to be the same for both sites). The assumption of equal D and E/D values for both sites is convenient but not crucial (see ref34) The g = 16 feature at X-band results from Δmi = 0 transitions between the members of a (quasi) doublet of m1,
m2 = ± 2 heritage which is split by Δ ≈ 9(E1/D1)4
D12/8J ≈ 0.2 cm−1. 34 The expression for Δ reveals that a minor structural change at the active site could change one of the relevant parameters such that Δ exceedes ≈ 0.3 cm−1, the energy of the microwave quantum at X-band, essentially abolishing the resonance. Given that J is already very small, a decline of J, for instance, by a mere 0.3 cm−1 would be sufficient to explain the disappearance of the EPR signal for P*. In principle, these changes could be probed at Q-band, but the interpretation of such experiments might be difficult as the signals from Hred and P* may overlap. Of course, in the transition from O to P*, the exchange coupling constant J may change sign to render an antiferromagnetically coupled system, with the consequence that the ground quasi doublet is EPR silent.
The Diferrous Nature of P* is not Consistent with a Superoxo or Peroxo Cluster
The analysis of the transient kinetics of the single turnover reaction of Hred in the presence of H33A MMOB and either presence or absence of furan allows prediction of the relative concentrations of O, P*, P, and Q at any point during the time course (Figure 4). Mössbauer spectra of samples taken at diagnostic points during the time course show that the concentrations of the previously characterized intermediates P and Q are predicted very accurately. This strongly suggests that P* is also present at the predicted concentrations. This being the case, the Mössbauer spectrum of the 320 ms sample shows that the only species present in sufficient concentration to be P* must have a diferrous cluster with parameters similar to those of Hred and O. Thus, P* can be neither an FeIIFeIII-superoxo intermediate nor an FeIIIFeIII-peroxo intermediate distinct from P. Considering that the spectroscopic characterization of P indicates that it is an FeIIIFeIII-peroxo intermediate of some sort, 18,20 the current spectroscopic and kinetic data suggest that the diiron cluster in MMOH proceeds directly from a diferrous state in P* to a bridged peroxo-bound diferric state in P without an intervening FeIIFeIII intermediate detectable on the millisecond time scale. This experimental conclusion is supported by density functional theory studies, which indicate that the binding of oxygen to one of the two iron atoms to form a FeIIFeIII -superoxo intermediate is thermodynamically unfavorable. 46,47
The Mössbauer Spectrum of P* Suggests that it is Structurally Similar to Hred
Hred from M. t. and M. c. have a characteristic Mössbauer spectrum in which one iron site exhibits a sharp quadrupole doublet while the spectrum of the second site has a distributed ΔEQ resulting in a broad quadrupole doublet. The distribution of ΔEQ must entail some form of a structural heterogeneity at site 2. However, as demonstrated by Stoian et al.,48a broadly distributed hyperfine parameter may possibly reflect a “soft” coordinate that vastly amplifies a minute structural heterogeneity, rather than a molecular structure with pronounced disorder. The distributed ΔEQ of site 2, while not yet understood, is valuable for the current study in that it becomes a sensitive monitor of the structure of the diferrous MMOH diiron cluster.
It is noteworthy that the zero field Mössbauer spectra of Hred and P* are essentially identical. There are very minor changes, but these changes are too small to be analyzed, and they may reflect subtle changes between preparations or even freezing effects. In particular, both Hred and P* seem to have a similar, if not identical, distributed ΔEQ for site 2. This suggests that the changes that occur during formation of P* and attendent loss of the g = 16 EPR signal have only small effects on the cluster structure and ligation.
Potential Structures of P*
The data available are not sufficient to determine the precise nature of the change occuring as O is converted to P*. These changes, however, result in the loss of the g = 16 signal without a change in oxidation state or major change in the cluster structure. One possibility, illustrated in Scheme 2, step a, is that O2 binds nearby or directly to the cluster, displacing the solvent water molecule that has been shown to bridge the iron atoms in the crystal structure of diferrous MMOH from the nearly identical Hred from M. c. Bath. 49 VTVH MCD and CD studies indicate that the iron atoms of the diferrous cluster are both primarily 5-coordinate, indicating that the solvent seen in the crystal structure is likely to be weakly bound and displaceable.46,50 The preceding intermediate O requires O2 for its formation but its decay rate monitored by the loss of the g = 16 EPR signal shows no O2 concentration dependence, suggesting that O2 binds essentially irreversibly in the active site. The formation of P* may involve the next step in O2 activation as solvent is released from the cluster to allow O2 to bind. If the conversion of O to P* involves actual association of O2 with the cluster, then O2 must bind with little electron transfer from the cluster iron atoms. While there is no precedent for this in O2 reactions with diiron clusters, we have recently shown that a weak FeII-O2 complex can form in the active site of a mononuclear FeII dioxygenase when an active site mutation prevents efficient electron transfer to the oxygen. 51 Computational studies for both nonheme and heme systems have shown that O2 binding to FeII is comparatively weak, a useful property in systems where modulation of O2 affinity is important. 52–55 Transfer of electron density from the FeII to the O2 is often far from complete, and it is strongly dependent on factors such as the state of hydrogen bonding or charge interactions in the active site, as well as the nature of the ligand trans to the metal bound O2.56,57 In the current case, displacing solvent and/or replacing it with weakly bound O2 in the coordination sphere would be consistent with the minor increase in the intensity of the optical spectrum of P* compared with that of O as well as the minor changes observed in the Mössbauer spectrum.
Scheme 2.

Proposed structures of MMOH single turnover cycle intermediates. The introduction of O2 in place of water in the cluster coordination sphere of intermediate O is speculative. However, the water must be displace and O2 bound at some point during process of formation of intermediate P.
Steps in O2 Activation
Three facts are known about intermediates P and/or Q that must be reconciled in considering the models for P* that arise from the discussion here and from DFT studies. 29–31,58–61 First, Mössbauer spectra reported here and elsewhere show that the two iron atoms in intermediate P are in very similar electronic environments, and the same is true of the two iron atoms in Q.18,20,21 Second, pH studies reported here and in previous studies of both M. t. and M. c. MMOH single turnover kinetics show that the O to P* transition does not require a proton transfer, whereas both the P* to P and P to Q transitions are pH dependent.16,19 A proton inventory study suggests that each of the pH dependent steps involve a single proton hop, although one essential and one nonessential proton translocation are predicted based on fitting the pH dependency for the P to Q transition in M. c. MMOH.16,18,19 Third, the Mössbauer spectrum of P exhibits an isomer shift of about 0.66 mm/s which is similar to that observed for deprotonated peroxo ligands. 62–65 This suggests that a proton is transferred to some moiety other than the bridging peroxo group as P is formed. It is also relevant to note here that metal centers in enzymes tend to maintain a constant net local charge, and this appears to be a net charge of zero for the structurally characterized states of MMOH. 66–68 For this to be true in the case of P formation from P*, the proton transfer must occur between groups bound to the diiron cluster, and charge balance at each iron requires that the donor and acceptor groups be associated with the same iron atom.
The intermediates shown in Scheme 2 steps a–c satisfy all of the experimental observations and maintain a neutral overall net charge for the cluster. Glu114 is proposed to play the role of accepting a proton from Fe1-bound solvent and eventually donating it to the peroxo moiety to promote O-O bond cleavage. 19 This maintains the local charge on Fe1 and satisfies the required one hop proton transfer process. The protonation of the carboxylate residue and its hydrogen bonding interaction with the bridging peroxide moeity is supported by information obtained from synthetic diiron model compound mimics of P and DFT studies of oxygen activation in RNR.69,70 This pathway also requires the minimum structural reorganizations in the O to P* to P sequence, consistent with the Mössbauer results reported here.
Comparison with Intermediate P* from M. capsulatus Bath
An intermediate occuring before Hperoxo in the M. c. Bath MMOH single turnover cycle (M. c. P*) has been observed that differs from the P* reported here in its reported oxidation state. 16 An FeIIIFeIII oxidation state assignment in the case of M. c. P* was based on the observation of an electronic absorption spectrum similar to that of Hperoxo. This was obtained by a global fit of the single turnover absorption data at two diagnostic wavelengths for a reaction of M. c. Hred with O2 in the presence of wild type M. c. MMOB and methane. While the presence of a substrate in both studies quenches the large absorption background arising from Q, the use of H33A MMOB in our study provides the additional advantages of increasing the yield of P* and reducing the background absorption from P. In our case, the oxidation state of P* was directly determined using Mössbauer spectroscopy, making it clear that there is insufficient FeIIIFeIII species present to account for P* at the time it maximizes. The kinetics of intermediate conversion also differ significantly in the M. t. OB3b and M. c. Bath enzymes. The P* formation rate constants at 4 °C and pH 7.0 are 26 s−1 and 6.7 s−1, for the M. t. and M. c. enzymes, respectively, in the presence of their specific WT MMOBs. Similarly, the P formation rate constants are reported to be 10 s−1 and 0.75 s−1 using wild type MMOB for M. t. and M. c. enzymes, respectively. The similarity of the formation rate constants of M. c. P* and M. t. P raises the possibility that M. c. P* is another form of M. c. P, accounting for its similar optical spectrum. Thus far, we have not observed evidence for this species in the M. t. system.
Conclusion
In summary, this study has elucidated the electronic absorption and Mössbauer spectra of intermediate P* in the single turnover cycle of M. t. MMOH. We should note that we were pleasantly surprised to find that the spectra-kinetic and Mössbauer data correlate exceedingly well, implying that either approach is likely to yield further useful results for eludidating the catalytic cycle of MMOH. Our results show that P* is a diferrous intermediate that is EPR silent at X-band (studies at Q-band might recover the g = 16 signal). One possibility for the formation of P* requiring minimal overall structural reorganization assumes association of O2 with the diferrous cluster as the first step in a multiphase binding process that follows an effectively irreversible O2 binding in the hydrophobic enzyme active site during the formation of intermediate O. A comparatively slow displacement of solvent from the metal center, and possibly formation of a weak FeII-oxygen complex, would overcome the most difficult steps in metal-O2 complex formation and facilitate formation of the strong peroxo complex needed for O-O bond cleavage in the next step.
Supplementary Material
Acknowledgments
Funding
This work is supported by the NIH grants GM40466 and GM100943 (to J. D. L.) and grant NSF Grant CHE-1012485 (to E.M.)
ABBREVIATIONS
- MMO
methane monooxygenase
- sMMO
soluble form of methane monooxygenase
- MMOH
sMMO hydroxylase component
- MMOB
sMMO component B
- MMOR
sMMO reductase
- WT-MMOB
wild-type MMOB
- MOPS
3-[N-Morpholino]propane-sulfonic acid
- Hox
Oxidized MMOH
- Hred
Reduced MMOH
- O
P*, P, Q, Compounds O, P*, P and Q from the MMOH catalytic cycle
- Hslred
unreactive form of Hred
- Hperoxo
peroxo intermediate from the Methylococcus capsulatus sMMO system equivalent to P
- RFQ
rapid freeze-quench
- FAD
flavin adenine dinucleotide
- ZFS
zero-field splitting
- CD
circular dichroism
- MCD
magnetic circular dichroism
- VTVH
variable-temperature variable-field
- DFT
density functional theory
- T4mo
toluene-4-monooxygenase
- RNR
ribonucleotide reductase
- EPR
electron paramagnetic resonance
- J
exchange-coupling constant
Footnotes
Supporting Information. A table describing the experimental basis for the intermediates occurring in the single turnover cycle of M. trichosporium OB3b MMOH and additional results of the Fourier transform Mössbauer data analysis are presented. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiol Rev. 1996;60:439–447. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruscic B, Litorja M, Asher RL. Ionization energy of methylene revisited: Improved values for the enthalpy of formation of CH2 and the bond dissociation energy of CH3 via simultaneous solution of the local thermochemical network. J Phys Chem A. 1999;103:8625–8633. [Google Scholar]
- 3.Hakemian AS, Rosenzweig AC. The biochemistry of methane oxidation. Annu Rev Biochem. 2007;76:223–241. doi: 10.1146/annurev.biochem.76.061505.175355. [DOI] [PubMed] [Google Scholar]
- 4.Hyman MR, Murton IB, Arp DJ. Interaction of ammonia monooxygenase from Nitrosomonas europaea with alkanes, alkenes, and alkynes. Appl Environ Microbiol. 1988;54:3187–3190. doi: 10.1128/aem.54.12.3187-3190.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox BG, Froland WA, Dege JE, Lipscomb JD. Methane monooxygenase from Methylosinus trichosporium OB3b. Purification and properties of a three-component system with high specific activity from a type II methanotroph. J Biol Chem. 1989;264:10023–10033. [PubMed] [Google Scholar]
- 6.Wallar BJ, Lipscomb JD. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 7.Baik MH, Newcomb M, Friesner RA, Lippard SJ. Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem Rev. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- 8.Tinberg CE, Lippard SJ. Dioxygen activation in soluble methane monooxygenase. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipscomb JD. Catalysis and regulation in the soluble methane monooxygenase system: Applications of isotopes and isotope effects, in. In: Kohen A, Limbach H-H, editors. Isotope Effects in Chemistry and Biology. CRC Press; Boca Raton, FL: 2006. pp. 931–953. [Google Scholar]
- 10.Que L, Jr, Tolman WB. Bis(m-oxo)dimetal “diamond” cores in copper and iron complexes relevant to biocatalysis. Angew Chem Int Edit. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Farquhar ER, Stubna A, Münck E, Que L., Jr A diiron(IV) complex that cleaves strong C-H and O-H bonds. Nature Chem. 2009;1:145–150. doi: 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Do LH, Lippard SJ. Evolution of strategies to prepare synthetic mimics of carboxylate-bridged diiron protein active sites. J Inorg Biochem. 2011;105:1774–1785. doi: 10.1016/j.jinorgbio.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SK, Nesheim JC, Lipscomb JD. Transient intermediates of the methane monooxygenase catalytic cycle. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 15.Brazeau BJ, Lipscomb JD. Kinetics and activation thermodynamics of methane monooxygenase compound Q formation and reaction with substrates. Biochemistry. 2000;39:13503–13515. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- 16.Tinberg CE, Lippard SJ. Revisiting the mechanism of dioxygen activation in soluble methane monooxygenase from M. capsulatus (Bath): Evidence for a multistep, proton-dependent reaction pathway. Biochemistry. 2009;48:12145–12158. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Nesheim JC, Lee SK, Lipscomb JD. Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J Biol Chem. 1995;270:24662–24665. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- 18.Liu KE, Valentine AM, Wang DL, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. Kinetic and spectroscopic characterization of intermediates and component interactions in reactions of methane monooxygenase from Methylococcus capsulatus (Bath) J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 19.Lee SK, Lipscomb JD. Oxygen activation catalyzed by methane monooxygenase hydroxylase component: proton delivery during the O-O bond cleavage steps. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 20.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr An Fe(IV)2O2 diamond core structure for the key intermediate Q of methane monooxygenase. Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 21.Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. A transient intermediate of the methane monooxygenase catalytic cycle containing an Fe(IV)Fe(IV) cluster. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 22.Nesheim JC, Lipscomb JD. Large isotope effects in methane oxidation catalyzed by methane monooxygenase: evidence for C-H bond cleavage in a reaction cycle intermediate. Biochemistry. 1996;35:10240–10247. doi: 10.1021/bi960596w. [DOI] [PubMed] [Google Scholar]
- 23.Brazeau BJ, Austin RN, Tarr C, Groves JT, Lipscomb JD. Intermediate Q from soluble methane monooxygenase hydroxylates the mechanistic substrate probe norcarane: Evidence for a stepwise reaction. J Am Chem Soc. 2001;123:11831–11837. doi: 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- 24.Zheng H, Lipscomb JD. Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling. Biochemistry. 2006;45:1685–1692. doi: 10.1021/bi051605g. [DOI] [PubMed] [Google Scholar]
- 25.Valentine AM, Stahl SS, Lippard SJ. Mechanistic studies of the reaction of reduced methane monooxygenase hydroxylase with dioxygen and substrates. J Am Chem Soc. 1999;121:3876–3887. [Google Scholar]
- 26.Priestley ND, Floss HG, Froland WA, Lipscomb JD, Williams PG, Morimoto H. Cryptic stereospecificity of methane monooxygenase. J Am Chem Soc. 1992;114:7561–7562. [Google Scholar]
- 27.Brazeau BJ, Wallar BJ, Lipscomb JD. Unmasking of deuterium kinetic isotope effects on the methane monooxygenase compound Q reaction by site-directed mutagenesis of component B. J Am Chem Soc. 2001;123:10421–10422. doi: 10.1021/ja016632i. [DOI] [PubMed] [Google Scholar]
- 28.Wallar BJ, Lipscomb JD. Methane monooxygenase component B mutants alter the kinetics of steps throughout the catalytic cycle. Biochemistry. 2001;40:2220–2233. doi: 10.1021/bi002298b. [DOI] [PubMed] [Google Scholar]
- 29.Basch H, Musaev DG, Mogi K, Morokima K. Theoretical studies on the mechanism of the methane -> methanol conversion reaction catalyzed by methane monooxygenase (MMO): The O-side vs N-side mechanisms. J Chem Phys A. 2001;105:3615–3622. [Google Scholar]
- 30.Siegbahn PEM, Crabtree RH, Nordlund P. Mechanism of methane monooxygenase - a structural and quantum chemical perspective. J Biol Inorg Chem. 1998;3:314–317. [Google Scholar]
- 31.Gherman BF, Baik MH, Lippard SJ, Friesner RA. Dioxygen activation in methane monooxygenase: a theoretical study. J Am Chem Soc. 2004;126:2978–2990. doi: 10.1021/ja036506+. [DOI] [PubMed] [Google Scholar]
- 32.Brazeau BJ, Lipscomb JD. Key amino acid residues in the regulation of soluble methane monooxygenase catalysis by component B. Biochemistry. 2003;42:5618–5631. doi: 10.1021/bi027429i. [DOI] [PubMed] [Google Scholar]
- 33.Froland WA, Andersson KK, Lee SK, Liu Y, Lipscomb JD. Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions. A novel role for protein-protein interactions in an oxygenase mechanism. J Biol Chem. 1992;267:17588–17597. [PubMed] [Google Scholar]
- 34.Hendrich MP, Münck E, Fox BG, Lipscomb JD. Integer-spin EPR studies of the fully reduced methane monooxygenase hydroxylase component. J Am Chem Soc. 1990;112:5861–5865. [Google Scholar]
- 35.Mitić N, Schwartz JK, Brazeau BJ, Lipscomb JD, Solomon EI. CD and MCD studies of the effects of component B variant binding on the biferrous active site of methane monooxygenase. Biochemistry. 2008;47:8386–8397. doi: 10.1021/bi800818w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang SL, Wallar BJ, Lipscomb JD, Mayo KH. Residues in Methylosinus trichosporium OB3b methane monooxygenase component B involved in molecular interactions with reduced- and oxidized-hydroxylase component: a role for the N-terminus. Biochemistry. 2001;40:9539–9551. doi: 10.1021/bi0103462. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Wallar BJ, Popescu CV, Renner DB, Thomas DD, Lipscomb JD. Methane monooxygenase hydroxylase and B component interactions. Biochemistry. 2006;45:2913–2926. doi: 10.1021/bi052256t. [DOI] [PubMed] [Google Scholar]
- 38.Fox BG, Froland WA, Jollie DR, Lipscomb JD. Methane monooxygenase from Methylosinus trichosporium OB3b. Methods Enzymol. 1990;188:191–202. doi: 10.1016/0076-6879(90)88033-7. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Lipscomb JD. Role of the C-terminal region of the B component of Methylosinus trichosporium OB3b methane monooxygenase in the regulation of oxygen activation. Biochemistry. 2006;45:1459–1469. doi: 10.1021/bi051721j. [DOI] [PubMed] [Google Scholar]
- 40.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Trapping and spectroscopic characterization of an FeIII-superoxo intermediate from a nonheme mononuclear iron-containing enzyme. Proc Natl Acad Sci U S A. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Münck E. Aspects of 57Fe Mössbauer spectroscopy. In: Que L Jr, editor. Physical Methods in Bioinorganic Chemistry. University Science Books; Sausalito, CA: 2000. pp. 287–319. [Google Scholar]
- 42.Fox BG, Surerus KK, Münck E, Lipscomb JD. Evidence for a μ-oxo-bridged binuclear iron cluster in the hydroxylase component of methane monooxygenase. Mössbauer and EPR studies. J Biol Chem. 1988;263:10553–10556. [PubMed] [Google Scholar]
- 43.Fox BG, Hendrich MP, Surerus KK, Andersson KK, Froland WA, Lipscomb JD, Münck E. Mössbauer, EPR, and ENDOR studies of the hydroxylase and reductase components of methane monooxygenase from Methylosinus trichosporium OB3b. J Am Chem Soc. 1993;115:3688–3701. [Google Scholar]
- 44.Dibar-Ure MC, Flinn PA. A technique for the removal of the “blackness” distortion of Mössbauer spectra. In: Gruverman IG, editor. Mössbauer Effect Methodology. Plenum Press; New York, N.Y: 1971. pp. 245–262. [Google Scholar]
- 45.Dunham WR, Harding LJ, Sands RH. Mössbauer spectroscopy of metalloproteins and the use of Fourier transforms. Eur J Biochem. 1993;214:1–8. doi: 10.1111/j.1432-1033.1993.tb17889.x. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz JK, Wei P-p, Mitchell KH, Fox BG, Solomon EI. Geometric and electronic structure studies of the binuclear nonheme ferrous active site of toluene-4-monooxygenase: Parallels with methane monooxygenase and insight into the role of the effector proteins in O2 activation. J Am Chem Soc. 2008;130:7098–7109. doi: 10.1021/ja800654d. [DOI] [PubMed] [Google Scholar]
- 47.Wei PP, Skulan AJ, Wade H, DeGrado WF, Solomon EI. Spectroscopic and computational studies of the de novo designed protein DF2t: correlation to the biferrous active site of ribonucleotide reductase and factors that affect O2 reactivity. J Am Chem Soc. 2005;127:16098–16106. doi: 10.1021/ja053661a. [DOI] [PubMed] [Google Scholar]
- 48.Stoian SA, Smith JM, Holland PL, Münck E, Bominaar EL. Mössbauer, electron paramagnetic resonance, and theoretical study of a high-spin, four-coordinate Fe(II) diketiminate complex. Inorg Chem. 2008;47:8687–8695. doi: 10.1021/ic8004568. [DOI] [PubMed] [Google Scholar]
- 49.Whittington DA, Lippard SJ. Crystal structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) demonstrating geometrical variability at the dinuclear iron active site. J Am Chem Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 50.Pulver S, Froland WA, Fox BG, Lipscomb JD, Solomon EI. Spectroscopic studies of the coupled binuclear non-heme iron active site in the fully reduced hydroxylase component of methane monooxygenase: Comparison to deoxy and deoxy-azide hemerythrin. J Am Chem Soc. 1993;115:12409–12422. [Google Scholar]
- 51.Mbughuni MM, Meier KK, Münck E, Lipscomb JD. Substrate-mediated oxygen activation by homoprotocatechuate 2,3-dioxygenase: intermediates formed by a tyrosine 257 variant. Biochemistry. 2012;51:8743–8754. doi: 10.1021/bi301114x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marti MA, Crespo A, Capece L, Boechi L, Bikiel DE, Scherlis DA, Estrin DA. Dioxygen affinity in heme proteins investigated by computer simulation. J Inorg Biochem. 2006;100:761–770. doi: 10.1016/j.jinorgbio.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Capece L, Marti MA, Crespo A, Doctorovich F, Estrin DA. Heme protein oxygen affinity regulation exerted by proximal effects. J Am Chem Soc. 2006;128:12455–12461. doi: 10.1021/ja0620033. [DOI] [PubMed] [Google Scholar]
- 54.Shibata T, Nagao S, Fukaya M, Tai H, Nagatomo S, Morihashi K, Matsuo T, Hirota S, Suzuki A, Imai K, Yamamoto Y. Effect of heme modification on oxygen affinity of myoglobin and equilibrium of the acid-alkaline transition in metmyoglobin. J Am Chem Soc. 2010;132:6091–6098. doi: 10.1021/ja909891q. [DOI] [PubMed] [Google Scholar]
- 55.Olea C, Boon EM, Pellicena P, Kuriyan J, Marletta MA. Probing the function of heme distortion in the H-NOX family. ACS Chemical Biology. 2008;3:703–710. doi: 10.1021/cb800185h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen H, Ikeda-Saito M, Shaik S. Nature of the Fe-O2 bonding in oxy-myoglobin: effect of the protein. J Am Chem Soc. 2008;130:14778–14790. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- 57.Jensen KP, Ryde U. How O2 binds to heme: reasons for rapid binding and spin inversion. J Biol Chem. 2004;279:14561–14569. doi: 10.1074/jbc.M314007200. [DOI] [PubMed] [Google Scholar]
- 58.Siegbahn PEM. O-O bond cleavage and alkane hydroxylation in methane monooxygenase. J Biol Inorg Chem. 2001;6:27–45. doi: 10.1007/s007750000184. [DOI] [PubMed] [Google Scholar]
- 59.Torrent M, Musaev Djamaladdin G, Basch H, Morokuma K. Computational studies of reaction mechanisms of methane monooxygenase and ribonucleotide reductase. J Comput Chem. 2002;23:59–76. doi: 10.1002/jcc.1157. [DOI] [PubMed] [Google Scholar]
- 60.Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. Intermediates in dioxygen activation by methane monooxygenase: A QM/MM study. J Am Chem Soc. 2007;129:3135–3147. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han WG, Noodleman L. Structural model studies for the high-valent intermediate Q of methane monooxygenase from broken-symmetry density functional calculations. Inorg Chim Acta. 2008;361:973–986. doi: 10.1016/j.ica.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neibergall MB, Stubna A, Mekmouche Y, Münck E, Lipscomb JD. Hydrogen peroxide dependent cis-dihydroxylation of benzoate by fully oxidized benzoate 1,2-dioxygenase. Biochemistry. 2007;46:8004–8016. doi: 10.1021/bi700120j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li FF, Meier KK, Cranswick MA, Chakrabarti M, Van Heuvelen KM, Münck E, Que L. Characterization of a high-spin non-heme FeIII-O intermediate and its quantitative conversion to an FeIV = O complex. J Am Chem Soc. 2011;133:7256–7259. doi: 10.1021/ja111742z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim K, Lippard SJ. Structure and Mössbauer spectrum of a (μ-1,2-peroxo)bis(μ-carboxylato)diiron(III) model for the peroxo intermediate in the methane monooxygenase hydroxylase reaction cycle. J Am Chem Soc. 1996;118:4914–4915. [Google Scholar]
- 65.Roelfes G, Vrajmasu V, Chen K, Ho RYN, Rohde JU, Zondervan C, la Crois RM, Schudde EP, Lutz M, Spek AL, Hage R, Feringa BL, Münck E, Que L., Jr End-on and side-on peroxo derivatives of non-heme iron complexes with pentadentate ligands: models for putative intermediates in biological iron/dioxygen chemistry. Inorg Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p. [DOI] [PubMed] [Google Scholar]
- 66.Orville AM, Elango N, Lipscomb JD, Ohlendorf DH. Structures of competitive inhibitor complexes of protocatechuate 3,4-dioxygenase: Multiple exogenous ligand binding orientations within the active site. Biochemistry. 1997;36:10039–10051. doi: 10.1021/bi970468n. [DOI] [PubMed] [Google Scholar]
- 67.Orville AM, Lipscomb JD, Ohlendorf DH. Crystal structures of substrate and substrate analog complexes of protocatechuate 3,4-dioxygenase: Endogenous Fe3+ ligand displacement in response to substrate binding. Biochemistry. 1997;36:10052–10066. doi: 10.1021/bi970469f. [DOI] [PubMed] [Google Scholar]
- 68.Dong Y, Yan S, Young J, VG, Que L., Jr The crystal structure of a synthetic nonheme diiron-O2 adduct: insight into oxygen activation. Angew Chem Int Edit. 1996;35:618–620. [Google Scholar]
- 69.Do LH, Hayashi T, Moenne-Loccoz P, Lippard SJ. Carboxylate as the protonation site in (peroxo)diiron(III) model complexes of soluble methane monooxygenase and related diiron proteins. J Am Chem Soc. 2010;132:1273–1275. doi: 10.1021/ja909718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jensen KP, Bell CB, III, Clay MD, Solomon EI. Peroxo-type intermediates in class I ribonucleotide reductase and related binuclear non-heme iron enzymes. J Am Chem Soc. 2009;131:12155–12171. doi: 10.1021/ja809983g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.