

Abstract
The proteasome has emerged as an important clinically relevant target for the treatment of hematologic malignancies. Since the Food and Drug Administration approved the first-in-class proteasome inhibitor bortezomib (Velcade®) for the treatment of relapsed/refractory multiple myeloma (MM) and mantle cell lymphoma, it has become clear that new inhibitors are needed that have a better therapeutic ratio, can overcome inherent and acquired bortezomib resistance and exhibit broader anti-cancer activities. Marizomib (NPI-0052; salinosporamide A) is a structurally and pharmacologically unique β-lactone-γ-lactam proteasome inhibitor that may fulfill these unmet needs. The potent and sustained inhibition of all three proteolytic activities of the proteasome by marizomib has inspired extensive preclinical evaluation in a variety of hematologic and solid tumor models, where it is efficacious as a single agent and in combination with biologics, che-motherapeutics and targeted therapeutic agents. Specifically, marizomib has been evaluated in models for multiple myeloma, mantle cell lymphoma, Waldenstrom’s macroglobulinemia, chronic and acute lymphocytic leukemia, as well as glioma, colorectal and pancreatic cancer models, and has exhibited synergistic activities in tumor models in combination with bortezomib, the immunomodulatory agent lenalidomide (Revlimid®), and various histone deacetylase inhibitors. These and other studies provided the framework for ongoing clinical trials in patients with MM, lymphomas, leukemias and solid tumors, including those who have failed bortezomib treatment, as well as in patients with diagnoses where other proteasome inhibitors have not demonstrated significant efficacy. This review captures the remarkable translational studies and contributions from many collaborators that have advanced marizomib from seabed to bench to bedside.
Keywords: Proteasome inhibitor, marizomib, bortezomib, NF-κB, multiple myeloma, pharmacodynamics, combination therapy
INTRODUCTION
The ubiquitin-proteasome system (UPS) is now a well established target in cancer therapy [1, 2]. By serving as the major pathway for regulating intracellular protein degradation in eukaryotic cells, the UPS plays a critical role in maintaining cellular homeostasis, the imbalance of which may trigger pathologies associated with cancer and inflammation. The enzymatic core engine of the UPS is the 26S proteasome, which recognizes polyubiquitin-tagged proteins for degradation and hydrolyzes them into short peptides (Fig. (1)). Degradation of abnormal or misfolded proteins by the 26S proteasome provides the cell with a mechanism for protein quality control, while blocking its function results in accumulation of unwanted proteins and cell death. This is particularly relevant to cancer cells, which proliferate at a greater rate than normal cells and therefore exhibit an increased rate of protein synthesis and degradation. Importantly, proteasome substrates include not only misfolded and aged proteins, but also those that regulate signaling pathways critical for cell growth, cell cycle progression and apoptosis. Thus, downstream effects of proteasome inhibition include the stabilization of proapoptotic proteins, including p53 and Bax, and the reduction of some antiapoptotic proteins, such as Bcl-2, collectively inducing a proapoptotic state [1, 3]. The critical observation that proteasome inhibitors attenuate growth and survival signaling by inhibiting the activation of nuclear factor-kappa B (NF-κB) helped to establish the initial rationale for targeting the 26S for the treatment of cancer [4–8]. These findings culminated in the development of the first-in-class proteasome inhibitor bortezomib (Velcade®; PS-341), which received Food and Drug Administration (FDA) approvals for the treatment of relapsed, relapsed/refractory, and newly diagnosed multiple myeloma (MM), as well as mantle cell lymphoma (MCL), based on significant objective clinical responses [9–15]. However, inherent and acquired resistance, together with side effects that include peripheral neuropathy, neutropenia and throm-bocytopenia [16, 17], have led to the search for unique proteasome inhibitors with the potential to treat patients who had failed, did not respond to, or were not candidates for treatment with bortezomib [2, 18]. One such agent is marizomib (NPI-0052; salinosporamide A), a novel marine-derived β-lactone-γ-lactam natural product that is active in multiple nonclinical tumor models and is currently in clinical trials for the treatment of patients with hematologic and solid tumor malignancies [19–21].
Fig. 1.
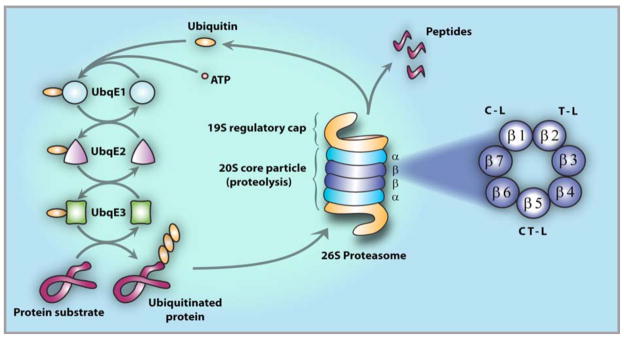
The Ubiquitin Protein System (UPS). Protein degradation by the UPS involves two distinct and successive steps: (i) polyubiquitination, i.e., covalent tagging of the target protein with multiple ubiquitin molecules; and (ii) degradation of the tagged protein by the 26S proteasome [3]. Conjugation of ubiquitin to the target protein substrate proceeds via a three-step mechanism, commencing with activation by ubiquitin-activating enzyme, E1, followed by transfer of ubiquitin (via one of several E2 enzymes) from E1 to a member of the ubiquitin-protein ligase family, E3, to which the substrate protein is specifically bound. In successive reactions, a polyubiquitin chain is synthesized by transfer of additional ubiquitin moieties to Lys48 of the previously conjugated molecule. The chain serves as a recognition marker for the 26S proteasome, which degrades the substrates to short peptides by the 20S proteasome and recycles ubiquitin via the action of isopeptidases. The 26S proteasome (center) comprises one or two 19S regulatory caps flanking the proteolytic 20S core particle [22, 23]. The 20S is a cylindrical structure formed by the stacking of two α-rings external to two β-rings, each of which contain seven β subunits, including catalytic subunits β1, β2 and β5 (right, expanded view).
This review offers the first comprehensive account of the preclinical and translational biology studies that provided the basis for the clinical evaluation of marizomib (Table 1). As a guide to the reader, the article commences with an introduction to the UPS pathway and the initial chemical and in vitro biological profiling of marizomib, followed by detailed pre-clinical findings in hematologic and solid tumor models, with descriptions of pharmacokinetics and pharmacodynamics, and concludes with results from Phase 1 clinical trials in patients with solid tumor and hematologic malignancies, as outlined below:
Table 1.
Preclinical Studies of Marizomib in Hematologic Malignancies and Solid Tumors
| Preclinical Investigation | Institute (Senior Investigators) | Selected References |
|---|---|---|
| Hematologic Malignancies | ||
| Multiple Myeloma: Single agent and combinations with Velcade, Revlimid, Thalomid, HDACis, Smac mimetics, thalomid. Drug resistant multiple myeloma cells. |
Dana Farber Cancer Institute (Anderson/Chauhan) | [49, 71] |
| Waldenstrom’s Macroglobulinemia and other B-cell malignancies (Burkitt’s lymphoma, and low grade IgM secreting lymphoma). | Dana Farber Cancer Institute (Anderson/Ghobrial) | [72] |
| B-Non-Hodgkin’s Lymphoma: Single agent and combination with Rituxan, CDDP in drug resistant cells. Mechanism of TRAIL resistance. Role of NF-κB, Snail, YY1, RKIP, DR5. |
University of California, Los Angeles (Bonavida) | [182, 188] |
| Leukemias (CLL, AML and ALL): Single agent and drug combinations (HDACis, VPA, entinostat, Zolinza. Rapidity of action versus Velcade. Regulation of NF-κB and related genes. |
MD Anderson Cancer Center (Chandra/McConkey) | [57, 104] |
| Mantle Cell Lymphoma: Single agent, combinations with Bcl-2 and HDAC inhibitors. Regulation of NF-κB and related genes. |
MD Anderson Cancer Center (Younes) MD Anderson Cancer Center (Aggarwal) |
[112] [65] |
| Solid tumors | ||
| Colon: Single agent and combination with CPT-11, Avastin, leucovorin, 5-FU and oxaliplatin. |
Massachusetts General Hospital (Cusack) | [120] |
| Pancreatic: Single agent and combination with Avastin, Tarceva, Gemzar, Iressa and Erbitux. Single agent and combination with TRAIL. |
Massachusetts General Hospital (Cusack) MD Anderson Cancer Center (McConkey) |
[131, 132] |
| Prostate: Single agent and combination with docetaxel and TRAIL. |
MD Anderson Cancer Center (McConkey) University of California, Los Angeles (Bonavida) |
[182] |
| Melanoma: Single agent and combination with HDACis. |
MD Anderson Cancer Center (McConkey) | Unpublished data |
| Glioma: Single agent and combination with radiation. Single agent |
University of California, Los Angeles (McBride/Pajonk) University of California, Irvine (Bota ) |
[167, 182] Unpublished data |
| Squamous Cell Carcinoma: Single agent. |
University of California, Los Angeles (Wang) | Unpublished data |
| Non-Small Cell Lung Carcinoma: Combination with HDACi (Zolinza). | University of South Carolina Medical School (Drabkin/Gemmil) | Unpublished data |
| Renal Carcinoma: Combination with HDACi (Zolinza). |
University of South Carolina Medical School (Drabkin/Gemmil) | Unpublished data |
-
Introduction
Ubiquitin-Proteasome Pathway and Proteasome Inhibitors
Marizomib (Origins, Mechanism of Binding to the Proteasome, Proteasome Inhibition Profile and Pharmacodynamics)
-
Marizomib in Hematologic Tumor Models (Preclinical Studies)
Multiple Myeloma
Waldenstrom’s Macroglobulinemia
Chronic Lymphocytic Leukemia
Acute Lymphocytic Leukemia
Mantle Cell and Hodgkin’s Lymphoma
-
Marizomib in Solid Tumor Models (Preclinical Studies)
Colorectal Carcinoma
Pancreatic Carcinoma
Glioma
Mechanism for reversal of tumor cell resistance to chemo- and immunotherapy
Pharmacokinetics and Absorption, Distribution, Metabolism and Excretion
Proteasome Activity as a Potential Biomarker
Clinical Trials with Marizomib
This account highlights the importance of collaborative research, which fostered the evolution of marizomib from a marine-derived natural product to a promising anticancer agent in clinical trials.
The Ubiquitin-Proteasome Pathway and Proteasome Inhibitors
The UPS pathway for protein degradation in eukaryotic cells comprises: 1) a series of enzymes [E1, activation; E2, conjugation; E3, ligation] that covalently modify proteins with a polyubiquitin tag for recognition and targeted degradation; and 2) the 26S proteasome, a 2.5 MDa multicatalytic enzyme complex that hydrolyzes the polyubiquitin-tagged proteins into short (4 – 25 residue) polypeptides, typically 7–9 amino acids in length [3] (Fig. (1)). Protein degradation by the UPS is a highly regulated process that is inherent to the molecular architecture of the 26S proteasome, which consists of one or two 19S regulatory caps flanking a 20S core particle (CP) in which substrate hydrolysis is executed [22, 23] (for reviews, see [3, 24, 25]). Upon recognition by the 19S regulatory caps, the polyubiquitin-tagged protein substrate is unfolded and translocated into the hydrolytic chamber of the 20S CP for degradation. In eukaryotes, the CP houses three pairs of catalytically active subunits, β1, β2 and β5, that exhibit protein substrate cleavage preferences referred to as caspase-like (C-L), trypsin-like (T-L) and chymotrypsin-like (CT-L), respectively, and which work in concert to degrade protein substrates. Substrate hydrolysis by the 20S CP commences with recognition of amino acid side chains (P1 – Pn) by sequential binding pockets (S1 - Sn) proximal to the proteolytic active site. It is the S1 “specificity pocket” adjacent to the active site that largely confers the CT-L, T-L, and C-L sites with their preferential binding to hydrophobic, positively-, and negatively-charged residues. Once bound, hydrolysis of the substrate peptide bond adjacent to S1 is catalyzed by the N-terminal threonine residue (Thr1), which employs the side chain Thr1Oγ and α-amino group as the nucleophile and general base, respectively, effectively classifying the proteolytic subunits among the N-terminal hydrolase family of enzymes.
The above mechanism for protein substrate hydrolysis provided strong rationale for the design of proteasome inhibitors comprising peptides that are derivatized with reactive functional groups at their C-termini to enable both the recognition of the peptide amino acid side chains by the proteasome S1 - Sn binding pockets and the formation of stable adducts with Thr1 [1]. It was later discovered that β-lactone-γ-lactam natural products bearing P1 moieties, which are recognized by S1, and reactive β-lactones, which covalently acylate Thr1Oγ, are highly selective proteasome inhibitors [26]. Peptidyl and β-lactone-γ-lactam proteasome inhibitors were instrumental in elucidating features of the proteasome active site that are critical to proteolysis [22, 23]. Both classes of inhibitor are currently under clinical evaluation (for structures, development status, proteasome inhibition profiles, routes of administration and treatment schedules, see Table 2 and supporting references). The unique identities of the P1 – Pn moieties and reactive functional head groups that stabilize the ligands at the proteolytic active site(s) (e.g., see Fig. (2)) impart each agent with a unique inhibition profile against the three proteolytic subunits of the proteasome, resulting in complementary and/or overlapping functional activities. These profile differences may provide opportunities to use these agents in combination and/or to overcome resistance, as described in this review in the context of marizomib.
Table 2.
Profiles and Treatment Regimens for Proteasome Inhibitors in Clinical Development
| Proteasome Inhibitor/Company | Structural Class | Structure | Pharmacodynamic Profile | Development Status | Route of Administration | Treatment Schedule | ||
|---|---|---|---|---|---|---|---|---|
| CT-L | T-L | C-L | ||||||
| Marizomib (NPI-0052, Salinosporamide A) Nereus | β-lactone-γ-lactam |
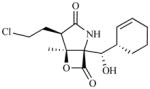
|
Sustaineda | Sustaineda | Sustaineda | Phase 1b | IV (Oralb and Subcutaneous efficacy in vivo) | Days 1,8,15 (28 day cycle) Days 1,4,8,11 (21 day cycle) |
| Bortezomib (Velcade®; PS-341) Takeda/Millennium | Peptide boronic acid |
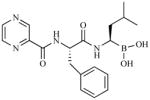
|
Slowly reversibleb | Sustainedb | Approved MM/MCL | IV (Active Subcutaneous in MM)c | Days 1,4,8,11 (21-day cycle) | |
| Carfilzomib (PR-171) Onyx/Proteolix | Peptide epoxyketone |
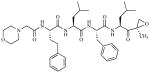
|
Sustainedd | Phase 2 | IV | Days 1,2,8,9,15,16 (28-day cycle)e | ||
| CEP-18770 Cephalon | Peptide boronic acid |
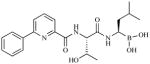
|
Slowly reversiblef | Phase 1 | IV (Oral efficacy in vivog) | Days 1,4,8,11 (21-day cycle)h | ||
| MLN9708 (MLN2238, active form) Takeda/Millennium | Peptide boronic acid |
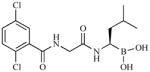
|
Reversiblei | Phase 1 | IV/Oral | Days 1,4,8,11 (21-day cycle) OR Days 1,8,15 (28- day cycle)j | ||
| ONX-0912 PR-047 Onyx/Proteolix | Peptide epoxyketone |
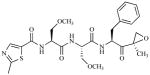
|
Sustainedk | Phase 1 | Oral | Days 1,2,3,4,5 (14-day cycle)l | ||
| PS-519 | β-lactone-γ-lactam |
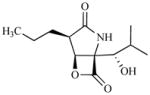
|
Slowly reversiblem | Discontinued after Phase 1 | IV | Day 1 or Days 1,2,3 (single cycle)n | ||
Patient PWB, ≥ 72 h; patient PBMCs, 48–72 h [61]; > 24 h in PWB and tumors from human MM.1S plasmacytoma xenograft murine model [49, 52]; < 24 h in liver, lung, spleen, kidney from human MM.1S plasmacytoma xenograft murine model [52].
Human MM.1S plasmacytoma xenograft murine model [49].
[203].
[60].
[204].
Slowly reversible in PWB and tissues (lung and liver), but sustained inhibition in tumor [205].
[205].
Clinicaltrials.gov: NCT00572637.
[206].
Clinicaltrials.gov: NCT00893464, NCT00830869, NCT00932698, NCT00963820.
[207].
Clinicaltrials.gov: NCT01129349.
Patient PWB; maximum inhibition at 2 h post-dosing and activity generally returned to normal within 24 h [34].
[34].
Fig. 2.

20S proteasome inhibition by bortezomib (left panels) and marizomib (right panels). A. Bortezomib forms a non-covalent adduct at the proteasome active site based on the high affinity of boronic acid for the hard oxygen nucleophile Thr1Oγ. The ligand is further stabilized by hydrogen bonding interactions between Thr1NH2 and B-OH, as well as non-covalent P1-P3 residue contacts with the proteasome S1-S3 binding pockets (see panels C, E); the collective binding modality results in slowly reversible proteasome inhibition. B. The β-lactone of marizomib acylates Thr1Oγ, followed by Thr1NH2-catalyzed nucleophilic displacement of chloride by C-3O to give a stable, irreversibly bound adduct. The binding mechanisms for these ligands were established via crystal structures of the yeast 20S proteasome CT-L site (subunit β5) in complex with bortezomib (C, E) and marizomib (D, F). Bortezomib residues P1 and P3 bind to the S1 and S3 pockets, respectively, while boron acts as an electron acceptor for the N-terminal threonine (T1) Thr1Oγ [55]. Marizomib residue P1 binds to the S1 pocket and is covalently bound to T1 via an ester linkage between Thr1Oγ and the carbonyl derived from the β-lactone ring [50]. T1, bortezomib and marizomib are presented as a ball and stick model. Electron density map (mesh) is contoured from 1σ around Thr1 and ligands with 2FO-FC coefficients (C, D). Surface representations of the CT-L active site complex with bortezomib (E) and marizomb (F).
Marizomib is a β-Lactone-γ-Lactam Proteasome Inhibitor Derived from the Marine Actinomycete Salinispora tropica
Marine microorganisms are being increasingly targeted as a resource for small molecule drug discovery [27]. Although not all taxa recovered from marine samples are unique to the marine environment, a number of chemically rich and taxonomically distinct marine groups have been discovered. Key among them is the genus Salinispora, which was the first obligate marine actinomycete genus to be formally described [28]. Initial chemical studies of strains in this group revealed high levels of cytotoxic activity [29] and quickly led to the isolation of salinosporamide A (USAN: marizomib; NPI-0052) from S. tropica [19]. Subsequent studies of this and two additional species led to the identification of many other new metabolites [30]. Marizomib exhibited a GI50 of < 10 nM across the National Cancer Institute (NCI) panel of 60 human tumor cell lines along with potent proteasome inhibitory activity [19]. An account of the early discovery and development of marizomib has been recently reported [20]. Genome sequencing of S. tropica led to the elucidation of the marizomib biosynthetic pathway [31] and the discovery of a new chlorination mechanism [32], as well as a unique starter unit in polyketide biosynthesis [33]. The collective biosynthetic machinery gives rise to a densely functionalized small molecule comprising a β-lactone-γ-lactam bicyclic core that is substituted with chloroethyl, methyl, and cyclohex-2-enylcarbinol groups at C-2, C-3 and C-4, respectively (Fig. (2)). This classifies marizomib among the β-lactone-γ-lactam superfamily of proteasome inhibitors, a distinct group of natural products derived from microbial sources and their derivatives that includes omuralide, marizomib, and the cinnabaramides (for a recent review, see [25]). These densely functionalized, low molecular weight ligands exhibit remarkable specificity for the 20S proteasome [26] that rivals peptidyl inhibitors. In fact, omuralide emerged as a gold standard among small molecule non-peptide based proteasome inhibitors in over a decade of research on proteasome structure and function; subsequently, the structurally related PS-519 (Table 2) was evaluated in Phase 1 clinical trials in young male volunteers for safety, tolerability and pharmacodynamics (PD) based on preclinical data demonstrating neuroprotective efficacy in models of cerebral ischemia [34]. The discovery of the marine-derived β-lactone-γ-lactam marizomib provided a next generation of proteasome inhibitors, which exhibit enhanced potency and increased breadth of proteasome inhibition compared to their terrestrially-derived counterparts [19, 25, 35].
The novel structure and biological activity of marizomib inspired production and analoging efforts using traditional fermentation, natural products chemistry, semi-synthesis, total synthesis, and mutasynthesis (for a recent review of the diverse methods of production and analogs, see [36]). Although over 50 analogs have been evaluated [35–45], it is the natural product that has entered clinical trials. Moreover, while the growing number of successful synthetic strategies may eventually result in the development of streamlined processes (currently, 13 published routes [36, 46]), the most efficient production of marizomib has been achieved through industrial marine microbiology, with large scale saline fermentation of S. tropica fueling the extensive preclinical studies and ongoing clinical trials described in this review [20, 21, 36]. Indeed, marizomib represents the first example of an active pharmaceutical ingredient (API) manufactured by saline fermentation for clinical trials. Extensive optimization of the original fermentation conditions was required to ensure consistent API production by a robust process that meets current Good Manufacturing Practice (cGMP) guidelines. The major process improvements to the original fermentation conditions for wild type strain S. tropica CNB476 included: 1) selection of a single colony isolate, S. tropica strain NPS21184, from strain CNB476, without mutation or genetic manipulation, to support higher and more selective production of marizomib; 2) extensive fermentation development to replace animal-derived media components and natural seawater with plant-derived nutrients and a chemically defined salt formulation, respectively, to be consistent with cGMP guidelines for the manufacture of APIs [47]; and 3) the addition of solid resin to the fermentation to bind, stabilize and capture marizomib from the aqueous media [48]. These and other process improvements led to > 100-fold improvement in the fermentation yield of marizomib in shake flask culture (from ~4 to 450 mg/L) and robust production of up to 360 mg/L in stainless steel fermentors. This saline fermentation process has been performed at up to 1000L scale. After purification from the crude extract, the final pharmaceutical grade cGMP API is obtained in >98% purity with overall ~50% recovery. Based on the potency of marizomib, both the production titer at fermentor scale and the recovery yield are suitable for both clinical development and commercial production [36].
Marizomib Regulates Proteasome CT-L, C-L and T-L Activities and Exhibits a Sustained Inhibition and Pharmacodynamic Profile
While marizomib shares a β-lactone-γ-lactam bicyclic core structure with omuralide and PS-519, its unique chloroethyl and cyclohex-2-enylcarbinol substituents give rise to mechanistically important interactions within the proteasome active sites that do not occur with other β-lactone inhibitors. These interactions contribute to the high affinity and specificity of marizomib for the proteasome, characterized by irreversible inhibition of all three proteolytic activities (CT-L, T-L, C-L) with IC50 values in the nM range [19, 35, 49]. The mechanism of inhibition of the 20S proteasome by marizomib has been well characterized via detailed kinetic studies [38] and crystal structures of the ligand in complex with the 20S CP [25, 50]. Binding commences with recognition of the cyclohexenyl P1 substituent by the proteasome S1 substrate specificity pocket. Once bound, the proteasome catalytic N-terminal Thr1Oγ forms an ester linkage with the carbonyl derived from the β-lactone ring of the inhibitor. This reaction sequence is analogous to that established for omuralide [23, 26, 51]; however, in the case of marizomib, this step is followed by Thr1NH2-catalyzed displacement of chloride by C-3O, giving rise to a tetrahydrofuran (THF) ring (Fig. (2)). Importantly, the chloride elimination step renders marizomib irreversibly bound to all proteolytic subunits [38, 50]. Irreversible binding at the molecular level translates to sustained inhibition of proteasome activity, the duration of which is dependent upon cell/tissue type [49, 52], and correlates with greater in vitro efficacy compared to slowly reversible β-lactone-γ-lactam congeners that are not structurally equipped to undergo this transformation (vide infra) [25, 53].
Other irreversible proteasome inhibitors that are currently under clinical evaluation include the peptide epoxyketones carfilzomib (PR-171) and ONX-0912 (PR-047), which form covalent morpholine adducts upon reaction with both Thr1Oγ and Thr1N, as elucidated by crystal structures of the natural product epoxomicin in complex with the 20S CP [54]. In contrast, peptide boronic acids, such as bortezomib and CEP-18770, form non-covalent adducts; their specificity for the proteasome is attributed to the high affinity of boronic acid for hard oxygen nucleophiles (i.e., Thr1Oγ) in contrast to soft cysteine nucleophiles, according to Lewis hard-soft acid-base principles. The ligand is further stabilized by hydrogen bonding interactions between Thr1NH2 and B-OH, as well as non-covalent P1-P3 residue contacts with the proteasome S1-S3 binding pockets (Fig. (2)) [55], and the collective binding modality results in slowly reversible proteasome inhibition. Thus, the binding kinetics and PD profiles of the various classes of proteasome inhibitors that are currently in clinical use (Table 2) are well established at the molecular level.
Marizomib exhibits high specificity for the proteasome as compared to other proteases [21] and also inhibits immuno-proteasomes [49], a specialized form of proteasomes that are induced by cytokines such as interferon-gamma and which are involved in the generation of antigenic peptides that are loaded onto major histocompatibility complex (MHC) class I proteins for eventual participation in the initiation of the immune response and generation of cytotoxic T-cells (CTL). Most CTLs express T-cell receptors (TCRs) that can recognize a specific antigenic peptide bound to Class I MHC molecules, which are present on nucleated cells, including tumors. Interestingly, immunoproteasomes are expressed in high levels predominantly in cells of hematopoetic origin, including MM cells [56], suggesting particular relevance to proteasome inhibitor therapy in hematologic malignancies and potential targets for new proteasome inhibitors under preclinical development.
Marizomib binds to and inhibits all three proteolytic subunits (β1, β2 and β5), despite being minimally substituted with a single moiety (P1) for recognition by the proteasome S1 specificity pocket. The non-covalent P1/S1 binding interactions of marizomib have been well characterized by enzyme inhibition kinetics, structural biology, and structure-activity relationship (SAR) studies, and the collective results are in good agreement [25, 38, 50]. The crystal structure of the marizomib:CP complex revealed that the inhibitor occupies all three pairs of proteolytic subunits [50], and when evaluated against purified 20S proteasomes, the IC50 rank order for inhibition is CT-L < T-L < C-L [35, 49]. However, cell-based studies reveal that this profile may vary depending upon the cell type [49, 57]. Although the P1 residue presents a relatively limited surface for binding to the proteasome S1 specificity pocket, the high affinity of marizomib for the proteasome is sufficient to induce broad and potent inhibitory activity compared to some peptidyl proteasome inhibitors, despite their ability to bind to several substrate binding pockets of the proteasome (S1 - Sn) (Fig. (2)). This nicely exemplifies the binding efficiency that is inherent to the dense functionality of the β-lactone-γ-lactam inhibitor designed by nature. The inhibition profiles of the different proteasome inhibitors in clinical use (Table 2) are distinguished by their relative binding affinities for the CT-L, T-L and C-L sites (associated with noncovalent interactions) as well as the duration of inhibition against isolated proteasomes in vitro (reversible or slowly reversible versus irreversible binding) and their PD profiles in vivo. Overall, the various proteasome inhibitors exhibit different inhibition profiles for the β1,β2 and β5 and immunoproteasome subunits that ultimately impart different potencies, cellular activities, target specificities and potentially different safety profiles.
The irreversible binding properties of marizomib result in lower IC50 values compared to structurally related yet slowly reversible β-lactone-γ-lactam inhibitors when measured against isolated proteasomes. The potential therapeutic benefit of this property may best be gauged by understanding the downstream consequences of irreversible binding in cells and tissues. Irreversible binding by marizomib has been correlated with markedly enhanced cytotoxicity in tumor cells; a PD profile characterized by prolonged proteasome inhibition in vivo; and sustained inhibition in tumor tissue and packed whole blood (PWB) associated with reduced tumor growth.
With respect to cytotoxicity, SAR studies indicate that irreversible binding imparts marizomib with potent cytotoxicity relative to slowly reversible inhibitors of the same structural class. This trend is consistent across various human tumor cell lines, including those of hematologic (RPMI 8226) and solid tumor (PC-3, HCT-116) origin, and is further supported by SAR studies of more structurally diverse salinosporamides and omuralide [25, 35, 39, 41, 53, 58]. While transport across the cell membrane may contribute to cytotoxicity, cell transport studies that directly compared [3H]-marizomib with the slowly reversible deschloro analog [3H]-salinosporamide B concluded that both compounds exhibit similar uptake characteristics in RPMI 8226 and PC-3 cells. However, marizomib exhibits slower efflux, greater extent of proteasome inhibition at lower concentrations, prolonged proteasome inhibition, and greater cytotoxicity, all of which can be attributed to irreversible binding to the proteasome [53]. Overall, it may be concluded that high affinity irreversible binding of marizomib results in inactivation of proteasome binding sites at low ligand concentrations as well as prolonged retention in cells, allowing the downstream consequences of sustained proteasome inhibition to play out to cell death.
Prolonged inhibition of proteasome activity in cells resulting from irreversible binding at the molecular level is readily observed in non-nucleated red blood cells (RBCs) that cannot generate new proteasomes. The PD profile of marizomib in animal models [49] and in clinical trials is characterized by sustained inhibition of proteasome activity (≥ 72 hours) in packed whole blood (PWB) lysates (containing > 98% RBCs that display a half-life of 15–17 weeks in humans) after a single intravenous (IV) administration of the drug, consistent with its irreversible binding profile in vitro. Moreover, in PWB samples obtained from ~90 patients treated with marizomib, a dose dependent inhibition of PWB 20S proteasome CT-L activity was observed, with increasing inhibition upon multiple administrations and only partial recovery between consecutive doses (Fig. (3)). The PD profile of marizomib is more durable than that of bortezomib, where recovery of proteasome activity was readily observed in both PWB and peripheral blood mononuclear cell (PBMC) lysates within 24 hours [49, 59], in agreement with its reversible binding mechanism (Fig. (2)). In contrast, the peptidyl epoxyketone carfilzomib, which forms an irreversible, covalent morpholine adduct with the proteasome (vide supra), exhibits a sustained PD profile in PWB lysates [60] (Table 2).
Fig. 3.
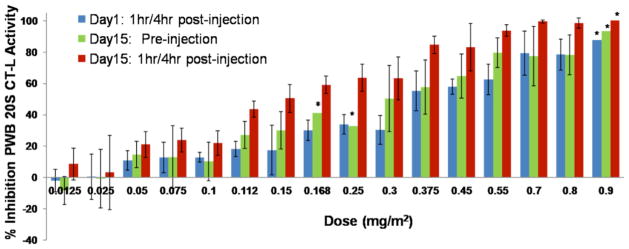
Inhibition of the packed whole blood (PWB) CT-L 20S proteasome activity in patient samples increases with dose and is more pronounced after the third administration of marizomib. Marizomib was administered IV on Days 1, 8 and 15 at the doses indicated. Proteasome activity does not restore to baseline levels, as indicated by the inhibition observed on Day 15 before the third administration of marizomib. Results are the average of 3 or more patients per cohort, except where indicated *, the average results of 2 patients is shown.
In nucleated cells, restoration of proteasome activity will reflect not only the ligand off-rate from the proteasome, but also the cell half-life and the rate of de novo proteasome synthesis, which can restore proteasome function in cells, even after treatment with an irreversible inhibitor. Indeed, recovery of proteasome activity after administration of marizomib is more rapid in patient PBMCs (48–72 h; nucleated cells with a half-life of a few days) compared to PWB (Fig. (4)) [61]. Interestingly, PD and efficacy studies in a human MM.1S plasmacytoma xenograft murine model similarly demonstrated rapid recovery of proteasome activity in normal tissues, including liver, lung, spleen and kidney (< 24 h), but more sustained inhibition in PWB and importantly, in tumors (> 24 h) (Fig. (5)). Notably, the prolonged proteasome inhibition in tumors correlated with reduced tumor growth in this model [52] (see Pharmacodynamics and efficacy of marizomib in a human MM.1S plasmacytoma xenograft murine model). Thus, it is important to distinguish irreversibility at the molecular level from the net biological effect on proteasome function in a given cell population or tissue type. The ability to monitor proteasome and immuno-proteasome inhibition and recovery profiles in different tissues may provide insights into tumor responses to proteasome inhibitors. Furthermore, initial research has shown that circulating proteasome protein levels and proteolytic activities may also be a potential biomarker that reflects the biology of the underlying disease and may serve as an independent prognostic factor for survival in MM and chronic lymphocytic leukemia (see Implications of Monitoring Proteasome Activity as a Potential Biomarker) [62, 63]. Studies to monitor proteasome activities in various tissues before and after marizomib treatment in the clinic are ongoing [61].
Fig. 4.

Marizomib pharmacodynamics (percent inhibition of CT-L activity), as monitored in patient PWB and PBMC lysates. Marizomib is administered IV at a dose of 0.55 mg/m2 on Days 1, 8 and 15 of a 28 day cycle. Proteasome activity is assessed before and after the first and third marizomib administration of cycle 1 and 2. Recovery of 20S proteasome CT-L activity between consecutive marizomib administrations is more rapid in PBMCs compared to PWB. Percent inhibition is calculated relative to the Cycle 1 baseline/Day 1 preinjection proteasome activity levels. Results are the average of 3 or more patients, except where indicated *, the average results of 2 patients is shown. Arrow indicates day of marizomib administration.
Fig. 5.
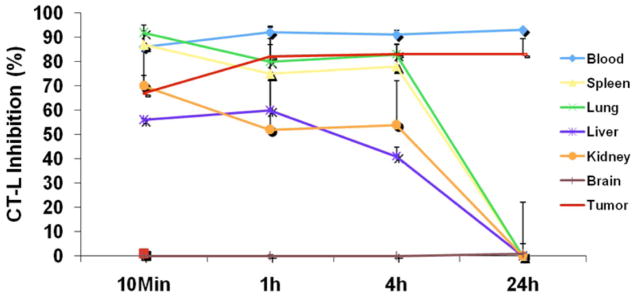
Inhibition of CT-L activity in tumors and various tissues after treatment with marizomib. [52]. MM.1S tumor-bearing mice were injected with three doses of marizomib (0.15 mg/kg, IV, Day 1, Day 4 and Day 8); euthanized at 10 min, 1, 4 and 24 hr after dosing; and PWB, liver, spleen, lung, kidney, brain and tumor were harvested. Protein extracts were prepared and analyzed for CT-L proteasome activity. Results are presented as percent inhibition compared to vehicle control. Data presented are means plus or minus SD (n = 3, p < 0.05).
MARIZOMIB IN PRECLINICAL HEMATOLOGIC TUMOR MODELS
The FDA approved bortezomib in 2003 as a treatment for relapsed/refractory MM and in 2006 for mantle cell lymphoma (MCL). This approval has validated the use of proteasome inhibitors in hematologic malignancies, particularly in B-cell cancers, and has since fostered the development of proteasome inhibitors with specificity profiles that may overcome both the cellular resistance patterns and toxicities to bortezomib. The ability of proteasome inhibitors to regulate NF-κB activation by inhibiting the degradation of IκBα, the cytoplasmic regulator of NF-κB activation, also expands the spectrum of tumors that are potential targets for these inhibitors. Marizomib, alone or in combination with other agents, may therefore fulfill the unmet need for new approaches to treat a broader spectrum of hematologic cancers. These concepts are further explored in the following sections, which highlight nonclinical studies of marizomib in hematologic tumor models, including MM, MCL, Waldenstrom’s macroglobulinemia, chronic and acute lymphocytic leukemias. These studies dissect specific mechanisms of action for marizomib, reveal synergies with bortezomib, histone deacetylase inhibitors and other agents in vitro, and demonstrate preclinical efficacy in vivo, providing the framework for ongoing clinical trials in patients with hematologic cancers.
Marizomib in Multiple Myeloma
Proteasome inhibitor therapy has proven to be a successful clinical strategy for the treatment of MM. Specifically, bortezomib is the standard of care for the treatment of relapsed, relapsed/refractory, and newly diagnosed MM [9–13]. However, clinical experience with bortezomib indicates possible off-target toxicities such as peripheral neuropathy, thrombocytopenia and neutropenia and the development of drug-resistance [16, 17]. In order to address these issues, recent research efforts have focused on the discovery and development of new proteasome inhibitors with equipotent anti-MM activity and fewer off-target activities. In this context, recent studies examined the efficacy of marizomib, which is orally active in MM models [19, 20]. Results from preclinical studies of marizomib in MM are highlighted below.
In Vitro and In Vivo Anti-Tumor Multiple Myeloma Activity of Marizomib
Initial screening of marizomib against the NCI panel of 60 human tumor cell lines showed a GI50 of < 10 nM for all cell lines [19]. In agreement with these observations, it was later demonstrated that 1) marizomib induces apoptosis in MM cells sensitive and resistant to both conventional and bortezomib therapies; and 2) the IC50 of marizomib for MM cells is within the low nanomolar concentration [49]. Examination of the effects of marizomib and bortezomib on normal PBMCs showed that marizomib does not significantly decrease normal lymphocyte viability at the IC50 for MM cells, with modest effects only at much higher concentrations. In contrast, bortezomib decreased the survival of PBMCs at concentrations close to the IC50 for MM cells. These data suggest a larger therapeutic index for marizomib than bortezomib. Importantly, marizomib induced apoptosis in tumor cells from MM patients relapsing after various prior therapies including bortezomib and/or thalidomide. The effectiveness of marizomib against tumor cells from bortezomib-refractory patients may be due, at least in part, to its ability to inhibit all three proteasome activities, i.e., CT-L, C-L and T-L, versus bortezomib, which predominantly affects CT-L activity. Indeed, studies using an in vitro protein model system demonstrated that simultaneous inhibition of multiple proteasome activities is a prerequisite for significant (i.e., > 50%) inhibition of proteolysis [64]. Therapeutic concentrations of bortezomib primarily target CT-L proteasome activity, and C-L as a secondary target. It is likely that the remaining proteolytic activity i.e., T-L, may compensate and allow proteasome functionality to be partially maintained. In contrast to bortezomib, marizomib inhibits all three proteolytic activities, thereby achieving maximal inhibition of proteolysis. Additionally, mechanisms conferring bortezomib-resistance may not be effective against marizomib (vide infra).
Mechanisms Mediating Marizomib-Induced Apoptosis in Multiple Myeloma Cells
Findings in MM models revealed that marizomib-induced MM cell death is associated with: 1) decrease in mitochondrial membrane potential; 2) increase in superoxide production; 3) activation of mitochondrial apoptogenic proteins cytochrome-c and second mitochondrial activator of caspases (Smac/Diablo); and 4) activation of caspase-9, caspase-8, caspase-3, and poly (ADP-ribose) polymerase (PARP) cleavage. Importantly, in MM cells, marizomib mediates apoptosis predominantly via caspase-8, whereas bortezomib-induced apoptosis requires both caspase-8 and caspase-9 activation. These findings indicate that marizomib is more dependent on FAS-associated via death domain (FADD)-caspase-8 apoptotic signaling pathway than bortezomib, suggesting differential action of marizomib versus bortezomib against MM cells. Furthermore, in contrast to marizomib, bortezomib-induced apoptosis requires activation of pro-apoptotic BH3-only family member proteins Bax and Bak [49]. The BH3-only members of the Bcl-2 protein family are essential for initiation of programmed cell death and stress-induced apoptosis.
Besides induction of apoptosis, marizomib downregulates various cell growth and survival signaling pathways in MM cells. In fact, the initial rationale for the therapeutic use of proteasome inhibitors as anticancer agents was, in part, based on their ability to inhibit growth and survival signaling via NF-κB [4–8, 49]. Indeed, marizomib, like bortezomib, targets NF-κB; importantly, marizomib is a more potent inhibitor of NF-κB and related cytokine secretion than bortezomib [49, 65]. A detailed study on the effects of marizomib on NF-κB regulated gene products demonstrated that marizomib potentiated apoptosis induced by tumor necrosis factor-alpha (TNF-α), bortezomib and thalidomide, and this correlated with down-regulation of gene products that mediate cell proliferation (cyclin D1, cyclooxygenase-2 (COX-2) and c-Myc), cell survival (Bcl-2, Bcl-xl, cFLIP, TRAF1, IAP1, IAP2 and survivin), invasion (matrix metalloproteinase-9; MMP-9) and ICAM-1 and angiogenesis (vascular endothelial growth factor (VEGF)). Marizomib also suppressed TNF-α-induced tumor cell invasion and receptor activator of NF-κB ligand (RANKL) induced osteoclastogenesis [65].
Several investigators have shown that the MM-host bone marrow microenvironment confers growth, survival, and drug resistance in MM cells [5, 66]. Adhesion of MM cells to bone marrow stromal cells (BMSCs) triggers transcription and secretion of MM cell growth and survival factor interleukin-6 (IL-6) [5, 66]. Marizomib significantly inhibits MM cell growth even in the presence of BMSCs. Furthermore, marizomib abrogates IL-6-induced proliferation of MM cells. In addition to NF-κB inhibition, marizomib overcomes survival and drug-resistance conferred by Bcl-2 in MM cells: overexpression of Bcl-2 provides more protection against bortezomib than marizomib [49]. Additional studies suggest that resistance to bortezomib, but not marizomib, involves heat shock proteins Hsp27 and Hsp70 [67, 68]. Marizomib also blocks VEGF-triggered migration of MM cells, suggesting that marizomib is an anti-migratory agent [49, 69].
Examination of the in vivo efficacy of marizomib using a human MM.1S plasmacytoma xenograft mouse model [70] shows potent oral anti-tumor activity [49]. Treatment of MM.1S-bearing mice with marizomib, but not vehicle, inhibits plasmacytoma growth and prolongs survival of these mice (Fig. (5)). Marizomib is well tolerated by mice, without significant weight loss or obvious neurological behavioral changes. Importantly, analysis at day 300 shows no recurrence of tumors in 57% of the marizomib-treated mice. A head-to-head examination of marizomib and bortezomib shows that both agents reduced tumor progression and prolonged survival.
Pharmacodynamics and Efficacy of Marizomib in a Human MM.1S Plasmacytoma Xenograft Murine Model
In vivo studies in mice using human MM.1S plasmacytoma xenografts demonstrate that IV administered marizomib is well tolerated, prolongs survival, and reduces tumor recurrence (vide supra). PD studies using the model described above demonstrated that marizomib: 1) rapidly leaves the vascular compartment and enters the tumors and other organs as the parent compound; 2) inhibits 20S proteasome CT-L, T-L, and C-L activities in extra-vascular tumors, PWB, liver, lung, spleen, and kidney, but not brain; and 3) triggers a more sustained (>24h) proteasome inhibition in tumors and PWB than in other organs (< 24h) [52] (Fig. (5)). These findings are consistent with earlier studies showing that marizomib targets all three 20S proteasomal activities [35, 49]. Indeed, the kinetics of proteasome inhibition differ between tumors and normal tissues. For example, the onset of marizomib-induced proteasome inhibition is rapid (within 10 min) in most tissues other than tumor, for which the onset of inhibition occurs at ~1h and is maximal at 24h. Intravenous injection of either a single or three doses of marizomib (0.15 mg/kg) blocks proteasome activities in peripheral organs, without inhibition of proteasome activity in the brain, indicating that marizomib does not cross the blood-brain barrier at this dose and schedule. The likely explanation for a sustained inhibition of proteasome activity in tumors may be the irreversible nature of marizomib; however, the cell half-life and rate of de novo proteasome synthesis in tumors may contribute. Importantly, marizomib-induced blockade of proteasome activity in liver, spleen, kidney, and lungs recovers by 24h, implying that de novo proteasome synthesis in these tissues may result in the rapid recovery of proteasome activity. An important conclusion of this study was that treatment of MM.1S bearing immunodeficient mice with marizomib reduces tumor proliferation without marked toxicity, which is associated with prolonged inhibition of proteasome activity in tumors and PWB, but not in normal tissues [52] (Fig. (5)).
Combination Studies of Marizomib with Bortezomib or the Immunomodulatory Agent Lenalidomide in Multiple Myeloma
Recent studies utilizing an in vitro protein model system have shown that simultaneous inhibition of multiple proteasome activities is a prerequisite for significant (i.e., > 50%) inhibition of proteolysis [64]. Since bortezomib predominantly inhibits proteasome CT-L, and more recently defined inhibition of C-L activities [49], it was hypothesized that marizomib, which blocks all three 20S proteasome activities, can be combined with bortezomib to confer a broader inhibition profile at lower and potentially safer doses. Indeed, combining marizomib and bortezomib induces synergistic anti-MM activity both in vitro using MM cell lines or patient bone marrow derived CD138+ MM cells and in vivo in the human MM.1S plasmacytoma xenograft murine model [71]. Combined marizomib and bortezomib-triggered apoptosis in MM cells is associated with: 1) activation of caspase-8, caspase-9, caspase-3, and PARP cleavage; 2) induction of endoplasmic reticulum (ER) stress response and c-Jun N-terminal kinase (JNK); 3) inhibition of migration of MM cells and angiogenesis; 4) suppression of CT-L, C-L and T-L proteasome activities; and 5) blockade of NF-κB signaling. Animal studies showed that administration of combined low doses of marizomib and bortezomib is well tolerated, and trigger synergistic inhibition of tumor growth, and CT-L, C-L and T-L proteasome activities in tumor cells. Histochemical analysis of the MM.1S plasmacytomas from marizomib plus bortezomib-treated mice showed growth inhibition, apoptosis, and a decrease in associated angiogenesis. Of note, it is clear from in vivo data that even 30–40% proteasome inhibition, albeit of all three activities, is sufficient to trigger significant anti-MM activity. The mechanisms mediating enhanced cytotoxicity of the combination regimen may likely result from greater and broader proteasome inhibition and/or differential apoptotic signaling pathways with the two-drug regimen. A similar synergistic cytotoxicity of marizomib plus bortezomib is reported in models of Waldenstrom’s macroglobulinemia - an incurable low-grade B-cell lymphoma (see Waldenstrom’s Macroglobulinemia below) [72]. Finally, the synergistic cytotoxicity of marizomib and bortezomib in lymphoma, leukemia, and solid tumor cells that are relatively resistant to bortezomib, suggests clinical applicability of this therapeutic regimen beyond MM (vide infra).
Combination therapeutic strategies have shown promise in reducing toxicities and overcoming drug resistance associated with bortezomib. For example, prior preclinical studies showed that lenalidomide (Revlimid®) - a novel immunomodulatory drug (IMiD) - triggered growth arrest or apoptosis in drug resistant MM cells. A Phase 1/2 clinical trial of bortezomib with lenalidomide and low dose dexamethasone demonstrated safety and remarkable efficacy in relapsed-refractory and newly diagnosed MM patients [73, 74]. The finding that the combined bortezomib and lenalidomide regimen has the ability to overcome clinical bortezomib resistance, coupled with findings that marizomib is a potent proteasome inhibitor, suggested that combining marizomib with lenalidomide may also trigger synergistic anti-MM activity. Consistent with this notion, combining low doses of marizomib and lenalidomide induced synergistic anti-MM activity [75]. Furthermore, marizomib plus lenalidomide-induced apoptosis correlates with: 1) activation of caspase-8, caspase-9, caspase-12, caspase-3, and PARP cleavage; 2) activation of BH3 protein Bim; 3) translocation of Bim to endoplasmic reticulum; 4) inhibition of migration of MM cells and angiogenesis; and 5) suppression of CT-L, C-L and T-L proteasome activities. Blockade of Bim using small interfering RNA (siRNA; sometimes known as short interfering RNA or silencing RNA, a class of double-stranded RNA molecules, 20–25 nucleotides in length) abrogated marizomib and lenalidomide-induced apoptosis. Biochemical studies demonstrate that marizomib plus lenalidomide-induced apoptosis is primarily dependent on caspase-8 signaling. The mechanistic studies suggest that the synergistic anti-MM activity of marizomib plus lenalidomide predominantly relies on the caspase-8 and Bim/ER/caspase-12 signaling axis. Importantly, low dose combinations of marizomib and lenalidomide are well tolerated, significantly inhibit tumor growth, and prolong survival in a human MM.1S plasmacytoma xenograft murine model.
Collectively, these preclinical studies demonstrate potent in vitro and in vivo anti-tumor activity of marizomib combined with either lenalidomide or bortezomib at doses that are well tolerated in a human MM.1S plasmacytoma xenograft murine model. These findings provide the framework for clinical trials of low dose combinations of marizomib with bortezomib or lenalidomide to increase response, overcome drug resistance, reduce side effects, and improve patient outcome in MM.
Marizomib in Waldenstrom’s Macroglobulinemia
Waldenstrom’s Macroglobulinemia (WM) is a biologically unique low grade B-cell lymphoma characterized by the presence of lymphoplasmacytic cells in the bone marrow and the secretion of immunoglobulin M (IgM) monoclonal protein in the serum, indicating that WM cells present with a high rate of protein turnover. Protein metabolism is a tightly regulated process, and inhibition of its turnover with proteasome inhibitors leads to apoptosis in malignant cells [76, 77]. A major activity of proteasome inhibitors in certain B-cell malignancies is the targeting of the IL-6 and NF-κB signalling pathways, both of which are critical regulators of survival and proliferation in B-cell malignancies, including WM [78–80]. Based on its activity in MM, bortezomib was tested in WM and achieved a 40–80% response rate in Phase 2 trials [81]. These striking clinical responses indicate that proteasome activity is critical for the survival of WM cells. While marizomib also inhibits the proteasome, it exhibits a complementary and partially overlapping proteasome inhibition profile and ultimately, a different mechanism of action than bortezomib; for example, as discussed above, apoptosis of MM cells induced by marizomib is mediated predominately through the caspase-8 cell death cascade [49]. The role of the proteasome in WM was therefore dissected using marizomib and bortezomib. It was demonstrated that marizomib inhibits proliferation and induces apoptosis in WM cell lines and CD19+ primary WM cells at doses consistent with previous studies and achievable in vivo [72, 82, 83]. It was later demonstrated that the combination of marizomib and bortezomib leads to synergistic cytotoxicity on WM human cell lines (BCWM.1, WM-WSU), IgM secreting cell lines (MEC-1, Namalwa) and bone marrow derived patient primary WM cells. These two agents lead to inhibition of nuclear translocation of p65 NF-κB, with activity on the canonical and non-canonical NF-κB pathway, and synergistic induction of caspase-3, -8 and -9 cleavage, as well as PARP cleavage and induction of Smac/Diablo. These findings begin to delineate the role of the canonical and non-canonical NF-κB pathways in WM.
Studies to further dissect the mechanism of synergy of marizomib and bortezomib demonstrated differential activities on both: 1) the Akt pathway; and 2) 20S proteasome CT-L, C-L and T-L functions. Marizomib induced cytotoxicity was completely abrogated in an Akt knockdown cell line (BCWM.1; established using lentavirus infection system), indicating that its major activity is mediated through this pathway, while bortezomib modestly activated Akt activity. Previous studies have demonstrated that activation of the Akt survival pathway may be one mechanism of bortezomib resistance in malignant B cells. It was subsequently demonstrated that the major activity of marizomib is mediated through inhibition and not activation of Akt and therefore, its combination with bortezomib may overcome resistance to bortezomib in vivo [72].
While little is known about the role of the bone marrow microenvironment in WM, the adhesion of WM cells to cytokines and/or fibronectin present in the bone marrow milieu was found to induce NF-κB activation and IL-6 induced Akt activation, which were both down-regulated in the presence of marizomib either alone, and more significantly in combination with bortezomib. Importantly, IL-6 and NF-κB induction by adhesion are two major pathways regulated by the proteasome, and marizomib and bortezomib overcome resistance induced by mesenchymal cells and the addition of IL-6 in a co-culture in vitro system. The combination of the two agents overcomes the protective effect of the bone marrow niches, without affecting the growth and differentiation of normal hematopoietic components. Homing, a complex process that is regulated by migration and adhesion of malignant cells to their specific bone marrow niches, is also influenced by the two agents: marizomib and bortezomib inhibit migration and adhesion of WM cells as well as their homing in vivo [72]. Together, these studies provide a stronger understanding of the biological role of the proteasome pathway in WM, and provide the preclinical framework for studying in clinical trials marizomib in WM and other low-grade lymphomas.
Marizomib in Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is the most common adult hematologic malignancy in the western world. The majority (>75%) of newly diagnosed CLL cases (about 16,000 in the US) occur in men over the age of 50. The disease is characterized by the accumulation of mature resting CD5+ B-cells in the peripheral blood and is thought to arise primarily as the result of defects in the regulation of apoptosis rather than proliferation [84, 85]. Thus, as a result of epigenic alterations in the regulation of the bcl-2 gene, CLL cells express very high levels of the anti-apoptotic protein Bcl-2 [86].
Chemotherapy regimens using combination approaches are effective in newly-diagnosed and relapsed CLL. Recent clinical trials have shown that combinations of the purine analog fludarabine (Fludara) with alkylating agents bendamustine (Treanda) or cyclophosphamide (Cytoxan) produce higher response rates and a longer progression-free survival than single agents. In addition, positive results have been obtained with monoclonal antibody regimens that include alemtuzumab (anti-CD52), rituximab (Campath-1H, anti-CD20), and ofatumumab (Arzerra, anti-CD20) [87, 88]. Although these approaches are effective, additional treatment regimens that are active as either a single agent or in combination with standard of care therapies are urgently needed. Preclinical studies confirmed that in CLL cells, bortezomib blocks activation of NF-κB and bypasses Bcl-2 mediated apoptosis resistance, possibly by activating the Bcl-2 inhibiting protein kinase, JNK [89, 90]. Based on these positive preclinical findings, bortezomib was evaluated in Phase 2 clinical trials in patients with refractory CLL. Unfortunately, only minimal (but suggestive) responses to bortezomib were obtained in these trials [91]. However, the preclinical biology strongly supported that a proteasome inhibitor should show clinical activity, supporting the evaluation of the second generation proteasome inhibitor marizomib in preclinical CLL models [57].
Why can marizomib succeed where bortezomib has failed to exhibit significant clinical activity in CLL? Findings using freshly isolated Ficoll-Paque fractionated peripheral blood from 37 newly diagnosed patients with CLL demonstrated some striking similarities and differences between these two inhibitors [57]. As discussed above, marizomib exhibits a more prolonged, broader proteasome inhibition profile compared to bortezomib. Although their steady-state IC50 values as inhibitors of proteasome CT-L activity were similar, marizomib exerted its effects more rapidly than bortezomib, and drug washout experiments showed that short exposures to marizomib (15 minutes) resulted in sustained (≥ 24 hours) proteasome inhibition. In contrast, proteasome CT-L activities recovered in CLL cells exposed to even a 10-fold higher concentration of bortezomib. In addition, prolonged exposure times with bortezomib of ≥ 8 hours were required for commitment to caspase activation and DNA fragmentation. It is currently believed that proteasome inhibitors prevent clearance of misfolded or damaged proteins leading to protein aggregation, ER stress and JNK and caspase-4 activation. Although there are differences in the activity profiles for marizomib and bortezomib, both involve caspase-4 as a central mediator of cell death.
Interestingly, there has been a recent report suggesting that plasma components, in particular the dietary flavinoid, quercetin, may inactivate bortezomib by binding to the boron moiety [92]. The findings provide an attractive potential explanation for why bortezomib did not display significant clinical activity in CLL and support testing of marizomib as a single agent or in combination with other standard of care therapies in this patient population. In relation to dietary intake, it has also been observed that the proteasome inhibitory and anticancer activity of bortezomib and other boronic acid-based proteasome inhibitors can be blocked by green tea polyphenols [93,94].
Marizomib in Acute Lymphocytic Leukemia
A Need for New Drugs and the Role of Proteasome Inhibitors in Acute Lymphocytic Leukemia
Although acute lymphocytic leukemia (ALL) boasts a cure rate of over 80%, patients that do not respond to the currently used combination chemotherapeutic regimen are left with few options [95]. In pediatric ALL cases, the cure rate is approaching 90%, however, late effects of the currently used therapy are a significant problem for survivors of childhood leukemia, since they live longer than their adult counterparts. Specific late effects of therapy such as cardiotoxicity stemming from anthracycline exposure or diminished neurocognitive function in patients that have received cranial irradiation for central nervous system disease are problematic and can be avoided by developing less pantoxic and more selective strategies for the treatment of ALL [96].
The clinical success of proteasome inhibitors in other hematologic malignancies such as MM and MCL suggested that leukemia patients may similarly benefit from this class of drugs [97]. Validation of this concept was carried out in a 2004 Phase 1 clinical trial of bortezomib, in which fifteen adult refractory/relapsed ALL patients were enrolled and a maximum tolerated dose was identified. Importantly, significant proteasome inhibition was observed at 1.3 mg/m2 and lower doses in peripheral blood specimens collected from the patients [98]. These data provided proof of concept that proteasome inhibitors can be utilized clinically in a leukemia population, thereby providing rationale for testing marizomib in leukemia populations.
Biochemical Effects of Marizomib in ALL
Evaluation of bortezomib and marizomib on proteasome CT-L, C-L and T-L activities in an array of leukemia cell lines indicated that marizomib more potently inhibited the CT-L and C-L activities. Importantly, the T-L activity was also inhibited by marizomib in stark contrast to bortezomib, which slightly stimulated T-L activity [83]. Another key difference between bortezomib and marizomib was in the mode of apoptosis induction in leukemia cells. Two biochemical markers of apoptosis, DNA fragmentation and caspase-3-like activity, were triggered more strongly by marizomib than bortezomib in ALL cells. Insight into the initiation of caspase activation was provided by using caspase-8 and -9 specific inhibitors. Caspase-8 inhibition significantly prevented cell death by marizomib whereas caspase-9 inhibitors had little to no effect. By using variants of the Jurkat ALL cell line that lack either FADD or caspase-8, a requirement for these two molecules in marizomib induced cell death was clearly determined for ALL cells [83].
Induction of oxidative stress has been observed in numerous cell systems after proteasome inhibition. Consistent with these reports, marizomib caused an increase in intracellular peroxide and superoxide levels. The antioxidant, N-acetylcysteine, abrogated cell death by marizomib but did not provide protection against caspase-8 activation by the drug. This highlighted a role for caspase-8 as upstream of oxidative stress generation by marizomib. The source of intracellular superoxide and peroxide upon treatment with marizomib remains unclear, and may arise from multiple processes which may be caspse-8 dependent or independent, since the peptide based chemical caspase-8 inhibitor, IETD-fmk, did not change levels of intracellular superoxide.
Marizomib in Combination with Histone Deacetylase Inhibitors in ALL and other Models
In leukemia therapy, as with most cancers, combining agents appears more efficacious than single agent therapies [99]. Proteasome inhibitors have been tested in vitro with numerous anti-leukemia drugs including nucleoside analogs [89], antibody based drugs [100], as well as alkylating agents [101]. One of the more promising combinations has been with a class of epigenetically targeted drugs: the histone deacetylases (HDAC) inhibitors [102]. One member of this class of compounds, vorinostat (suberoylanilide hydroxamic acid (SAHA), Zolinza®), has been FDA approved for the treatment of cutaneous T cell lymphoma, indicative of activity in hematologic malignancies. Bortezomib has been tested in combination with numerous HDAC inhibitors and found to exert synergistic effects [103]. Marizomib also synergizes with HDAC inhibitors in ALL [83, 104] and MM (Chauhan et al. unpublished results), however, quantification of the degree of synergy using isobologram based analyses reveals that the marizomib and HDAC inhibitor combinations are more potent than bortezomib combinations in leukemia cell lines and primary acute leukemia specimens [104, 105]. Since HDAC inhibitors are divided into classes based on their chemical structure and based on which HDAC family members they inhibit [102], it was interesting to note that diverse HDAC inhibitor compounds (hydroxamic acids, benzamide and aliphatic acids) were strongly synergistic with marizomib, and more so than with bortezomib.
Investigating the mechanism of action of the synergies between proteasome inhibitors and HDAC inhibitors revealed surprising and overlapping mechanisms, as highlighted in Fig. (6). In ALL, the HDAC inhibitors were found to repress mRNA expression of the three 20S proteasomal β subunits that are responsible for the enzymatic activity. An unexpected yet distinguishing feature between bortezomib and marizomib was the ability of marizomib to cause an increase in total histone H3 and acetylation of histone H3. This histone modification is a hallmark of HDAC inhibitor action but had never before been reported as occurring as a consequence of proteasome inhibition. This epigenetic consequence of marizomib action may provide insight into the stronger synergy of marizomib with HDAC inhibitors than bortezomib and is currently under investigation. Other potential synergies between HDAC and proteasome inhibitors have been observed in pancreatic tumor cells, involving aggresome formation and HDAC6 inhibition (see Marizomib in Pancreatic Cancer Models). More recently, bortezomib and marizomib demonstrated synergistic effect with the class I HDAC inhibitor MGCD0103 in Hodgkin’s lymphoma cell lines by inhibiting TNF-α-induced NF-κB activation [106] (see below). Moreover, regulation of NF-κB by both proteasome inhibitors and HDAC inhibitors represents a point of convergence for these drugs that may contribute to their synergistic activities in MM (Chauhan et al. unpublished observations) and solid tumors, such as non-small cell lung carcinoma (NSCLC) (Drabkin et al. unpublished observations).
Fig. 6.
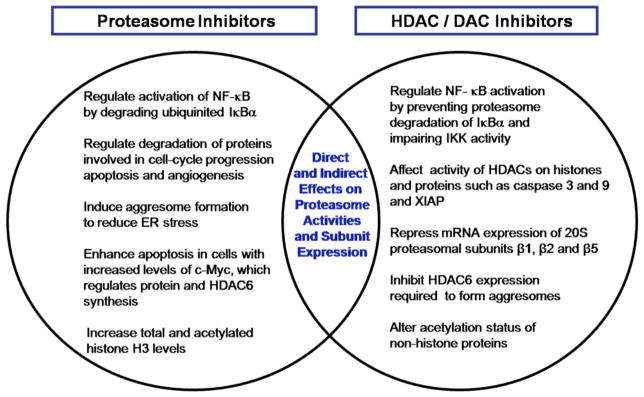
Potential distinct and overlapping mechanisms for synergies between HDAC and proteasome inhibitors.
The consequences of combining these classes of drugs have the potential to impact many cellular processes. In support of this point, a recent comprehensive analysis of the acetylome revealed many unexpected non-histone acetylation targets for various HDAC inhibitors including vorinostat, entinostat (SNDX-275, previously called MS-275) and romidepsin (Istodax®) [107]. The HDAC inhibitors vorinostat and romidepsin were initially approved by the FDA in 2006 and 2009, respectively, for the treatment of cutaneous T-cell lymphoma. The recent preclinical findings and increasing knowledge of the distinct and overlapping mechanisms of action for proteasome inhibitors and HDAC inhibitors paved the way for testing these drug combinations in a broader spectrum of lymphoid and solid tumor malignancies [104,108].
Marizomib in Mantle Cell Lymphoma and Hodgkin’s Lymphoma
In addition to MM, CLL and WM, preclinical studies have identified additional B-cell malignancies that express constitutively activated NF-κB and are therefore considered targets for proteasome inhibitor therapy. Subsequent clinical studies lead to the FDA approval in 2006 of bortezomib as a treatment for MCL [109–111], an aggressive and one of the rarest forms of non-Hodgkin’s lymphoma. Bortezomib was also tested clinically in an additional B-cell malignancy, Hodgkin’s lymphoma. However in this patient population, bortezomib showed minimal single agent activity [109,110]. In light of the different proteasome inhibition profile for marizomib, it was considered of interest to evaluate its activity in preclinical models for both these B-cell malignancies.
Marizomib was shown to be active as a single agent in Hodgkin’s lymphoma cell lines (HD-LM2, L-428 and KM-H2) and MCL cell lines (Jeko1, Mino and SP53) [112]. The antiproliferative activity of marizomib was observed in all cell lines tested in a time- and concentration-dependent manner. The effect was observed in as early as 24 hours, and lasted for up to 72 hours at a dose range of 5 nM to 50 nM. The activity was comparable to bortezomib at the same conditions. The antiproliferative activity occurred through induction of apoptosis and was enhanced by treating cells in combination with the HDAC inhibitor vorinostat (SAHA) [110] or with GX15-070, a small-molecule antagonist of the BH3-binding groove of the Bcl-2 family proteins [113]. More recently, marizomib was shown to have a synergistic antiproliferative activity in combination with the class I HDAC inhibitor MGCD0103, suggesting that synergy between HDAC inhibitors and proteasome inhibitors (Fig. (6)) can also be maintained through an HDAC 6-independent mechanism [106]. The potent activity of marizomib in MCL and Hodgkin’s lymphoma as a single agent and in combination with HDAC inhibitors support additional clinical testing in these diseases.
MARIZOMIB IN PRECLINICAL SOLID TUMOR MODELS
Although proteasome inhibitors have demonstrated clinical activity in hematologic, and in particular B-cell, malignancies, the clinical results with bortezomib in solid tumor malignancies have not demonstrated appreciable benefits [114–116]. Why bortezomib did not exhibit activity in patients with solid tumors may be explained in part by its proteasome inhibition profile, onset and duration of activity. Given the noted differences in mechanisms of action for bortezomib and marizomib, it is possible that marizomib may yield greater clinical efficacy in solid tumors as a single agent or in combination with clinically relevant drugs. The following sections provide preclinical findings in solid tumor models that address these issues.
Marizomib in Colorectal Carcinoma
Many early studies assessing the efficacy of proteasome inhibitors in cancer treatment were based upon the assumption that inhibition of the NF-κB pathway was the predominant anti-tumor mechanism, as discussed above in the context of various hematologic malignancies. High levels of basal NF-κB are also common in colorectal cancer (CRC) samples [117], and the clinically used chemotherapy treatments 5-fluorouracil (5-FU) and irinotecan (Camptosar, CPT-11) have been shown to activate NF-κB signaling leading to chemoresistance [118, 119]. Furthermore, increased NF-κB activity is predictive of poor response and reduced survival time in patients with CRC. These and similar studies have been used as a rationale for treating CRC with proteasome inhibitors.
Marizomib treatment blocks the activation of NF-κB by SN-38 (the active metabolite of irinotecan) in CRC cells and results in the accumulation of the phosphorylated form of IκBα (a marker of inhibited NF-κB activity). In this setting, marizomib is a 2-fold more potent inhibitor of TNFα-induced NF-κB activation than bortezomib [120]. As single agent therapies in preclinical studies, bortezomib, MG132 and marizomib have been shown to decrease proliferation and induce apoptosis in CRC cells [120–123]. While p53, p21, PUMA and Bax have all been implicated in the induction of apoptosis by proteasome inhibitors in CRC, a consensus on the mechanism behind this response is yet to be reached [118, 121, 124].
Several preclinical studies have identified targeted therapies that show synergy with bortezomib in CRC, including vorinostat (HDAC inhibitor) [125], ABT-737 (Bcl-2 inhibitor) [126] and TNF-α [127]. However, the study of bortezomib in combination with traditional cytotoxic therapies in CRC cells has been limited. Marizomib increases the apoptotic response of CRC cells to various chemotherapy combinations, including the clinically relevant drugs 5-FU, leukovorin, oxaliplatin (Eloxatin®) and SN-38 (the active metabo-lite of irinotecan) [120]. Combining marizomib with chemotherapy was also shown to increase the levels of various cell cycle regulatory proteins, including p21, p27 and p53. This was associated with an increase in cells arrested at the G1/S cell cycle checkpoint [120]. The efficacy of marizomib as part of a combination regimen in human CRC cell lines was recapitulated in a murine subcutaneous xenograft CRC model, where reduction in tumor growth rates by combinations of 5-FU and leucovorin with or without irinotecan (CPT-11), oxaliplatin and bevacizumab (Avastin) were consistently improved by the addition of oral marizomib (Fig. (7)). Indeed, the combined treatment with 5-FU, leukovorin, irinotecan, bevacizumab and marizomib not only slowed tumor growth in mice but actually decreased tumor size over four weeks of treatment [120].
Fig. 7.

Marizomib treatment reduces tumor burden when added to three conventional colon cancer therapy regimens in a human colon carcinoma (LoVo) xenograft model (n = 6 mice per treatment group, error bars represent the standard error of the mean). CPT-11 (irinotecan); Leuk (leukovorin); Oxa (oxaliplatin); Ava (Avastin; bevacizumab). Adapted with permission from [120].
Taken together, these results clearly highlight that marizomib is able to induce apoptosis and reduce tumor burden in models of CRC, particularly when used in conjunction with traditional standard of care chemotherapeutics and biologics. Despite showing some promise in the pre-clinical setting of CRC, clinical trials using bortezomib alone [128] or in combination with irinotecan [129] or the FOLFOX-4 regimen [130] consistently failed to show any appreciable benefit of bortezomib treatment in patients with advanced colorectal disease. Given the noted differences in response of other cancer models to bortezomib and marizomib and the above highlighted efficacy of marizomib in the preclinical colon cancer setting, it is conceivable that marizomib may yield greater clinical efficacy than bortezomib in combination treatment for advanced CRC.
Marizomib in Pancreatic Carcinoma
Recent studies analyzing the molecular sequelae of marizomib treatment in models of pancreatic cancer have shown that marizomib treatment effectively induces apoptosis in pancreatic cancer cell lines [131,132]. This finding is in agreement with results for bortezomib [133]. The current standard of care for patients with advanced pancreatic cancer is the nucleoside analogue gemcitabine (Gemzar®). Marizomib has been shown to be significantly more effective at inducing apoptosis than gemcitabine both in vitro and in an in vivo xenograft model of pancreatic cancer [131]. Furthermore, addition of marizomib to either gemcitabine or a combination of gemcitabine and the anti-epithelial growth factor receptor (EGFR) monoclonal antibody cetuximab (Erbitux) significantly improved the efficacy of these regimens in vivo. These data suggest that marizomib may improve treatment response when combined with gemcitabine in pancreatic cancer patients.
Proteasome inhibition has been shown to sensitize many tumor types to traditional cytotoxic therapies, radiation treatment and certain targeted therapies. In pancreatic cancer models, bortezomib has been shown to increase cancer cell sensitivity to irinotecan [134], gemcitabine [135, 136] and docetaxel [137] treatment, as well as HDAC inhibitors (SAHA or trichostatin-A) or the TNF related apoptosis inducing ligand (TRAIL) [138–140] (also see “Marizomib-induced sensitization to immunotherapy). Analysis of the signaling pathways involved in cellular responses to proteasome inhibitor treatment has provided insight into the success of certain combination therapies. For example, HDAC inhibitors synergize with proteasome inhibitors through prevention of the formation of aggresomes (vide supra; Fig. (6)), an essential part of the aggresome pathway which can be utilized as an alternative to the 26S proteasome to degrade poly-ubiquitinated proteins [140]. Several other distinct anti-apoptotic responses to proteasome inhibition have been also described. In breast cancer cells, proteasome inhibitor-induced apoptosis was dependent on activation of JNK. Proteasome inhibitors also increased cellular levels of mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1), a specific deactivator of pro-apoptotic JNK activity, thereby inhibiting the apoptotic response [141, 142]. In other models, including lymphoma and MM, proteasome inhibition increased the expression of the anti-apoptotic heat shock proteins Hsp27, Hsp70 and Hsp90 [68, 143–146]. Suppression of these various anti-apoptotic mechanisms by small molecule inhibitors or siRNA interference significantly increased the apoptotic response of tumor cells to proteasome inhibition [68, 141–146]. In agreement with these findings, analysis of the molecular pathways activated in response to marizomib treatment of pancreatic carcinoma cells showed that marizomib treatment activates EGFR, extracellular signal-regulated kinase (ERK), Akt and JNK with varying kinetics [131]. Targeted inhibition of these pathways in vitro and in vivo enhanced the apoptotic response of pancreatic cancer cells to marizomib treatment.
Bortezomib as a single agent [147] or in combination with other chemotherapies [130, 148–150] has been tested in Phase 1 trials to determine the maximum tolerated dose (MTD) in patients with solid tumors. These trials formed the basis of a Phase 2 trial in which bortezomib was used for the treatment of advanced pancreatic cancer both alone and in combination with gemcitabine [151]. Despite the promising results from the various Phase 1 trials, the authors concluded that there is no significant benefit when bortezomib was combined with gemcitabine for the treatment of pancreatic cancer [151]. Due to the fundamental differences in the mechanisms between the various proteasome inhibitors, it is difficult to predict the clinical utility of second generation proteasome inhibitors currently in development. However, encouraging responses in the preclinical models of pancreatic cancer, such as increased tumoricidal response to marizomib compared to bortezomib [131], warrant further evaluation of these potent inhibitors of the proteasome in the treatment of patients with refractory solid organ malignancies.
Effects of Marizomib on Angiogenesis and Autophagy in Pancreatic Carcinoma Cells
Proteasome inhibitors have heterogeneous effects on apoptosis in human pancreatic cancer cells [133]. For example, some lines (L3.6pl, BxPC-3, and CFPac-1) are highly sensitive to low nanomolar concentrations of marizomib or bortezomib, whereas others (MiaPaCa-2, AsPC-1 and HS766T) are highly resistant [133]. Furthermore, xenografts derived from cell lines that are sensitive to proteasome inhibitor-induced apoptosis also display increased apoptosis in response to proteasome inhibitors in vivo, whereas xenografts derived from proteasome inhibitor-resistant lines do not [137]. Resistance to proteasome inhibitors appears to correlate with resistance to gemcitabine and other DNA damaging agents and with epithelial-to-mesenchymal transition (EMT) [152]. Based on a number of different comparisons of the effects of marizomib and bortezomib in human pancreatic cancer cell lines, it was concluded that they exhibit very similar effects at concentrations that produce equivalent effects on 20S proteasome CT-L inhibition. Specifically, it was not possible to overcome resistance to bortezomib-mediated apoptosis using either marizomib alone or in combination with low concentrations of bortezomib (R. Andtbacka, unpublished observations).
However, in vivo studies in orthotopic xenografts demonstrated that proteasome inhibitors also have broad, potent anti-angiogenic effects that contribute to tumor growth inhibition, even in tumors that are completely resistant to direct drug-induced apoptosis [133, 137]. These anti-angiogenic effects have also been observed in other preclinical solid tumor models [153, 154] and produce dose-dependent down-regulation of VEGF expression and tumor microvessel densities [133, 137, 153, 154] and central necrosis [133]. These observations are counterintuitive given what is known about the molecular mechanisms that control VEGF expression, which primary involve hypoxia-inducible factor-1 (and -2) alpha (HIF-1α), a transcription factor that is dynamically regulated by proteasome-mediated degradation [155]. Under conditions of normoxia (in the presence of oxygen), HIF-1α is hydroxylated by a prolyl hydroxylase, promoting recognition by a complex that includes the von Hippal-Lindau (VHL) ubiquitin ligase, resulting in polyubiquitylation and HIF-1α degradation. Because of this, one might expect proteasome inhibitors to promote HIF-1α accumulation and downstream VEGF production. However, proteasome inhibitors appear to uncouple HIF-1α from VEGF transcription via mechanisms that are still poorly understood [128, 156, 157]. Thus, proteasome inhibitor-mediated accumulation of HIF-1α is not associated with increased VEGF transcription.
A second explanation for the VEGF blockade induced by marizomib and bortezomib has been identified. Both drugs actually caused HIF-1α downregulation in some cancer cell lines within 4 h of drug exposure. These effects were linked to phosphorylation of the translation initiation factor eIF2α and downregulation of HIF-1α translation. Thus, cells that displayed proteasome inhibitor-induced eIF2α phosphorylation also displayed HIF-1α downregulation, whereas other cells (including knock-in mouse embryonic fibroblasts (MEFs) expressing a phosphorylation-deficient form of eIF2α) were deficient in both responses, and proteasome inhibitors directly downregulated HIF-1α translation [69]. These results are consistent with other emerging evidence indicating that HIF-1α is also subject to tight translational control through the PI3kinase-Akt-mammalian target of ra-pamycin (mTOR) pathway [158].
From a translational perspective, the anti-angiogenic effects of proteasome inhibitors may be less important in pancreatic cancer than they are in other solid tumors. Pancreatic cancer is characterized by a dense desmoplastic response that may impair angiogenesis and overall tumor blood flow [159], and more specific anti-angiogenic agents like bevacizumab and sunitinib (Sutent, previously known as SU11248) that have produced excellent clinical activity in other models have failed to provide any benefit in pancreatic cancer patients [160]. However, there may be a major role for proteasome inhibitors in clear cell renal cell cancer (RCC), a disease that is characterized by VHL mutations, constitutive HIF-1α and VEGF expression, and high level VEGF production [158, 161]. Although direct VEGF-VEGFR inhibitors and mTOR inhibitors have excellent clinical activity in RCC patients, a significant subset displays de novo resistance and in others therapeutic resistance emerges over time. A Phase 2 trial has been designed to evaluating the effects of combining bortezomib with bevacizumab in patients with RCC to test this possibility (G. Falchook, Principal Investigator). There is clearly an opportunity to better understand the molecular mechanisms that govern sensitivity or resistance to proteasome inhibitor-mediated angiogenesis inhibition and then to use this information to identify the subsets of patients with RCC and other tumors that are highly dependent on VEGF-mediated angiogenesis.
Recent work has also shown that marizomib- and bortezomib-induced phosphorylation of eIF2α promotes autophagy [162], an evolutionarily conserved “recycling” system that directs protein aggregates, other macromolecules, and organelles to lysosomes for degradation and energy liberation [163, 164]. Although some studies suggested that autophagy might act as an alternative programmed cell death pathway in tumors possessing defective apoptotic control mechanisms, more recently, greater consensus has emerged implicating autophagy in tumor cell survival under conditions of low glucose and oxygen [163, 164]. Thus, the anti-angiogenic effects of proteasome inhibitors may be undermined by proteasome inhibitor-induced autophagy. Consistent with this idea, knockdown of critical autophagy pathway genes (Atg5, Atg7) or exposure to chemical inhibitors of autophagy (chloroquine, 3-methyladenine) enhanced marizomib- and bortezomib-induced tumor cell death [162]. These data support the further evaluation of proteasome and autophagy inhibitor combinations in preclinical models to determine their effects on tumor cell death, angiogenesis, and systemic toxicity.
Marizomib in Glioma
Although bortezomib has excellent anti-tumor activity in preclinical in vitro and in vivo models, it has shown minimal clinical efficacy against solid cancers as a single agent or in combination with standard of care treatment protocols [114, 115]; therefore, marizomib has opened new possibilities for the treatment of these challenging malignancies. Glioblastoma multiforme (GBM), the most common astrocytoma and one of the most aggressive solid tumors in humans, has a median survival of about 12 months, after debulking surgery and radiotherapy. The only drug shown to be effective in combination with radiotherapy is temozolomide (Temodal®) [165], therefore, there is a clear need for novel treatment options and drug combinations for this devastating disease. Bortezomib has shown promising results in vitro against glioma cell lines, as a single agent or in combination with chemotheraputic drugs [166–168] however, these results did not translate into an anti-tumor effect in preclinical models of glioma [169]. A recent Phase 1 trial in glioma combining bortezomib with temozolomide and radiation did not show any additional benefit for patients treated with bortezomib [116].
The ability of marizomib to target all three activities of the 26S proteasome [35, 49] makes it a more effective inducer of cancer cell death than bortezomib [57]. Marizomib has already shown to be effective in preclinical models of solid tumors (vide supra) [131, 149], thus making it a good candidate for the treatment of glioma. Besides the potent cytotoxic effects of this drug, potentiated by the prolonged inhibition of all three proteolytic activities of the proteasome, one of the attractive features of marizomib is its low in vivo toxicity, including low toxicity to the brain [52]. Similar to bortezomib [170], a recent study on the PD of marizomib in a preclinical MM.1S murine model demonstrated that marizomib does not cross the blood brain barrier at the doses and schedule tested [52]. However, it is believed that the presence of a malignant glioma in the brain can disrupt the blood-brain barrier, thus allowing cytotoxic drugs that cannot cross a healthy blood-brain barrier, to reach the tumor [171]. Unfortunately, to date, the number of studies that have analyzed the effect of marizomib on preclinical models of glioma are limited. A recent study by Vlashi et al. analyzed the efficacy of marizomib as a single agent or in combination with temozolomide and radiation in a murine glioma xenograft model [167]. In vitro, marizomib showed a dose-dependent toxicity in five different glioma cell lines. Interestingly, its combination with temozolomide resulted in radiosensitization of only the cell lines with a mutated p53 status. It has been demonstrated that proteasome inhibitors sensitize cancer cells to radiation [172]; therefore, given that radiation remains one of the main treatment modalities in glioma, combining a proteasome inhibitor with radiation could result in the improvement of current glioma therapy. In the study by Vlashi et al. the effect of marizomib as a single agent on glioma xenografts in vivo was only modest in controlling tumor growth, and it failed to consistently radiosensitize glioma cells in vitro and did not synergize with radiation in vivo [167]. Overall, these results may indicate that while gliomas in general may be sensitive to marizomib-induced cell death, its combination with radiation may have only little effect in a subgroup of gliomas. Moreover, a recent study showed that glioma stem cells are resistant to proteasome inhibitors [173], further suggesting that proteasome inhibitors have only limited use in the radiotherapy of gliomas. However, the extent of sensitization of cells by proteasome inhibitors (possibly in combination) from cytotoxic agents may depend on the sequence of application [174] and the microenvironment [175]. Given the limited number of preclinical studies performed on this particularly challenging malignancy, it is too early to rule out the possibility that the right drug administration schedule, dose and optimal drug combinations could lead to potential improvement in glioma therapy.
MARIZOMIB SENSITIZES RESISTANT TUMOR CELLS TO APOPTOSIS BY CHEMO- AND IMMUNO-THERAPEUTIC DRUGS
Current treatments for the majority of cancers consist of chemotherapy, radiation, hormonal therapy and immunotherapy and combinations thereof. While most cancer patients initially respond to chemotherapeutic drug treatment, resulting in the inhibition of tumor cell proliferation and survival as well as induction of apoptosis, a subset of patients does not respond initially to such treatment modalities and/or develops cross-resistance following treatment with cytotoxic agents. Several underlying molecular mechanisms have been proposed for the acquisition of tumor cell resistance that help tumor cells evade drug-induced apoptosis. For example, various anti-apoptotic mechanisms result from the hyperactivation of cell survival pathways (such as the MAPK, NF-κB and phosphatidylinositol 3-kinase (PI3K)/Akt pathways) that regulate the transcription and expression of gene products involved in the apoptotic process [176]. However, inhibition of such survival/anti-apoptotic pathways may prevent or reverse tumor cell resistance.
The activation of NF-κB is proposed to be a major pathway for the development of drug resistance. Proteasome inhibitors, which inhibit NF-κB activity (vide supra), may also inhibit the expression and activity of several anti-apoptotic gene products that are under the regulation of NF-κB; thus, marizomib may also sensitize resistant tumor cells to apoptosis. This hypothesis was tested using both carcinoma and lymphoma cell lines for the ability of marizomib to sensitize the resistant tumor cells to immunotherapy (TRAIL as a momodel) and chemotherapy (cisplatin (cis-diamminedichloroplatinum(II) (CDDP)) as a model).
Marizomib-Induced Sensitization to Immunotherapy
Cytotoxic cells (i.e., cytotoxic T-cells (CTL) and natural killer (NK) cells) mediate their cytotoxic activities on tumor cells by several mechanisms, including perforin/granzyme and death receptor signaling. Death receptors become activated after ligation with the corresponding ligands, which constitute TNF family members (TNF-α, FasL, TRAIL) [177]. TRAIL is a type-2 transmembrane protein and induces cell death by apoptosis against a variety of sensitive tumor cell lines after binding to functional death receptors 4 and 5 (DR4, DR5) [178]. The role of marizomib in the response to TRAIL in TRAIL-resistant tumor cell lines was examined. TRAIL was selected based on its poor toxicity to normal tissues and its current evaluation against different tumors in clinical trials [179]. However, most tumors in vivo are resistant to TRAIL-induced apoptosis, and resistance can be reverted by the use of sensitizing agents to modify the anti-apoptotic pathways.
TRAIL-resistant carcinoma and lymphoma tumor cell lines may be sensitized to TRAIL apoptosis via marizomib-induced inhibition of the constitutively activated NF-κB pathway. This hypothesis was tested in prostate (PC-3) and non-Hodgkin’s B-cell lymphoma (B-NHL) tumor cell lines, where marizomib sensitized both cell lines to TRAIL-induced apoptosis in a concentration-dependent manner via direct inhibition of NF-κB and its downstream targets, and behaved similarly to treatment with the specific NF-κB inhibitor, dehydroxymethylepoxyquinomycin (DHMEQ). Marizomib-mediated inhibition of NF-κB activation resulted in inhibition of the NF-κB transcriptional target, Yin Yang 1 (YY1) [180]. YY1 acts as a transcriptional repressor of the TRAIL receptor, DR5, and, thus, the marizomib-mediated inhibition of YY1 resulted in upregulation of DR5 expression [181, 182]. Furthermore, the direct inhibition of YY1 by siRNA mimicked marizomib in its ability to sensitize tumor cells to TRAIL apoptosis through DR5 upregulation. In addition to marizomib-mediated activation of the extrinsic apoptotic pathway, tumor sensitization to TRAIL apoptosis by marizomib also involved activation of the intrinsic apoptotic pathway via increase of the mitochondrial membrane depolarization, inhibition of several anti-apoptotic gene products (such as survivin, Bcl-2, Bcl-xL, XIAP) and induction of pro-apoptotic proteins, such as Bax and Bid. Although the above gene modifications induced after single agent treatment with marizomib were not sufficient for the induction of apoptosis, the combination treatment with TRAIL resulted in a significant synergistic effect for induction of apoptosis through cooperation of both extrinsic and intrinsic apoptotic cascades [182].
An additional mechanism by which marizomib sensitizes tumor cells to TRAIL was examined. Yeung et al. [183, 184] reported that a novel gene product, Raf-1 Kinase Inhibitor Protein (RKIP), inhibits the activation state of both the MAPK and NF-κB pathways. It was recently reported that RKIP transcription is under the negative regulation of Snail, a transcription factor that is positively regulated by NF-κB [185, 186]. Since RKIP induction reverses tumor cell resistance to TRAIL-mediated apoptosis [187], it was hypothesized that marizomib-mediated inhibition of NF-κB may result in the inhibition of Snail and derepression of RKIP. Therefore, RKIP induction by marizomib might play a pivotal role in tumor cell sensitization to TRAIL. Indeed, treatment of tumor cells with marizomib resulted in significant induction of RKIP mRNA and protein expression concomitant with the inhibition of both NF-κB and Snail. The direct role of RKIP induction by marizomib in TRAIL sensitization was corroborated in tumor cells overexpressing RKIP. Such cells were rendered sensitive to TRAIL apoptosis and mimicked marizomib-induced sensitization. In addition, treatment of tumor cells with Snail siRNA resulted in the upregulation of RKIP and sensitization to TRAIL [188]. These studies demonstrate that marizomib sensitizes TRAIL-resistant carcinoma and lymphoma tumor cells to TRAIL-induced apoptosis. The results also demonstrate that marizomib dysregulates the NF-κB/Snail/YY1/DR5/RKIP loop (Fig. (8)). The findings suggest that marizomib may also sensitize resistant tumor cells to ligands other than TRAIL, such as TNF-α and FasL, as well as sensitize the tumor cells to cytotoxic effector cells expressing such ligands.
Fig. 8.
Schematic representation of the involvement of the NF-κB/YY1/Snail/RKIP/DR5 circuit in the regulation of tumor cell resistance to apoptotic stimuli in both chemotherapy and immunotherapy.
Marizomib-Induced Sensitization of Tumor Cells to Chemotherapy
As discussed above for the response to immunotherapy, tumor cells develop cross-resistance to multiple apoptotic stimuli, including chemotherapy. Marizomib-mediated inhibition of major constitutively activated survival pathways, such as the NF-κB pathway, may sensitize resistant tumor cells to chemotherapy. This hypothesis was tested using in vitro CDDP-resistant tumor cell lines and the chemotherapeutic drug CDDP as models. Treatment with marizomib followed by treatment with CDDP resulted in sensitization of CDDP-resistant DU-145 and LNCaP prostate carcinoma and M202 melanoma cell lines to apoptosis. The sensitization and the extent of apoptosis were a function of both the concentrations used by each agent. The combination treatment resulted in synergistic cytotoxicity [188]. The molecular mechanism underlying marizomib-mediated sensitization to CDDP was examined. The direct role of marizomib-induced inhibition of NF-κB in the sensitization to CDDP was corroborated by the use of the NF-κB inhibitor DHMEQ. Cell treatment with DHMEQ sensitized the tumor cells to CDDP-apoptosis, similarly to marizomib-induced sensitization. Marizomib-induced inhibition of NF-κB and the downstream inhibition of the RKIP repressor Snail resulted in upregulation of RKIP expression. Although cell treatment with marizomib or CDDP as single agents resulted in modest activation of the type II apoptotic pathway via measurable inhibition of several anti-apoptotic gene products, the above apoptotic stimuli, separately, were not sufficient to induce a significant potentiation of apoptosis. In contrast, the combination treatment significantly enhanced in the activation of the type II apoptotic pathway and synergistic activity in apoptosis [188]. These and supporting studies demonstrate that marizomib, as shown above for TRAIL, dysregulates the NF-κB/Snail/RKIP circuit which plays a critical role in the regulation of apoptosis by chemotherapeutic drugs.
Mechanism for Reversal of Tumor Cell Resistance to Chemo- and Immunotherapy
The above findings clearly demonstrate that treatment of both solid and hematologic malignances with marizomib results in the reversal of tumor cell resistance to both chemo- and immunotherapy. The reversal of resistance and sensitization by marizomib result in dysregulation of the NF-κB/YY1/Snail/RKIP circuitry, as schematically diagrammed in Fig. (8). Briefly, marizomib treatment results in the inhibition of NF-κB activity through the inhibition of the proteasome and concommitant inhibition of IκBα degradation. The inhibition of NF-κB results downstream in the inhibition of both YY1 and Snail transcription and expression, leading to RKIP induction. Since YY1 is a transcriptional repressor of the TRAIL receptor DR5, its inhibition by marizomib results in the upregulation of DR5 and sensitization to TRAIL apoptosis. In addition, the inhibition of Snail and the induction of RKIP by marizomib further result in tumor sensitization to TRAIL- and CDDP-apoptosis. Furthermore, the marizomib-mediated tumor cell sensitization to CDDP- or TRAIL-apoptosis may also result from the partial activation of the mitochondrial type II apoptotic pathway and inhibition of several anti-apoptotic gene products that play a direct role in the regulation of tumor cell sensitivity to chemo- and immunotherapy. Likewise, the inhibition of Snail and induction of RKIP sensitizes cells to CDDP. Overall, the above findings suggest the clinical application of subtoxic doses of marizomib in combination with conventional and/or new therapeutic drugs in the reversal of both chemo- and immuno-resistance of solid and hematologic tumors.
PHARMACOKINETICS AND ABSORPTION, DISTRIBUTION, METABOLISM AND EXCRETION (ADME) PROPERTIES OF MARIZOMIB
During the course of the preclinical and clinical studies, the pharmacokinetics of marizomib have been described in the cynomolgus monkey and in humans. The ADME characteristics have been described in the rat and mouse, using [3H]-marizomib. Marizomib is well behaved from a pharmacokinetic perspective, and due to the short half-life must be measured in whole blood rather than plasma. The following is an overview of the pharmacokinetic and ADME characteristics of marizomib following IV administration.
Pharmacokinetics
The pharmacokinetic profile of IV-administered marizomib is similar in cynomolgus monkeys and in man, and is characterized by dose-dependent increases in Cmax and area under the total response curve (AUCtotal), with a short half-life, rapid clearance and large volume of distribution. Cmax and AUCtotal have a highly significant linear correlation with dose, with similar values for both monkeys and humans within the same dose range (e.g., Fig. (9) for dosing in the cynomolgus monkey). Clinical studies indicate that exposure (maximum and total) to marizomib does not change upon repeated dosing.
Fig. 9.

Marizomib pharmacokinetics. Cmax and AUCtotal dose relationship after single IV administration to cynomolgus monkeys.
The whole blood concentration-time curves for marizomib from both preclinical and clinical studies clearly distinguish distribution and elimination phases at doses ≥ 0.3 mg/m2, with a terminal half-life in the range of 10–15 minutes. At low doses, the duration of measurable compound in the blood is too short to distinguish these phases. In parallel with the short half-life, the clearance of marizomib is very rapid (e.g. 10–20 L/min in man) and does not show any dose-related trend. The volume of distribution in both monkeys and humans is similar to, or exceeds, total body mass.
ADME
As marizomib is administered by the IV route in clinical studies, the absorption is considered to be 100% with Tmax occurring at the end of the infusion (1–10 minutes, depending on dose). Preclinical studies have demonstrated the potential for an oral route of administration, with a bioavailability of approximately 30–40% in cynomolgus monkeys (when the IV formulation was administered orally).
The distribution of [3H]-marizomib has been determined in normal male Sprague-Dawley rats (quantitative whole body autoradiography (QWBA) study) and in MM.1S tumor-bearing male CB-17 SCID-mice (also see Pharmacodynamics and efficacy of marizomib in a human MM.1S plasmacytoma xenograft murine model). The QWBA study demonstrated that radioactivity is rapidly distributed from the central compartment, with highest concentrations occurring mainly in organs of excretion and metabolism. Interestingly, at the dose studied (0.6 mg/m2), there is good penetration into bone marrow (the site of MM tumor cells) and very little penetration (well below blood levels) into brain regions, spinal cord and peripheral nerves. This latter finding is consistent with the lack of proteasome inhibition following marizomib administration to mice at doses which exhibit a clear anti-tumor activity [52] (Fig. (5)).
The human MM.1S plasmacytoma xenograft murine model study with [3H]-marizomib demonstrated that the parent compound distributes rapidly from the central compartment and enters tumors within minutes of IV administration. This is followed by inhibition of proteasome activity in the tumor within an hour (Fig. (5)). In addition to the extravascular distribution, [3H]-marizomib also binds to cellular blood elements, with maximum binding occurring within one minute [52]. This is consistent with the rapid inhibition of proteasome functions in RBCs and other blood elements.
The metabolism of marizomib has not been defined. The excretion of marizomib occurs via both urinary and fecal routes following IV administration (QWBA study in the rat), without evidence of excretion via expired air. The levels of urinary and fecal excretion are similar in the rat, whereas this has not been assessed in clinical studies.
MONITORING PROTEASOME ACTIVITY AS A POTENTIAL BIOMARKER
Proteasome inhibition profiles have been monitored in multiple clinical trials with marizomib, bortezomib and carfilzomib, and have demonstrated a rapid inhibition of CT-L activities in PWB using fluorogenic assays [189]. However, due to the different properties of these inhibitors and due to the long half-life and inability of RBCs to regenerate proteasomes, it has become clear that PWB while useful, may not be the only cell type of interest to assess clinical activity or response to these types of agents (also see Marizomib Regulates Proteasome CT-L, C-L and T-L Activities and Exhibits a Sustained Inhibition and Pharmacodynamic Profile).
In addition to the importance of the cell type, the assays utilized to determine inhibition profiles can also influence monitoring of proteasome activities. Three different assay methodologies, each with distinct advantages, have been developed to monitor proteasome activities: 1) fluorogenic assays [189]; 2) small molecule based activity assays [190, 191]; and 3) recombinant reporter based assays [192–194]. Although useful in various tissue and cell types, some small molecule and fluorogenic based assays cannot be used to probe living cells, while reporter-based assays are confined to genetically altered cells.
A multi-pronged approach was adopted to evaluate whether monitoring changes in proteasome activity has a potential use as a biomarker. The first approach utilized a fluorogenic-based assay to monitor proteasome activities in plasma and serum. The second approach utilized a fluorescent vinylsulfone based proteasome binding assay, which allows for the detection of labeled proteasome subunits in both live cells and cell extracts by SDS-PAGE, followed by scanning the gel for fluorescent emission [191]. The following describes these different methodologies and their potential usefulness to monitor proteasome activity changes as a potential biomarker.
Profiling Free Circulating Proteasome Activities in Plasma and Serum
It is frequently difficult to access patient cancer cells for analysis, especially when a pure population is desired with minimal or no dilution by normal cells. Increasing evidence suggests that in a patient with a tumor, plasma is enriched by tumor-specific DNA, RNA and protein [195–197]. This enrichment in plasma is believed to be due to the high turnover of tumor cells [197], and may include apoptotic bodies, microvessels, protein complexes or simple debris. In contrast, the products of normal cells are systematically cleared by the reticuloendothelial system and less likely to circulate [195–199]. Despite their size and complex structure, proteasomes circulate in plasma/serum in functional forms; importantly, they can be quantified and their activities easily measured [62, 63, 200]. Furthermore, the circulating proteasomes respond to inhibitors in a fashion similar to that of cell proteasomes [200], which may have some implication for use of proteasome inhibitors in patients with high levels of circulating proteasomes. For example, it is possible that the circulating proteasomes may sequester a portion of the drug, such that higher doses might be needed for patients with high levels of circulating proteasomes.
One of the most important aspects of measuring proteasome protein levels and enzymatic activities in plasma or serum as compared to cells is the ability to express these levels in a specific quantity of plasma or serum, which helps in standardization. By measuring both proteasome protein levels and their enzymatic activities in a specific quantity of plasma or serum, the specific enzymatic activities of the individual proteasome can be determined. This can be calculated by simply dividing the level of an enzymatic activity by the level of proteasome protein in the same quantity of plasma or serum to derive three new measurements: CT-L activity/proteasome protein, C-L activity/proteasome protein, and T-L activity/proteasome protein. These values are important because absolute plasma values of enzymatic activities and proteasomes reflect tumor load, while normalizing activities to quantity of proteasome reflects the specific activity of the proteasome in the cell of origin.
Recent data have shown that circulating proteasome protein levels and their enzymatic activities can be used as biomarkers reflecting the biology of the underlying disease [62]. Therefore, studies were initiated to monitor plasma proteasome activities after treatment with marizomib. When proteasome levels and enzymatic activities were compared in plasma of patients with various cancers treated with low (0.025 or 0.05 mg/m2) and higher doses (0.075 or 0.112 mg/m2) of marizomib, there were significant changes in median proteasome levels at 1h and 4h. Therefore, the plasma proteasome activity was normalized to proteasome levels. Significant changes were observed in the median normalized levels of CT-L, C-L and T-L activity at 1h in low-dose and higher-dose groups that remained detectable at 4h in the higher-dose for CT-L and C-L, but not the low-dose groups. Normalized T-L activity returned to pre-therapy levels at 4h in both groups. These preliminary results support that with increasing doses of marizomib, proteasome activities for CT-L, C-L and T-L were reduced compared to pretreatment levels and that further investigation is warranted to determine the utility of these types of measurements [61].
The fact that each tissue or cell type has a specific number of proteasomes and specific enzymatic activities suggest that profiling the enzymatic activities (CT-L, T-L, and C-L), along with determining the proteasome protein levels in plasma, may be useful for the diagnosis and prediction of the clinical response of a specific cancer. Studies are needed to further explore these possibilities.
Profiling Proteasome Activity in Tissues with Fluorescent Probes
As the second approach, a fluorescent activity-based proteasome probe [191] was developed to profile proteasome activities in PWB and PBMC preparations after treatment with marizomib or bortezomib. Using this probe, we demonstrated that marizomib targeted the β1, β2 and β5 proteasome subunits in rat PBMC lysates, while bortezomib targeted only the β1 and β5 subunits. As a result, a single dose of marizomib (0.05 or 0.10 mg/kg) reduced total proteasome activity by 70–80% while a single dose of bortezomib (0.15 mg/kg) reduced activity by 40%.
The fluorescent proteasome probe was also utilized to profile PWB- and PBMC-derived preparations from cancer patients treated with marizomib. The results indicated that PWB, which contains >98% RBCs, has a different active proteasome subunit composition and recovers proteasome activity markedly slower than PBMCs, consistent with independent results of PD studies (see Marizomib Regulates Proteasome CT-L, C-L and T-L Activities and Exhibits a Sustained Inhibition and Pharmacodynamic Profile). Due to the high proteasome concentration in blood cells, intravenous administration of proteasome inhibitors may rapidly saturate proteasomes in the RBCs before peripheral sites are reached. In addition, studies using the fluorescent activity-based probe demonstrated that proteasome activity levels prior to treatment differed between patients, and that the initial proteasome activity level may be an important determinant of the sensitivity to proteasome inhibitors. Furthermore, it was demonstrated that PBMCs are able to rapidly change their proteasomal subunit composition in response to marizomib treatment, and that this ability may be another factor mediating sensitivity to proteasome inhibition.
In summary, the results suggest that PWB preparations probably do not fully represent the proteasome inhibition profiles in other tissues. Together, the data indicate that fluorescent proteasome probes can be used to profile patient blood samples and may enable prediction of patient responses to proteasome inhibitors, which can contribute to a more personalized proteasome inhibition strategy to improve the therapeutic potential of this class of drugs.
CLINICAL TRIALS WITH MARIZOMIB
The extensive body of preclinical data presented above suggest that marizomib, with its novel structure, produces unique signal transduction, safety and efficacy profiles compared to other proteasome inhibitors, and led to the initiation of clinical trials. The ability of marizomib to synergize with bortezomib and other chemotherapeutics (vide supra) and overcome bortezomib resistance, together with marizomib’s greater therapeutic index and different toxicology profile (lacking neutropenia, thrombocytopenia and neuropathy) suggested that marizomib could be developed and provide unique benefits to patients, particularly those that had failed or were not candidates for treatment with bortezomib. Pre-clinical data showing efficacy in cancers such as CLL and solid tumour malignancies, where bortezomib has not shown efficacy in clinical trials, suggested additional potential.
The clinical evaluation of marizomib has consisted of 4 clinical trials, including 3 single agent Phase 1 studies in patients with solid tumors, lymphomas, leukemias and MM, and one study in combination with the HDAC inhibitor vorinostat in patients with selected advanced malignancies. Each study consists of a dose escalation to a recommended Phase 2 dose (RP2D), followed by a RP2D cohort or Phase 2 portion to gain additional data in specified indications. At the time of writing, over 150 patients have been treated with marizomib at doses ranging from 0.0125 to 0.9 mg/m2, administered on a Days 1, 8, and 15 schedule in 28-day cycles (weekly) or Days 1, 4, 8 and 11 in 21-day cycles (twice weekly).
Clinical development of marizomib began with a Phase 1 dose escalation first-in-human study in patients with solid tumors or lymphomas [201]. As the duration of proteasome inhibition induced by marizomib in PWB is markedly longer than that of bortezomib [49], marizomib was administered once weekly instead of twice weekly. Clinical trials in patients with other diagnoses such as MM and leukemia were subsequently initiated based on preclinical and clinical data. Dose escalation was carried out through a dose of 0.9 mg/m2, with 0.6–0.7 mg/m2 being selected as the RP2D variously in these studies. The most common adverse events reported in marizomib studies included fatigue, nausea, headache, diarrhea, vomiting, constipation, dizziness, infusion site pain, back pain, anorexia, anemia and dyspnea [59]. Proof-of-mechanism, with proteasome inhibition levels (inhibition of CT-L activity in PWB increasing with time and dose, up to 100%; Fig. (3)) reaching and exceeding those reported with therapeutic doses of bortezomib, was attained at lower doses than for bortezomib, supporting the potential for a significantly improved therapeutic ratio. Cumulative or new toxicities did not appear to be elicited with prolonged treatment, with most events occurring in early cycles of therapy. Importantly, marizomib did not appear to induce the limiting toxicities associated with bortezomib, such as peripheral neuropathy, neutropenia and thrombocytopenia, in spite of eliciting levels of proteasome inhibition that equal or exceed those produced by bortezomib. Improvements in tumor assessments have indicated the anti-tumor activity of marizomib in patients, including many with MM previously treated with bortezomib. Together with the preclinical data with other standard of care oncology agents, such as cytotoxic agents, immunomodulatory drugs and HDAC inhibitors, combination clinical trials have been initiated with marizomib. The combination of marizomib with vorinostat has been assessed in a clinical trial, which to date has not revealed adverse drug-drug interactions, or other significantly different safety findings [202].
Further analysis of the PD data demonstrated inhibition of all three proteasome activities in a dose, cycle and schedule dependent manner. Treatment with marizomib resulted in a dose related inhibition of proteasome CT-L activity in PWB as determined 1–4 hours after treatment on days 1 and 15 (Fig. (3)). Maximum inhibition of CT-L activity in PWB reached ~88% at Day 1 and 100% at Day 15 at the highest dose assessed; in contrast, bortezomib given at 1.3 mg/m2 is reported to inhibit 65% of the CT-L activity. At high doses, or after multiple low doses, T-L and C-L activities are also inhibited. Interestingly, as initially reported with marizomib in preclinical models [49], minimal recovery of proteasome inhibition between doses in PWB (principally non-nucleated RBC without the ability to synthesize new proteasomes) was seen, but recovery was rapid in PBMC (nucleated cells that can regenerate proteasomes) (Fig. (4)). Thus, marizomib administered on the same schedule as bortezomib (Day 1, 4, 8 and 11) is now being assessed as well. When administered on this twice weekly schedule, low doses of marizomib administered have been seen to result in increasing inhibition of all three 20S proteasome proteolytic activities that is both dose- and cycle-dependent. No unexpected toxicities have yet been revealed with this alternate schedule, and anti-tumor activity has been seen in patients with MM. Clinical evaluation of marizomib alone and in combination with standard of care cancer therapies continues towards determining the characteristics and optimal uses of this novel agent.
CONCLUSIONS
As the demands for new targeted anti-cancer therapies have increased, it has become important to maximize drug related research and development efforts. The approach to developing marizomib involved an industrial principal, Nereus Pharmaceuticals, collaborating with university laboratories with expertise in chemistry, proteasome biology, hematologic and solid tumor malignancies, and ultimately conducting clinical trials. Marizomib, a β-lactone-γ-lactam that is structurally distinct from bortezomib and carfilzomib, answered the need for chemically and biologically distinct proteasome inhibitors. Marizomib is unique due to its broader, faster acting and durable inhibition of all three 20S proteasome proteolytic activities in various in vitro models, and is efficacious in vivo against hematologic malignancies, such as MM and WM, and solid tumor models, including colon and pancreatic carcinomas.
Marizomib and bortezomib can exhibit synergistic anti-tumor activity in models of MM and WM, which may be attributed, in part, to complementary proteasome inhibition profiles and activation of downstream signally pathways, such as caspase-8 and caspase-9. In contrast, while these agents can synergize in these models, marizomib can overcome resistance to: 1) bortezomib in MM cells that can involve over-expression of Hsp27 and Hsp70; and 2) TRAIL and CDDP in lymphoma and prostate cancer cells, in part by regulating the NF-κB, RKIP, Snail and YY1 circuitry pathway. As in the case of bortezomib, marizomib can regulate multiple NF-κB controlled genes that mediate tumor progression, angiogenesis, metastasis, and apoptosis and that contribute to drug resistance in many tumor types. In addition to inhibiting NF-κB regulated anti-apoptotic pathways, marizomib overcomes survival and drug-resistance conferred by Bcl-2 in MM cells. Marizomib also synergizes with immunomodulatory agents such as Revlimid® in MM and with various HDAC inhibitors such as vorinostat and entinostat in MM, NSCLC and ALL.
The collective preclinical and translational biology studies from these laboratories provided the framework for ongoing clinical trials in patients with MM, lymphoma, leukemia and solid tumors. The findings have been published and/or presented in over 120 different venues and capture the remarkable translational studies that have advanced marizomib from seabed to bench to bedside.
Acknowledgments
We gratefully acknowledge the contributions of the many individuals whose research findings are captured in this review and whose efforts have contributed to the extensive body of knowledge on marizomib. We thank SR Potts for contributing Fig. (1). This work was supported by NIH grants SPORE-P50100707, PO1-CA078378, and NIH RO1CA050947 to DC and KCA; NIH R01CA98871 to JCC; F31 CA123645 to CPM; NIH R01CA115811 to JC; and NCI R37CA044848 to WF.
ABBREVIATIONS
- ADME
absorption, distribution, metabolism and excretion
- ALL
acute lymphocytic leukemia
- API
active pharmaceutical ingredient
- AUCtotal
area under the total response curve
- Bax
Bcl-2-associated X protein
- Bak
Bcl-2 antagonist/killer
- Bcl-2
B-cell lymphoma 2 gene product
- Bcl-xl
B-cell lymphoma x gene product (long form)
- BH3
Bcl-2 homology domain 3
- Bid
BH3-interacting domain death agonist
- BMSCs
bone marrow stromal cells
- B-NHL
B-cell non-Hodgkin’s lymphoma
- CD
cluster of differentiation (or cluster of designation)
- CDDP
cis-diamminedichloroplatinum(II)
- c-FLIP
cellular FLICE-like inhibitory protein
- cGMP
current Good Manufacturing Practice
- C-L
caspase-like
- CLL
chronic lymphocytic leukemia
- Cmax
maximum concentration of drug observed in blood after its administration
- COX-2
cyclooxygenase-2
- CP
core particle
- CRC
colorectal cancer
- CT-L
chymotrypsin-like
- CTL
cytotoxic T-cells
- Diablo
direct IAP binding protein with low pI
- DHMEQ
dehydroxymethylepoxyquinomycin
- DR
death receptor
- EGFR
epithelial growth factor receptor
- EMT
epithelial-to-mesenchymal transition
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FADD
FAS-associated via death domain
- FDA
Food and Drug Administration
- FOLFOX
folinic acid, fluorouracil and oxaliplatin chemotherapy regimen
- 5-FU
5-fluorouracil
- GBM
glioblastoma multiforme
- GI50
concentration of drug required for 50% growth inhibition
- HIF
hypoxia-inducible factor
- Hsp
heat shock protein
- IAP
inhibitor of apoptosis
- IC50
concentration of drug required for 50% inhibition
- ICAM
inter-cellular adhesion molecule
- IETD-fmk
benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylketone
- IgM
immunoglobulin M
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- IL-6
interleukin-6
- IMiD
immunomodulatory drug
- IV
intravenous
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MCL
mantle cell lymphoma
- MEF
mouse embryonic fibroblast
- MHC
major histocompatibility complex
- MKP
MAPK phosphatase
- MM
multiple myeloma
- MMP
matrix metalloproteinase
- MTD
maximum tolerated dose
- mTOR
mammalian target of rapamycin
- NCI
National Cancer Institute
- NF-κB
nuclear factor-kappa light-chain-enhancer of deactivated B Cell
- NK
natural killer
- NSCLC
non-small cell lung carcinoma
- PARP
poly (ADP-ribose) polymerase
- PBMC
peripheral blood mononuclear cell
- PD
pharmacodynamics
- PI3K
phosphatidylinositol 3-kinase
- PUMA
p53 upregulated modulator of apoptosis
- PWB
packed whole blood
- QWBA
quantitative whole body autoradiography
- RANKL
receptor activator of NF-κB ligand
- RBC
red blood cell
- RCC
renal cell cancer
- RKIP
Raf-1 kinase inhibitor protein
- RP2D
recommended Phase 2 dose
- SAHA
suberoylanilide hydroxamic acid
- SAR
structure-activity relationship
- siRNA
small interfering RNA
- Smac
second mitochondria-derived activator of caspases
- TCRs
T-cell receptors
- THF
tetrahydrofuran
- Thr1
N-terminal threonine residue
- T-L
trypsin-like
- Tmax
time of maximum concentration of drug observed in blood after its administration
- TNF
tumor necrosis factor
- TRAF
TNF receptor-associated factor
- TRAIL
TNF related apoptosis inducing ligand
- Ub
ubiquitinated
- UPS
ubiquitinated-proteasome system
- USAN
United States adopted name
- VEGF
vascular endothelial growth factor
- VHL
von Hippal-Lindau
- WM
Waldenstrom’s macroglobulinemia
- XIAP
X-linked inhibitor of apoptosis protein
- YY1
Yin Yang 1
References
- 1.Adams J. Proteasome Inhibitors in Cancer Therapy. Humana Press; Totowa, NJ: 2004. [Google Scholar]
- 2.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 3.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 4.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 5.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, Anderson KC. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–1112. [PubMed] [Google Scholar]
- 6.Russo SM, Tepper JE, Baldwin AS, Jr, Liu R, Adams J, Elliott P, Cusack JC., Jr Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF-kappaB. Int J Rad Onc Biol Phys. 2001;50:183–193. doi: 10.1016/s0360-3016(01)01446-8. [DOI] [PubMed] [Google Scholar]
- 7.Adams J. Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr Opin Chem Biol. 2002;6:493–500. doi: 10.1016/s1367-5931(02)00343-5. [DOI] [PubMed] [Google Scholar]
- 8.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dong L, Castro A, Palombella V, Adams J, Anderson KC. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 9.Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 10.Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O’Connor OA, Soignet SL. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol. 2002;20:4420–4427. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- 11.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 12.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 13.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine DL, Liu K, Cakana A, van de Velde H, Richardson PG. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 14.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O’Connor OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 15.Paoluzzi L, O’Connor OA. Mechanistic rationale and clinical evidence for the efficacy of proteasome inhibitors against indolent and mantle cell lymphomas. BioDrugs. 2006;20:13–23. doi: 10.2165/00063030-200620010-00002. [DOI] [PubMed] [Google Scholar]
- 16.Richardson PG, Briemberg H, Jagannath S, Wen PY, Barlogie B, Berenson J, Singhal S, Siegel DS, Irwin D, Schuster M, Srkalovic G, Alexanian R, Rajkumar SV, Limentani S, Alsina M, Orlowski RZ, Najarian K, Esseltine D, Anderson KC, Amato AA. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J Clin Oncol. 2006;24:3113–3120. doi: 10.1200/JCO.2005.04.7779. [DOI] [PubMed] [Google Scholar]
- 17.Lonial S, Waller EK, Richardson PG, Jagannath S, Orlowski RZ, Giver CR, Jaye DL, Francis D, Giusti S, Torre C, Barlogie B, Berenson JR, Singhal S, Schenkein DP, Esseltine DL, Anderson J, Xiao H, Heffner LT, Anderson KC. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood. 2005;106:3777–3784. doi: 10.1182/blood-2005-03-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15:243–249. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew Chem Int Ed Engl. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 20.Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Discovery and development of the anticancer agent salinosporamide A (NPI-0052) Bioorg Med Chem. 2009;17:2175–80. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam KS, Lloyd GK, Neuteboom ST, Palladino MA, Sethna KM, Spear MA, Potts BC. From natural product to clinical trials: NPI-0052 (salinosporamide A), a marine actinomycete-derived anticancer agent. Royal Society of Chemistry; Cambridge, UK: 2010. pp. 355–373. Vol. Biomolecular Science. [Google Scholar]
- 22.Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3. 4 A resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 23.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2. 4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 24.Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 25.Groll M, Potts BC. Proteasome structure, function, and lessons learned from beta-lactone inhibitors. Curr Top Med Chem. doi: 10.2174/156802611798281320. in press. [DOI] [PubMed] [Google Scholar]
- 26.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 27.Bull AT, Stach JEM. Marine actinobacteria: new opportunities for natural product search and discovery. Trends Microbiol. 2007;15:491–499. doi: 10.1016/j.tim.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Maldonado LA, Fenical W, Jensen PR, Kauffman CA, Mincer TJ, Ward AC, Bull AT, Goodfellow M. Salinispora arenicola gen. nov., sp. nov. and Salinispora tropica sp. nov., obligate marine actinomycetes belonging to the family Micromonosporaceae. Int J Syst Evol Microbiol. 2005;55:1759–1766. doi: 10.1099/ijs.0.63625-0. [DOI] [PubMed] [Google Scholar]
- 29.Mincer T, Jensen P, Kauffman C, Fenical W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl Environ Microbiol. 2002;68:5005–5011. doi: 10.1128/AEM.68.10.5005-5011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fenical W, Jensen PR. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 31.Udwary D, Zeigler L, Asolkar R, Singan V, Lapidus A, Fenical W, Jensen P, Moore B. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci U S A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eustaquio AS, Pojer F, Noe JP, Moore BS. Discovery and characterization of a marine bacterial SAM-dependent chlorinase. Nat Chem Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eustaquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethyl-malonyl-coenzyme A from S-adenosyl-L-methionine. Proc Natl Acad Sci USA. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah IM, Lees KR, Pien CP, Elliott PJ. Early clinical experience with the novel proteasome inhibitor PS-519. Br J Clin Pharmacol. 2002;54:269–276. doi: 10.1046/j.1365-2125.2002.01638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao TH, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom ST, Lam KS, Palladino MA, Potts BC. Structure-activity relationship studies of salino-sporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J Med Chem. 2005;48:3684–3687. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- 36.Potts BC, Lam KS. Generating a generation of proteasome inhibitors: from microbial fermentation to total synthesis of salinosporamide a (NPI-0052) and other salinosporamides. Mar Drugs. 2010;8:835–880. doi: 10.3390/md8040835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nett M, Gulder TA, Kale AJ, Hughes CC, Moore BS. Function-oriented biosynthesis of beta-lactone proteasome inhibitors in Salinispora tropica. J Med Chem. 2009;52:6163–6167. doi: 10.1021/jm901098m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manam RR, McArthur KA, Chao TH, Weiss J, Ali JA, Palombella VJ, Groll M, Lloyd GK, Palladino MA, Neuteboom ST, Macherla VR, Potts BC. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. J Med Chem. 2008;51:6711–6724. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 39.Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical W. New cytotoxic salinosporamides from the marine actinomycete Salinispora tropica. J Org Chem. 2005;70:6196–6203. doi: 10.1021/jo050511+. [DOI] [PubMed] [Google Scholar]
- 40.Manam RR, Macherla VR, Tsueng G, Dring CW, Weiss J, Neuteboom ST, Lam KS, Potts BC. Antiprotealide is a natural product. J Nat Prod. 2009;72:295–297. doi: 10.1021/np800578e. [DOI] [PubMed] [Google Scholar]
- 41.Potts BC, Macherla VR, Mitchell SS, Manam RR, Reed KA, Lam KS, Neuteboom STC, Chao T-H, Nicholson B, Billstrom C. WO 2006/028525 A2. [3.2.0] Heterocyclic compounds and methods of using the same. 2006
- 42.McGlinchey RP, Nett M, Eustaquio AS, Asolkar RN, Fenical W, Moore BS. Engineered biosynthesis of antiprotealide and other unnatural salinosporamide proteasome inhibitors. J Am Chem Soc. 2008;130:7822–7823. doi: 10.1021/ja8029398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macherla VR, Potts BC, Manam RR, McArthur KA, Chao T-H, Neuteboom STC. US 2009/0298906 A1. Proteasome inhibitors. 2009
- 44.Reddy LR, Fournier JF, Reddy BVS, Corey EJ. An efficient, stereocontrolled synthesis of a potent omuralide-salinosporin hybrid for selective proteasome inhibition. J Am Chem Soc. 2005;127:8974–8976. doi: 10.1021/ja052376o. [DOI] [PubMed] [Google Scholar]
- 45.Reed KA, Manam RR, Mitchell SS, Xu J, Teisan S, Chao TH, Deyanat-Yazdi G, Neuteboom ST, Lam KS, Potts BC. Salinosporamides D-J from the marine actinomycete Salinispora tropica, bromosalinosporamide, and thioester derivatives are potent inhibitors of the 20S proteasome. J Nat Prod. 2007;70:269–276. doi: 10.1021/np0603471. [DOI] [PubMed] [Google Scholar]
- 46.Ling T, Potts BC, Macherla VR. Concise formal synthesis of (−)-salinosporamide a (marizomib) using a regio- and stereoselective epoxidation and reductive oxirane ring opening strategy. J Org Chem. 2010;75:3882–3885. doi: 10.1021/jo100432g. [DOI] [PubMed] [Google Scholar]
- 47.Tsueng G, Teisan S, Lam KS. Defined salt formulations for the growth of Salinispora tropica strain NPS21184 and the production of salinosporamide A (NPI-0052) and related analogs. Appl Microbiol Biotechnol. 2008;78:827–832. doi: 10.1007/s00253-008-1358-9. [DOI] [PubMed] [Google Scholar]
- 48.Tsueng G, Lam KS. Stabilization effect of resin on the production of potent proteasome inhibitor NPI-0052 during submerged fermentation of Salinispora tropica. J Antibiot. 2007;60:469–472. doi: 10.1038/ja.2007.61. [DOI] [PubMed] [Google Scholar]
- 49.Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao TH, Neuteboom ST, Richardson P, Palladino MA, Anderson KC. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell. 2005;8:407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 50.Groll M, Huber R, Potts BC. Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 51.Dick LR, Cruikshank AA, Grenier L, Melandri FD, Nunes SL, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin: a central role for clasto-lactacystin beta-lactone. J Biol Chem. 1996;271:7273–7276. doi: 10.1074/jbc.271.13.7273. [DOI] [PubMed] [Google Scholar]
- 52.Singh AV, Palladino MA, Lloyd GK, Potts BC, Chauhan D, Anderson KC. Pharmacodynamic and efficacy studies of the novel proteasome inhibitor NPI-0052 (marizomib) in a human plasmacytoma xenograft murine model. Br J Haematol. 2010;149:550–559. doi: 10.1111/j.1365-2141.2010.08144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chao T-H, Wahlgren B, Obaidat A, Weiss J, Manam RR, McArthur KA, Macherla VR, Palladino MA, Hagenbuch B, Potts BC, Enna SJ, Lloyd GK, Neuteboom STC. The chlorine leaving group of the proteasome inhibitor NPI-0052 is key to its potency, durability of proteasome inhibition and may influence its efflux rate from tumor cells. Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; 2009. p. 4539. [Google Scholar]
- 54.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin : 20S proteasome reveals a molecular basis for selectivity of alpha ‘,beta ‘-epoxyketone proteasome inhibitors. J Am Chem Soc. 2000;122:1237–1238. [Google Scholar]
- 55.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 56.Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, Shenk KD, Bennett MK. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood. 2009;114:3439–3447. doi: 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- 57.Ruiz S, Krupnik Y, Keating M, Chandra J, Palladino M, McConkey D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol Cancer Ther. 2006;5:1836–1843. doi: 10.1158/1535-7163.MCT-06-0066. [DOI] [PubMed] [Google Scholar]
- 58.Hale R, Miller C, Manam RR, Macherla VR, Palladino M, Potts B, Chandra J. Reactive oxygen species (ROS) generation and caspase-8 activation by analogs of proteasome inhibitor NPI-0052. Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; 2009. p. 5634. [Google Scholar]
- 59.Townsend AR, Millward M, Price T, Mainwaring P, Spencer A, Longenecker A. Clinical trial of NPI-0052 in advanced malignancies including lymphoma and leukemia (advanced malignancies arm) J Clin Oncol. 2009;27:15s. #3582. [Google Scholar]
- 60.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67:6383–6391. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 61.Palladino MA, Chao T, Neuteboom ST, Spear M, Ma W, Albitar M. Preclinical and clinical monitoring of cell-and circulating plasma specific proteasome biomarkers after treatment with the proteasome inhibitor NPI-0052. Eur J Cancer Suppl. 2008;6:235. [Google Scholar]
- 62.Ma W, Kantarjian H, Bekele B, Donahue AC, Zhang X, Zhang ZJ, O’Brien S, Estey E, Estrov Z, Cortes J, Keating M, Giles F, Albitar M. Proteasome enzymatic activities in plasma as risk stratification of patients with acute myeloid leukemia and advanced-stage myelodysplastic syndrome. Clin Cancer Res. 2009;15:3820–3826. doi: 10.1158/1078-0432.CCR-08-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jakob C, Egerer K, Liebisch P, Türkmen S, Zavrski I, Kuckelkorn U, Heider U, Kaiser M, Fleissner C, Sterz J, Kleeberg L, Feist E, Burmester GR, Kloetzel PM, Sezer O. Circulating proteasome levels are an independent prognostic factor for survival in multiple myeloma. Blood. 2007;109:2100–2105. doi: 10.1182/blood-2006-04-016360. [DOI] [PubMed] [Google Scholar]
- 64.Kisselev AF, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem. 2006;281:8582–8590. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- 65.Ahn KS, Sethi G, Chao TH, Neuteboom STC, Chaturvedi MM, Palladino MA, Younes A, Aggarwal BB. Salinosporamide A (NPI-0052) potentiates apoptosis, suppresses osteoclastogenesis and inhibits invasion through down-regulation of NF-κB -regulated gene products. Blood. 2007;110:2286–2295. doi: 10.1182/blood-2007-04-084996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson KC. Targeted therapy of multiple myeloma based upon tumor-microenvironmental interactions. Exp Hematol. 2007;35:155–162. doi: 10.1016/j.exphem.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 67.Chauhan D, Hideshima T, Anderson KC. Proteasome inhibition in multiple myeloma: therapeutic implication. Annu Rev Pharm Tox. 2005;45:4655–4476. doi: 10.1146/annurev.pharmtox.45.120403.100037. [DOI] [PubMed] [Google Scholar]
- 68.Chauhan D, Li G, Shringarpure R, Podar K, Ohtake Y, Hideshima T, Anderson KC. Blockade of Hsp27 overcomes bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res. 2003;63:6174–6177. [PubMed] [Google Scholar]
- 69.Zhu K, Chan W, Heymach J, Wilkinson M, McConkey DJ. Control of HIF-1alpha expression by eIF2 alpha phosphorylation-mediated translational repression. Cancer Res. 2009;69:1836–1843. doi: 10.1158/0008-5472.CAN-08-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 71.Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, Munshi N, Palladino MA, Anderson KC. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111:1654–1664. doi: 10.1182/blood-2007-08-105601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roccaro AM, Leleu X, Sacco A, Jia X, Melhem M, Moreau AS, Ngo HT, Runnels J, Azab A, Azab F, Burwick N, Farag M, Treon SP, Palladino MA, Hideshima T, Chauhan D, Anderson KC, Ghobrial IM. Dual targeting of the proteasome regulates survival and homing in Waldenstrom macroglobulinemia. Blood. 2008;111:4752–4763. doi: 10.1182/blood-2007-11-120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richardson P, Lonial S, Jakubowiak AJ, Jagannath S. Lenalidomide, Bortezomib, and Dexamethasone in patients with newly diagnosed multiple myeloma: Encouraging efficacy in high risk groups with updated results of a phase I/II study. Blood. 2008;112:92. [Google Scholar]
- 74.Richardson P, Jagannath S, Jakubowiak AJ, Lonial S. Lenalidomide, Bortezomib, and Dexamethasone in patients with relapsed or relapsed/refractory multiple myeloma: Encouraging response rates and tolerability with correlation of outcome and adverse cytogenetics in a phase II study. Blood. 2008;112:1742. [Google Scholar]
- 75.Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S, Burger P, Rumizen MJ, Podar K, Chauhan D, Hideshima T, Munshi NC, Richardson P, Clark A, Ogden J, Goutopoulos A, Rastelli L, Anderson KC, Tai YT. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol. 2010 doi: 10.1111/j.1365-2141.2010.08127.x. [Epub a head of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev. 2003;29:3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 77.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 78.Panwalkar A, Verstovsek S, Giles F. Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer Biol Ther. 2004;100:1578–1589. doi: 10.1002/cncr.20182. [DOI] [PubMed] [Google Scholar]
- 79.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 80.Hatzimichael EC, Christou L, Bai M, Kolios G, Kefala L, Bourantas KL. Serum levels of IL-6 and its soluble receptor (sIL-6R) in Waldenstrom’s macroglobulinemia. Eur J Haematol. 2001;66:1–6. doi: 10.1034/j.1600-0609.2001.00152.x. [DOI] [PubMed] [Google Scholar]
- 81.Treon SP, Hunter ZR, Matous J. Multicenter clinical trial of bortezomib in relapsed/refractory waldenstrom’s macroglobulinemia: results of WMCTG trial 03–248. Clin Cancer Res. 2007;13:3320–3325. doi: 10.1158/1078-0432.CCR-06-2511. [DOI] [PubMed] [Google Scholar]
- 82.Ahn KS, Sethi G, Aggarwal BB. Embelin, an inhibitor of X chromosome-linked inhibitor-of-apoptosis protein, blocks nuclear factor-kappaB (NF-kappaB) signaling pathway leading to suppression of NF-kappaB-regulated antiapoptotic and metastatic gene products. Mol Pharmacol. 2007;71:209–219. doi: 10.1124/mol.106.028787. [DOI] [PubMed] [Google Scholar]
- 83.Miller CP, Ban K, Dujka ME, McConkey DJ, Munsell M, Palladino M, Chandra J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood. 2007;110:267–277. doi: 10.1182/blood-2006-03-013128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keating MJ, Chiorazzi N, Messmer B, Damle RN, Allen SL, Rai KR, Ferrarini M, Kipps TJ. Biology and Treatment of Chronic Lymphocytic Leukemia. Hematology. 2003;2003:153–175. doi: 10.1182/asheducation-2003.1.153. [DOI] [PubMed] [Google Scholar]
- 85.Chiorazzi N, Rai KR, Ferrarini M. Mechanisms of disease: chronic lymphocytic leukemia. N Engl J Med. 2005;352:804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 86.Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood. 1993;82:1820–1828. [PubMed] [Google Scholar]
- 87.Czuczman MS, Gregory SA. The future of CD20 monoclonal antibody therapy in B-cell malignancies. Leuk Lymphoma. 2010 doi: 10.3109/10428191003717746. Epub, April 2010. [DOI] [PubMed] [Google Scholar]
- 88.Lundin J, Kimby E, Bjorkholm M, Broliden PA, Celsing F, Hjalmar V, Mollgard L, Rebello P, Hale G, Walderman H, Mellstedt H, Osterborg A. Phase II trials of subcutaneous anti-CD52 monoclonal antibody alemtuzumab ( Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL) Blood. 2002;100:768–773. doi: 10.1182/blood-2002-01-0159. [DOI] [PubMed] [Google Scholar]
- 89.Duechler M, Linke A, Cebula B, Shehata M, Schwarzmeier JD, Robak T, Smolewski P. In vitro cytotoxic effect of proteasome inhibitor bortezomib in combination with purine nucleoside analogues on chronic lymphocytic leukaemia cells. Eur J Haematol. 2005;74:407–417. doi: 10.1111/j.1600-0609.2004.00406.x. [DOI] [PubMed] [Google Scholar]
- 90.Pahler JC, Ruiz S, Calvert LR, Andreeff M, Keating M, Faderl S, McConkey DJ. Proteasome inhibitor, bortezomib, on apoptosis in isolated lymphocytes obtained from patients with chronic lymphocytic leukemia. Clin Cancer Res. 2003;9:4570–4577. [PubMed] [Google Scholar]
- 91.Faderl S, Rai K, Gribben J, Byrd JC, Flinn IW, O’Brien S, Sheng S, Esseltine DL, Keating MJ. Phase II study of single-agent bortezomib for the treatment of patients with fludarabine-refractory B-cell chronic lymphocytic leukemia. Cancer. 2006;107:916–924. doi: 10.1002/cncr.22097. [DOI] [PubMed] [Google Scholar]
- 92.Liu FT, Agrawal SG, Movasaghi Z, Wyatt PB, Rahman IU, Gribben JG, Newland AC, Jia L. Dietary flavonoids inhibit the anticancer effects of proteasome inhibitor bortezomib. Blood. 2008;112:3835–3846. doi: 10.1182/blood-2008-04-150227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Golden EB, Lam PY, Kardosh A, Gaffney KJ, Cadenas E, Louie SG, Petasis NA, Chen TC, Schonthal AH. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113:5927–5937. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- Kim TY, Park J, Oh B, Min HJ, Jeong TS, Lee JH, Suh C, Cheong JW, Kim HJ, Yoon SS, Park SB, Lee DS. Natural polyphenols antagonize the antimyeloma activity of proteasome inhibitor bortezomib by direct chemical interaction. Br J Haematol. 2009;146:270–281. doi: 10.1111/j.1365-2141.2009.07752.x. [DOI] [PubMed] [Google Scholar]
- 95.Cortes JE, Kantarjian HM. Acute lymphoblastic leukemia. A comprehensive review with emphasis on biology and therapy. Cancer. 1995;76:2393–2417. doi: 10.1002/1097-0142(19951215)76:12<2393::aid-cncr2820761203>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 96.Haddy TB, Mosher RB, Reaman GH. Late effects in long-term survivors after treatment for childhood acute leukemia. Clin Pediatr (Phila) 2009;48:601–608. doi: 10.1177/0009922809332680. [DOI] [PubMed] [Google Scholar]
- 97.Yang H, Zonder JA, Dou QP. Clinical development of novel proteasome inhibitors for cancer treatment. Expert Opin Investig Drugs. 2009;18:957–971. doi: 10.1517/13543780903002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cortes J, Thomas D, Koller C, Giles F, Estey E, Faderl S, Garcia-Manero G, McConkey D, Ruiz SL, Guerciolini R, Wright J, Kantarjian H. Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res. 2004;10:3371–3376. doi: 10.1158/1078-0432.CCR-03-0508. [DOI] [PubMed] [Google Scholar]
- 99.Lewis BJ, DeVita VT., Jr Combination chemotherapy of acute leukemia and lymphoma. Pharmacol Ther. 1979;7:91–121. doi: 10.1016/0163-7258(79)90026-3. [DOI] [PubMed] [Google Scholar]
- 100.O’Connor OA. Marked clinical activity of the proteasome inhibitor bortezomib in patients with follicular and mantle-cell lymphoma. Clin Lymphoma Myeloma. 2005;6:191–199. doi: 10.3816/CLM.2005.n.046. [DOI] [PubMed] [Google Scholar]
- 101.O’Connor OA, Smith EA, Toner LE, Teruya-Feldstein J, Frankel S, Rolfe M, Wei X, Liu S, Marcucci G, Chan KK, Chanan-Khan A. The combination of the proteasome inhibitor bortezomib and the bcl-2 antisense molecule oblimersen sensitizes human B-cell lymphomas to cyclophosphamide. Clin Cancer Res. 2006;12:2902–2911. doi: 10.1158/1078-0432.CCR-05-0308. [DOI] [PubMed] [Google Scholar]
- 102.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dai Y, Chen S, Kramer LB, Funk VL, Dent P, Grant S. Interactions between bortezomib and romidepsin and belinostat in chronic lymphocytic leukemia cells. Clin Cancer Res. 2008;14:549–58. doi: 10.1158/1078-0432.CCR-07-1934. [DOI] [PubMed] [Google Scholar]
- 104.Miller CP, Rudra S, Keating MJ, Wierda WG, Palladino M, Chandra J. Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: a mechanism for synergy in leukemia cells. Blood. 2009;113:4289–4299. doi: 10.1182/blood-2008-08-174797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;122:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 106.Buglio D, Mamidipudi V, Khaskhely NM, Gloghanni A, Carbone A, Heise C, Besterman JM, Martelli RE, MacBeth KJ, Younes A. The histone deacetylase inhibitor MGCD0103 down regulates CD30. Activates NF-kB and synergizes with proteasome inhibitors by HDAC6 independent mechanism in Hodgkin lymphoma. Blood. 2009;114:3735. [Google Scholar]
- 107.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 108.Place RF, Noonan EJ, Giardina C. HDAC inhibition prevents NF-kB activation by suppressing proteasome activity: down regulation of proteasome subunit expression stabilizes IkBa. Biochem Pharmacol. 2005;70:394–406. doi: 10.1016/j.bcp.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 109.Goy A, Younes A, McLaughlin P, Pro B, Romaguera JE, Hagemeister F, Fayad L, Dang NH, Samaniego F, Wang M, Broglio K, Samuels B, Gilles F, Sarris AH, Hart S, Trehu E, Schenkein D, Cabanillas F, Rodriguez AM. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncology. 2005;23:667–675. doi: 10.1200/JCO.2005.03.108. [DOI] [PubMed] [Google Scholar]
- 110.O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, Dumetrescu O, DE, Trehu E, Adams J, Schenkein D, Zelenetz AD. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncology. 2005;23:676–684. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 111.Kane RC, Dagher R, Farrell AT, Ko CW, Sridhara RM, RJ, Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 112.Buglio D, Georgios GVY, Chao T-H, Neuteboom S, Palladino MAY. A novel proteasome inhibitor, NPI-0052 is active in Hodgkin and Mantle cell lymphoma cell lines. Proceedings of the American Association for Cancer Research Annual Meeting; 2007. p. 1454. [Google Scholar]
- 113.Yazbeck VY, Georgakis GV, Li Y, McConkey D, Andreeff M, Younes A. Inhibition of the pan-Bcl-2 family by the small molecule GX15-070 induces apoptosis in mantle cell lymphoma (MCL) cells and enhances the activity of two proteasome inhibitors (NPI-0052 and bortezomib), and doxorubicin chemotherapy. Blood. 2006;108:253. [Google Scholar]
- 114.Lieu C, Chow L, Pierson AS, Eckhardt SG, O’Bryant CL, Morrow M, Tran ZV, Wright JJ, Gore L. A phase I study of bortezomib, etoposide and carboplatin in patients with advanced solid tumors refractory to standard therapy. Invest New Drugs. 2009;27:53–62. doi: 10.1007/s10637-008-9154-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pugh TJ, Chen C, Rabinovitch R, Eckhardt SG, Rusthoven KE, Swing R, Raben D. Phase I trial of bortezomib and concurrent external beam radiation in patients with advanced solid malignancies. Int J Rad Onc Biol Phys. 2010 doi: 10.1016/j.ijrobp.2009.07.1715. Epub 2010/02/06. [DOI] [PubMed] [Google Scholar]
- 116.Kubicek GJ, Werner-Wasik M, Machtay M, Mallon G, Myers T, Ramirez M, Andrews D, Curran WJ, Jr, Dicker AP. Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys. 2008;74:433–439. doi: 10.1016/j.ijrobp.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kojima M, Morisaki T, Sasaki N, Nakano K, Mibu R, Tanaka M, Katano M. Increased nuclear factor-kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res. 2004;24:675–681. [PubMed] [Google Scholar]
- 118.Cusack JC, Jr, Liu R, Baldwin AS., Jr Inducible chemoresistance to 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothe cin (CPT-11) in colorectal cancer cells and a xenograft model is overcome by inhibition of nuclear factor-kappaB activation. Cancer Res. 2000;60:2323–2330. [PubMed] [Google Scholar]
- 119.Voboril R, Hochwald SN, Li J, Brank A, Weberova J, Wessels F, Moldawer LL, Camp ER, MacKay SL. Inhibition of NF-kappa B augments sensitivity to 5-fluorouracil/folinic acid in colon cancer. J Surg Res. 2004;120:178–188. doi: 10.1016/j.jss.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 120.Cusack JC, Jr, Liu R, Xia L, Chao TH, Pien C, Niu W, Palombella VJ, Neuteboom ST, Palladino MA. NPI-0052 enhances tumoricidal response to conventional cancer therapy in a colon cancer model. Clin Cancer Res. 2006;12:6758–6764. doi: 10.1158/1078-0432.CCR-06-1151. [DOI] [PubMed] [Google Scholar]
- 121.Ding WX, Ni HM, Chen X, Yu J, Zhang L, Yin XM. A coordinated action of Bax, PUMA, and p53 promotes MG132-induced mitochondria activation and apoptosis in colon cancer cells. Mol Cancer Ther. 2007;6:1062–1069. doi: 10.1158/1535-7163.MCT-06-0541. [DOI] [PubMed] [Google Scholar]
- 122.Loeffler-Ragg J, Mueller D, Gamerith G, Auer T, Skvortsov S, Sarg B, Skvortsova I, Schmitz KJ, Martin HJ, Krugmann J, Alakus H, Maser E, Menzel J, Hilbe W, Lindner H, Schmid KW, Zwierzina H. Proteomic identification of aldo-keto reductase AKR1B10 induction after treatment of colorectal cancer cells with the proteasome inhibitor bortezomib. Mol Cancer Ther. 2009;8:1995–2006. doi: 10.1158/1535-7163.MCT-08-0987. [DOI] [PubMed] [Google Scholar]
- 123.Uddin S, Ahmed M, Bavi P, El-Sayed R, Al-Sanea N, AbdulJabbar A, Ashari LH, Alhomoud S, Al-Dayel F, Hussain AR, Al-Kuraya KS. Bortezomib (Velcade) induces p27Kip1 expression through S-phase kinase protein 2 degradation in colorectal cancer. Cancer Res. 2008;68:3379–3388. doi: 10.1158/0008-5472.CAN-07-6109. [DOI] [PubMed] [Google Scholar]
- 124.Yu J, Tiwari S, Steiner P, Zhang L. Differential apoptotic response to the proteasome inhibitor bortezomib [VELCADE, PS-341] in Bax-deficient and p21-deficient colon cancer cells. Cancer Biol Ther. 2003;2:694–699. [PubMed] [Google Scholar]
- 125.Pitts TM, Morrow M, Kaufman SA, Tentler JJ, Eckhardt SG. Vorinostat and bortezomib exert synergistic antiproliferative and proapoptotic effects in colon cancer cell models. Mol Cancer Ther. 2009;8:342–349. doi: 10.1158/1535-7163.MCT-08-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Okumura K, Huang S, Sinicrope FA. Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin Cancer Res. 2008;14:8132–3142. doi: 10.1158/1078-0432.CCR-08-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nowis D, McConnell EJ, Dierlam L, Palamarchuk A, Lass A, Wojcik C. TNF potentiates anticancer activity of bortezomib (Velcade) through reduced expression of proteasome subunits and dysregulation of unfolded protein response. Int J Cancer. 2007;121:431–441. doi: 10.1002/ijc.22695. [DOI] [PubMed] [Google Scholar]
- 128.Mackay H, Hedley D, Major P, Townsley C, Mackenzie M, Vincent M, Degendorfer P, Tsao MS, Nicklee T, Birle D, Wright J, Siu L, Moore M, Oza A. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin Cancer Res. 2005;11:5526–5533. doi: 10.1158/1078-0432.CCR-05-0081. [DOI] [PubMed] [Google Scholar]
- 129.Kozuch PS, Rocha-Lima CM, Dragovich T, Hochster H, O’Neil BH, Atiq OT, Pipas JM, Ryan DP, Lenz HJ. Bortezomib with or without irinotecan in relapsed or refractory colorectal cancer: results from a randomized phase II study. J Clin Oncol. 2008;26:2320–2326. doi: 10.1200/JCO.2007.14.0152. [DOI] [PubMed] [Google Scholar]
- 130.Caponigro F, Lacombe D, Twelves C, Bauer J, Govaerts AS, Marreaud S, Milano A, Anthoney A. An EORTC phase I study of Bortezomib in combination with oxaliplatin, leucovorin and 5-fluorouracil in patients with advanced colorectal cancer. Eur J Cancer. 2009;45:48–55. doi: 10.1016/j.ejca.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 131.Sloss CM, Wang F, Liu R, Xia L, Houston M, Ljungman D, Palladino MA, Cusack JC., Jr Proteasome inhibition activates epidermal growth factor receptor (EGFR) and EGFR-independent mitogenic kinase signaling pathways in pancreatic cancer cells. Clin Cancer Res. 2008;14:5116–5123. doi: 10.1158/1078-0432.CCR-07-4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sloss CM, Wang F, Palladino MA, Cusack JC., Jr Activation of EGFR by proteasome inhibition requires HB-EGF in pancreatic cancer cells. Oncogene. 2010;29:3146–3152. doi: 10.1038/onc.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nawrocki ST, Bruns CJ, Harbison MT, Bold RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J, McConkey DJ. Effects of the proteasome inhibitor PS-341 on apoptosis and angiogenesis in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2002;1:1243–1253. [PubMed] [Google Scholar]
- 134.Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP. 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J Cell Biochem. 2001;82:110–122. doi: 10.1002/jcb.1150. [DOI] [PubMed] [Google Scholar]
- 135.Bold RJ, Virudachalam S, McConkey DJ. Chemosensitization of pancreatic cancer by inhibition of the 26S proteasome. J Surg Res. 2001;100:11–17. doi: 10.1006/jsre.2001.6194. [DOI] [PubMed] [Google Scholar]
- 136.Chandler NM, Canete JJ, Callery MP. Caspase-3 drives apoptosis in pancreatic cancer cells after treatment with gemcitabine. J Gastrointest Surg. 2004;8:1072–1078. doi: 10.1016/j.gassur.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 137.Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ. The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2004;3:59–70. [PubMed] [Google Scholar]
- 138.Bai J, Demirjian A, Sui J, Marasco W, Callery MP. Histone deacetylase inhibitor trichostatin A and proteasome inhibitor PS-341 synergistically induce apoptosis in pancreatic cancer cells. Biochem Biophys Res Commun. 2006;348:1245–1253. doi: 10.1016/j.bbrc.2006.07.185. [DOI] [PubMed] [Google Scholar]
- 139.Khanbolooki S, Nawrocki ST, Arumugam T, Andtbacka R, Pino MS, Kurzrock R, Logsdon CD, Abbruzzese JL, McConkey DJ. Nuclear factor-kappaB maintains TRAIL resistance in human pancreatic cancer cells. Mol Cancer Ther. 2006;5:2251–2260. doi: 10.1158/1535-7163.MCT-06-0075. [DOI] [PubMed] [Google Scholar]
- 140.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K, Jr, Pal A, Bornmann WG, Chiao PJ, Huang P, Xiong H, Abbruzzese JL, McConkey DJ. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–2781. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- 141.Shi YY, Small GW, Orlowski RZ. Proteasome inhibitors induce a p38 mitogen-activated protein kinase (MAPK)-dependent anti-apoptotic program involving MAPK phosphatase-1 and Akt in models of breast cancer. Breast Cancer Res Treat. 2006;100:33–47. doi: 10.1007/s10549-006-9232-x. [DOI] [PubMed] [Google Scholar]
- 142.Small GW, Shi YY, Edmund NA, Somasundaram S, Moore DT, Orlowski RZ. Evidence that mitogen-activated protein kinase phosphatase-1 induction by proteasome inhibitors plays an antiapoptotic role. Mol Pharmacol. 2004;66:1478–1490. doi: 10.1124/mol.104.003400. [DOI] [PubMed] [Google Scholar]
- 143.Hideshima T, Podar K, Chauhan D, Ishitsuka K, Mitsiades C, Tai YT, Hamasaki M, Raje N, Hideshima H, Schreiner G, Nguyen AN, Navas T, Munshi NC, Richardson PG, Higgins LS, Anderson KC. p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene. 2004;23:8766–8776. doi: 10.1038/sj.onc.1208118. [DOI] [PubMed] [Google Scholar]
- 144.Meriin AB, Gabai VL, Yaglom J, Shifrin VI, Sherman MY. Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis. J Biol Chem. 1998;273:6373–9. doi: 10.1074/jbc.273.11.6373. [DOI] [PubMed] [Google Scholar]
- 145.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, Munshi NC, Richardson PG, Hideshima T, Anderson KC. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci USA. 2002;99:14374–14379. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Robertson JD, Datta K, Biswal SS, Kehrer JP. Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem J. 1999;344(Pt 2):477–485. [PMC free article] [PubMed] [Google Scholar]
- 147.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 148.Dudek AZ, Lesniewski-Kmak K, Shehadeh NJ, Pandey ON, Franklin M, Kratzke RA, Greeno EW, Kumar P. Phase I study of bortezomib and cetuximab in patients with solid tumours expressing epidermal growth factor receptor. Brit J Cancer. 2009;100:1379–1384. doi: 10.1038/sj.bjc.6605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ryan DP, Appleman LJ, Lynch T, Supko JG, Fidias P, Clark JW, Fishman M, Zhu AX, Enzinger PC, Kashala O, Cusack J, Jr, Eder JP. Phase I clinical trial of bortezomib in combination with gemcitabine in patients with advanced solid tumors. Cancer. 2006;107:2482–2489. doi: 10.1002/cncr.22264. [DOI] [PubMed] [Google Scholar]
- 150.Ryan DP, O’Neil BH, Supko JG, Rocha Lima CM, Dees EC, Appleman LJ, Clark J, Fidias P, Orlowski RZ, Kashala O, Eder JP, Cusack JCA., Jr Phase I study of bortezomib plus irinotecan in patients with advanced solid tumors. Cancer. 2006;107:2688–2697. doi: 10.1002/cncr.22280. [DOI] [PubMed] [Google Scholar]
- 151.Alberts SR, Foster NR, Morton RF, Kugler J, Schaefer P, Wiesenfeld M, Fitch TR, Steen P, Kim GP, Gill S. PS-341 and gemcitabine in patients with metastatic pancreatic adenocarcinoma: a North Central Cancer Treatment Group (NCCTG) randomized phase II study. Ann Oncol. 2005;16:1654–1661. doi: 10.1093/annonc/mdi324. [DOI] [PubMed] [Google Scholar]
- 152.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Papageorgiou A, Kamat A, Benedict WF, Dinney C, McConkey DJ. Combination therapy with IFN-alpha plus bortezomib induces apoptosis and inhibits angiogenesis in human bladder cancer cells. Mol Cancer Ther. 2006;5:3032–3041. doi: 10.1158/1535-7163.MCT-05-0474. [DOI] [PubMed] [Google Scholar]
- 154.Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001;7:1419–1428. [PubMed] [Google Scholar]
- 155.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 156.Kaluz S, Kaluzova M, Stanbridge EJ. Proteasomal inhibition attenuates transcriptional activity of hypoxia-inducible factor 1 (HIF-1) via specific effect on the HIF-1alpha C-terminal activation domain. Mol Cell Biol. 2006;26:5895–5907. doi: 10.1128/MCB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Birle DC, Hedley DW. Suppression of the hypoxia-inducible factor-1 response in cervical carcinoma xenografts by proteasome inhibitors. Cancer Res. 2007;67:1735–1743. doi: 10.1158/0008-5472.CAN-06-2722. [DOI] [PubMed] [Google Scholar]
- 158.Hudes GR, Berkenblit A, Feingold J, Atkins MB, Rini BI, Dutcher J. Clinical trial experience with temsirolimus in patients with advanced renal cell carcinoma. Semin Oncol. 2009;36(Suppl 3):S26–36. doi: 10.1053/j.seminoncol.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 159.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cartwright T, Richards DA, Boehm KA. Cancer of the pancreas: are we making progress? A review of studies in the US Oncology Research Network. Cancer Control. 2008;15:308–313. doi: 10.1177/107327480801500405. [DOI] [PubMed] [Google Scholar]
- 161.Kaelin WG., Jr Treatment of kidney cancer: insights provided by the VHL tumor-suppressor protein. Cancer. 2009;115:2262–2272. doi: 10.1002/cncr.24232. [DOI] [PubMed] [Google Scholar]
- 162.Zhu K, Dunner K, Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 29:451–462. doi: 10.1038/onc.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 164.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 165.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 166.Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM) Oncogene. 2005;24:344–354. doi: 10.1038/sj.onc.1208225. [DOI] [PubMed] [Google Scholar]
- 167.Vlashi E, Mattes M, Lagadec C, Donna LD, Phillips TM, Nikolay P, McBride WH, Pajonk F. Differential effects of the proteasome inhibitor NPI-0052 against glioma cells. Transl Oncol. 2010;3:50–55. doi: 10.1593/tlo.09244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pedeboscq S, L’Azou B, Passagne I, De Giorgi F, Ichas F, Pometan JP, Cambar J. Cytotoxic and apoptotic effects of bortezomib and gefitinib compared to alkylating agents on human glioblastoma cells. J Exp Ther Oncol. 2008;7:99–111. [PubMed] [Google Scholar]
- 169.Labussiere M, Pinel S, Delfortrie S, Plenat F, Chastagner P. Proteasome inhibition by bortezomib does not translate into efficacy on two malignant glioma xenografts. Oncol Rep. 2008;20:1283–1287. [PubMed] [Google Scholar]
- 170.Phuphanich S, Supko JG, Carson KA, Grossman SA, Burt Nabors L, Mikkelsen T, Lesser G, Rosenfeld S, Desideri S, Olson JJ. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol. 2010 doi: 10.1007/s11060-010-0143-7. Epub 2010 Mar 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zunkeler B, Carson RE, Olson J, Blasberg RG, DeVroom H, Lutz RJ, Saris SC, Wright DC, Kammerer W, Patronas NJ, Dedrick RL, Herscovitch P, Oldfield EH. Quantification and pharmacokinetics of blood-brain barrier disruption in humans. J Neurosurg. 1996;85:1056–1065. doi: 10.3171/jns.1996.85.6.1056. [DOI] [PubMed] [Google Scholar]
- 172.Pajonk F, van Ophoven A, Weissenberger C, McBride WH. The proteasome inhibitor MG-132 sensitizes PC-3 prostate cancer cells to ionizing radiation by a DNA-PK-independent mechanism. BMC Cancer. 2005;5:76. doi: 10.1186/1471-2407-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vlashi E, Kim K, Dealla Donna L, Lagadec C, McDonald T, Eghbali M, Sayre J, Stefani E, McBride W, Pajonk F. In-vivo imaging, tracking, and targeting of cancer stem cells. J Natl Cancer Inst. 2009;101:350–359. doi: 10.1093/jnci/djn509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mortenson MM, Schlieman MG, Virudachalam S, Bold RJ. Effects of the proteasome inhibitor bortezomib alone and in combination with chemotherapy in the A549 non-small-cell lung cancer cell line. Cancer Chemother Pharmacol. 2004;54:343–353. doi: 10.1007/s00280-004-0811-4. [DOI] [PubMed] [Google Scholar]
- 175.Pajonk F, Grumann T, McBride WH. The proteasome inhibitor MG-132 protects hypoxic SiHa cervical carcinoma cells after cyclic hypoxia/reoxygenation from ionizing radiation. Neoplasia. 2006;8:1037–1041. doi: 10.1593/neo.06634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giamas G, Man YL, Hirner H, Bischof J, Kramer K, Khan K, Lavina Ahmed SS, Stebbing J, Knippschild U. Kinases as targets in the treatment of solid tumors. Cell Signal. 2010;22:984–1002. doi: 10.1016/j.cellsig.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 177.Sheikh MS, Fornace AJ., Jr Death and decoy receptors and p53-mediated apoptosis. Leukemia. 2000;14:1509–1513. doi: 10.1038/sj.leu.2401865. [DOI] [PubMed] [Google Scholar]
- 178.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 179.Yagita H, Takeda K, Hayakawa Y, Smyth MJ, Okumura K. TRAIL and its receptors as targets for cancer therapy. Cancer Sci. 2004;95:777–783. doi: 10.1111/j.1349-7006.2004.tb02181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang H, Hertlein E, Bakkar N, Sun H, Acharyya S, Wang J, Carathers M, Davuluri R, Guttridge DC. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol Cell Biol. 2007;27:4374–4387. doi: 10.1128/MCB.02020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Baritaki S, Huerta-Yepez S, Sakai T, Spandidos DA, Bonavida B. Chemotherapeutic drugs sensitize cancer cells to TRAIL-mediated apoptosis: up-regulation of DR5 and inhibition of Yin Yang 1. Mol Cancer Ther. 2007;6:1387–1399. doi: 10.1158/1535-7163.MCT-06-0521. [DOI] [PubMed] [Google Scholar]
- 182.Baritaki S, Suzuki E, Umezawa K, Spandidos DA, Berenson J, Daniels TR, Penichet ML, Jazirehi AR, Palladino M, Bonavida B. Inhibition of Yin Yang 1-dependent repressor activity of DR5 transcription and expression by the novel proteasome inhibitor NPI-0052 contributes to its TRAIL-enhanced apoptosis in cancer cells. J Immunol. 2008;180:6199–6210. doi: 10.4049/jimmunol.180.9.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 184.Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21:7207–7217. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Beach S, Tang H, Park S, Dhillon AS, Keller ET, Kolch W, Yeung KC. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008;27:2243–2248. doi: 10.1038/sj.onc.1210860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L. Activation of NF-kappaB by Akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- 187.Baritaki S, Katsman A, Chatterjee D, Yeung KC, Spandidos DA, Bonavida B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 up-regulation. J Immunol. 2007;179:5441–5453. doi: 10.4049/jimmunol.179.8.5441. [DOI] [PubMed] [Google Scholar]
- 188.Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009;69:8376–8385. doi: 10.1158/0008-5472.CAN-09-1069. [DOI] [PubMed] [Google Scholar]
- 189.Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott PJ. Proteasome inhibition measurements: clinical application. Clin Chem. 2000;46:673–683. [PubMed] [Google Scholar]
- 190.Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods. 2005;2:357–362. doi: 10.1038/nmeth759. [DOI] [PubMed] [Google Scholar]
- 191.Berkers C, van Leeuwen FWB, Groothuis TA, Peperzak V, van Tilburg EW, Borst J, Neefjes JJ, Ovaa H. Profiling proteasome activity in tissue with fluorescent probes. Mol Pharm. 2007;4:739–748. doi: 10.1021/mp0700256. [DOI] [PubMed] [Google Scholar]
- 192.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 193.Lindsten K, Menendez-Beito V, Masicci MG, Dantuma NP. A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol. 2003;21:897–902. doi: 10.1038/nbt851. [DOI] [PubMed] [Google Scholar]
- 194.Luker GD, Pica CM, Song J, Luker KE, Piwnica-worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 195.Rogers A, Joe Y, Manshouri T, Dey A, Jilani I, Giles F, Estey E, Freireich EJ, Keating M, Kantarjian H, Albitar M. Relative increase in leukemia-specific DNA in peripheral blood plasma from patients with acute myeloid leukemia and myelodysplasia. Blood. 2004;103:2799–2801. doi: 10.1182/blood-2003-06-1840. [DOI] [PubMed] [Google Scholar]
- 196.Manshouri T, Do KA, Wang X, Giles FJ, O’Brien SM, Saffer H, Thomas D, Jilani I, Kantarjian HM, Keating MJ, MA Circulating CD20 is detectable in the plasma of patients with chronic lymphocytic leukemia and is of prognostic significance. Blood. 2003;101:2507–2513. doi: 10.1182/blood-2002-06-1639. [DOI] [PubMed] [Google Scholar]
- 197.Giles FJ, Albitar M. Plasma-based testing as a new paradigm for clinical testing in hematologic diseases. Expert Rev Mol Diagn. 2007;7:615–623. doi: 10.1586/14737159.7.5.615. [DOI] [PubMed] [Google Scholar]
- 198.Yeh CH, Tseng R, Albitar M. Plasma-based detection of clonality in lymphoid malignancies. Eur J Haematol. 2009;82:450–453. doi: 10.1111/j.1600-0609.2009.01231.x. [DOI] [PubMed] [Google Scholar]
- 199.Ma W, Kantarjian H, Zhang X, Sun W, Buller AM, Jilani I, Schwartz JG, Giles F, Albitar M. Higher detection rate of JAK2 mutation using plasma. Blood. 2008;111:3906–3907. doi: 10.1182/blood-2008-02-139188. [DOI] [PubMed] [Google Scholar]
- 200.Ma W, Kantarjian H, O’Brien S, Jilani I, Zhang X, Estrov Z, Ferrajoli A, Keating M, Giles F, MA Enzymatic activity of circulating proteasomes correlates with clinical behavior in patients with chronic lymphocytic leukemia. Cancer. 2008;112:1306–1312. doi: 10.1002/cncr.23301. [DOI] [PubMed] [Google Scholar]
- 201.Hamlin PA, Aghajanian C, Younes A, Hong DS, Palladino MA, Longenecker AM, Lloyd GK, Hannah AL, Spear MA, Kurzrock R. First-in-human phase I study of the novel structure proteasome inhibitor NPI-0052. J Clin Oncol. 2009;27:15s, 3516. [Google Scholar]
- 202.Millward M, Spear MA, Townsend A, Sweeney C, Sukumaran S, Longenecker A, Palladino MA, Lloyd GK, Neuteboom STC, Price T. Clinical trial combining proteasome (NPI-0052) and HDAC (vorinostat) inhibition in melanoma, pancreatic and lung cancer. Mol Cancer Ther. 2009;8:Abstr. 107. [Google Scholar]
- 203.Moreau P, Coiteux V, Hulin C, Leleu X, van de Velde H, Acharya M, Harousseau JL. Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. Haematologica. 2008;93:1908–1911. doi: 10.3324/haematol.13285. [DOI] [PubMed] [Google Scholar]
- 204.Vij R, Siegel DS, Kaufman JL, Jakubowiak AJ, Stewart AK, Jagannath S, Kukreti V, Le MH, Bennett MK, Wang M. Results of an ongoing open-label, phase II study of carfilzomib in patients with relapsed and/or refractory multiple myeloma (R/R MM) J Clin Oncol. 2010;28:7s, 8000. [Google Scholar]
- 205.Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, Giai V, Coscia M, Peola S, Massaia M, Pezzoni G, Allievi C, Pescalli N, Cassin M, di Giovine S, Nicoli P, de Feudis P, Strepponi I, Roato I, Ferracini R, Bussolati B, Camussi G, Jones-Bolin S, Hunter K, Zhao H, Neri A, Palumbo A, Berkers C, Ovaa H, Bernareggi A, Inghirami G. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood. 2008;111:2765–2775. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 206.Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70:1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 207.Zhou HJ, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, Ho MN, Jiang J, Kirk CJ, Laidig GJ, Lewis ER, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk KD, Shields J, Shwonek PJ, Stanton T, Sun CM, Sylvain C, Woo TM, Yang J. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047) J Med Chem. 2009;52:3028–3038. doi: 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]