

1. Introduction: MCR Space, Shape and Diversity
Multicomponent reactions (MCRs) are one-pot reactions employing more than two starting materials, e.g. 3, 4, … 7, where most of the atoms of the starting materials are incorporated in the final product.1 Several descriptive tags are regularly attached to MCRs (Fig. 1): they are atom economic, e.g. the majority if not all of the atoms of the starting materials are incorporated in the product; they are efficient, e.g. they efficiently yield the product since the product is formed in one-step instead of multiple sequential steps; they are convergent, e.g. several starting materials combine in one reaction to form the product; they exhibit a very high bond-forming-index (BFI), e.g. several non-hydrogen atom bonds are formed in one synthetic transformation.2 Therefore MCRs are often a useful alternative to sequential multistep synthesis.
Figure 1.

Above: multistep syntheses can be divergent (sequential) or convergent; below: in analogy MCR reactions are convergent and one or two component reactions are divergent or less convergent.
Many basic MCRs are name reactions, e.g. Ugi,3 Passerini,4 van Leusen,5 Strecker,6 Hantzsch7, Biginelli8 or one of their many variations. E.g. in the Ugi reaction the primary scaffold is mostly dictated by the type of acid component (and to a less degree by the amine component), e.g. carboxylic acid, carbonic acid, thiocarboxylic acids,9 HN3, H2O, H2S, HNCO, HNCS, and phenol, which is one of the few recent innovations regarding primary scaffold diversity in Ugi reactions,10 leading to α-acylaminocarboxamides, carbamates, α-acylaminothiocarbonamides, tetrazoles, α-aminoamides, α-aminothioamides, hydantoines, thiohydantoines and α-aminoarylamides.11 Additionally, since MCRs are often highly compatible with a range of unprotected orthogonal functional groups - on a second level - the scaffold diversity of MCR can be greatly enhanced by the introduction of orthogonal functional groups into the primary MCR product and reacting them in subsequent transformations, e.g. ring forming reaction. This two layered strategy has been extremely fruitful in the past leading to a great manifold of scaffolds now routinely used in combinatorial and medicinal chemistry for drug discovery purposes (Fig. 2).12
Figure 2.

The immense scaffold diversity based on MCR is derived from primary (often “classical”) MCRs and secondary reactions made possible by the great functional group compatibility of MCRs (Reprinted with permission from Reference 44. Copyright 2009 ACS.).
Thus the initial MCR derived product can be considered as a synthetic hub to a vast diversity of novel cyclic or acyclic scaffolds by employing different secondary transformations. Typically, only 1-3 synthetic steps are needed to synthesize libraries of drug-like advanced compounds. A versatile example of this strategy are the UDC-procedures (Ugi-Deprotection-Cylization) leading to a great scaffold diversity, e.g. benzimidazoles (1, 2, 3), benzodiazepinedione (4), tetrazolodiazepinone (5), quinoxalinones (6), γ-lactames (7), piperazines (8) (Scheme 1).13
Scheme 1.

The UDC-strategy allows for the great scaffold diversification of an initial Ugi reaction by using orthogonal protected bifunctional starting materials.
The rapid and easy access to biologically relevant compounds by MCRs and the scaffold diversity of MCRs has been recognized by the synthetic community in industry and academia as a preferred method to design and discover biologically active compounds. MCR chemistry has been reviewed multiple times in the past in journals and books, however focusing mostly on diverse synthetic and structural aspects.12,13n,14 The biological activities of MCR derived molecules has been review in the past12,13j,14t,15. However there has never been an extensive summary of the biological properties and potential of MCR derived molecules in one review.15 The biological chemistry of MCRs however is very rich and provides great opportunities for drug hunters and researchers interested in small molecular weight compounds with biological activity. Therefore we want to fill a gap writing this dedicated review on MCR's chemistry and biology. Due to the overwhelming number of published examples of compounds with bioactivity and synthesized by MCR chemistry, however this contribution intends to give an overview based on a personal selection of recent and significant examples rather than a comprehensive review.
Chemical space is the ensemble of all possible molecules, which is believed to contain at least 1060 organic molecules below 500 Da of possible interest for drug discovery.16 This number is mindboggling and impossible to even enumerate or screen. In addition the majority of the compounds - likely - would be very difficult to synthesize or even unstable. An interesting – because synthetically largely amenable – chemical subspace is the MCR chemical space. In the following we therefore define the MCR chemical space as the ensemble of possible molecules which can be synthesized by the multitude of MCR chemistry. This practical definition of chemical space has the advantage of synthetic feasibility which is important to test the computationally driven hypothesis (e.g. similarity, pharmacophore, docking searches). Ultimately, the success of small molecule drug discovery projects depends on the sector of chemical space chosen for discovery, optimization and development. Current design efforts are therefore directed towards target class specific compound libraries.17 The 3-dimensional shape of ligands in addition to electrostatic complementarity between receptor and ligand is one of the most crucial descriptors of bioactive compounds as it determines its interaction with its target(s).18 This has been recently taken into account by designing topography-biased compound libraries using MCR chemistry (Fig. 3).19 Indeed, it can be shown that the 3D-shape space of MCR scaffolds differ considerably from other scaffold spaces. For example some of these MCR libraries are more diffuse than others and conventional backbones, which can be understood based on their different shape and their higher substituent density. The high density of atoms of MCR-based compounds seems to play an important role in their propensity for specific target classes where traditional non-MCR compounds seem to have lower screening hit rates, e.g. protein-protein interactions (PPIs). With this in mind strong emphasis is put on examples with structural and mechanistic information.
Figure 3.

Distributions of MoI-derived shapes for Ro5 compliant libraries deriving from the corresponding color-coded scaffolds (Reprinted with permission from Dr. Akritopoulou-Zanze, Abbott Laboratories).
Chemical transformations towards rare scaffold types annotated with unusual physicochemical properties are amenable by MCR in a straight forward, short manner. E.g. recently, the construction of libraries of bicyclic lactam with bridgehead amide nitrogen (9 and 10) has been reported by the synthesis sequential Ugi/RCM/Heck.20 X-ray diffraction studies revealed that the bicyclic products contain varying degrees of pyramidalization of the bridgehead nitrogen atom (Fig. 4). Such compounds cannot be easily accessed by other chemical methods and certainly not in such a high number and diversity.
Figure 4.
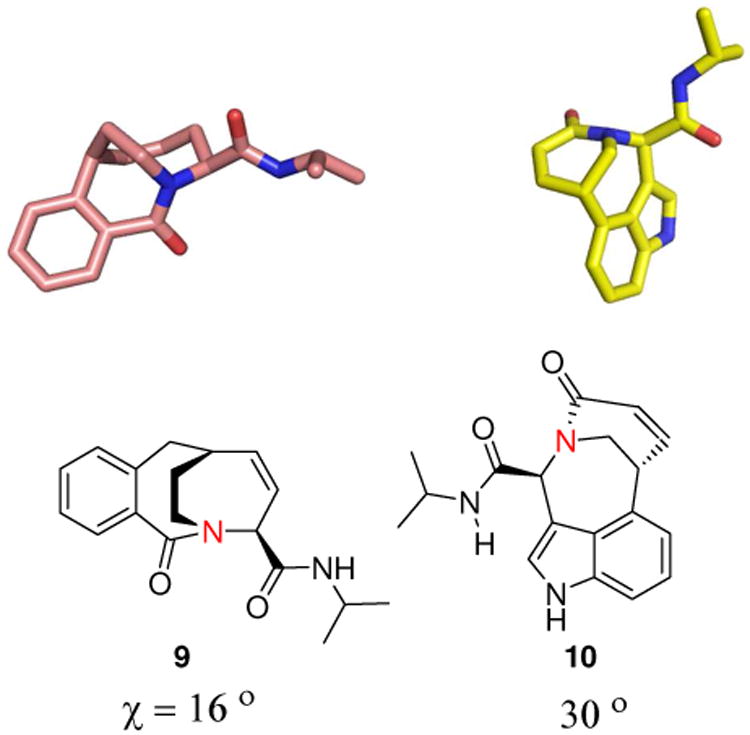
Two examples (9 and 10) of the 3D structure of unususal pyramidalized nitrogen in bicyclic bridgeheaded amides accessible by a 3-step sequence Ugi/RCM/Heck. The pyramidalization χ of planar formamide is 0° and 60° for a fully pyramidalized sp3 atom and is calculated from the X-ray structures.
Another uniquely shaped scaffold, 3-azabicyclo[4.2.0]octan-4-one derivative (15), can be synthesized by combining the Ugi multicomponent reaction with [2+2] enone-olefin photochemical transformations (Fig. 5). During this transformation up to five stereocenters are formed; however in most cases only two diastereomers are observed.21 This scaffold displays a very stiff tricyclic ring system with only minor degrees of rotation. The number of rotatable bonds is a very important parameter in compound optimization as it has major influence on orally bioavailability of drugs and on binding affinity.
Figure 5.
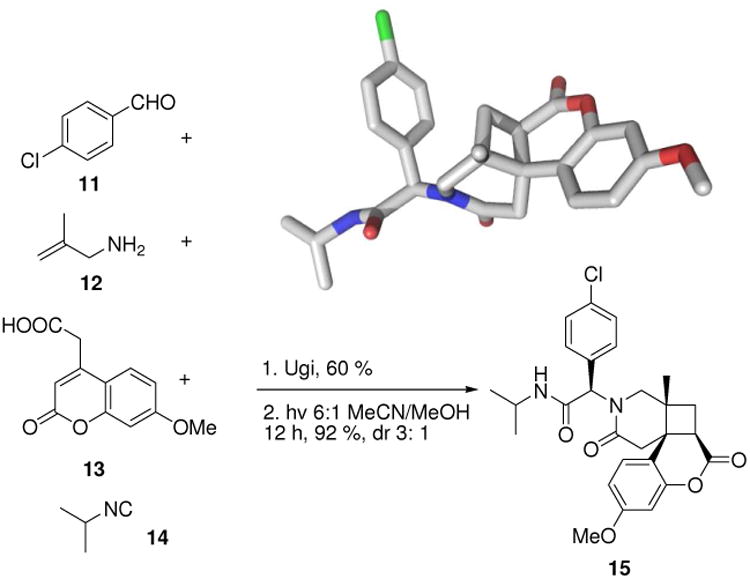
An Ugi MCR involving orthogonal coumarine 13 and allyl moieties 12 followed by a [2+2] photocyclisation leads to unusual densely functionalized scaffolds and libraries thereof.
A third example is the recently described assembly of polycyclic indole alkaloid-type libraries (19) by the combination Ugi/Pictet Spengler reaction (Fig. 6).22 Notable, in this scaffold is the ease of formation of a quaternary carbon stemming from the cyclic oxo carboxylic acid input.
Figure 6.
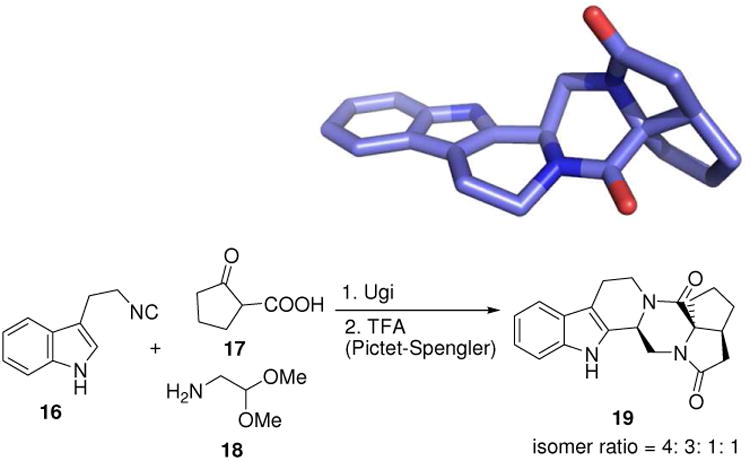
Complex indole natural product-like polycyclic compounds 19 made in two steps from simple commercial starting materials, involving a U-4CR and a subsequent Pictet-Spengler cyclization (CCDC ID: 749252).
A tricyclic scaffold with unusual shape provided by MCRs is the biomimetic transformation of 2-deoxyribose, aryl amine and acetyl acetone under InCl3 catalysis, stereospecifically leading to aminols (23, Fig. 7).23 The reaction typically leads to 1:1 mixtures of two diastereomers and shows considerable scope in the nature of the substitutents of the aniline component (20).
Figure 7.
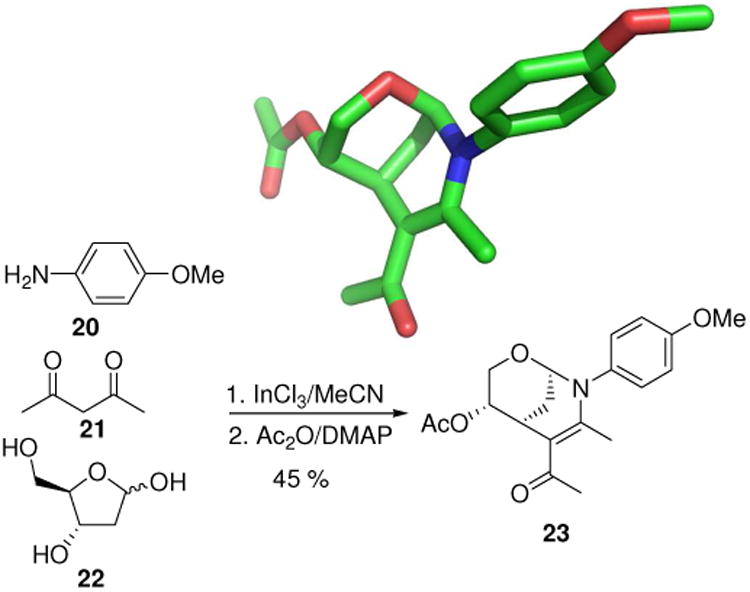
Unusual bicyclic aminol scaffold 23 and 3D structure as determined by X-ray structure analysis (CCDC ID: 675996).
A fragment of repetitive occurrence in investigational drugs is the cyclopropyl group. In addition the cyclopropyl group widely occurs in natural products with often interesting biological activities. Through the synthesis of cyclopropylisocyanides (26) from isocyanoacetic acid esters libraries of cyclopropyl containing compounds (28) can be easily generated under very mild conditions (Fig. 8).24
Figure 8.
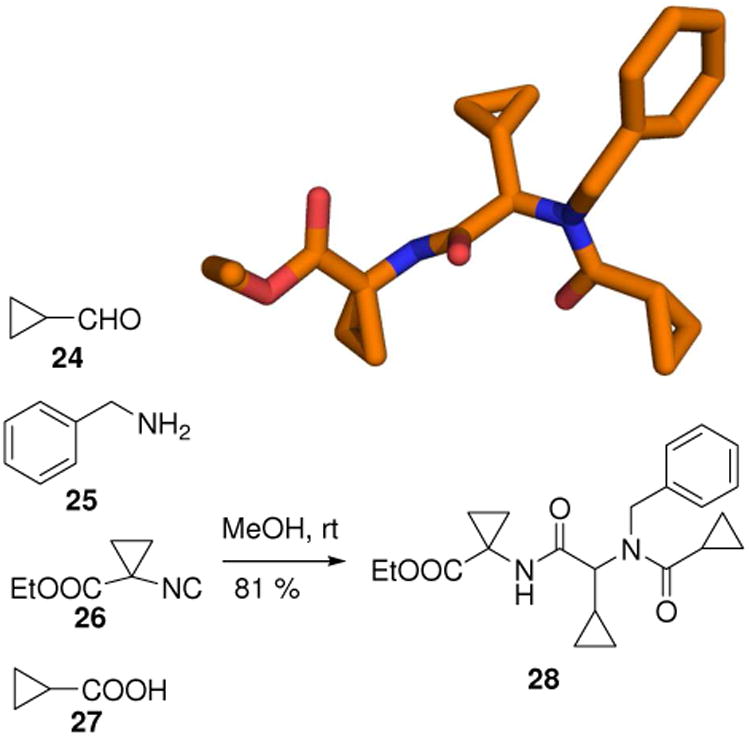
Compounds 28 with three cyclopropyl groups can be easily assembled using a mild and convergent U-MCR (CCDC ID: 604792).
Spirocompounds are considered privileged structures and often show interesting biological activity. They are frequently occurring fragments in drugs and natural products. Spiroheterocycle synthesis can be accomplished using different classes of MCRs. A popular access to stiff spirocycles with indole fragments starts from isatin (29) and cyanoacetic ethyl ester (30) and different classes of bisnucleophiles such as 31. E.g. tetracyclic heterospiro compound 32 can be isolated in 72% yield.25 Additionally, very elegant enantioselective approaches towards spirooxindoles with p53-mdm2 anti-cancer activity using distinct organocascade reactions have been recently published.26
Natural product-like macrocycles have been generated in an efficient sequence involving MCR and different ring closure techniques.27 For example the 22-membered ring compound 38 can be made in three steps from commercially available starting materials using a Passerini-3CR (intermediate 37) followed by a RCM (Fig. 10). It contains several attributes reminiscent to natural products: the different stereo elements, atropisomerism generated by the biphenyl axis, a double bond, a tertiary amide, an ester moiety and a stereogenic carbon. The macrocycle features reduced flexibility due to an intramolecular hydrogen bond. Similar to many natural macrocycles the molecule displays a hydrophobic and a hydrophilic face. In fact synthetic macrocycles are a highly underexploited structural class for drug discovery.28
Figure 10.
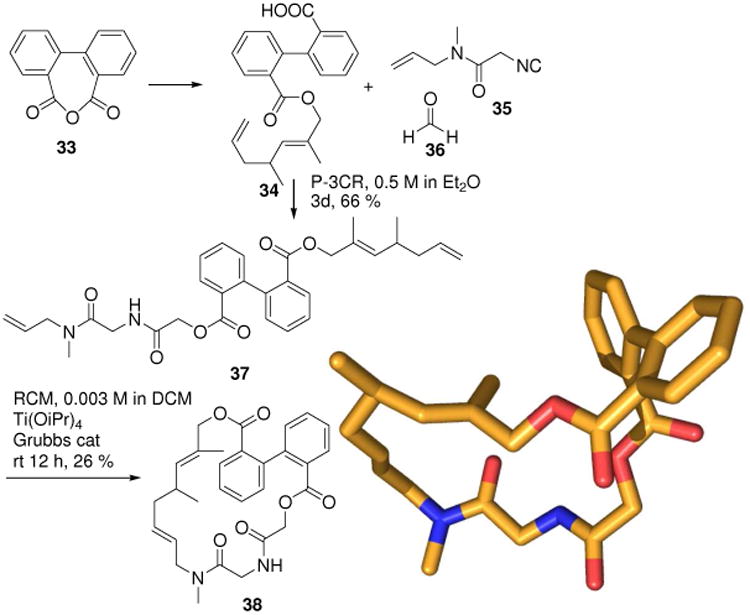
Macrocyclic compounds 38 featuring natural product-like properties can be assembled by an efficient and short three-step sequence involving a Passerini-3CR (CCDC ID: 200226).
An extended bicyclic “flat-land” chemotype 42 can be exemplified in great diversity by employing a three component reaction of 5- and 6-membered (hetero-) aromatic amidines (39), aldehydes (40) and isocyanides (41), an MCR discovered at the same time by three different groups (Fig. 11).29 Clearly, such heterocycles have potential as GPCR and kinase directed agents and several examples will be discussed later on. This very popular MCR has been recently extensively reviewed.30
Figure 11.
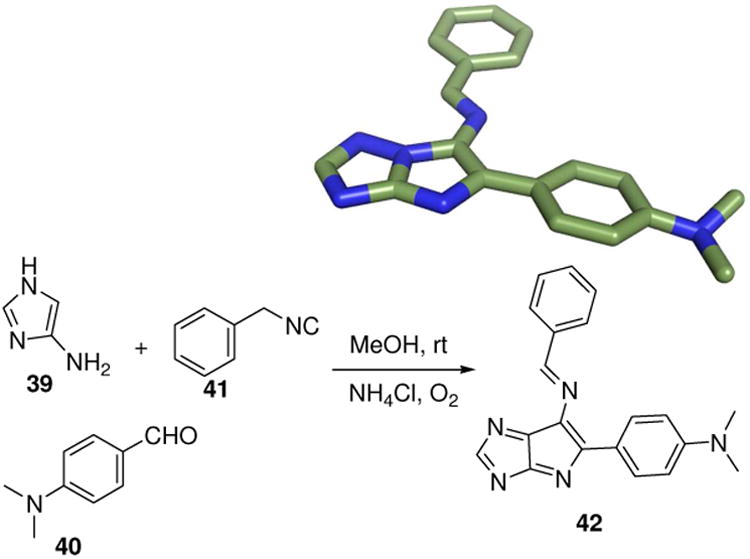
A flat heteroaromatic bicyclic chemotype (42) by the Groebke-Blackburne-Bienaymé-MCR (GBB-MCR) (CCDC ID: 614188).31
The 3D shape, the special arrangement of the H-bond donor and acceptor moieties, the charge distribution of the lead compound and its binding into the target pocket are of great importance for the primary compound-biological target interaction. It also forms the basis of a drug discovery process called scaffold hopping.32 During scaffold hopping an existing biological active scaffold is transformed into a chemically unrelated scaffold with similar biological activity and similar binding features to its biological target. Scaffold hopping is an essential process in order to improve binding, selectivity and ADMET properties but also to create new intellectual property (IP) and to overall improve the chances to successfully manoeuvre projects through development towards the market. In this context it is important to be aware of the diversity of scaffolds offered by a certain type of chemistry. For example in Figure 12 fifteen different piperazines are depicted which can be reportedly accessed by IMCR.33 Optimal leverage of the chemical space offered by MCR chemistry by drug design requires the knowledge of the 2D parameters of the different scaffold as well as their 3D pharmacophore. 2D descriptors for example are the connectivity, the quality and quantity of H-bond donors and acceptors, whereas 3D descriptors are the 3D structure, shape, the 3D H-bond donor and acceptor distribution and directionality.
Figure 12.
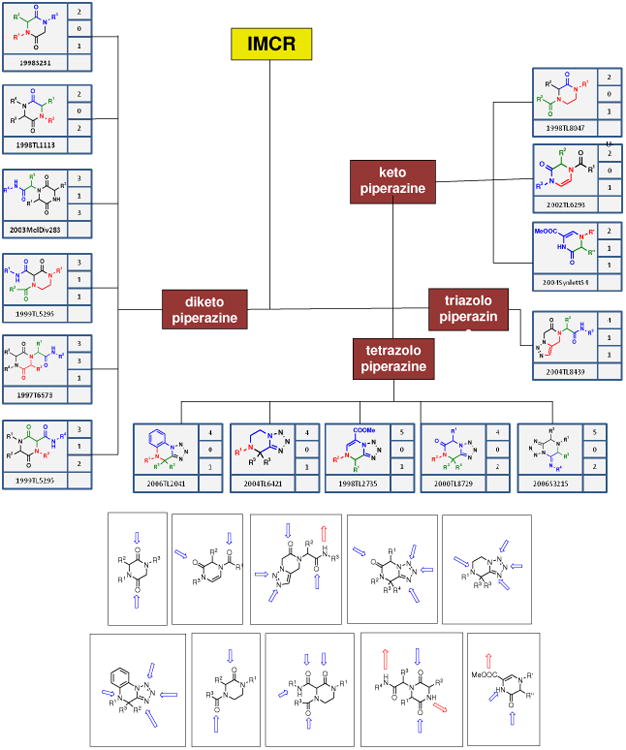
Sector of the piperazine scaffold space offered by IMCR. Above the relationship of 15 different piperazine scaffolds based on different heterocyclic systems and hydrogen-bond donor-acceptor features is depicted. Below several piperazine scaffolds are shown with their imminent 2D hydrogen bond donor acceptor propensity (blue and red arrows, H-bond acceptors and donors, respectively).
Currently, the majority of bioactive compounds based on MCR chemistry belong to only a few scaffold classes. The reason for this is the rapid pace by which the MCR field is moving. Consequently, many new scaffolds have only been recently discovered; therefore the general knowledge about their chemistry and biology is yet poor. For example there are 36 piperazine scaffolds described to be accessible only using isocyanide-based MCR chemistry.33 The majority of these backbones have not been exploited in drug discovery yet. In fact the majority of bioactive molecule reported in this review is based only on a small number of MCRs. These major MCRs are summarized in Table 1.
Table 1.
The majority of bioactive compounds reported here belong to a relatively small number of MCRs.
|
|
Ugi-4CR |
|
|
Ugi-4CR tetrazole |
|
|
Ugi-4CR hydantoine |
|
|
Ugi-5C-4CR |
|
|
Ugi-3CR |
|
|
Groebke- Bienayme- Blackburn- 3CR |
|
|
Passerini-3CR |
|
|
Orru-3CR |
|
|
Van Leusen-3CR |
|
|
Gewald-3CR |
|
|
Hantzsch-3CR |
|
|
Povarov-3CR34 |
|
|
Doebner-3CR35 |
|
|
Biginelli-3CR |
|
|
Betti-3CR |
|
|
Petasis-3CR |
|
|
Staudinger-3CR |
2. MCRs By Target Class
Currently, the number of drug targets is surprisingly low compared to the number of human genes and posttranslational modifications thereof as revealed by the human genome project and work based upon. Thus it has been reviewed that current target counts are of the order of hundreds, whereas estimations of the number of potential drug targets are an order of magnitude higher. Specifically the number of targets for current drugs on the market is only 218.36 Estimates of the total number of targets suitable for drug discovery have been published often referred to as the druggable genome and are between 3,000 and 5,000 depending on the metric.37 Whatever the hypothetical number of targets is, the fundamental question arising is how to connect the chemical space with the biological space to efficiently generate bioactive compounds. In the following we will discuss biological activity of compounds based on MCRs categorized by the different drug targets classes and aim to elaborate the connectivity of chemical and biological space.
2.1. Proteases
Of the >500 known human proteases, >10% are under investigation as drug targets in pharmaceutical industry.38 Additionally, many parasite, bacterial and viral proteases represent important targets for drug discovery.39 Proteases cleave biological material into smaller fragments for metabolic or anabolic purposes. They are involved in all fundamental biological and in many pathogenic processes. Clearly, based on the number of different protease inhibitors in therapeutic use, proteases are druggable, that is small molecular weight inhibitors with suitable pharmacological properties can be developed. An archetypical, highly efficacious and successful class of drugs in this area is the β-lactam antibiotics. The design of protease inhibitors relays often on the powerful idea of transition state mimics. The fundamental idea is to design non cleavable molecular fragments resembling the transition state of the enzyme mechanism and otherwise mimicking the shape and pharmacophore of the central part of the substrate. In another successful approach the active side amino acids or other functional moieties, e.g. metals, are captured by the inhibitor in a covalent or non-covalent manner. These moieties are often called “warheads” since they provide initial inhibitory and mechanism-based activity, whereas potency and selectivity to related targets can be achieved by targeting specific substrate pockets in the proteases. Thus protease inhibitors often contain α-ketoamide, (nor)statine or hydroxamic acid moieties. MCRs are very useful for the rapid assembly of diverse protease-type compound libraries. Already in the 1960s Hagedorn and Eholzer prepared α-hydroxy acid amides and Ugi prepared α-hydroxy tetrazoles by developing special Passerini conditions thus providing the foundation for such powerful protease inhibitor synthesis strategies.40
The most efficient way to access complex, structurally advanced and “screening-ready” α-keto-amide and hydroxymethyl-amide based protease inhibitors scaffolds is the so called Passerini-Reaction-Amine-Deprotection-Acyl-Migration strategy (PADAM) which was independently described by two groups (Scheme 2).41 This elegant 2-3 step sequence involves an initial Passerini reaction of a (chiral) N-protected amino acid derived aldehyde. Upon deprotection of the P-3CR intermediate an O=>N transacylation occurs yielding a hydroxymethyl-amide which eventually can be oxidized to the keto-amide. For e.g. compound 47 comprising a prolyl endopeptidase inhibitor can be assembled in only 3 steps from commercially available starting materials isocyanide 43, aldehyde 44 and carboxylic acid 45, using the PADAM strategy.42 Classical sequential synthesis of compound 47 likely requires many more synthetic steps. Similarly impressively the complex thrombin inhibitor natural product cyclotheonamide C (IC50 = 2.9-200 nM), isolated from the marine sponges Theonella swinhoei and Theonella ircinia, has been assembled with hitherto unreported elegance using PADAM.43 Cyclotheonamide C has been cocrystallized with thrombin representing a model compound for the understanding of the molecular interaction in the complex and the requirements for compounds to effectively inhibit the serine protease (Fig. 13).44 The α-ketoamide fragment derived from the aldehyde component during the P-3CR is covalently attached to the active site Ser195. Respective PADAM sequences of thrombin inhibitors have been performed on a kg scale to obtain material for (pre)clinical development.43b
Scheme 2.

Above: The generalized scheme as an archetypical example to illustrate the synthetic power of MCR chemistry. Middle: In a sequence of only 2-3 steps molecular diversity of high relevance for protease inhibitors (47) is assembled. Below: the complex natural product thrombin inhibitor cyclotheonamide C has been synthesized using this strategy as a key transformation in an unprecedented efficient and convergent approach.
Figure 13.
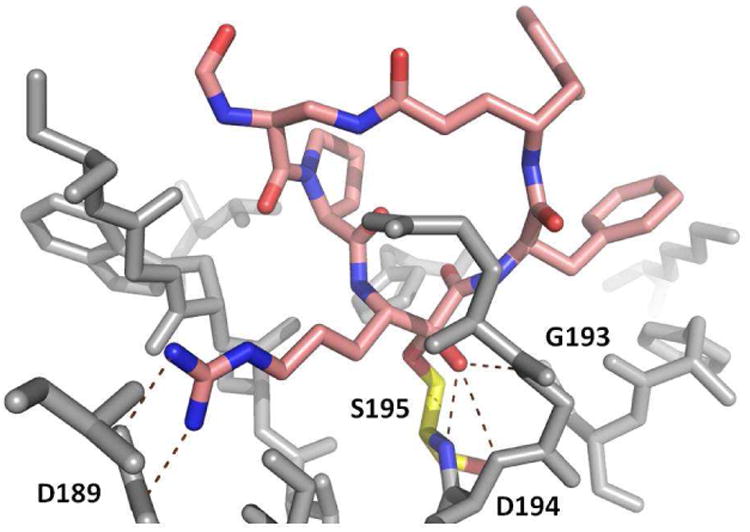
Cyclotheonamide C in complex with human thrombin (PDB ID: 1TYN). Thrombin receptor is shown as grey sticks (several amino acids have been omitted for clarity). Highlighted in pink cyclotheoamide C and in yellow the active side Ser195 forming a covalent hemi acetale bond with the α-ketoamide moiety of cyclotheonamide C. Additionally, the structure is stabilized by a hydrogen bond network of the hydroxyl group of the hemi acetale and backbone amide Gly193, Asp194 and Ser195, the so called oxy anion hole.
Protease inhibitor-type compound libraries have been designed based on the initial discovery by Ugi of the access to hydroxymethyl tetrazoles using a variant of the Passerini reaction: a 3-step short sequence performed with α-amino aldehydes, followed by deprotection and N-functionalization (Scheme 3).45 This reaction sequence has been elaborated for the automated synthesis of ten thousands of compounds, e.g. yielding compounds 51, 52 and 53. Cleary these constitute Asp-protease biased libraries comprising norstatine type motifs. Significantly, recently, several enantio- and diastereoselective approaches towards this important class of biological active compounds have been described, the most efficient one using catalytic amounts of a chiral Al-salen complex.46
Scheme 3.

2-Hydroxy-3-amino-ethyltetrazoles (51-53) as targeted Asp-protease library accessible in high number and diversity by the 3-step sequence Passerini reaction, deprotection and acylation.
A two component Passerini type yields products containing oxazole norstatine-type motifs (56, Scheme 4) in typically very good chemical yield.47 Clearly, this backbone has considerable potential for the design and synthesis of enzyme inhibitors. In addition the oxazole ring hides the otherwise ubiquitary isocyanide secondary amide, thus reducing the number of H-bond donors and acceptors. Recently, a catalytic, highly enantioselective variation of this MCR has been described using a heterobimetallic Ga(OiPr)3/Yb(OTf)3/chiral Schiff base complex.48
Scheme 4.
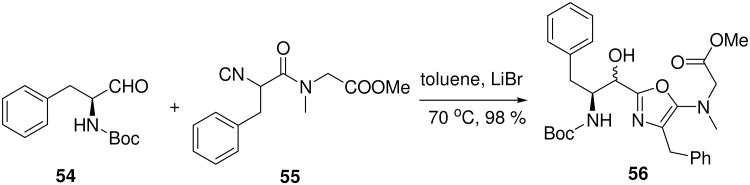
Heterocyclic norstatine 56 accessible by an intramolecular Passerini variation of isocyanoacetamides 55 and α-amino acid derived aldehydes 54.
Other heterocyclic protease inhibitor backbones (57-60) with proteases inhibitory potential, having reduced isocyanide-dependant amide character and being amenable by isocyanide chemistry in just 1-2 steps are shown in scheme 5.49 All these examples have a reduced number of amide bonds as compared to the parent Ugi or Passerini backbone by replacing the amide group by a heterocyclic motif. Clearly, such bioisosteric replacements can potentially greatly enhance the pharmacodynamic and pharmacokinetic properties of their non-heterocyclic isocyanide chemistry parents. Clearly, the secondary hydroxyl function also has potential as protease inhibitor needle.50
Scheme 5.

Various heterocyclic motifs combined with a secondary alcohol amenable by different (intramolecular) isocyanide chemistry variations.
The influence of the α-amino acid N-protecting groups on the degree of racemization during P-3CR and U-4CR was only recently investigated. Their influence turns out to be crucial and is also not constant when the amino acid is changed. After optimization, the Passerini reaction product 63 was obtained with 99% yield and >98% de from cyclohexanone 62 as the carbonyl component (Scheme 6).51 Similar results can be obtained with the Ugi reaction involving chiral α-amino acid derived isocyanides if specific precautions are taken.52 Despite recent innovations, in fact, reliable syntheses of chiral isocyanoacetates have been invented by Ugi and can be accomplished by careful selection of dehydration conditions.53 Also it is well known that dipeptide derived or longer isocyanides are configurationally stable.54 Additionally, orthoesters have been recently introduced as new racemisation free protecting groups for α-amino acid derived isocyanides. These materials have the additional advantage of being solid and odour less.55
Scheme 6.

Ugi and Passerini reaction can be performed under retention of stereochemistry using chiral α-amino acid derived isocyanides.
2.1.1. Serine Proteases
The catalytic mechanism of serine proteases is comparatively well established.56 Serine proteases display a key nucleophilic serine in the active site responsible for cleaving the substrate. Other features characterizing serine proteases include the oxy anion hole, a site nearby the active site serving to stabilise the negatively charged transition state during the nucleophilic attack of the serine onto the cleavable bond. Human and infectious organism derived serine proteases are major targets for pharmaceutical interventions.57 For example, the NS3 protease has been recognized as an essential target to develop treatments for hepatitis C, on which several compounds are currently undergoing advanced clinical trials. Hepatitis C virus is a major worldwide health problem leading to chronic infections in ∼200 million people in addition to the fact that a major fraction of population is a silent carrier of the virus.
However, HCV NS3 protease inhibitor discovery is very challenging since it requires rather large fragments of the natural substrate making the inhibitor molecules quite large, with many chiral centers and thus difficult to synthesize. An often reoccurring key element in many HCV NS3 protease inhibitors, the α-ketoamide structure can be synthesized using the classical Passerini reaction or the PADAM strategy (Scheme 7). During the discovery of α-ketoamide HCV NS3 protease inhibitors, for example this reaction was instrumental in order to optimize the C-terminal part of the inhibitors residing near the active site.58 Cyclic and acyclic HCV NS3 protease have been described and synthesized using key Passerini transformations.59 For example, the exocyclic α-ketoamide unit in compound 64 and similar compounds has been synthesized using a P-3CR followed by oxidation of the secondary hydroxyl group.
Scheme 7.

Use of Passerini reactions to convergently synthesize the α-ketoamide fragment, which is essential in many classes of serine protease inhibitors.
Numerous co-crystal structures between α-ketoamide inhibitors and the HCV NS3 protease have been recently solved and show key molecular interactions with the different functional moieties (Fig. 14). A macrocyclic HCV inhibitor 64 and 65 features a 16-membered ring encircling Ala156 in a “donut-shaped” conformation thus providing many hydrogen bonds and additional van der Waals contacts.60 The n-propyl norvaline side chain fits very well into the S1 pocket. This side chain is introduced via the aldehyde component in the P-3CR. Boceprevir is the first-in-class recently approved HCV NS3 inhibitors which showed excellent clinical trial results.61 It is a linear and primary α-ketoamide with oral bioavailability (Fig. 14). The keto moiety forms a reversible covalent adduct with the active site Ser139. An extended network of hydrogen bonds of the peptidic backbone to the HCV NS3 protease is formed. Additionally, strong hydrogen bond interactions are made by the oxy-anion hole amino acids Ser138 and Gly137. By forming a covalent adduct the enzyme mechanism is inhibited.
Figure 14.

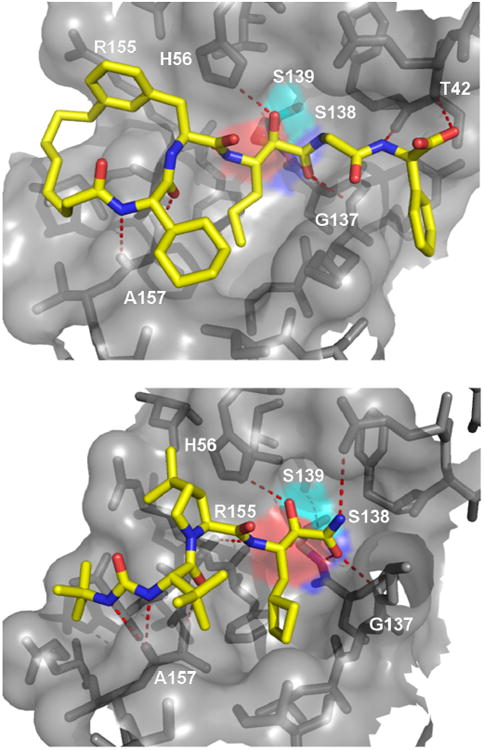
Atomic details of a macrocyclic (64, above) and linear (Boceprevir, below) α-ketoamide HCV NS3 protease inhibitors (PDB-IDs: 2A4Q and 2OC8). The active Ser is marked by a cyan surface, the inhibitor by yellow sticks and the binding surface of the protease is shown as grey surface and sticks.
Factor-Xa (FXa) is an important blood coagulation medicinal chemistry target. Non-covalent FXa inhibitors based on the phenylglycine backbone have been disclosed (Scheme 8).62 The Ugi chemistry represented an intriguing approach to this scaffold and offered the potential advantage of enabling to draw upon the commercial availability of a wide variety of aryl aldehydes as the requisite starting materials. It has been noted that despite the moderate yield (not optimized 24%) of the U-4CR to form racemic compound 70, the Ugi route was found to be superior to alternate approaches involving the synthesis of 2-thiazolyl glycine for multigram preparation of compound 72. Additionally, it has to be noted that recently, the very mild cleavable chiral 4-methoxy-1-ethylamino group has been introduced in Ugi chemistry as a chiral auxiliary.63 This method makes the synthesis of even very racemisation-prone chiral N-acylaminoamides possible.63
Scheme 8.

Convergent FXa inhibitor (72) synthesis by U-4CR.
An interesting approach to screen the immensely large chemical space of MCR chemistry, however physically synthesizing only a small fraction of possible compounds is the genetic algorithm (GA).64 GA is an optimization method that uses techniques inspired by evolutionary biology such as inheritance, fitness, mutation, selection, and crossover (also called recombination). GAs are advantageously applied in complex systems whenever exact solutions cannot be generated, e.g. drug discovery. In one application of GAs potent thrombin inhibitors (220 nM) have been found within a chemical space of 320,000 U-3CR and U-4CR products based on 10 isocyanides, 40 aldehydes, 10 amines and 40 carboxylic acids. The starting material classes represent the different gene classes. 20 Starting compounds based on the theoretical MCR space have been generated randomly in a first generation. These are screened for their inhibitory activity against thrombin (fitness function). The best compounds are computationally stored and are also allowed to undergo recombination and mutation, thus ensuring survival of the most active structures and “breading” of even more active structures in the next generation. After only 16 generations of evolution the average effective inhibitory activity of the 20 best products at each generation was submicromolar. In generation 18 after physically synthesizing only 400 products out of a theoretic space of 320,000 compounds the highly active compound 73 was found (Fig. 15). This approach is highly significant as it can systematically and effectively search very large chemical spaces provided by MCR chemistry while having to synthesize only a small number of compounds. It does not require structural insight into the target nor does it require target knowledge at all (e.g. using a phenotypic assay).
Figure 15.
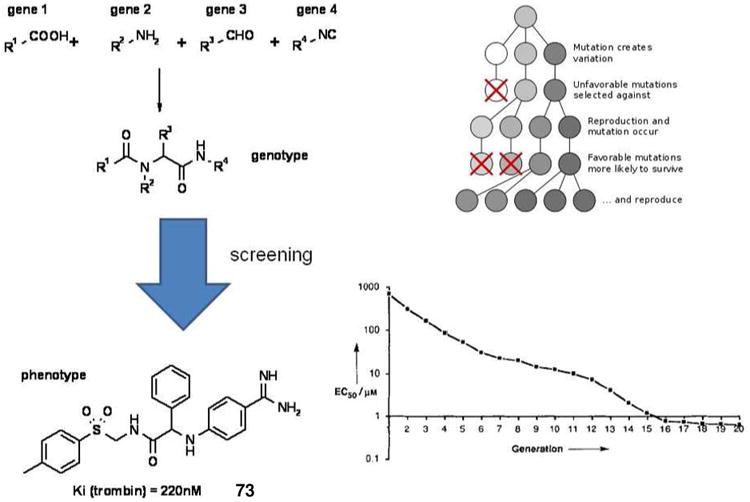
U-4CR and U-3CR based generation of potent and selective thrombin inhibitors (73) using genetic algorithm techniques. In the graph the evolution of active compounds (EC50) over the number of generations is shown.
Factor VIIa (FVIIa), another key intervention point of the blood coagulation cascade has been extensively targeted with MCR chemistry. A potential advantage of targeting FVIIa over FXa is that specific inhibition of the TF/FVIIa complex results in an antithrombotic effect without enhancing bleeding propensity, a possible side effect of coagulation inhibitors.65 Synthesis of the N-aroyl phenylglycine derivatives 77 involves a BF3-catalyzed addition of the diaroyl Schiff base in ethanol onto a suitable isocyanide (benzyl or morpholinoethyl 74) (Scheme 9). The intermediate ethyloxyimidine 77 has to be extensively hydrolyzed and the isocyanide only contributes the carbon resulting in the carboxylic carbonyl.66
Scheme 9.

Synthesis of an oral bioavailable, highly potent and selective FVIIa inhibitors 78 involves a U-3CR variation.
An advanced compound 78 had good potency and selectivity, was oral active as a double prodrug in the guinea pig and showed a dose-dependent antithrombotic effect in an established model of arterial thrombosis without prolonging bleeding time. This compound has also be crystallized with its target (Fig. 16).67 The amidine group forms a strong charge charge complex with the Asp189 at the bottom of the S1 pocket. The aniline NH forms a hydrogen bond to Ser195 and the carboxylate of the amino acid another favourable charge charge interaction to Lys192. Selectivity to the related thrombin pocket can be accomplished by the introduction of the m-ethoxy group which cannot be accommodated easily in thrombin.
Figure 16.
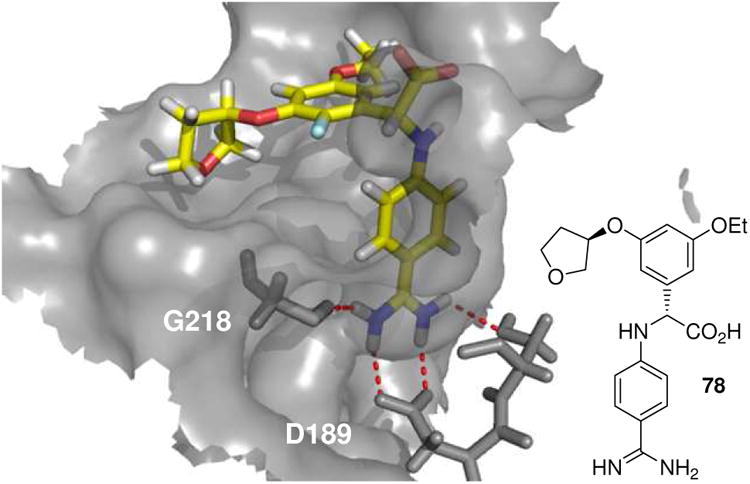
Phenylglycine derivative 78 co-crystallized with FVIIa (PDB ID: 2BZ6).
The human cytomegalovirus (HCMV) protease catalyzes the maturational process of the herpes virus assembly protein and plays a key role during the manufacture of the viral capsid. It is an attractive target for potential anti-herpes-virus agents with novel structures and new mechanisms. A chemical library containing 32 compounds with different substitutions on the U-4CR skeleton and incorporating an α-ketoamide moiety was prepared by the oxidation of a precursor α-hydroxylamide library, which was constructed from the four types of building blocks: 4 carboxylic acids, 2 amines, 2 aldehydes and 2 hydroxyl group containing isocyanides based on a U-4CR following liquid phase strategies.68
The natural product and proteasome inhibitor omuralide has been synthesized in a stereo controlled manner using a intramolecular U-4CR of the ketocarboxylic acid 79 as a key step (Scheme 10).69 Herein a novel convertible isocyanide, 1-isocyano-2-(2,2-dimethoxyethyl)benzene (80) was used, which was introduced independently by two groups.70 The p-methoxybenzylamine 81 is used as an ammonia surrogate. The indole acyl of the intermediate 82 resulting from the convertible isocyanide can be cleaved under very mild conditions.
Scheme 10.

Synthesis of proteasome inhibitor and natural product omuralide by an elegant short sequence involving an intermolecular and highly stereoselective U-4CR using a new cleavable isocyanide.
Dipeptidyl peptidase IV (DPP-IV) is a serine protease that degrades the incretin hormone glucagon-like peptide 1 (GLP-1), a peptide required for the glucose-dependent regulation of insulin. Inhibition of DPP-IV is a very successful therapeutic principle: Vildagliptin (Scheme 11), FDA approved for diabetes type-2 treatment, increases the level of active GLP-1, resulting in improved glucose tolerance. The common pharmacophore of many current DPP-IV inhibitors is an α-amino nitrile. On the basis of the crystal structures of chemically related pyrrolidine nitriles with DPP-IV, it is believed that the α-amino nitrile forms a reversible covalent imidate ester adduct with the active site serine (Ser610).71 Interestingly, α-amino nitriles are accessible in two different ways using Ugi-type MCRs. First, the reaction of amino acid derived α-amino amides with oxocomponents and isocyanides, surprisingly yield α-aminoacyl nitriles (compound 83).72 Second, the reaction of amino acid derived α-amidoisocyanides also yields α-aminonitriles (compound 84).73 Both reactions are clearly complementary since they represent different scaffolds and populate different areas of the chemical space of α-amino nitriles. Additionally, different starting materials are utilized in both reactions.
Scheme 11.

The marketed DPP-IV inhibitor vildagliptin and two complementary MCR approaches towards the pharmacophore α-amino nitrile.
2.1.2. Aspartyl Proteases
Aspartyl proteases – disproportionally underrepresented in the proteasome as compared to serine proteases, however are a very important and successful class of targets.74 In fact more drugs against Asp proteases are approved than for all other protease classes together. For e.g. renin is a major target for cardiovascular diseases. The renin-angiotensine-aldosterone system (RAS) has a key role in the regulation of blood pressure and has yielded already three important drug classes, the aldosterone receptor antagonists, the AT1 receptor blocker and the ACE inhibitors.75 Renin inhibitors are expected to partly replace the therapeutic importance of the ACE inhibitors. Currently, the one renin inhibitor approved is aliskiren, a secondary hydroxyl transition state mimic. Notably, aliskiren is a rather complex molecule incorporating 4 stereocenter and has to be synthesized by a lengthy 20 step synthesis.76 Most of the currently described renin inhibitors incorporate similar hydroxyl needles. A decade ago, however, 3,4,5-trisubstituted piperidines (85) have been described as renin inhibitors. It was shown by X-ray structure analysis that this class of compounds induce a major rearrangement in the active site.77 Recently, a piperazine-imidazole class of Asp protease inhibitors, for e.g. compound 87 was described which is convergently amenable by van Leusen's MCR from substituted TOSMICs, aldehydes and 4-aminopiperidine (86) under protecting group free conditions (Scheme 12).78
Scheme 12.

Approved renin inhibitor aliskiren, an early piperidine inhibitor (85) and general one-pot synthesis of 1,4,5-trisubstituted imidazole using van Leusen's 3CR of TOSMICs, aldehydes and unprotected 4-aminopiperidine (86).
The binding mode of aliskiren and the piperidine inhibitors (85 and 87) is quite different. Aliskiren acts as a classical substrate mimic.79 The X-ray structure of a piperazine inhibitor together with a modelled representative piperazine-imidazole 87 is shown in Figure 17.
Figure 17.
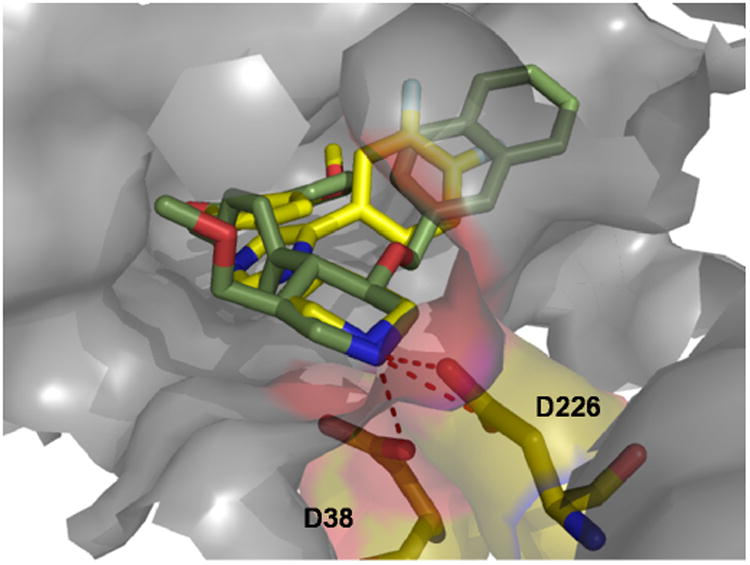
Inhibitor 85 (green sticks) (PDB ID: 1PR8) and a docked piperazine imidazole inhibitor 87 (yellow sticks) bound into a very deep cleft in renin. The piperazine-N is sandwiched between the two active side Asp38 and Asp226 (pink sticks) and replacing the active water.
The chemistry of tosylmethyisocyanide (TOSMIC) and derivatives was started by the Dutch chemist van Leusen.80 TOSMICs display a high functional group density. Thus TOSMIC chemistry is determined by three distinct properties: the isocyanide reactivity, the strong α-acidity of the adjacent methylene group embedded between the two electron withdrawing sulfone and isocyanide group (N, S-acetal) and the leaving group ability of the sulfone group (Figure 18). As a result TOSMIC chemistry is very versatile and is now widely used for the synthesis of many different heterocyclic systems. An outstandingly useful MCR is the vL-3CR which can lead to 1,4,5-trisubstituted, 4,5-, 1,4-and 1,5-disubstituted or 1-, 4- and 5-monosubstituted imidazoles. The mechanism involves Schiff base formation, addition of the isocyanide carbanion to the imine and subsequent ring closure and sulfinic acid elimination. This reaction likely can be considered as the most versatile to substituted imidazoles. Additionally, due to the availability of many α-substituted TOSMICs the accessible imidazole chemical space is very large.80a,81 The imidazole scaffold is incorporated in quite a number of drugs.
Figure 18.

TOSMIC is a densely functionalized reagent, which accounts for its versatility in different reaction pathways.
Cerebral deposition of amyloid β-peptide (Abeta) is an early and critical feature of Alzheimer's disease. Abeta generation in the brain depends on proteolytic cleavage of the amyloid precursor protein (APP) by two proteases: β-secretase (BACE) and γ-secretase. These proteases are prime therapeutic targets.82 β-Secretase belongs to the small class of human aspartyl proteases. Recent inhibitors are mostly of complex, peptide-like structure enriched in asymmetric carbons and in amide bonds, build around a warhead statine motif.83 Additionally, development of β-secretase inhibitors is challenging since the target protein is compartmented in the brain; thus inhibitors must penetrate the blood-brain-barrier (BBB). Recently, hydantoine based inhibitors (88-90) have been described which can be synthesized in a 3-step sequence involving a one-pot MCR using a variation of the classical Ugi MCR.84 In this reaction, a primary amine a piperidine-4-one, and isocyanide and potassium cyanate react to yield iminohydantoine (Scheme 13).
Scheme 13.

Synthesis of spiropiperidine-hydantoine-4-imides (88-90) by Ugi-MCR and representative BACE inhibitors with their bioactivity.
An X-ray structure analysis of a cocrystal of the small molecular weight inhibitor 90 and BACE-1 revealed a novel mode of binding whereby the inhibitor interacts with the catalytic aspartates via bridging water molecules (Fig. 19). Libraries of spirocyclic heterocycles have been prepared in a one-pot fashion using a variation of the Ugi MCR. Noteworthy is the ease of formation of the quaternary carbon center at room temperature, which is a general consequence of using ketones in the Ugi reaction. The design and synthesis of spirocycles is a challenging task because it involves the creation of a quaternary center, which itself is considered to be one of the most difficult tasks among synthetic transformations. Iminohydantoins in principle can exist in different tautomeric forms, however analysis of the hydrogen bonding pattern in the cocrystal structure of 90 favours one tautomer.
Figure 19.
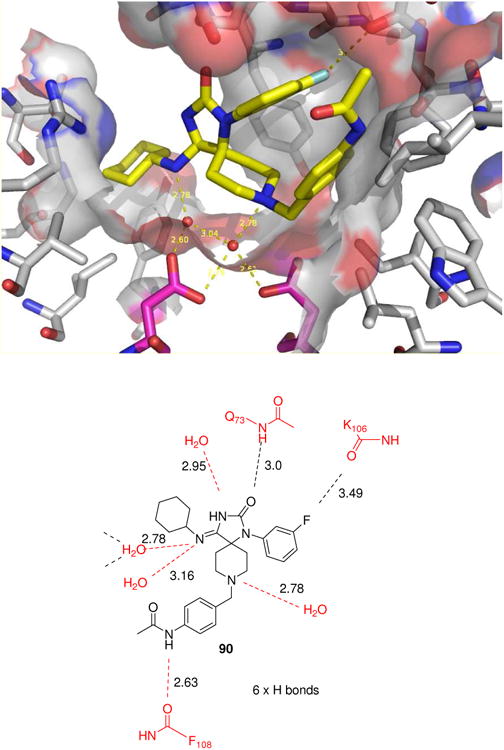
A new MCR derived scaffold showing promising BACE-1 activity. Above: synthesis of the general scaffold involving a key Ugi-4CR and representative inhibitors (90) with enzyme and cellular activity. Below: Binding mode of compound 90 (PDB ID: 3E3W) and schematic representation of the major short contacts to the BACE-1 receptor and to water molecules. Noteworthy there is no direct contact of the ligand to the two catalytic asp, it is however mediated by two crystal water molecules. Also noteworthy is a short contact between the fluorine of the ligand and a backbone carbonyl-O with the aromatic plane almost perpendicular to the amide group (α (CF-OC) = 177°). The distance of 3.49 Å, however is more than the sum of the atom radii (r (F) 1.47 Å + r (O) 1.52 Å = 2.99 Å). The two central Asp residues are marked pink.
Although the initially described compounds are not highly potent they show several noteworthy features. The best compound 90 shows an in vitro enzyme based IC50 of 2 μM and the activity in cell based assays only worsened by a factor of 4. Additionally, the compound shows nice plasma and brain concentrations and is no phospho-glyco-protein (PGP) efflux pump substrate.
A different Passerini-MCR involving strategy towards BACE inhibitors has been reported providing weak inhibitors (96) which might form a starting point for further optimization (Scheme 14).85 These examples clearly show how challenging it is to target the flat and spatially extensive BACE active site with useful activity and at the same time accomplish oral bioavailability and entrance through the BBB.
Scheme 14.

Introduction of MCR chemistry into the total synthesis of complex pharma products can potentially lead to a considerable shortage of steps and thus to lower cost-of-goods as exemplified here with the HIV protease inhibitor crixivan (indinavir).
The third Asp protease of high pharmaceutical interest is the HIV protease. Of the currently available HIV medications 7 drugs are HIV protease inhibitors. Similar to the above mentioned HCV NS3 protease inhibitors the described inhibitors are quite large and have a peptide-like appearance (e.g. 96 and indinavir). Often they have to be synthesized by sequential up to 20 step synthesis. Therefore it is worthwhile to consider alternative synthesis approaches involving MCRs. E.g. the key intermediate piperazine of indinavir can be advantageously and stereoselectively synthesized using a key and quantitative U-4CR followed by an enantioselective hydrogenation (Scheme 14).86 The introduction of the MCR into the total synthesis can lead to a considerable shorter synthesis and eventually reduced cost-of-goods.
Another research group asked the question if HIV protease inhibitors can also be de novo designed using convergent MCR chemistry.87 The design of a 2-step reaction sequence involving a Passerini reaction with α-oxocarboxylicacid esters and a subsequent Dieckmann ring closure indeed leads to low μM hits resulting also in an unprecedented MCR scaffold: tetronic acid (Scheme 15). A cocrystal structure of a molecule 97 with HIV protease underscores the validity of this synthesis design concept (Fig. 20). This de novo MCR approach seems to be quite promising and the initial hits can be potentially further optimized for potency and selectivity.
Scheme 15.

The sequence P-3CR – Dieckmann condensation leads into a tetronic acid backbone with HIV protease inhibitor activity.
Figure 20.
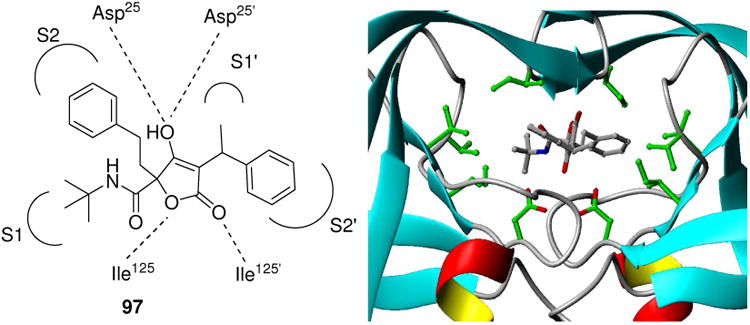
Cocrystal of a tetronic acid MCR-derivative (97) bound into HIV protease. The enol group is sandwiched between the two active site aspartates.
2.1.3. Metallo Proteases
The recent FDA approval of the histone deacylate (HDAC) inhibitor SAHA as an anti-cancer drug for the treatment of the manifestations of cutaneous T-cell lymphoma spurred the search for novel, improved and more selective compounds not only for cancer therapy but also for application for the treatment of human brain disorders such as Rubinstein–Taybi syndrome, Rett syndrome, Friedreich's ataxia, Huntington's disease and multiple sclerosis.88 Popular mechanism based warheads found in metallo protease inhibitors are hydroxamic acids and thiols which form complexes with the active side metal (usually Zn) and thus stop the catalytic cycle. The challenge with these strongly metal complexating functional groups is to introduce selectivity and thus to potentially reduce side effects. Recently, o-phenylendiamine monoamides were discovered as a novel warhead for metal proteases (Scheme 16).89 Thus compound 98 was synthesized by a U-3CR and showed good activity and selectivity. A complementary approach using the U-4CR and subsequent hydroxylamination also yields active hydroxamic acids (99) of unprecedented variability.90
Scheme 16.

Top: the recently approved metalloproteinase inhibitor SAHA; bottom: a U-3CR product with a novel type of metal binding war head, mono-acyl-o-phenylendiamine (98) and hydroxamic acids (99).
2.1.4. Cysteine Proteases
Cysteine protease inhibitors typically depend on potent warhead moieties which are often covalently and irreversibly reacting with the nucleophilic active site cysteine, e.g. epoxides, nitriles, α-ketoamides, α-ketoheterocycles, halo-ketones, diazo-ketones, peptidyl aldehydes, or epoxy-succinyl derivatives.91 Several of these warheads have been already discussed to be accessible in great diversity and numbers by Passerini- and Ugi-type MCRs. Remaining challenges for the clinical development of cysteine protease inhibitors include i.e. metabolic e.g. protease and chemical stability, selectivity of the highly reactive warhead units, solubility and cellular penetrability.
Calpains are calcium-activated neutral proteases belonging to the papain superfamily of cysteine proteases; several of these calpains have implications in diseases such as Alzheimer, brain and cardiac ischemia, spinal cord injury, muscular dystrophy, and cataract. Recently, compounds have been described targeted the orphan X-chromosome-linked inherited Duchenne muscular dystrophy (DMD). The compounds are prepared by PADAM and exhibit impressive enzyme and muscle cellular activity (Scheme 17).92 The non-polar lipophilic residue, lipoyl of compound 100 is believed to provide muscle cell targeting properties to selectively shuttle compound into disease tissue.93 Selected inhibitors of this series have been tested as well in a mouse model and showed significantly improved relevant histopathological parameters demonstrating their potential as a treatment for this devastating disease.
Scheme 17.

Selected reported cysteine protease inhibitors.
The pathways of apoptosis involve a cascade of initiator and effector caspases. Caspase-3 is known to be the main executioner of apoptosis through cleavage of protein substrates that leads to irreversible cell death.94 4-Aryl-4H-chromene (104), for example is a multicomponent condensation product of malonodinitrile (101), benzaldehyde (103) and 8-hydroxyindole (102) effectively inhibiting caspases and comprising a non-peptide backbone.95
Amongst non IMCRs those of cyanoacetic acid derivatives are extremely versatile regarding the multiplicity of scaffolds (Scheme 18). (For a recent comprehensive review see: 96) Often these MCRs involve primary Knoevenagel-type condensations of the cyanoacetic acid derivative with an aldehyde or ketone, followed by a Michael attack of a nucleophile and a subsequent ring closure via a second nucleophile through attack of the nitrile. A disadvantage of those MCRs is the current low variability of the cyanoacetic acid input. A recent combinatorial access to cyanoacetamides, however is enhancing the value by greatly expanding the large MCR scaffold space of cyanoacetic acid derivatives.97 A well-known MCR of this class is the Gewald-3CR (G-3CR) which has recently gained ground by the usage of cyanoacetamides.98
Scheme 18.

MCR scaffold diversity of malonodinitrile derivatives.
2.2. Kinases
Kinases have emerged over the last two decades as one of the most prolific therapeutic targets with many drugs under clinical evaluation or in clinical practice.99 They are a large class of enzymes dephosphorylating hydroxyl containing amino acids in target proteins. According to their substrate specificity one broadly distinguishes Ser/Thr from (receptor) Tyr kinases. They are involved in many different pathophysiological processes and are amongst the most popular contemporary target classes in pharmaceutical industry. Most kinase inhibitors currently under development are ATP mimics. They display an often heterocyclic aromatic flat topology mimicking the adenosine heterocycles of ATP and an adjacent hydrogen donor acceptor moiety mimicking the amidine substructure of ATP. Many opportunities exist to employ MCR chemistry in the kinase field. A p38 kinase inhibitor SB220025 was recently clinically evaluated in phase III for rheumatoid arthritis. The synthesis of SB220025 involves a vL-3CR and the corresponding α-4-fluorophenyl substituted tosylmethylisocyanide (105) has been produced in 500 kg batches.100 A cocrystal of SB220025 and the p38 kinase has been published and can serve to understand the crucial features of kinase inhibitors and their connection to this MCR scaffold (Fig. 21).101
Figure 21.
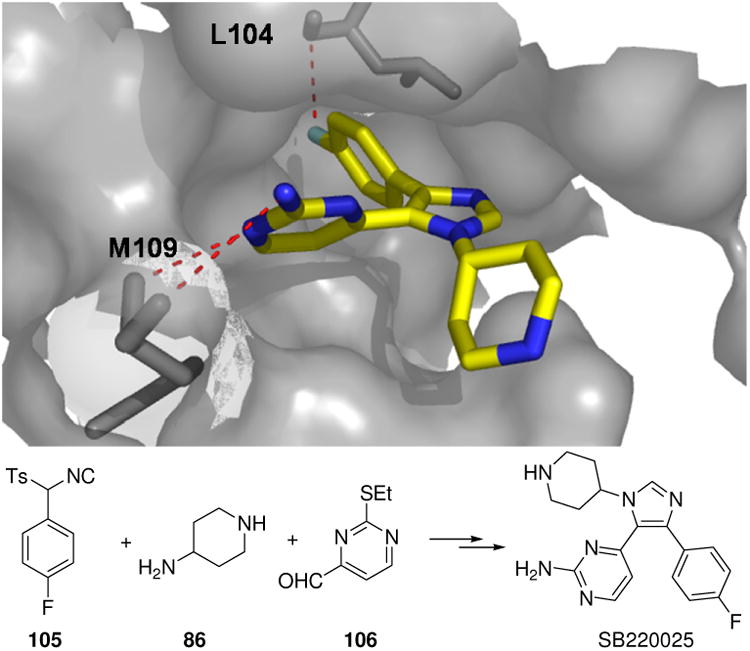
vL-3CR compound SB220025 is a potent p38 inhibitor. Above: Binding of SB220025 into p38 active site (PDB ID: 1BL7). The 2-amino portion forms hydrogen bonds, the kinase hinge region and the fluorine of the ligand is involved in a short contact to the backbone carbonyl-C (3.1 Å). Below: reaction scheme of the vL-3CR.
Substituted 2-aminofuranes could be active as kinase inhibitors as they show the hallmarks: they are flat aromatic heterocycles and they incorporate an adjacent hydrogen donor acceptor moiety which is suited to undergo a hydrogen bond network with the hinge region of the active site of kinases (Fig. 22). Recently, a multitude of new MCR approaches have been published resulting in this scaffold.
Figure 22.

MCR derived 2-aminofuranes are potential kinase scaffolds displaying kinase inhibitor specific pharmacophores: they are flat heteroaromatic and display a vicinal H-bond donor/acceptor moiety (shown in comparison to the ATP bond to the hinge region of kinases).
This versatile MCR chemistry is based on the acetylene isocyanide adduct first described in a seminal paper by Winterfeld.102 This reactive intermediate can be described as a zwitterionic or carbine-type mesomeric form and is the starting point of a rich MCR chemistry resulting in a diversity of scaffolds (Scheme 19). E.g. the reaction of isocyanides with acetylendicarboxylic acid methyl esters (107, DMAD) and suitable acids yields highly substituted 2-aminofuranes (108).103 Acidic components described are N,N-dimethylbarbituric acid,104 3,6-dihydroxypyridazine,105 (iso)nicotinic acid,106 4-hydroxycoumarins,107 vicinal tricarbonyl systems,108 2-pyridinecarboxaldehyde,109 isatin,110 4-arylurazoles,111 phenols,112 4,5-diphenyl-1,3-dihydro-2H-imidazol-2-one,113 3-methylcyclopentane-1,2,4-trione, yielding 4H-pyrano[3,2-d]pyrimidine (109),114 3-amino-5,8-dioxo-5,8-dihydro-1H-pyrazolo[1,2-a] pyridazines (118),105 2,3-dihydro-1,3-dioxo-1H,5H-pyrazolo[1,2-a][1,2,4]triazoles (119),1115H-imidazo[2,1-b][1,3]oxazine derivatives (106),113 annulated 2-amino-4H-pyrans (123),1074H-chromene derivatives (121) respectively.112 A facile and direct synthetic entry to 4-hydroxy-1H-pyrrole-2,3-dicarboxylic acid derivatives (117) based on the reaction of DMAD, α-amino acids with isocyanides or carbodiimide (DCC) as condensation agents under neutral conditions was reported.115 In an extension of these synthetic ideas, it was described recently, that isocyanide, aldehyde, dimedone and ammonium acetate react in a 4-CR fashion to highly substituted 1H-indole-4(5H)-ones.116 DMAD can also be reacted with benzoic acid derivatives and isocyanides in the presence of triphenylphosphine to yield highly substituted 2-aminofuranes.117 The same scaffold is available by the reaction of benzoylchloride, DMAD and isocyanide.118 However, whereas electron-withdrawing groups in para position of the benzoylchloride yield 2-aminofuranes, others result in 2,5-dihydro-1H-pyrroles.118 Aliphatic α-acidic carboxylic acids under the same conditions react with DMAD and isocyanides to form 2,5-diaminofurans.119 Similarly, N-(2-pyridyl)amides, isocyanides, and DMAD undergo cyclization to 4H-pyrido[1,2-a]pyrimidines, which after N-deprotection can yield kinase inhibitory signature.120 These DMAD incorporating MCRs are very interesting regarding their structural diversity and taking into account that the two ester functionalities can be further regioselectively functionalized, e.g. by amidation thus also providing a large chemical space. Another additional benefit of these reactions is that they often are performed under very mild conditions and the products are easily purified.
Scheme 19.

MCR chemistry of acetylenedicarboxylicacid diesters (DMAD) leads to multiple scaffolds (108-123) with hydrogen donor acceptor fragments thus potentially providing a kinase inhibitor pharmacophore.
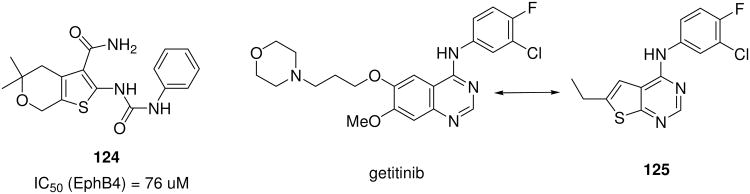
Eph (erythropeitin-producing hepatoma) tyrosine kinase cell surface receptors are the largest tyrosine kinase family with therapeutic implications in e.g. cancer and nerve regeneration.121 Active site EphB4 inhibitors were discovered by a virtual docking/fragmentation approach of a large 730,000 collection amongst them high ranking G-3CR compound 124 (Scheme 20).122
Scheme 20.
Kinase inhibitors by Gewald MCR.
The Gewald 3-CR (G-3CR) of cyanoacetic acid derivatives, methylene active carbonyls and elemental sulphur is a popular MCR often used in drug discovery yielding 2-amino-3-carbonyl thiophenes (e.g. 126-136) (Scheme 21).123 These reactions are quite versatile and can lead to a large number of substituted thiophenes otherwise difficult to access. The interest in Gewald products also steams from the fact that the thiophene moiety is bioisosteric to phenol. Thus Gewald products can also be considered as bioisosteric to anthranilic acid derivatives. As opposed to the difficulty in accessing substituted anthranilic acids, however, Gewald thiophenes are available in great numbers. Additionally, Gewald products can be easily transformed into further scaffolds by secondary transformations (Scheme 21).124 For example, condensation of Gewald products with formamide opens a versatile synthetic avenue to thiopheno-2-aminopyrimidine type kinase inhibitors (125, 126) (Scheme 20). 126 Is a moderate potent KDR inhibitor (IC50 = 4.6 μM), while derivatives display low nM activity, significant oral efficacy and favourable pharmacokinetic profiles. 125
Scheme 21.

A diversity of products based on secondary reaction of the initial G-3CR.125a,127
Applying the isostery concept thienopyrimidine based derivatives 125 of the marketed anti-cancer drug gefitinib have been synthesized based on G-3CR (Scheme 20).126
5- And 6-membered (hetero)aromatic amidines react with aldehydes and isocyanides to form bicyclic imidazo[1,2-x]-heterocycles derivatives (GBB-3CR).[27,28] o-Formyl benzoic acid esters (138) input together with tert-butylisocyanide (93) leads in a straight forward manner into polycyclic heteroaromatic ring systems (Fig. 23) displaying in addition a vicinal H-bond donor/acceptor fragment.128 These compounds clearly incorporate the kinase pharmacophore. A library of compounds (139-144) has been profiled against a panel of diverse kinases and potent and selective inhibitors have been discovered (Fig. 23). Potent compounds with differential selectivity have been obtained, which can be further optimized using secondary transformations addressing different binding regions in the active site of kinases.
Figure 23.
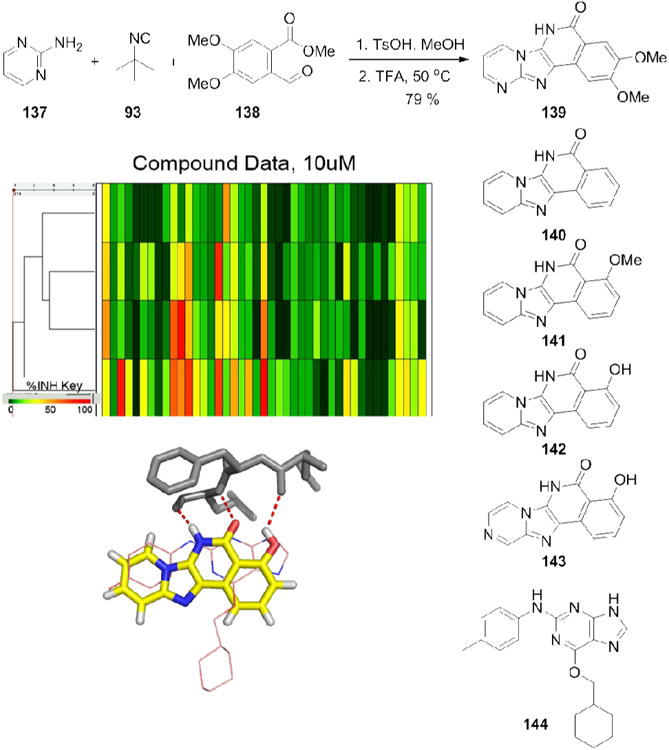
MCR kinase inhibitors. Above: 2-Step synthesis of kinase inhibitors (139) using the GBB-3CR. Middle: Selectivity profile of some representative compounds (140-143) against a panel of kinases. Below: Docking of compound 142 (yellow sticks) into the active site of CDK2 together with the cocrystallized anilino-purine compound 144 (pink lines) (PDB ID: 1OI9).129 Compound 142 forms a strong hydrogen bond network with the hinge region of CDK2 (grey sticks, red dotted lines), a prerequisite for a potent kinase inhibitor.
Rho-associated kinase isoform 1 (ROCK1a) is an enzyme involved in diverse cellular signalling functions such as smooth muscle contraction, cytoskeleton rearrangement, cell migration, and proliferation.130 This compound is accessible by a 3-CR of acetoacetamide (145), benzamidine (146) and pyridinecarbaldehyde (147). The compound 148 has been cocrystallized with Rho kinase.
An elegant synthesis of the highly active marine natural product meridianin isolated from the ascidian Aplidium meridianum was reported using a four-component pyrimidine synthesis.131 The 2,4,6-trisubstituted pyrimidines are synthesized based upon an elegant consecutive carbonylative coupling–cyclocondensation sequence (Fig. 24). Several derivatives are highly active multi kinase inhibitors. Cocrystal structure of several derivatives and SAR have been reported.132
Figure 24.
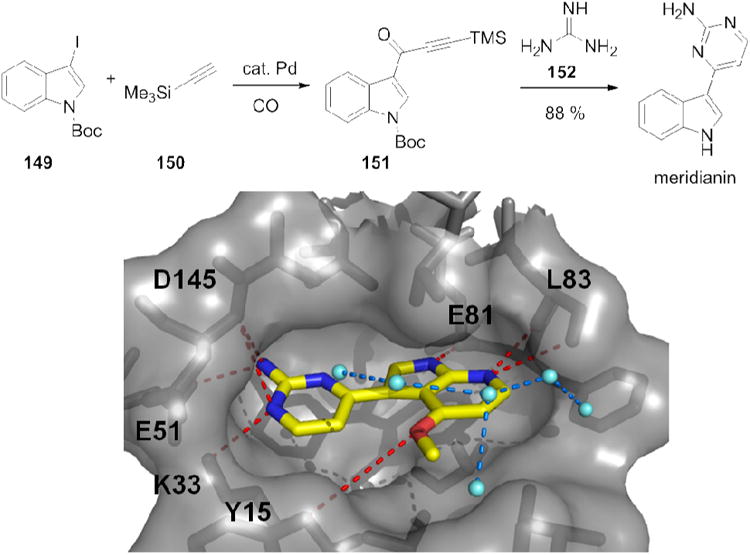
Kinase inhibitory natural product meridianin in a short and efficient MCR synthesis and its natural origin, an Aplidium sp. sponge. Below: The cocrystal structure of the 7-aza meridianin in complex with CDK2 (PDB ID: 3BHT). Shown with red dotted lines is the extensive H-bond network of the natural products with the hinge region and other amino acid side chains of the receptor. A tight water network on top of meridianin is shown as turquoise balls and blue dotted lines.
2.3. Phosphatases
Whereas kinases have been extremely successful as drug targets leading to many clinical and preclinical drugs, phosphatases are rather difficult to target by small molecules while retaining an acceptable PKPD profile. Glucose-6-phosphate translocase (G6PT) is a promising diabetes type-II target.133 By using the above described GA strategy new, potent and selective G6PT inhibitors have been discovered in iterative rounds of evolutionary optimization (Fig. 25).134 Different scaffold spaces based on vL-3CR and reductive amination/acylation chemistries were investigated. Within the performed evolutionary cycles of synthesis, analytics, screening, and library design, promising lead structures were found. In a second step the best compounds from the first phase served as structural prototypes for a similarity-triggered genetic algorithm to select molecules for focused compound libraries around these lead structures. Maintaining the reaction scheme, a refinement of the used building blocks was achieved and compounds with high activity were identified. Finally, the preferred substituents were transferred into a new chemical backbone, using the advantage of one-step MCR chemistry while maintaining the biological activity. In the shown cases the genetic algorithm has proven its capability as a library design tool to select diverse compounds from a given large chemical space based either on measured biological activities or on chemical similarity.
Figure 25.
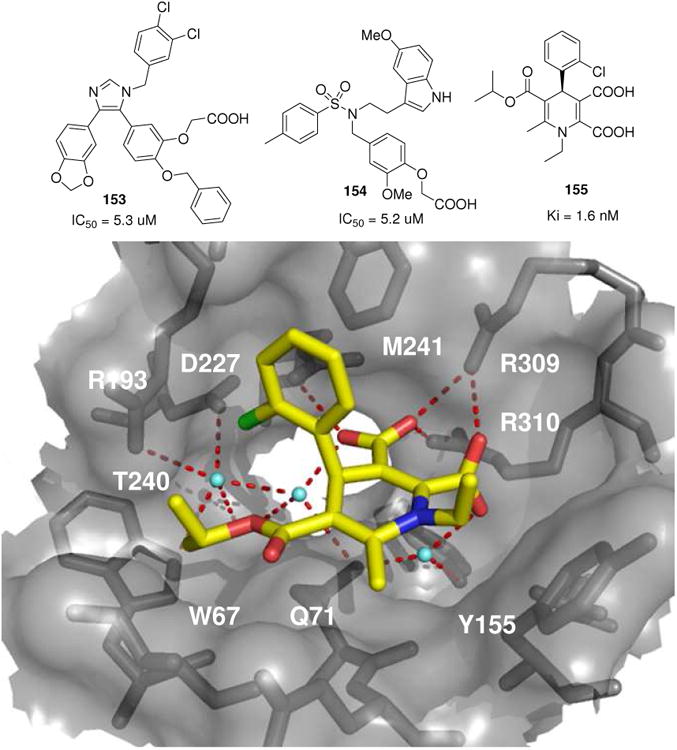
Above: Structure of different MCR derived phosphatase inhibitors. Below: Glycogen phosphatase (PDB ID: 2AMV) in complex with a Hantzsch MCR derived dihydropyrimidine 155. Note the typical boat conformation of the central heterocycl
The Hantzsch dihydropyridine synthesis is a classical MCR discovered by Arthur Hantzsch in 1881.7 It is the four component reaction between ammonia or a primary amine, a benzaldehyde derivative and two equivalents of a 1,3-dioxo derivative (H-4CR). The proposed mechanism involves a Knoevenagel condensation of one oxo component and an enamine formation of the other oxo component followed by a Michael-type addition and subsequent ring formation under dehydration conditions. Many improvements using different catalysts have been described, including Montmorillonite K10 clay, sulfonic acid on silica gel, ultrasound on silica gel absorbed starting materials or different solvent systems, e.g. water or ionic liquids. The H-4CR has led to potent glycogen phosphorylase b inhibitors (155, Fig. 25).135 The dihydropyridine-5,6-dicarboxylate groups mimic the phosphate group of ligands that bind to the allosteric site and contact three arginine residues (Arg309, 310 and 193). Several crystallographic water molecules play a crucial role in mediating a strong hydrogen bond network.
Synthesis of compound libraries based on the tandem aza [4+2] cycloaddition/allylboration multicomponent reaction between 1-aza-4-boronobutadienes (156), maleimides (157), and aldehydes (158) have been described (161, Scheme 23). They involve and use multiple strategies, including liquid phase synthesis with resin capture and two solid phase variants.136 The compounds were screened against several phosphatases, including PTP1B, MPTPA, MPTPB, VEPTP, and PP1 and the dual-specificity phosphatases Cdc25A and VHR and two examples (162-163) showed activity.
Scheme 23.

MCR library synthesis of phosphatase inhibitors involving hetero Diels Alder of α,β-unsaturated Schiff bases and allylboration.
2.4. Other Enzymes
Dihydroorotate dehydrogenase (DHODH) is a key enzyme of the de novo pyrimidine biosynthesis, converting dihydroorotate to orotate. DHODH inhibitors are believed to have implications for the control of inflammatory processes but have been also investigated for other indications, e.g. cancer and malaria.
A DHODH inhibitor, brequinar has been synthesized by the Doebner-3CR of α-ketoacid (165), substituted benzaldehyde (166) and substituted aniline (164) and has undergone multiple clinical trials for cancer and immunosuppression.137 A cocrystal structure has been published.138 The inhibitor is situated in a long hydrophobic channel and makes an important charge charge interaction with the Arg136 (Fig. 26).
Figure 26.
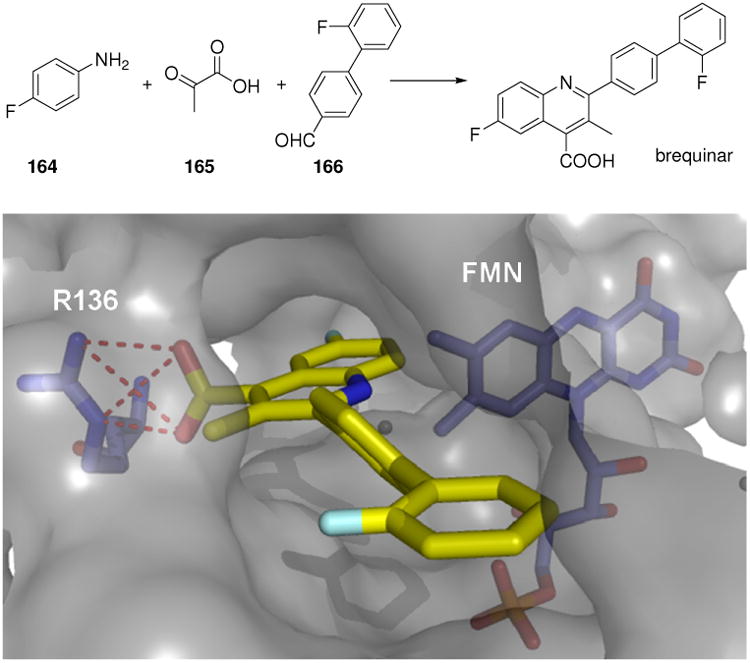
Above: The DHODH inhibitor brequinar synthesized by Doebner-3CR. Below: The Doebner-MCR product is located in a deep and hydrophobic protein binding site (PDB ID: 1UUO). The key interaction is the salt bridge between the carboxylic acid and the guanidine unite of Arg136. Noteworthy the tight interaction of the two fluorine atoms located at the isoquinoline and the external biphenyl ring with the hydrophobic protein environment.
Cyclooxygenase (COX) is an enzyme responsible for the synthesis of prostanoids and represents a major inflammation and pain target. The group of non-steroidal anti-inflammatory drugs, such as the well-known aspirin and ibuprofen are COX inhibitors. Recently, imidazo[1,2-a]pyridine derivatives were designed as novel COX-2 inhibitors, 10-fold more potent than celecoxib as an analgesic and an anti-inflammatory agent in several disease relevant animal models (Fig. 27).139 Docking studies were used to rationalize the results. The compound 170 is orally bioavailable. Compound 170 is a product of the GBB-3CR variation of the Ugi reaction and can be synthesized in one step from the isocyanide (167), benzaldehyde (169) and 2-aminopyridine (168) in 60% yield. Interestingly, the same class of compounds was also found by an unrelated approach. A ligand based virtual screening cascade of a commercially available library involving 2D similarity, shape and 3D pharmacophore similarity served to find new and potent 5-lipoxygenase inhibitors (Fig. 27).140 Several of the high ranking hits are MCR reaction products, including G-3CR (173) and GBB-3CR (172). Clearly, such an approach is suited to economically screen large MCR libraries and to produce different hits based on different MCR scaffolds sic “scaffold-hopping”.
Figure 27.
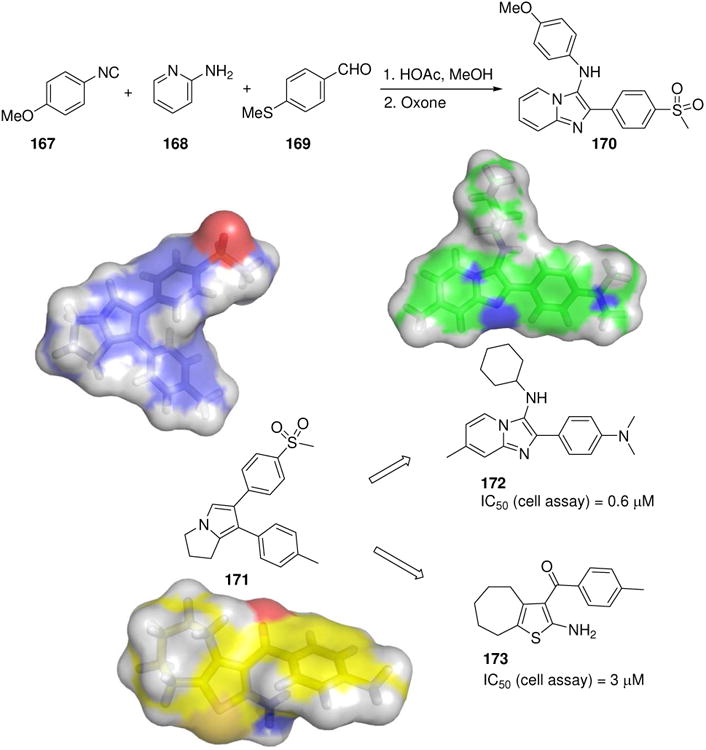
Scaffold hopping via virtual screening towards discovery of novel COX inhibitors. Compound 171 served as a template to screen a 1323 compound library using a screening cascade. Amongst the hits several scaffolds based on MCR were discovered. The most potent hit, compound 172 showed nM activity in a cell based assay.
3′,5′-Nucleotide phosphodiesterase enzymes (PDE) play dominant therapeutic roles in depression, emetic response and inflammation showing a distinct subtype specificity. A tetrahydrobenzothiophene bisamide (174) was recently discovered as a potent and modestly PDE4B-over 4D-selective inhibitor and has emerged from an HTS based on docking models.141 The compound has been synthesized using a three step procedure involving a key Gewald-3CR. Co-crystal structure of PDE4 with Gewald compounds (174) revealed that the compounds are rather rigid in forming an intramolecular hydrogen bridge between the 2-amide and the 3-carboxy group (Fig 28). This is in agreement with numerous small molecule x-ray structures of the Gewald scaffold.124a Additionally, the co-crystal structure of 174 with the receptor was surprising since a considerable induced fit was observed; this is in contrast to dozens of previous apo and co-crystal structures. These results can be helpful in designing subtype specific PDE inhibitors.
Figure 28.
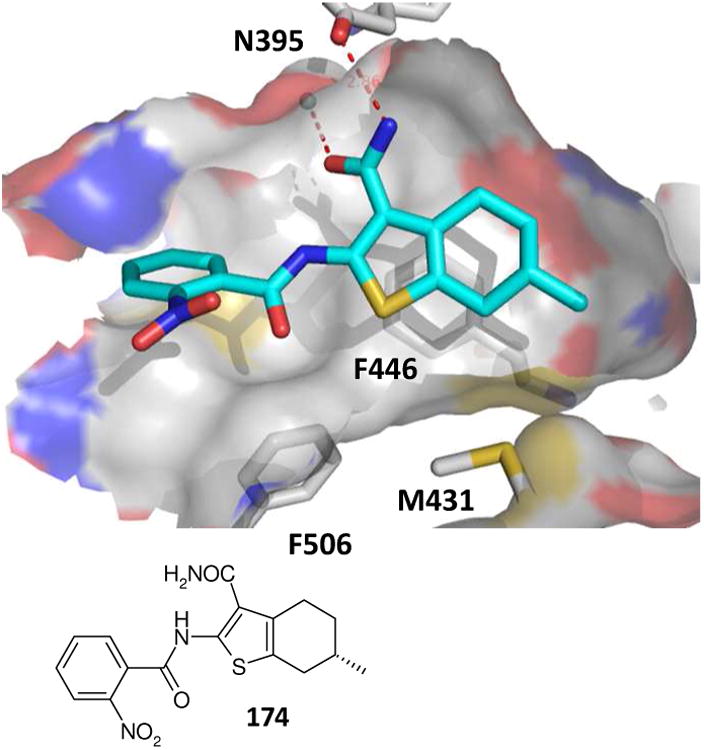
Gewald-3CR product 174 (turquoise sticks) as subtype specific 3′,5′-nucleotide phosphodiesterase enzyme inhibitor bound to PDE4B (PDB ID: 3HMV). The inhibitor pocket is shown in a cut-off view. Several amino acid side chains are removed for clarity. The primary amide of the inhibitor makes a hydrogen bonding contact to Asn395 and an adjacent water molecule. A π-π interaction can be observed between the thiophene ring and Phe446. Additionally there are hydrophobic contacts to Phe506 and Met431.
2.5. G-Protein Coupled Receptors
GPCR ligands derived from MCR chemistry are particular popular as indicated by the wealth of patent applications, compounds in development and on the market. In fact GPCRs are the single largest drug target class, representing 25-50% of marketed drugs.142 GPCR drug discovery in the past was dominated by HTS, however the recent structure elucidation of several novel GPCRs in addition to rhodopsin provides the foundation to complementary techniques, e.g. homology modelling and structure-based design.143 The orexin receptor was discovered during an effort to de-orphanize brain related GPCRs. Orexins, also called hypocretins, are a pair of highly excitatory neuropeptide hormones that are produced by a very small population of cells in the lateral and posterior hypothalamus and they send projections throughout the brain. The orexin system is involved into a range of basic physiological states, including wakefulness and food intake and is therefore an important new target area for drug discovery.144 Almorexant is a first-in-class orexin receptor antagonist, currently undergoing phase III clinical development for insomnia.145 The tetrahydroisoquinoline derivative was originally discovered from a series of Ugi/Pictet-Spengler reaction products (Scheme 24).146
Scheme 24.

Structure of almorexant, a first in class orexin I antagonists currently in advanced clinical trials for sleeping disorders.
Preterm labour is the major reason for neonatal morbidity and occurs in 10% of all birth worldwide. Currently, antagonistic derivatives of the neurohypophyseal nonapeptide hormone oxytocin are used to control preterm labours, however they are associated with the typical disadvantages of peptide drugs, such as lacking oral bioavailability, short half live time and potential immunogenicity. The diketopiperazine scaffold (175) has been discovered in a HTS campaign and developed to the first clinical class of small molecular weight oxytocin antagonists (Fig. 29). The optimized derivative GSK221149A is undergoing advanced clinical trials to study safety, tolerability and metabolism.147 GSK221149A is a very potent (Ki 650 pM) and selective oxytocin antagonist and has been shown to inhibit oxytocin-induced uterine contractions in the anaesthetised rat. Interestingly, the compound (MW 495 Dalton) is a >20 fold more potent receptor antagonist than the current clinically used peptide derivative Atosiban (MW 994 Dalton). Moreover GSK221149A displays a far superior selectivity profile over the peptide drug with respect to the related vasopressin receptors (>1400-fold).148 In addition, GSK221149A is orally bioavailable, in contrast to the peptide derivative.
Figure 29.
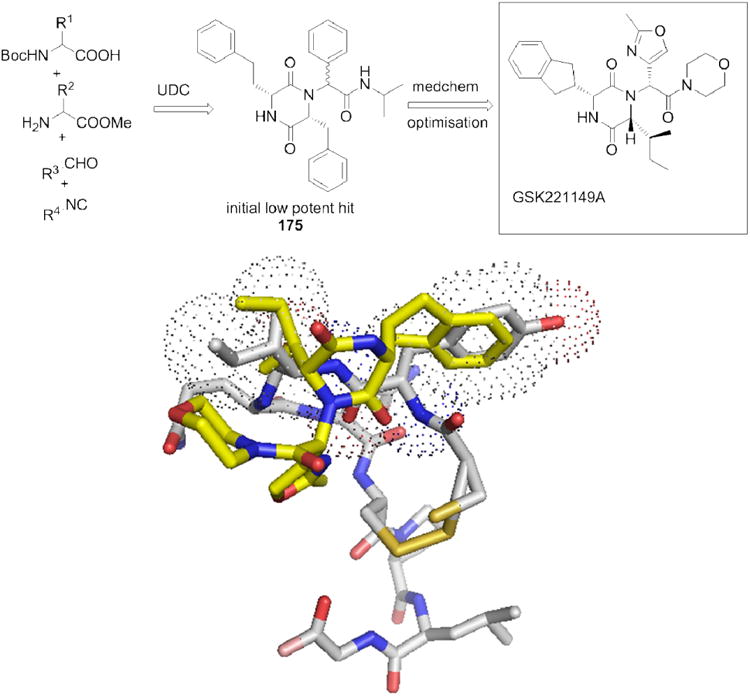
Above: Retrosynthesis of the oxytocin antagonist (compound 175 and GSK221149A). Below: X-ray structure of oxytocin (grey sticks, PDB ID: 1XY2) and an energy minimized model of GSK221149A (yellow sticks).149 It is hypothesized that the indane part of GSK221149A mimics Tyr2 and the Ile fragment Ile3 of oxytocin. The oxazole fragment imparts a conformational lock and the morpholine water solubility, respectively.
Due to the convergent and efficient nature of the MCR chemistry detailed SAR has been performed.150 In order to rapidly establish SAR and the optimal stereochemistry all 8 stereoisomers of this Ugi DKP backbone had to be synthesized. In a landmark paper all 8 different stereoisomers have been synthesized using different strategies, however all involving Ugi chemistry (Scheme 25 and 26).151 E.g. reaction of the chiral N- and C-protected amino acid derivatives (177), respectively with tert-butylisocyanide (93) and benzaldehyde (176) yields the Ugi product 179. N-deprotection and cyclisation under basic conditions yields the two stereosisomers 180(RRR) and 181(RRS) differing in the benzaldehyde derived stereocenter (Scheme 25). The two diastereomers can be conveniently separated using silica chromatography.
Scheme 25.

Synthesis of the (RRR)-180 and (RRS)-181 stereoisomers of oxytocin antagonist derivatives.
Scheme 26.

Alternative MCR synthesis of the RRR and SSS stereoisomers of oxytocin antagonist derivatives.
The RRR stereoisomer 180 can be prepared alternatively using an initial U-5C-4CR employing unprotected L-Leu HCl salt, benzaldehyde (176) and tert-butylisocyanide (93), yielding the iminodicarboxylic acid mono amide derivative 182 in very good yields and diastereoselectivity (Scheme 26). Saponification, acylation (183), N-deprotection and subsequent cyclisation yields the expected stereoisomer on a multi mg scale. The other stereoiosmers were synthesized using similar strategies and enantiomerically pure amino acids as starting materials. Attempts to simplify the DKPs e.g. by removing the Ugi side chain and providing “classical” DKPs did lead to inactive compounds. Clearly, such highly substituted DKPs are not readily available by other synthetic strategies involving 2-CRs.33,152
Corticotropin releasing factor (CRF) is a 41-amino acid peptide hormone involved in stress response. It exerts its activity through binding to the GPCR receptor CRF1-r. Antagonists are under investigation for generalized anxiety disorder and for the potential treatment of alcoholism. A novel series of CRF1 antagonists was discovered by using a computational library design strategy and differing much from previous CRF antagonist pharmacophores.153 The N-phenylphenylglycine amides, such as 184, were synthesized in a two-step process involving a boronic acid Mannich (Petasis) MCR followed by amidation (Scheme 27).154 These compounds were synthesized as racemic mixtures and separated rapidly using chiral super-critical CO2 fluid chromatography (SFC). Generally, only one enantiomer showed activity. Additionally, preliminary pharmacokinetic studies showed encouraging results. An alternative pathway to this compound class consists of the U-3CR. Based on the different availabilities of the starting materials of the two approaches different chemical spaces can be investigated. Recently, a major improvement of the U-3CR has been reported using phenylphosphinic acid in toluene under refluxing conditions.155
Scheme 27.

CRF receptor antagonist (R)-185 optimisation by Petasis or Ugi-3CR.
The 3-CR product of two equivalents of 5,5-dimethylcyclohexane-1,3-dione (191) and salicylaldehyde (192) yielding a xanthene derivative (193) has been shown to potently antagonize NPY, a 36 amino acid peptide with potent, centrally mediated orexigenic (stimulates food intake) effects (Fig. 30). The lead compound 193 is a selective and orally active neuropeptide Y5 receptor antagonist and has an advantageous PKPD profile, including penetration of the blood-brain barrier. Thus compound 193 and its derivatives will serve as valuable tools to study biology of NPY receptor in cell-based systems as well as in vivo.156
Figure 30.
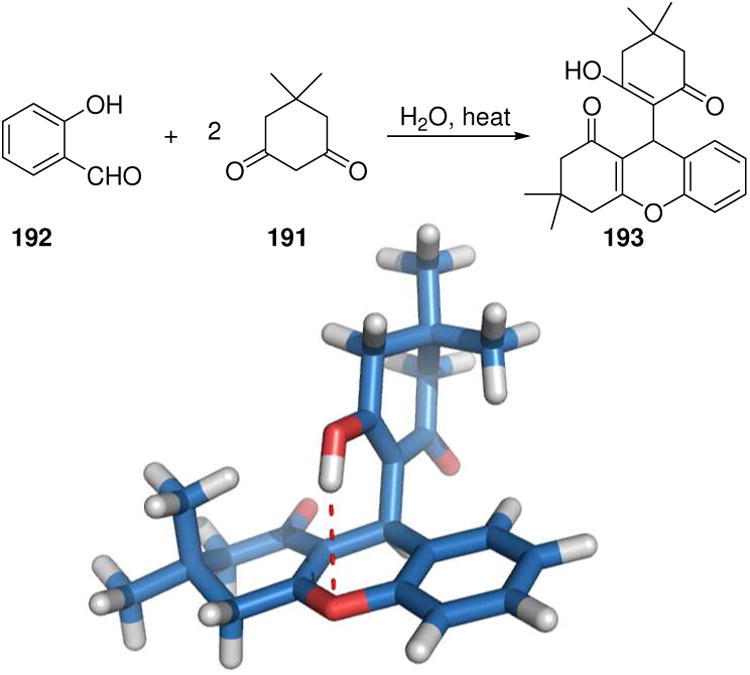
Potent NPY antagonist 193 made by an old157 and experimentally simple MCR.
α-Amino acid derived isocyano esters - but interestingly not the amides – react with aldehydes or ketones and primary amines to yield stereospecifically the corresponding syn-imidazoline as a major product. This Orru-3CR is useful because three independent starting materials which are all available abundantly allow the access to a very large chemical space.158 The reaction has been recently used to discover m-opioid receptor selective inhibitors (194, Scheme 28).159
Scheme 28.

The Orru-3CR and a biologically active m-opioid receptor antagonist (194) thereof.
Melanin-concentrating hormone (MCH) is orexigenic and thus represents an important pharmaceutical target. Chiral dihydropyrimidone inhibitor (SNAP-7941) currently undergoes preclinical evaluation as an anorectic, antidepressant and anxiolytic agent. The compound can be produced by the Biginelli-MCR and recently two enantioselective routes towards its synthesis have been published, both employing again MCRs (Scheme 29).160 The first route uses an asymmetric Mannich reaction of ethyl acetoacetate 195 and imine 196 in the presence of cinchona alkaloid catalyst 197. The second route employs an asymmetric Biginelli reaction catalyzed by chiral binapthol derived phosphoric acid 203. Thus Biginelli intermediate 204 can be formed in 96% yield in an e.r. of 95:5. The heterocycle was purified by recrystallization to provide DHPM enantiomerically pure. The asymmetric Mannich reaction catalyzed by cinchona alkaloids and the asymmetric Biginelli reaction catalyzed by chiral phosphoric acids were equally effective at producing the desired heterocycle.
Scheme 29.

Two scale-up asymmetric routes towards a preclinical MCH inhibitor SNAP-7941.
The complement system is comprised of a cascade of interrelated proteases that are activated in response to immunoglobins binding to a foreign antigen. Activation of the complement systems leads to a stepwise hierarchy of proteolytic cleavage events ultimately leading to the release of bioactive fragments (C3a, C4a and C5a) known as anaphylatoxins. C5a is recognised as a potent mediator of inflammation by recruiting inflammatory cells to the site of infection or injury. Novel C5a receptor antagonists 208 based on U-4CR have been disclosed and found useful as a tool for the rapid identification of antagonists with low in vitro clearance.161 A large number of compounds with ‘lead-like’ potency were prepared but these had poor metabolic stability. Thus rapid MCR chemistry helped to identify weaknesses of a lead series and consequently it was not progressed into lead optimisation (Fig. 31).
Figure 31.
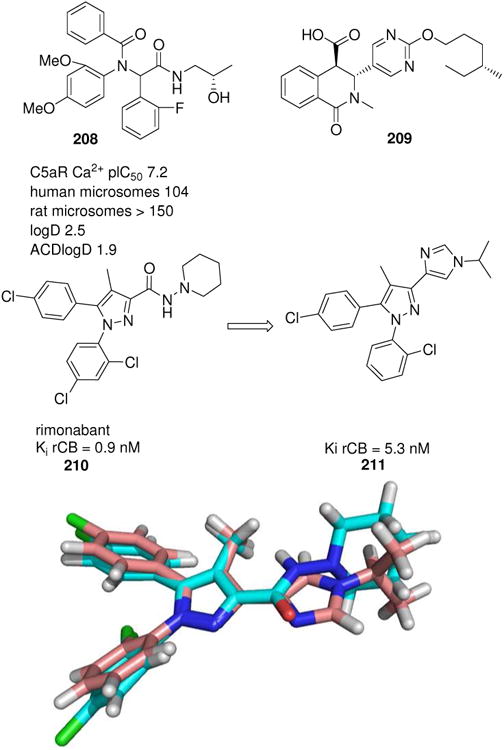
Various GPCR MCR-receptor binders. Below: Overlap of the CB1 receptor antagonist rimonabant with an imidazole isostere 211 synthesized by vL-3CR.
A recently characterized G-protein coupled receptor, GRP40 is believed to represent a selective target for type 2 diabetes. GPR40 is preferentially expressed in the pancreas with elevated levels reported in the islets and also in the pancreatic β-cell lines. A HTS screening identified MCR products 209 of homophtalicacid anhydride and primary amines and aldehydes (Fig. 31).162 Few rounds of optimization revealed a candidate with GPR40 activity and satisfactory PK parameters.
The endocannabinoid system (ECS), and specifically the cannabinoid type 1 (CB1) receptor, plays a pivotal role in energy homeostasis and is a major obesity target. Recent clinical trials however revealed that several CB1 receptor inverse agonists/antagonists were associated with major side effects. In order to potentially overcome these side effects compounds are synthesized to have an improved profile. Thus bioisosteric replacement of the hydrazide functionality with a suitably substituted imidazole using van Leusen's MCR was recently proposed (211).163 Equally potent compounds could be achieved showing an excellent overlap of the different pharmacophore elements and being orally bioactive (Fig. 31).
Gonadotropin-releasing hormone (Gn-RH) is secreted from the hypothalamus and its action on the pituitary gland then leads to the release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). Their involvement in the biology of reproduction made them key targets for drug discovery. The bulky hydrophobic amino acid residue in position 6 appears to be very important for the high potency of the analogues. An efficient method for the synthesis of some Gn-RH analogues based on Ugi reaction has been developed (Scheme 30).164 Four-component reaction of N- and C-terminus peptides, aromatic aldehydes and isocyanides affords novel Gn-RH analogues derived from triptorelin and gonadorelin. Mild ligation of two peptide fragments is one of the earliest applications of the Ugi MCR in biological chemistry and has been recently named the Ugi-ligation.165 Potential advantages of such modified peptides could be their enhanced protease stability, the easy tunability of hydrophobicity/hydrophilicity properties via the other components and their different biological activity. Moreover, one can imagine that certain bioactive conformation of otherwise flexible peptides could be frozen.
Scheme 30.

Ugi-ligation of Gn-RH analogues.
2.6. Ion Channels/Transporter
Channels are the gates of charged and uncharged small molecules between the inside and the extracellular world of cells. They play an eminent role in the transduction of information. Malfunctional channels on the other hand play an outstanding role in many diseases.166 With a lot of recent information available on structures of channels a rational approach to channel drug discovery is now feasible besides HTS.166b,167 Specifically, chloride channels are involved in a wide range of biological functions and thus are an important class of drug targets.168 Interestingly, however, chloride channels are relatively under-explored as a target class for drug discovery as elucidation of their physiological roles has lagged behind that of many other channels. They are involved for example in epithelial fluid secretion, cell-volume regulation, neuroexcitation, smooth-muscle contraction and acidification of intracellular organelles. Diseases associated with chloride channels are cystic fibrosis, macular degeneration, myotonia, kidney stones, renal salt wasting, secretory diarrhoeas, polycystic kidney disease, osteoporosis and hypertension and hyperekplexia, just to name a few.
For example, mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel cause cystic fibrosis. cylaminocarboxamides 212 has been identified by high-throughput screening and can be accessed synthetically by a classical Ugi 4-CR (Scheme 31).169 This phenylglycine derivative can correct defective gating in a number of CF-causing CFTR mutants. Thus compound 212 could display a lead structure for the development of a drug for cystic fibrosis.
Scheme 31.

Examples of chloride channel interacting MCR products.
Calcium-activated chloride channels (CaCCs) are widely expressed in mammalian tissues, including intestinal epithelia, where they facilitate fluid secretion. Potent, selective CaCC inhibitors have not been available. Recent small molecule screening to identify inhibitors of human intestinal CaCC(s), using a halide influx assay, identified several classes of CaCC inhibitors.170 The most potent inhibitors identified were of the Gewald scaffold, e.g. 3-acyl-2-aminothiophene 213 (Scheme 31). SAR studies based on several derivatives were performed and yielded insight into optimal potent compounds. Interestingly, cylohexanone derived compounds are active whereas cyclopentanone derived Gewald heterocyles with one carbon less were inactive. Small-molecule CaCC inhibitors may be useful in pharmacological dissection of CaCC functions and in reducing intestinal fluid losses in CaCC-mediated secretory diarrheas.170
The Hantzsch reaction has attracted a lot of interest due to a block buster drug based on this scaffold: nifedipine (Scheme 32, Fig 32).171 This drug comprises anti-hypertensive properties, targets heart specific Ca2+ channels and represented a major breakthrough in the treatment of heart diseases.
Scheme 32.

Channel blockers derived from the classical Hantzsch-4CR.
Figure 32.
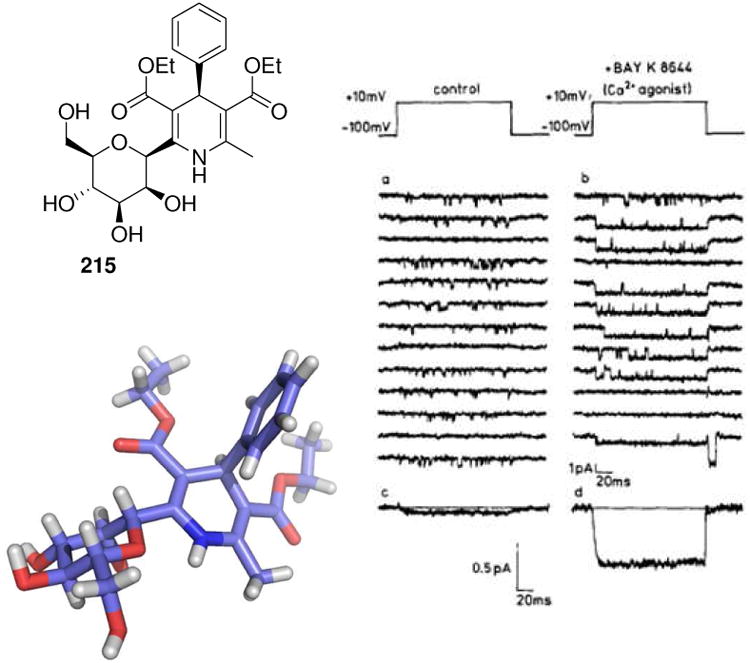
Left: Structure and X-ray structure of glycosylated dihydropyridine 215 in its typical bioactive boat conformation (CCDC ID: 182892); right: Patch-clamp recording for individual Ca2+ channels in the absence (left) and in the presence (right) of a dihydropyridine calcium antagonist.
Diydropyridines can be easily oxidized to the corresponding pyridine derivatives, e.g. using ammonium nitrate/Montmorillonite K10 Clay during the H-4CR.172 Alternatively, the Hantzsch products can be separated and oxidized to e.g. 214 with all kinds of oxidizers, e.g. DDQ.173 The unsymmetrical Hantzsch reaction using two different β-keto-esters has been optimized for a plant-scale manufacture of the potassium-channel opener ZD0947.174 The Hantzsch MCR is also nicely working with C-glycosylated reagents as displayed in 2,4-dihydropyridine 215 (Fig 32). 175
The reaction of isocyanides, oxo components and primary or secondary amines yields α-amino carbonamides, as disclosed by Ugi in 1959.176 The reaction has been employed by Ugi to synthesize the local anesthetic Xylocaine and many derivatives thereof (Scheme 33). Xylocaine alters depolarization in neurons, by blocking the fast voltage gated sodium (Na+) channels in the cell membrane.
Scheme 33.

U-3CR scheme and the structure of xylocaine which can be advantageously synthesized by it.
Philanthotoxin-433 (PhTX-433), a low molecular weight natural polyamine toxin that originally isolated from the venom of wasp, showed potential non-competitive inhibitory effects on various types of ionotropic receptors in the central nervous system such as ionotropic glutamate receptors (iGluRs) and nicotinic acetylcholine receptors (nAChRs) in mammalians and in insects. Polyamines have been recently deemed to be “universal templates” in drug discovery.177 Libraries of PhTX-433 analogs were synthesized using Ugi's MCR (Scheme 34).178 It is well known that Ugi reactions often are faster and higher yielding if performed under high concentration. Here the MCR was performed under solventless conditions and 20 reactions were analyzed with no solvent or using the standard solvent methanol. In average 10% higher yields have been obtained under solventless conditions and the reaction time could be reduced to less than 1 h. A typical example is compound 216 (Scheme 34). Additionally, recently a concise synthesis of polyamines using Ugi MCR and subsequent exhaustive reduction was described giving now easy access to this “universal template”, e.g. 217.179
Scheme 34.

Different MCR derived channel modulators and the natural product philanthotoxin-433.
Compound 220 was found to be a selective T-type Ca-channel blocker equipotent to the marketed compound milbefradil.180 This morpholin-2-one-5-carboxamide and derivatives were prepared by using the one-pot Ugi MCR of glycolaldehyde (219), an isocyanide (41) and an α-amino acid (218). The use of the non-nucleophilic polar trifluoroethanol as a solvent is essential to suppress intermolecular reactions. The voltage-dependent Ca2+ channels (VDCC) are the primary route for translating electrical signals into biochemical events underlying key processes such as enzyme activity, neurotransmitter release, neuronal excitability, neurite outgrowth and gene transcription.
Atrial fibrillation (AF) and flutter are the very common cardiac arrhythmias encountered in clinical practice. The ultra-rapid delayed rectifier potassium current plays a significant role in the repolarization of the atrial action potential and selective inhibition of this current in human atrial myocytes prolongs action potential duration. Prolongation of the action potential is believed to prolong the atrial effective refractory period; therefore inhibition of the respective potassium channel Kv1.5 would produce an appropriate antiarrhythmic effect. Dihydropyrazolopyrimidine is a potent and selective inhibitor of the potassium channel Kv1.5 (Scheme 34).181 The Biginelli-3CR of benzaldehyde, 3-aminopyrazole and β-ketoester and two more subsequent reactions yielded dihydropyrazolopyrimidines, e.g. 221 with an IC50 for Kv1.5 block of 30 nM without significant block of other cardiac ion channels. The orally bioavailable compound 221 undergoes development for AF.182
The discovery of the first class of subtype-selective inhibitors of the human excitatory amino acid transporter subtype 1 (EAAT1) is reported. An SAR of 25 analogues was presented that addresses the influence of substitutions at the 4- and 7-positions of the parental skeleton 2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile. The most potent analogue 222 (Scheme 34) displays high nanomolar inhibitory activity at EAAT1 and a >400-fold selectivity over EAAT2 and EAAT3, making it a highly valuable pharmacological tool. Corresponding chromene derivatives can be conveniently accessed by a 3-CR of malonodinitrile (101), 4-methoxy-benzaldehyde (158) and 5-(naphthalen-1-yl)cyclohexane-1,3-dione (Scheme 34).186
The P2X7 receptor is a ligand-gated ion-channel and expressed on different lineages of cells, including macrophages, microglia, mast cells and T- and B-lymphocytes. Activation of the P2X7 receptor has been implicated in giant cell formation, regulation of cell proliferation, release of proinflammatory cytokines to name a few. Recent preclinical in vivo studies suggest implications of P2X7 receptor for inflammatory, neuropathic and visceral pain treatments.183 Several scaffold classes have been disclosed as modulators of P2X7 receptor, piperidinone, pyrrole and isoindole carboxamide derivatives (Scheme 35).184 Corresponding compound classes, e.g. 225 can be convergently synthesized by isocyanide-based MCRs using bifunctional and reactive oxocarboxylic acids 223.185
Scheme 35.
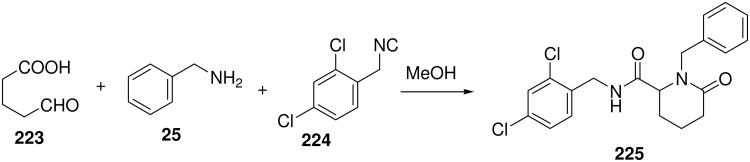
Intramolecular Ugi-MCR leading into activators of P2x7 receptor (225).
Alantrypinone is an insecticidal alkaloid that acts as a selective antagonist for housefly (vs rat) GABA receptors, and is considered to be a lead compound for the development of safer insecticides. The natural product and a library of derivatives thereof have been elegantly synthesized using a key one-pot MCR under microwave conditions and a subsequent hetero-Diels-Alder reaction (Scheme 36).187 The first step constitutes a condensation between anthranilic acid 226 and Boc-protected alanine in the presence of triphenylphosphite. Then the glycine methylester was added and treated under microwave conditions to yield the dihydro-quinazoline. Upon treatment with Borontrifluoride etherate and final oxidation the quinazoline (227) can be isolated. A Diels Alder reaction with the isatine derived in situ formed dienophile 228 finally yields alantrypinone. Detailed SAR based on substituted anthranilic acids (226), indones (228) and different amino acids is described.
Scheme 36.

Natural product GABA antagonist alantrypinone MCR synthesis. In 227 the Ala and Gly fragments are shown in red and blue, respectively.
2.7. Protein Protein Interactions
Protein protein interactions (PPIs) are a rather complex group of pharmaceutical targets being systematically studied only in recent years. Often PPIs are mediated by large interfaces, don't show deep and spatially confined binding isles (“hot spots”) and thus are difficult to target by small molecules. In fact it has been reported over and over that PPI modulator identification is challenging with today's HTS libraries.188 On the other hand PPIs sometimes are suitable for small molecule binding especially in the presence of deep and rather small binding grooves. For example PPIs targetable by small molecules have been classified by the dimensions and electrostatics of their interfaces.189
CCR5 is a chemokine receptor that is highjacked by the HIV to enter the cell and with the recent approval of maraviroc it consists a validated and new target to fight AIDS. Taking up the privileged structure idea pharma company scientists synthesized spirodiketopiperazines 231 using Ugi reactions (Scheme 37).190 Amongst all synthetic pathways to (di)(keto)piperazines IMCRs certainly are the most versatile ones.33,152 Several hundred spiroketopiperazines have been synthesized using solid and liquid phase techniques. Very potent, however poorly water soluble compounds have been discovered. Interestingly, a metabolite was found to be even more active and also more water soluble. An exemplary synthesis is shown in Scheme 37. Also to mention is the use of the commercial morpholinoethylisocyanide as a cleavable isocyanide.190 An advanced compound, aplaviroc is undergoing clinical trials.
Scheme 37.

Potent protein-protein interaction antagonists 231 of the entrance of HIV into cells have been assembled using Ugi-4CR.
The PPI between the transcription factor p53 and its negative regulator protein mdm2 has been reported to play an important role in the chemo and radiation resistance of cancers.191 The interaction has been described in molecular detail and the dimension and character of the binding site indicate a suitable small molecule target.192 One of the first potent antagonists of this interaction described is the imidazolidine class of nutlins.193 Numerous biological studies indicate their potential usefulness in cancer therapy.194 In additions to the nutlins several other small molecular weight compounds amenable by MCR chemistry have been recently described. The first classes of compounds discovered by a high throughput screening exercise are highly substituted benzodiazepindiones (238).195 The compound class is generally accessible by a Ugi-4CR of anthranilic acids, cyclohexenyl isocyanides (237) as a representative of the convertible isocyanides, aldehydes and primary amines.196 Cyclisation via a Münchnone intermediate results in the target class. Due to the general, efficient and versatile access in excess of 20.000 derivatives have been produced and screened. Detailed SAR has been published and a high resolution X-ray structure of a representative benzodiazepinone in the mdm2 binding site has been reported (Fig. 33).
Figure 33.
Synthesis and cocrystal structure of potent small molecular weight p53-mdm2 antagonists 238. The synthesis involves a U-4CR of N-protected anthranilic acid, a primary amine, and aldehyde and the convertible isocyanide cyclohexenylisocyanide, followed by acid deprotection and cyclisation via a Münchone intermediate. A highly affine benzodiazepindione derivative 238 bound to mdm2 is shown below (PDB ID: 1T4F). The 4-chlorophenyl glycine, the 4-chlorophenyl and the 7-iodophenyl moieties occupy the Leu26, Trp23 and Phe19 binding pockets in mdm2, respectively.
The scaffold of 4-carboxy tetrahydroquinolines 243 has been reported as mdm2 binder as shown by detailed 2D-NMR studies.197 In addition, the ability to dissociate the preformed p53/mdm2 complex was reported by a new NMR experiment called antagonist induced dissociation assay (AIDA).198 E.g. compound 243 antagonizes the complex with a KD of 1 μM. The compound class was discovered by a computational chemistry approach using a ligand based scaffold-hopping compound selection. The same approach yielded 245 as a novel p53/mdm2 antagonist. Both classes can be efficiently synthesized by appropriate MCRs. The first tetrahydroisoquinoline derivative 243 is the product of 3-CR of homophtalicacid anhydride (239) an appropriate aldehyde (240) and primary amine (241), following the amidation of free carboxylic acid and amine (242); whereas the second pyrrolidone-derivative 245 can be accessed by a variation of the Döbner MCR (Scheme 38). Both classes of compounds show mechanism-based activities in cellular assays.
Scheme 38.

MCR compounds 243 and 245 antagonizing p53-mdm2.
A novel drug discovery technique based on the tight interplay of computational and MCR chemistry, docking and high content screening yielded 10 unprecedented scaffolds predicted to bind into the p53 binding site of mdm2 and have been subsequently shown to bind as predicted by HSQC NMR experiments and cocrystal structure analysis.199 The key steps of the approach are as follows: The interface of a particular PPI is analyzed and certain amino acid side chains are classified as anchor residues according to their high burriedness. The assumption is that the more a side chain is buried in the receptor the higher its energetic contribution. Next the anchor side chain is imposed on many different MCR scaffolds and virtual libraries are generated, in a way that all compounds contain the anchor residue. Next the virtual library is docked into the PPI interface in a way that the anchor of the compounds is overlapping with the corresponding amino acid side chain using the freeware ANCHOR.QUERY (http://anchorquery.ccbb.pitt.edu/). From the corresponding docking lists compounds are chosen for synthesis and screening based on shape complementarity, electrostatic interactions and practical aspects such as ease of synthesis based on available starting materials. Although this approach resembles a fragment-based approach, however it overcomes one of its current limitations, the fragment optimisation, by combining the fragment with a very large and efficiently accessible chemical space: MCR (Fig. 34).200
Figure 34.
Schematic process of discovery of PPI antagonists, based on structural information, hot spot anchors and rapid MCR chemistry.
Several predicted compound classes (246-254) showed potent cellular activity and could be optimized from initial μM to nM affinity due to the convergent MCR chemistry approach (Scheme 39).199 The binding mode of a van Leusen indoloimidazole into the p53 binding site in mdm2 is shown in Fig. 35 and 36 as revealed by X-ray structure analysis and as predicted by the above approach.201 This approach makes advantageous use of MCR chemistry since several backbones are predicted at the same time and could be optimized in parallel thus reducing the effect of attrition of a particular scaffold due to inferior properties. Additionally, the scaffolds are intrinsically optimization friendly since they are based on MCR chemistry. This parallel drug discovery approach seems to have high predictive power. Significantly, this approach can be an alternative to current drug discovery techniques in this area namely high throughput screening (HTS). A freely accessible web server was build up performing this analysis for any given protein protein interactions (http://anchorquery.ccbb.pitt.edu/).
Scheme 39.

P53-mdm2 antagonists (246-254) accessible by MCR and predicted by a new approach, ANCHOR-QUERY.
Figure 35.
Interaction of vL-indoloimidazole 253 with the p53 binding islet of the mdm2 receptor (PDB ID: 3LBK). The anchor residue chloro-indole occupies the Trp23 binding site, whereas the 4-chlorobenzyl mimics the Leu26 and the phenyl moiety the Phe19 site. Notably, the indole forms a nice hydrogen bridge to Leu54 backbone carbonyl of the mdm2 receptor similar to the Trp23p53 mdm2 interaction.
Figure 36.
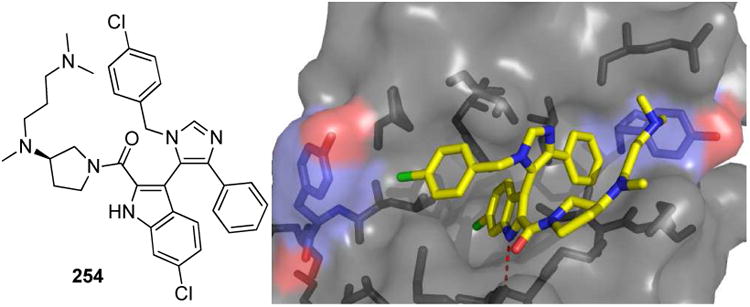
Interaction of vL-indoloimidazole 254 with the p53 binding site of the mdm4 receptor (PDB ID: 3LBJ). The anchor residue chloro-indole occupies the Trp23 binding site, whereas the 4-chlorobenzyl mimics the Leu26 and the phenyl moiety the Phe19 site. Notable, the indole forms a nice hydrogen bridge to Leu54 backbone carbonyl of the mdm2 receptor similar to the Trp23p53 mdm2 interaction.
Most of the scaffolds resulting from this approach are drug like and straight forward to optimize since they are MCR derived. As an example the imidazoline scaffold derived from the Orru-3CR with initial double digit μM Ki could be optimized to 1 μM compounds with high water solubility.202 One of the discovered scaffolds, imidazolindoles, has been previous described as anti-cancer active and some derivatives show high affinity to mdm2.203 E.g. compound 253 has a Ki = 400 nM and could be cocrystallized with mdm2 (Figure 35).201
The improved amide 254 has a Ki = 4 nM to mdm2 and, interestingly also shows low uM affinity to mdm4. The first X-ray cocrystal structure of a small molecule binding to mdm4 could be subsequently solved (Fig. 36).201 Clearly, the indole moiety of the indoloimidazoles overlaps with the p53-Trp23 almost perfectly in both structures which nicely validate the above described process. The phenyl group points into the Phe19 and the p-chlorobenzyl group into the Leu26 pockets, respectively.
Inhibitors of apoptosis proteins (IAP) are an eight-membered family, defined by the presence of a baculovirus IAP repeat (BIR) protein domain, and they are key regulators of apoptosis.204 XIAP is unique among IAP proteins, because of its ability to inhibit and directly bind to activated caspases. Through its BIR2 domain with its N-terminal linker, XIAP binds to the active site of effectors caspase-3 or -7 and prevents substrate binding and subsequent catalysis sic apoptosis.205 Using the known tetrapeptide AlaValPheIle, specifically, the N-terminal Ala-Val anchor several new scaffolds (255-259) based on MCR chemistry have been discovered using the above described anchor based drug discovery approach (Scheme 40).206
Scheme 40.

Above: the pharmacophore of XIAP antagonists. The N-terminus is very important in forming a tight charge charge interaction with Glu314. In addition to the tight network of hydrogen bonds addressing the hydrophobic pockets, the central heterocycle with cis-geometry is of importance for inhibitor design. Below: several MCR scaffolds with μM XIAP activity and accessible by a rather short synthesis sequence.
Clinical development of the antagonist of antiapoptotic Bcl family proteins by ABT-737 is a success story of the new fragment-based drug discovery approach.207 Simplified derivatives (260) have been synthesized using a very fast and convergent access: U-4CR followed by SnAr substitution, thereby introducing an isosteric replacement of the central N-acylsulfonamide for an α-acylaminocarboxamides (Ugi backbone) (Scheme 41).208
Scheme 41.

Antagonists of antiapoptotic Bcl2.
Heat shock proteins (Hsps) are a family of highly conserved molecular chaperones responsible for the folding of nascent protein chains, for the refolding of misfolded proteins, and for the degradation of polypeptide substrates that are unable to achieve their native conformations. They have recently become important molecular targets for cancer, malaria and stroke. Several Hsp90 inhibitors undergo clinical trials for cancer. The first small molecular weight compounds targeting the less known Hsp70 brother are products of two MCRs, the Ugi and Biginelli reaction.81a,209 The active compounds alter the ATP hydrolytic rate, an event that is catalyzed by the N-terminal, ATPase domain in Hsp70s. The binding and hydrolysis of ATP, and the release of ADP are linked to the binding and entrapment of polypeptide substrates in the C-terminal half of Hsp70. Some of these agents also inhibit the proliferation of transformed cell lines and the growth of the malaria parasite, which—like cancer cells—requires high levels of diverse Hsp70s for its survival.210 Additionally, the in silico design of compounds interacting with the Hsp70 peptide recognition site has been reported.211 These compounds were designed to mimic the tri-leucine motif of Hsp70 peptides, specifically based on the anchor residue Leu and with built-in water solubility. They have been synthesized by a U-4CR (Scheme 42).
Scheme 42.

HSP-70 inhibitors by MCR.
The discovery of a new MCR subsequently also leads to a new class of protein protein interaction antagonists. Thus, the three-component synthesis of diversely substituted and fused amino-pyrrolo-heterocycles by the condensation of activated methylene compounds, aldehydes and isonitriles was recently reported (Scheme 43). This efficient 3CR leads to a diversity of heterocycles in a one-pot fashion and is useful for the synthesis of tens of thousands of discrete compounds.212 By a high throughput screening approach 3-alkyl-2-phenethylindolizine-1-carbonitriles (268) were found to be potent inhibitors of the protein - protein interaction between vascular endothelial growth factor (VEGF) and neuropilin-1, a process which is believed to be involved in the invasion of tumour cells into human prostate.
Scheme 43.

A new MCR lead to a new class of VEGF receptor antagonists.
The RGD (arginine-glycine-aspartic acid) loop contains peptides that are the molecular attachment points of many cellular and extracellular matrices. Along with the integrins, their receptors constitute a major system for cell adhesion,213 which is crucial in many pathological processes, such as tumour metastasis, angiogenesis, osteoporosis, and thrombosis. Drug-like RGD mimetic development is challenging due to the receptor imposed zwitterionic requirements for the ligands. Two groups independently reported RGD mimics using Ugi MCRs (269-270, Scheme 44).214 Although the molecules display rather large molecular weight and abundant peptide character, these works nicely shows the advantages of MCR chemistry in providing fast, efficient and convergent access to biologically relevant screening compounds.
Scheme 44.

Peptide-like RGD mimetics 269 and 270 and a viral capsid assembly inhibitor 271 by MCRs.
Heteroaryldihydropyrimidines have been reported to inhibit Hepatitis B virus replication by drug-induced depletion of nucleocapsids although the exact mechanism-of-action is unknown.215 For example compound 271 possess potent in vitro and in vivo antiviral activity. Such compounds have been synthesized by multicomponent condensation of a suitable amidine, benzaldehyde and acetoacetate by a Biginelli variation.
It is intriguing to note that numerous molecules amenable by MCR chemistry have been described in the past to antagonize PPIs. MCR-derived molecules represent a significant fraction of currently described PPI (ant)agonists and support the notion that MCR space is especially suitable for PPIs. A hypothesis why MCR reaction products are more suitable to (ant)agonize than “traditional” compounds libraries relates to their general higher atom density. Protein protein interfaces contain mostly a very dense array of interactions, including van der Waals, hydrogen bonds and charge charge interactions. A typical small drug-like molecule only allows for rather few interactions to a target structure due to the very much reduced amount of atoms, functional groups and substituents per volume around a given scaffold. MCR scaffolds, however, are known to be much more densely functionalized than other scaffolds. In fact MCR scaffolds have been often described as peptide-mimetics with the advantage, however of much reduced secondary amide bonds and thus more drug likeness.
2.8. Miscellaneous
The proportion of engineered antibodies approved for diagnostics and human therapy has increased significantly during the last decade. At present, 17 human therapeutic monoclonal antibodies (mAbs) are on the market; additionally multiple other mAbs are currently undergoing final clinical trials and they are representing nearly a quarter of all biologics undergoing trials. To date, six Fab molecules have been also approved by the FDA for human use. These monovalent immunoglobulin fragments provide therapeutic alternatives to their parental relatives, by retaining their antigenic specificity, whilst being produced more economically. Such Fabs have to be large scale produced and purified using affinity chromatography. A novel use of the U-4CR to generate a solid-phase library suitable for the purification of immunoglobulins and their fragments by affinity chromatography has been reported.216 An optimized candidate for production purposes was obtained and also docked into a human Fab fragment to rationalize the binding interaction (Fig. 37). The Ugi scaffold offers an alternative route to the well-defined triazine chemistry for generating synthetic ligands. The final ligand 272 clearly suggests the potential of the Ugi scaffold in the development of potent ligands. Due to its synthetic nature, compound 272 is expected to be inexpensive to produce.
Figure 37.
Sepharose solid-support Ugi products (272) for the affinity purification of therapeutic Fab fragments. Docking of the best Ugi ligand (blue sticks) into human Fab fragment (PDB ID: 1AQK).217
The farnesoid X receptor (FXR), is a nuclear hormone receptor with activity similar to that seen in other steroid receptors such as estrogen or progesterone. FXR is expressed at high levels in the liver and intestine. FXR modulators are believed to be useful for the treatment of increased lipid and cholesterol levels. A recently disclosed FXR modulator is composed of a highly substituted benzimidazole 276 which can be accessed by UDC (Scheme 45).218 For example, compound 276 shows an affinity for FXR of 13 nM.
Scheme 45.

FXR nuclear hormone receptor modulators by UDC.
MCR have been frequently described for the synthesis of bioactive compounds to treat neglected tropical diseases (NTDs). Drug discovery for NTD is not a high priority for pharma companies due to the financially unattractive market and the prohibiting high costs of development.219 This application seems to be perfectly suited for MCRs since the costs of the early discovery chemistry and the cost-of-goods (COG) of the drug production are potentially very low. Praziquantel, for example, is a member of the 12 drugs comprising the WHO list of essential medicines.220 It is used to treat the parasitical disease schistosomiasis also called bilharziose. Schistosomiasis is one of the largest burden of mankind affecting more than 200 million people worldwide.221 Importantly, there is evidence for a strong correlation between schistosomiasis and HIV infection in Africa. Thus, the urinary form of schistosomiasis, which affects up to 50 per cent of women in parts of Africa, damages the lining of the vagina, the first defensive barrier against HIV. An affordable $0.32 (US) solution per treatment for preventing HIV/AIDS has thus been recently proposed based on the highly effective and low-cost anti-schistosomal drug praziquantel (PZQ).222 The tetrahydroisoquinoline derivative PZQ is the major drug to treat this disease due to its advantageous properties, including efficiency, safety and low cost-of-goods to potentially reach a very large number of infected patients.223 Current technical syntheses involve sequential 5-7 step sequences. Recently, a considerably shorter and scalable synthesis including an Ugi and subsequent Pictet-Spengler approach has been described which has the potential to further reduce the COG of this life saving essential drug.224 COG is a key factor for the development of drugs neglected tropical diseases. Moreover this approach allows for the synthesis of many analogs based on the central MCR chemistry to overcome potentially upcoming occurrence of resistance.225
MCR reactions have been described several times to discover novel agents to treat malaria.226 E.g. 4-aminoquinoline 2-imidazolines have been recently described to be active against the malaria parasites against two strains of Plasmodium falciparum and Trypanosoma brucei.226 Compound 280 was the most active across all parasites with ED50 = 3.3 nM against a chloroquine sensitive strain, ED50 = 33 nM against a chloroquine-resistant strain and ED50 = 70 nM against T. brucei and can be synthesized by the Orru-3-CR.
Aryloxy cyclohexyl imidazoles (281) which can be beneficially synthesized by a key α-aminoalkylation of cyclohexanone, 2 equivalents of formaldehyde and pyrrolidine and subsequent transformations have been described as a novel class of antileishmanial agents (Scheme 47).227 These compounds are superior than the existing drugs, sodium stibogluconate and pentamidine in respect to IC50 and SI values. Promising compounds were tested further in vivo. Among all, compound 281 exhibited significant in vivo inhibition of 79%, thus providing new structural lead for antileishmanials.
Scheme 47.

MCR compounds for different NTDs.
Novel nucleoside analogues, e.g. compound 282 based on the approved antiviral drug Cidofovir have been synthesized as potential antiviral and antileishmanial agents via different variations of the Ugi MCR. Several synthetic products showed antileishmanial activity in the 10−5 M range.228
Glutamine synthetase is required by M. tuberculosis for nitrogen metabolism and mycobacterial cell-wall biosynthesis and has emerged as a potential target for antibiotics against TB. Functionalized 3-amino-imidazo[1,2-a]pyridines – products of the GBB-3CR have been discovered as a novel class of drug-like Mycobacterium tuberculosis glutamine synthetase inhibitors with impressive activity. Compound 283, for example is much more active than the so far known inhibitors L-methionine-SR-sulfoximine and phosphinothricin.229
New infectious diseases appear regularly in diverse parts of the globe, most recently swine flu, creating new global health threats. The upcoming of new multiple drug resistance and highly infectious and deadly influenza is of great concern. Current weaponry to fight influenza can only build on a handful of chemotherapeutic options besides immunisation. The anti-influenza neuramidase inhibitor (-)-oseltamivir is one of them and has been synthesized by a remarkably short and high-yielding asymmetric synthesis taking advantage of a one-pot MCR involving an asymmetric Michael addition of aldehyde 284 to nitro compound 286 subsequent second Michael addition/intramolecular Horner–Wardsworth–Emmons reaction with vinylphosphonate 285.230 Subsequent treatment with p-toluenethiol 287 afforded the heavily functionalized ethylcyclohexanecarboxylate 288 in good yield (70%) in a single-pot operation (Scheme 48). This work represents a landmark of efficiency in organic synthesis: In only nine reactions, a total of three separate one-pot operations, and one purification by column chromatography the drug is stereoselectively amenable in overall excellent yields (from nitroalkene 57%). All the reagents are inexpensive and the synthesis compares very favourably with the current technical synthesis.231
Scheme 48.

Enantioselective and short oseltamivir synthesis.
Of considerable interest is the anti-cancer activity described for BG-3CR products binding to the emerging cancer target kinesin motor spindle protein. A potent inhibitor, monastrol, which was synthesized from ethyl 3-oxobutanoate (195), thiourea (289) and 3-hydroxybenzaldehydehas (290), has been first discovered by a phenotypical cell-based screening (Fig 39).232 Several high resolution X-ray structures have been reported and the role of the BG-scaffold in their binding can be studied.233 Another cocrystallized MCR derived molecule with atomic resolution is the Gewald thiophene 291.234 Recent evidence supports a mechanism by which monastrol and similar compound weaken the interaction of the motor kinesin Eg5 and the microtubule by an allosteric mechanism.223,235 Both molecules bind into a deep hydrophobic allosteric pocket, however establishing different molecular interactions.
Figure 39.
Synthesis of Biginelli product monastrol and a Gewald thiophene 291.
Crystal structure of the motor protein KSP in complex with monastrol (above: yellow sticks, PDB ID: 1Q0B) and the Gewald thiophene (below; PDB ID: 2UYM). The thiourea and 3-hydroxy benzaldehyde portion of the Biginelli backbone is buried deeply in an induced-fit binding site some 12 Å apart from the ATP binding site. The phenolic hydroxyl group forms a hydrogen bond to the backbone carbonyl of Glu118 and to Arg119. The thiourea sulfur undergoes extensive van der Waals contacts to aliphatic amino acids. Note the planar structure of the Biginelli backbone and the orthogonal exit of the phenol substituent. The Gewald backbone, doesn't make any direct hydrogen bond contacts to the protein, however they are mediated by two water molecules (aquamarine ball). The carbonyl component and the cyanoacetamide component side chain of the Gewald product form strong van der Waals interactions with excellent shape complementarity to the binding pocket. In both X-ray structures tightly bound water play a prominent role.
A product (295) of the Povarov-3CR from benzaldehyde (176), aniline (292) and electron-rich olefin (293) were found to be a kinesin-5 inhibitor (Fig 40).236 The compound showed promising potency in an in vivo xenograft model of colo 205 cells and is currently undergoing early investigation in clinical cancer trials.
Figure 40.
Cocrystal structure of 295 with kinesin-5. The dilemma of target-required hydrophobicity and inherent bad water solubility, poor PK properties and metabolic instability was solved by adding the solubilizing dimethylaminoethylamine urea moiety onto the tetrahydropyrane ring which points into the solvent space. The tetrahydroquinoline NH is involved into a hydrogen bond to backbone carbonyl Glu116 as is the urea carbonyl forming a hydrogen bond to a water molecule and backbone carbonyl Arg119.
Coenzyme A is a ubiquitous cofactor in many different enzymes. Many of these are involved in pathogenic processes. For example, malonyl-CoA transferase (FabD) is an essential enzyme involved in the assembly of fatty acids. Due to the considerable difference of the human enzyme form, the bacterial one FabD consists an antibacterial target.237 An approach to inhibit FabD could be for example by modified CoA derivatives. Recently, glutathione, and homoglutathione derivatives (296 and 297) were synthesized by the Ugi four-component reaction using various benzylthio aldehydes and ketones as carbonyl building blocks (Scheme 49).81b
Scheme 49.

(Homo) glutathione derivatives by U-4CR and the structure of Acetyl CoA.
FTY720 is a clinically investigated immunosuppressive and it also shows very promising clinical results in multiple sclerosis treatment. This fungal natural product myriocin-derived agent seems to work on lymphocyte trafficking by antagonizing the sphingosine-1-phosphate after being phosphorylated by sphingosine kinase. A short two step synthesis using the Petasis reaction (Pt-3CR, a boronic acid Mannich reaction variation) of dihydroxyacetone 298, benzylamine 25 and vinylboronic acid 299 was reported (Scheme 50).238
Scheme 50.

Immunosuppressive and anti-MS drug FTY720 synthesized by Petasis-3CR.
Another recently approved compound the cholesterol absorption inhibitor Zetia ™ (ezetimibe) is produced by a Staudinger-3CR (Scheme 51).239 During the Staudinger reaction a methylene active acylchloride (ketene precursor) reacts with a Schiff base formed by aldehyde and amine, likely in a stepwise cycloaddition process.240 Although the reaction cannot be performed by the simultaneous addition of all starting materials at once a convenient one pot protocol exists.241
Scheme 51.

Staudinger-3CR product Zetia ™ and a bioactive aza-steroid.
Steroids are ubiquitous often highly potent hormones involved in most aspects of health and disease. Historically, steroids have played an extraordinary role in the collection of drug and still many steroids are used in different therapeutic areas. Azasteroids can be easily synthesized in high diversity and numbers using MCR (Scheme 51).242
Potentially antiviral 4-(3H)-quinazolinone N-nucleosides (302) have been elegantly assembled by the MCR of anthranilic acid, ribosylamine and a substituted/unsubstituted benzoic acid in a one-pot reaction under MW irradiation and solvent-free conditions (Scheme 52).243
Scheme 52.

Antiviral quinazolinone N-nucleoside (302).
The progesterone receptor (PR), is an intracellular steroid nuclear receptor that specifically binds progesterone. α-Aminotetrazoles amenable by U-4CR have been recently disclosed as potent and selective partial agonists and have potential as a new treatment for endometriosis.244 Compound 305, for example, optimized for potency, selectivity and P450 inhibition, has excellent oral half life time and is suitable for in vivo pharmacology studies (Scheme 53).
Scheme 53.

Classical synthesis of a progesterone receptor agonist and a corresponding MCR synthesis.
A 3-CR of an isocyanide, a dialkyl acetylenedicarboxylate, and tetronic acid in dichloromethane at room temperature afforded 4H-furo[3,4-b]pyran derivatives (308).245 These compounds are structurally closely related with some natural products, e.g. TAN-2483B and fusidilactones with several reported biological activities, including strong c-src kinase inhibitory action, in vivo bone protection and a broad spectrum of activity against cultured tumor cell lines, including adriamycin-resistant HL-60 cells. A related MCR of alkyl isocyanides various aldehydes and 3-hydroxy-1H-phenalene-1-one yields 9-(alkyl or arylamino)-7H-phenaleno[1,2-b]furan-7-one (311) derivatives which are reminiscent to the furophenalenone scaffold of many natural products, such as atrovenetin with multiple described biological activities (Scheme 54).246 Combinatorial applications where described and scope and limitations are reported.
Scheme 54.

MCRs leading into natural product-like furopyrans.
Discovery and development of plant protecting and other agrochemical materials also seems to be an important application of MCR chemistry, since the COG of the active ingredient is a key parameter in this area. For example, a successful case of the application of MCR for the generation of valuable bioactive compounds is the recent market approval of mandipropamide a plant protecting agent discovered and made by isocyanide based MCRs including Passerini and Ugi reactions.247 Noteworthy, in this elegant synthetic scheme is the one-pot isocyanide formation / TiCl4-mediated Passerini reaction from the formamide precursor 312 without isolation of the isocyanide. The intermediate Passerini product 313 is then alkylated by propargylbromide to yield the marketed product (Fig. 41). The compound is highly active against a variety of economically important plant pathogens leading to crop destruction of potato, tomato late and grape. For example, the effective concentration to kill 80% of the pathogen Phytophthora infestans (grape downy mildew) is only 100 μg/l (EC80). Another agrochemical application of MCR is the short synthesis of novel avermectin derivatives as insecticital agents through the diastereoselective Ugi reaction to an phenylsulfinimide intermediate.248 Fipronil is a new fluorinated pyrazole with high insecticide activity and derivatives thereof have been synthesized by the Mannich reaction of hydrazones coupled with a [4+1] cycloaddition with isocyanides.249
Figure 41.
Marketed mandipropamide as an agrochemical application of MCRs. It is used to protect wine grapes from fungal infections. Below: Infected and healthy grapes (Phytophthora infestans)
Key diketopiperazine moieties in the DNA targeting anticancer natural products naphthyridinomycin, lemonomycin and the clinical liposarcoma compound ecteinascidin have been assembled using Ugi-MCRs as key steps (Scheme 55).250
Scheme 55.

Napthyridinomycin, lemonomycin and ecteinascidin's piperazine moiety all has been assembled using one-pot U-4CR.
The recent discovery of 2,4-diphenylthiazolyl-5-amides 315 and 316 as antiprion agents lead to a straight forward and general access towards this scaffold class involving a short sequence of U-4CR involving ammonia equivalent and the acid cleavable Walborsky reagent 314, followed by acid amine deprotection and thiazole formation and finally acidic amide deprotection.251 The substituents introduced at the 2-and 4-positions are derived from simple and widely variable building blocks, carboxylic acids and aldehydes respectively. Though yields are modest, the route offers access to a large number of diverse new compounds, based around this pharmaceutically relevant substructure, which would otherwise be considerably more difficult to prepare by alternative routes.
4. Summary and Outlook
MCRs are a useful class of reactions for the never-ending hunt for biologically active compounds and complementarily add into the large arsenal of tool boxes available to the modern chemist. How do MCR derived molecules differ from the others? One distinguishing feature is the densely functionalisation of MCR derived molecules. Due to the fact that several ligands are introduced around a common scaffold, typically the ligand density as well as the number of functional groups can be very high. Based on the densely functionalized scaffolds and their often non-flat, sometimes spirocyclic nature the 3D shape of MCR derived molecules is different from the rest. An ever-increasing body of data suggests that in fact MCR derived molecules might be more suitable for certain drug discovery areas than other type of molecules. For example, the high number of MCR derived molecules in the area of protein protein interactions is striking. An advantage of MCR chemistry is the very large chemical space, probably the largest available chemical space for discovery and medicinal chemistry purposes. This also poses very high demands for the right choice of the discovery strategy, e.g. high throughput screening or structure-based design. Clear financial and technical limits are given for the screenable library size in traditional HTS.223 A promising and complementary strategy which leverages the strength of MCR chemistry is the use computation screening and e.g. genetic algorithms.
Twenty years ago MCR chemistry was broadly unrecognized and only considered of use for the synthesis of specific classes of compounds. Only recently its broad applicability and values were recognized by the synthetic community, including the short and highly efficient synthetic access to a plethora of scaffold with very large numbers of compounds per scaffold. Access to many different types of pharmacophores exemplified in different MCRs backbones turned out to be of particular value for the discovery of bioactive compounds. Additionally, many MCR can be performed in an enantioselective manner. Often MCR chemistry suites well the discovery phase and later on the production of the candidate use different chemistry. In other cases, however, MCR chemistry can be advantageously used during discovery chemistry as well as in the production phases. Different large scale technical productions of advanced compounds have been described using MCR. The growing number of compounds on the market and in clinical evaluation discovered and synthesized by MCR technologies manifests their growing importance. Whereas in the past we witnessed only few examples of MCRs in natural product total synthesis, the efficiency and convergence of these reactions will certainly become of great value in future natural product synthesis. A final aspect of MCR chemistry should not be kept secret: MCR chemistry is intellectually stimulating and can be very aesthetic (Scheme 57).252 MCR chemistry & biology certainly has a bright future!
Scheme 57.

Who can solve the jigsaw? One pot 8-CR to compound 323 based on three sequential MCRs with an 85% yield per bond formation.
Figure 9.
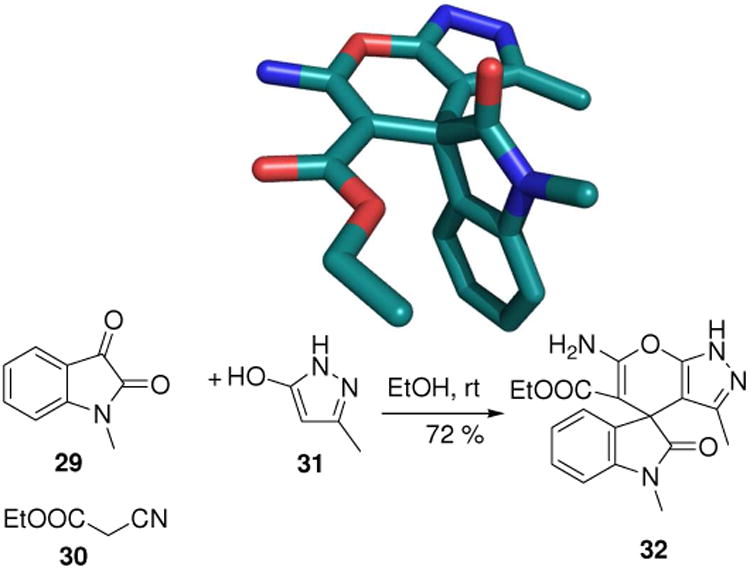
Spiroheterocycles of great diversity, e.g. 32 can be accessed by different MCRs (CCDC ID: 643526).
Figure 38.
Alignment of the cocrystal structures of S-276 (yellow sticks) and optimized S-278 (marine sticks) to hFXR (PDB ID: 3OKI, 3OMM). The FXR binding site of 3OKI is shown as grey surface and selected amino acids as sticks. The inhibitors are encapsulated almost fully into the receptor. The highly conserved Tyr373 is making a hydrogen bridge to the scaffold benzimidazole-3N and is key to the efficient ligand binding (red dotted line). π-Stacking can be seen between Phe333 and the carboxylic acid derived p-chlorophenol. Ser336 (not shown) is engaged into hydrogen binding to the amide-NH resulting from the isocyano component. Additionally, in the hydrophilicity-optimized structure 277 a p-carboxyphenyl moiety at the mouth of the binding site mimics two tight waters (grey balls), forming an extensive hydrogen bond network with Arg335 and Gln267. The o-fluoro substituent of the isocyanide derived phenyl of 277 is accommodated in a hydrophobic bulb formed by the two Met332 and 294 (not shown) forming short hydrophobic contacts.
Scheme 22.

Synthesis of the potent Rho kinase inhibitors 148 by a 3-CR.
Scheme 46.

The Schistosomiasis drug PZQ can be convergently assembled using a key U-4CR and PS-2CR.
Scheme 56.

Versatile assembly of 5-aminothiazoles (315 and 316) based on the U-4CR leading to antiprion-active compounds.
Acknowledgments
AD acknowledge generous financial support by the University of Pittsburgh. This work has been partially supported by the grants 1R21GM087617, 1R01GM097082 and 1P41GM094055 from the National Institute of Health.
Abbreviations
- Abeta
amyloid β-peptide
- AIDA
antagonist induced dissociation assay
- AF
atrial fibrillation
- APP
myloid precursor protein
- BACE
β-secretase
- BBB
blood-brain-barrier
- Bcl
B-cell lymphoma
- BG-3CR
Biginelli three component reaction
- BIR
baculovirus IAP repeat protein domain
- CaCCs
Calcium-activated chloride channels
- CB1
cannabinoid type 1 receptor
- Cdc25A
cell division cycle 25 homolog A
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator chloride channel
- CoA
coenzyme A
- COG
cost-of-goods
- COX
cyclooxygenase
- CRF
corticotropin releasing factor
- DCC
dicyclohexylcarbodiimide
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DHODH
dihydroorotate dehydrogenase
- DMAD
acetylenedicarboxylicacid diester
- DMAD
dimethylaminopyridine
- DMD
Duchenne muscular dystrophy
- DKP
diketopiperazine
- DPP-IV
dipeptidyl peptidase IV
- ECS
endocannabinoid system
- EAAT1
excitatory amino acid transporter subtype 1
- EphB4
ephrin type-B receptor 4
- Fab
fragment antibody
- FabD
malonyl-CoA transferase
- FDA
food and drug administration
- FSH
follicle-stimulating hormone
- FVIIa
blood coagulation factor VIIa
- FXa
blood coagulation factor-Xa
- FXR
farnesoid X receptor
- G-3CR
Gewald three component reaction
- GA
genetic algorithm
- GABA
γ-amino butyric acid
- GBB-MCR
Gröbcke-Blackburne-Bienaymé-MCR
- Gn-RH
gonadotropin-releasing hormone
- GLP-1
incretin hormone glucagon-like peptide 1
- GPCR
G-protein coupled receptor
- G6PT
glucose-6-phosphate translocase
- IAP
inhibitor of apoptosis proteins
- H-4CR
Hantzsch four component reaction
- HDAC
histone deacylase enzyme
- HIV
human immunosuppressive virus
- HL-60
human promyelocytic leukemia cells
- Hsp
heat shock protein
- HSQC
heteronuclear single quantum coherence
- HTS
high throughput screening
- iGluR
ionotropic glutamate receptors
- IMCR
isocyanide-based multicomponent reaction
- LH
luteinizing hormone
- mAb
monoclonal antibody
- MC
multicomponent reaction
- MDM2
mouse double minute 2, negative regulator of p53
- MDM4
mouse double minute 4, negative regulator of p53
- MCH
melanin-concentrating hormone
- MoI
Program suite to calculate e.g. shapes of molecules: Pipeline Pilot, SciTegic, Inc. 10188 Telesis Court, Suite 100, San Diego, CA 92121, SA, http://www.scitegic.com/products_services/pipeline_pilot.htm.
- MPTPA/B
Mycobacterium tuberculosis protein tyrosine phosphates A and B
- MW
micro wave
- nAChRs
nicotinic acetylcholine receptors
- NPY
neuropeptide Y
- NTDs
neglected tropical diseases
- PADAM
Passerini-reaction-Amine-Deprotection-Acyl-Migration
- PDE
3′,5&prime-nucleotide phosphodiesterase
- P-3CR
Passerini three component reaction
- P53
transcription factor
- PGHS-2
prostaglandin H synthase-2
- PGP
phosphor-glyco-protein
- PhTX-433
philanthotoxins-433
- PKPD
pharmacokinetic pharmacodynamic
- PPI
protein protein interaction
- PR
progesterone receptor
- Pt-3CR
Petasis three component reaction
- PTP1B
tyrosine-protein phosphatase non-receptor type 1
- PS
Pictet-Spengler reaction
- PZQ
praziquantel
- RCM
ring closing metathesis
- RAS
renin-angiotensine-aldosterone system
- RGD
arginine-glycine-aspartic acid motif
- Ro5
rule of five
- ROCK1a
Rho-associated kinase isoform 1
- SAHA
suberoylanilide hydroxamic acid, Vorinostat
- SAR
structure activity relationship
- SFC
super critical carbondioxide fluid chromatography
- TF
tissue factor
- U-4CR
Ugi four component reaction
- U-5C-4CR
Ugi five center four component reaction
- UDC
Ugi-deprotection-cyclisation
- VDCC
voltage-dependent Ca2+ channels
- VEGF
vascular endothelial growth factor
- vL-3CR
van Leusen three component reaction
- XIAP
x-linked inhibitor of apoptosis protein
Biographies

Alexander Dömling (borne 1964) studied Chemistry & Biology at the Technical University Munich (TUM). He performed his PhD under the surveillance of the late Ivar Ugi working on the “Seven Component Reaction”. As a Feodor Lynen Fellow of the Alexander von Humboldt foundation he performed his postdoc in the laboratory of the Nobel laureate Barry Sharpless working on novel multicomponent reactions (MCRs) of hydrazines, epoxides and carboxylic acid derivatives. In 1996 he started the biotech company Morphochem and served as vice president chemistry and board member till 2004. During this time several drug candidates have been discovered at Morphochem and are currently in late preclinical or clinical trials. In 2003 he performed the Habilitation at the TUM and received the “Lehrerlaubniss in Chemie”. Since 2004 he is faculty member at the TUM. In 2006 he accepted an professor position in the School of Pharmacy (Drug Discovery Institute) at the University of Pittsburgh with secondary appointments in the department of chemistry and computational and systems biology. Recently he accepted a position as chair for drug design at the University of Groningen, The Netherlands. His research interest are centred around MCRs, including new MCR, stereoselective MCRs, chemoinformatic of MCRs and its applications to medicinal and combinatorial chemistry. Specifically, he is interested in the rational design protein protein interactions (ant)agonists, protease inhibitors and drugs for neglected tropical diseases (NTD). His therapeutic interests include cancer, NTD, COPD, diabetes and infectious diseases. He is offering his expertise in MCR chemistry to pharma and agro companies and to Universities by performing in-house short courses.
Kan Wang was born in Anhui, China in 1975. He obtained his B.S. in Applied Chemistry from University of Science and Technology of China in 1997. He went to Shanghai Institute of Organic Chemistry and obtained M.S. in 2000 under Prof. Jianxun Wen focused in liquid crystal material. In 2001 he went to University of Pittsburgh and obtained his Ph.D. in organic chemistry in 2007 under Prof. Scott G. Nelson focused in organometallic and organo catalysis methodology and synthesis. Currently he is working with Prof. Alexander Doemling on the design and synthesis of new drugs and combinatorial chemistry research. He has produced >20 peer reviewed papers.
Wei Wang was born in Hubei, China in 1980. He obtained his B.S. in chemistry from Wuhan University in 2000. He completed his Ph. D. thesis under guidance of Prof. Yuanyin Chen and Prof. Shuling Gong in Wuhan University in 2005. He moved to USA and took a researcher position to study the carbene and super base catalyst at Southern Illinois University after one year as synthetic chemist at a pharmaceutical company in Shanghai. In 2008, he joined the Dömling laboratory at the University of Pittsburgh. His interest focuses around anti cancer drug discovery based on the protein-protein interactions in the ubiquitin-proteasome system. Recently he accepted a position as assistant professor in college of pharmaceutical science in Wuhan University, China. His synthetic research currently targets the p53-HDM2/HDMX system and combinational and parallel synthesis methodologies based on multicomponent reactions
References
- 1.Ugi I, Dömling A, Horl W. Endeavour. 1994;18:115. [Google Scholar]
- 2.Tietze LF. Chem Rev. 1996;96:115. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- 3.Ugi I, Meyr R, Fetzer U, Steinbrückner C. Angew Chem. 1959;71:386. [Google Scholar]
- 4.Passerini M. Gazz Chim Ital. 1921;51:126. [Google Scholar]
- 5.van Leusen D, van Leusen AM. Org React (NY) 2003;57:419. [Google Scholar]
- 6.Strecker A. Annalen der Chemie und Pharmacie. 1854;91:349. [Google Scholar]
- 7.Hantzsch A. Chem Ber. 1881;14:1637. [Google Scholar]
- 8.(a) Biginelli P. Chem Ber. 1891;24:1317. [Google Scholar]; (b) Biginelli P. Chem Ber. 1891;24:2962. [Google Scholar]; (c) Kappe CO. Tetrahedron. 1993;49:6937. [Google Scholar]
- 9.Heck S, Dömling A. Synlett. 2000;2000:424. [Google Scholar]
- 10.(a) El Kaïm L, Grimaud L, Oble J. Angew Chem Int Ed. 2005;44:7961. doi: 10.1002/anie.200502636. [DOI] [PubMed] [Google Scholar]; (b) El Kaïm L, Grimaud L. Mol Divers. 2010;14:855. doi: 10.1007/s11030-009-9175-3. [DOI] [PubMed] [Google Scholar]
- 11.Ugi I, Steinbrückner C. Angew Chem. 1960;72:267. [Google Scholar]
- 12.Dömling A. Chem Rev. 2006;106:17. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 13.(a) Keating TA, Armstrong RW. J Am Chem Soc. 1996;118:2574. [Google Scholar]; (b) Szardenings AK, Burkoth TS, Lu HH, Tien DW, Campbell DA. Tetrahedron. 1997;53:6573. [Google Scholar]; (c) Hulme C, Ma L, Cherrier MP, Romano JJ, Morton G, Duquenne C, Salvino J, Labaudiniere R. Tetrahedron Lett. 2000;41:1883. [Google Scholar]; (d) Hulme C, Ma L, Romano J, Morrissette M. Tetrahedron Lett. 1999;40:7925. [Google Scholar]; (e) Golebiowski A, Klopfenstein SR, Shao X, Chen JJ, Colson AO, Grieb AL, Russell AF. Org Lett. 2000;2:2615. doi: 10.1021/ol006145s. [DOI] [PubMed] [Google Scholar]; (f) Tempest P, Ma V, Thomas S, Hua Z, Kelly MG, Hulme C. Tetrahedron Lett. 2001;42:4959. [Google Scholar]; (g) Nixey T, Tempest P, Hulme C. Tetrahedron Lett. 2002;43:1637. [Google Scholar]; (h) Faggi C, Marcaccini S, Pepino R, Cruz Pozo M. Synthesis. 2002;2002:2756. [Google Scholar]; (i) Zhang W, Tempest P. Tetrahedron Lett. 2004;45:6757. [Google Scholar]; (j) Hulme C, Dietrich J. Mol Divers. 2009;13:195. doi: 10.1007/s11030-009-9111-6. [DOI] [PubMed] [Google Scholar]; (k) Tejedor D, Garcia-Tellado F. Chem Soc Rev. 2007;36:484. doi: 10.1039/b608164a. [DOI] [PubMed] [Google Scholar]; (l) Isambert N, Lavilla R. Chem -Eur J. 2008;14:8444. doi: 10.1002/chem.200800473. [DOI] [PubMed] [Google Scholar]; (m) Willy B, Mueller TJJ. Arkivoc. 2008;2008:195. [Google Scholar]; (n) Simon C, Constantieux T, Rodriguez J. Eur J Org Chem. 2004;2004:4957. [Google Scholar]
- 14.(a) Ugi I. Angew Chem. 1962;74:9. [Google Scholar]; (b) Ugi I. Angew Chem Int Ed Engl. 1982;21:810. [Google Scholar]; (c) Armstrong RW, Combs AP, Tempest PA, Brown SD, Keating TA. Acc Chem Res. 1996;29:123. [Google Scholar]; (d) Montgomery J. Acc Chem Res. 2000;33:467. doi: 10.1021/ar990095d. [DOI] [PubMed] [Google Scholar]; (e) Dömling A. Curr Opin Chem Biol. 2000;4:318. doi: 10.1016/s1367-5931(00)00095-8. [DOI] [PubMed] [Google Scholar]; (f) Ulaczyk-Lesanko A, Hall DG. Curr Opin Chem Biol. 2005;9:266. doi: 10.1016/j.cbpa.2005.04.003. [DOI] [PubMed] [Google Scholar]; (g) Mironov MA. QSAR Comb Sci. 2006;25:423. [Google Scholar]; (h) Tempest P. Curr Opin Drug Discov Dev. 2005;8:776. [PubMed] [Google Scholar]; (i) Ramón DJ, Yus M. Angew Chem Int Ed. 2005;44:1602. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]; (j) Kappe CO. QSAR Comb Sci. 2003;22:630. [Google Scholar]; (k) Orru RVA, de Greef M. Synthesis. 2003;2003:1471. [Google Scholar]; (l) Ugi I, Werner B, Dömling A. Molecules. 2003;8:53. [Google Scholar]; (m) Balme G, Bossharth E, Monteiro N. Eur J Org Chem. 2003;2003:4101. [Google Scholar]; (n) Zhu J. Eur J Org Chem. 2003;2003:1133. [Google Scholar]; (o) Hulme C, Gore V. Curr Med Chem. 2003;10:51. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]; (p) Masciadri R, Kamer M, Nock N. Eur J Org Chem. 2003;2003:4286. [Google Scholar]; (q) Dömling A. Curr Opin Chem Biol. 2002;6:306. doi: 10.1016/s1367-5931(02)00328-9. [DOI] [PubMed] [Google Scholar]; (r) Weber L, Illgen K, Almstetter M. Synlett. 1999;1999:366. [Google Scholar]; (s) Lockhoff O, Frappa I. Comb Chem High Throughput Screen. 2002;5:361. doi: 10.2174/1386207023330219. [DOI] [PubMed] [Google Scholar]; (t) Akritopoulou-Zanze I, Djuric S. In: Synthesis of Heterocycles via Multicomponent Reactions II. Orru RVA, Ruijter E, editors. Vol. 25. Springer Berlin/Heidelberg; 2010. p. 231. [Google Scholar]
- 15.Akritopoulou-Zanze I. Curr Opin Chem Biol. 2008;12:324. doi: 10.1016/j.cbpa.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 16.(a) Bohacek RS, McMartin C, Guida WC. Med Res Rev. 1996;16:3. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; (b) Ertl P. J Chem Inf Comput Sci. 2002;43:374. doi: 10.1021/ci0255782. [DOI] [PubMed] [Google Scholar]
- 17.Orry AJW, Abagyan RA, Cavasotto CN. Drug Discov Today. 2006;11:261. doi: 10.1016/S1359-6446(05)03717-7. [DOI] [PubMed] [Google Scholar]
- 18.Fischer E. Chem Ber. 1894;27:2985. [Google Scholar]
- 19.Akritopoulou-Zanze I, Metz JT, Djuric SW. Drug Discov Today. 2007;12:948. doi: 10.1016/j.drudis.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 20.Ribelin TP, Judd AS, Akritopoulou-Zanze I, Henry RF, Cross JL, Whittern DN, Djuric SW. Org Lett. 2007;9:5119. doi: 10.1021/ol7023373. [DOI] [PubMed] [Google Scholar]
- 21.Akritopoulou-Zanze I, Whitehead A, Waters JE, Henry RF, Djuric SW. Org Lett. 2007;9:1299. doi: 10.1021/ol070164l. [DOI] [PubMed] [Google Scholar]
- 22.Wang W, Herdtweck E, Dömling A. Chem Comm. 2010;46:770. doi: 10.1039/b917660h. [DOI] [PubMed] [Google Scholar]
- 23.Yadav JS, Reddy BVS, Satheesh G, Narasimhulu G, Portier Y, Madhavi C, Kunwar AC. Tetrahedron Lett. 2008;49:3341. [Google Scholar]
- 24.Osipova A, Yufit DS, de Meijere A. Synthesis. 2007;2007:131. [Google Scholar]
- 25.Redkin RG, Shemchuk LA, Chernykh VP, Shishkin OV, Shishkina SV. Tetrahedron. 2007;63:11444. [Google Scholar]
- 26.Bencivenni G, Wu LY, Mazzanti A, Giannichi B, Pesciaioli F, Song MP, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2009;48:7200. doi: 10.1002/anie.200903192. [DOI] [PubMed] [Google Scholar]
- 27.Beck B, Larbig G, Mejat B, Magnin-Lachaux M, Picard A, Herdtweck E, Dömling A. Org Lett. 2003;5:1047. doi: 10.1021/ol034077e. [DOI] [PubMed] [Google Scholar]
- 28.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Nat Rev Drug Discovery. 2008;7:591. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 29.(a) Groebke K, Weber L, Mehlin F. Synlett. 1998;1998:661. [Google Scholar]; (b) Bienaymé H, Bouzid K. Angew Chem Int Ed. 1998;37:2234. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2234::AID-ANIE2234>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; (c) Blackburn C, Guan B, Fleming P, Shiosaki K, Tsai S. Tetrahedron Lett. 1998;39:3635. [Google Scholar]
- 30.Hulme C, Lee YS. Mol Divers. 2008;12:1. doi: 10.1007/s11030-008-9072-1. [DOI] [PubMed] [Google Scholar]
- 31.Parchinsky VZ, Koleda VV, Shuvalova O, Kravchenko DV, Krasavin M. Tetrahedron Lett. 2006;47:6891. [Google Scholar]
- 32.(a) Zhao H, Akritopoulou-Zanze I. Expert Opin Drug Discovery. 2010;5:123. doi: 10.1517/17460440903584874. [DOI] [PubMed] [Google Scholar]; (b) Zhao H. Drug Discov Today. 2007;12:149. doi: 10.1016/j.drudis.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Dömling A, Huang Y. Synthesis. 2010;2010:2859. [Google Scholar]
- 34.(a) Povarov LS, Mikhailov BM, Akad I. Nauk SSR, Ser Khim. 1963;1963:953. [Google Scholar]; (b) Povarov LS. Russ Chem Rev. 1967;36:656. [Google Scholar]
- 35.Döbner O. J Lieb Ann Chem. 1887;242:265. [Google Scholar]
- 36.(a) Imming P, Sinning C, Meyer A. Nat Rev Drug Discovery. 2006;5:821. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]; (b) Zheng C, Han L, Yap CW, Xie B, Chen Y. Drug Discov Today. 2006;11:412. doi: 10.1016/j.drudis.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 37.Hopkins AL, Groom CR. Nat Rev Drug Discovery. 2002;1:727. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 38.(a) Southan C. Drug Discov Today. 2001;6:681. doi: 10.1016/s1359-6446(01)01793-7. [DOI] [PubMed] [Google Scholar]; (b) Turk B. Nat Rev Drug Discovery. 2006;5:785. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 39.(a) Aguero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, Carmona S, Carruthers IM, Chan AWE, Chen F, Crowther GJ, Doyle MA, Hertz-Fowler C, Hopkins AL, McAllister G, Nwaka S, Overington JP, Pain A, Paolini GV, Pieper U, Ralph SA, Riechers A, Roos DS, Sali A, Shanmugam D, Suzuki T, Van Voorhis WC, Verlinde CLMJ. Nat Rev Drug Discovery. 2008;7:900. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tong L. Chem Rev. 2002;102:4609. doi: 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- 40.(a) Hagedorn I, Eholzer U. Chem Ber. 1965;98:936. doi: 10.1002/cber.19650980125. [DOI] [PubMed] [Google Scholar]; (b) Ugi I, Meyr R. Chem Ber. 1961;94:2229. [Google Scholar]
- 41.(a) Banfi L, Guanti G, Riva R. Chem Comm. 2000;2000:985. [Google Scholar]; (b) Semple JE, Owens TD, Nguyen K, Levy OE. Org Lett. 2000;2:2769. doi: 10.1021/ol0061485. [DOI] [PubMed] [Google Scholar]; (c) Banfi L, Basso A, Guanti G, Riva R. Mol Div. 2003;6:227. doi: 10.1023/b:modi.0000006778.42751.7f. [DOI] [PubMed] [Google Scholar]
- 42.Banfi L, Guanti G, Riva R, Basso A, Calcagno E. Tetrahedron Lett. 2002;43:4067. [Google Scholar]
- 43.(a) Faure S, Hjelmgaard T, Roche SP, Aitken DJ. Org Lett. 2009;11:1167. doi: 10.1021/ol900048r. [DOI] [PubMed] [Google Scholar]; (b) Owens TD, Semple JE. Org Lett. 2001;3:3301. doi: 10.1021/ol0165239. [DOI] [PubMed] [Google Scholar]
- 44.Lee AY, Hagihara M, Karmacharya R, Albers MW, Schreiber SL, Clardy J. J Am Chem Soc. 2002;115:12619. [Google Scholar]
- 45.Nixey T, Hulme C. Tetrahedron Lett. 2002;43:6833. [Google Scholar]
- 46.(a) Kusebauch U, Beck B, Messer K, Herdtweck E, Dömling A. Org Lett. 2003;5:4021. doi: 10.1021/ol035010u. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Fan Y. J Org Chem. 2005;70:9667. doi: 10.1021/jo050549m. [DOI] [PubMed] [Google Scholar]; (c) Andreana PR, Liu CC, Schreiber SL. Org Lett. 2004;6:4231. doi: 10.1021/ol0482893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Frey R, Galbraith SG, Guelfi S, Lamberth C, Zeller M. Synlett. 2003;2003:1536. [Google Scholar]; (e) Yue T, Wang MX, Wang DX, Zhu J. Angew Chem Int Ed. 2008;47:9454. doi: 10.1002/anie.200804213. [DOI] [PubMed] [Google Scholar]
- 47.Cuny G, Gámez-Montaño R, Zhu J. Tetrahedron. 2004;60:4879. [Google Scholar]
- 48.Mihara H, Xu Y, Shepherd NE, Matsunaga S, Shibasaki M. J Am Chem Soc. 2009;131:8384. doi: 10.1021/ja903158x. [DOI] [PubMed] [Google Scholar]
- 49.(a) Henkel B, Beck B, Westner B, Mejat B, Dömling A. Tetrahedron Lett. 2003;44:8947. [Google Scholar]; (b) Kobayashi K, Takagoshi K, Kondo S, Morikawa O, Konishi H. Bull Chem Soc Jpn. 2004;77:553. [Google Scholar]; (c) Kobayashi K, Irisawa S, Matoba T, Matsumoto T, Yoneda K, Morikawa O, Konishi H. Bull Chem Soc Jpn. 2001;74:1109. [Google Scholar]; (d) Krishna PR, Lopinti K. Synlett. 2007;2007:83. [Google Scholar]
- 50.Oaksmith JM, Peters U, Ganem B. J Am Chem Soc. 2004;126:13606. doi: 10.1021/ja0450152. [DOI] [PubMed] [Google Scholar]
- 51.Berlozecki S, Szymanski W, Ostaszewski R. Tetrahedron. 2008;64:9780. [Google Scholar]
- 52.Berłożecki S, Szymański W, Ostaszewski R. Synth Commun. 2008;38:2714. [Google Scholar]
- 53.(a) Skorna G, Ugi I. Angew Chem Int Ed Engl. 1977;16:259. [Google Scholar]; (b) Owens TD, Araldi GL, Nutt RF, Semple JE. Tetrahedron Lett. 2001;42:6271. [Google Scholar]; (c) Banfi L, Basso A, Guanti G, Merlo S, Repetto C, Riva R. Tetrahedron. 2008;64:1114. [Google Scholar]; (d) Kamer PCJ, Cleij MC, Nolte RJM, Harada T, Hezemans AMF, Drenth W. J Am Chem Soc. 2002;110:1581. [Google Scholar]; (e) Eckert H, Forster B. Angew Chem Int Ed Engl. 1987;26:894. [Google Scholar]; (f) Zhu J, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:577. doi: 10.1016/j.tetlet.2008.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urban R, Eberle G, Marquarding D, Rehn D, Rehn H, Ugi I. Angew Chem. 1976;88:644. [Google Scholar]
- 55.Zhdanko AG, Nenajdenko VG. J Org Chem. 2008;74:884. doi: 10.1021/jo802420c. [DOI] [PubMed] [Google Scholar]
- 56.(a) Hedstrom L. Chem Rev. 2002;102:4501. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]; (b) Dodson G, Wlodawer A. Trends Biochem Sci. 1998;23:347. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 57.(a) Rosenblum JS, Kozarich JW. Curr Opin Chem Biol. 2003;7:496. doi: 10.1016/s1367-5931(03)00084-x. [DOI] [PubMed] [Google Scholar]; (b) Sakanari JA, Staunton CE, Eakin AE, Craik CS, McKerrow JH. Proc Natl Acad Sci U S A. 1989;86:4863. doi: 10.1073/pnas.86.13.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(a) Velázquez F, Venkatraman S, Blackman M, Pinto P, Bogen Sp, Sannigrahi M, Chen K, Pichardo J, Hart A, Tong X, Girijavallabhan V, Njoroge FG. J Med Chem. 2009;52:700. doi: 10.1021/jm801201u. [DOI] [PubMed] [Google Scholar]; (b) Venkatraman S, Velazquez F, Wu W, Blackman M, Chen KX, Bogen S, Nair L, Tong X, Chase R, Hart A, Agrawal S, Pichardo J, Prongay A, Cheng KC, Girijavallabhan V, Piwinski J, Shih NY, Njoroge FG. J Med Chem. 2008;52:336. doi: 10.1021/jm800940u. [DOI] [PubMed] [Google Scholar]
- 59.Velazquez F, Venkatraman S, Wu W, Blackman M, Prongay A, Girijavallabhan V, Shih NY, Njoroge FG. Org Lett. 2007;9:3061. doi: 10.1021/ol0711265. [DOI] [PubMed] [Google Scholar]
- 60.Chen KX, Njoroge FG, Prongay A, Pichardo J, Madison V, Girijavallabhan V. Bioorg Med Chem Lett. 2005;15:4475. doi: 10.1016/j.bmcl.2005.07.033. [DOI] [PubMed] [Google Scholar]
- 61.Prongay AJ, Guo Z, Yao N, Pichardo J, Fischmann T, Strickland C, Myers J, Weber PC, Beyer BM, Ingram R, Hong Z, Prosise WW, Ramanathan L, Taremi SS, Yarosh-Tomaine T, Zhang R, Senior M, Yang RS, Malcolm B, Arasappan A, Bennett F, Bogen SL, Chen K, Jao E, Liu YT, Lovey RG, Saksena AK, Venkatraman S, Girijavallabhan V, Njoroge FG, Madison V. J Med Chem. 2007;50:2310. doi: 10.1021/jm060173k. [DOI] [PubMed] [Google Scholar]
- 62.Sheehan SM, Masters JJ, Wiley MR, Young SC, Liebeschuetz JW, Jones SD, Murray CW, Franciskovich JB, Engel DB, Weber WW, Marimuthu J, Kyle JA, Smallwood JK, Farmen MW, Smith GF. Bioorg Med Chem Lett. 2003;13:2255. doi: 10.1016/s0960-894x(03)00462-1. [DOI] [PubMed] [Google Scholar]
- 63.Wang W, Joyner S, Khoury KAS, Dömling A. Org Biomol Chem. 2010;8:529. doi: 10.1039/b918214d. [DOI] [PubMed] [Google Scholar]
- 64.Weber L, Wallbaum S, Broger C, Gubernator K. Angew Chem Int Ed Engl. 1995;34:2280. [Google Scholar]
- 65.(a) Himber J, Refino CJ, Burcklen L, Roux S, Kirchhofer D. Thromb Haemostasis. 2001;85:475. [PubMed] [Google Scholar]; (b) Himber J, Kirchhofer D, Riederer M, Tschopp TB, Steiner B, Roux SP. Thromb Haemostasis. 1997;78:1142. [PubMed] [Google Scholar]
- 66.(a) Zbinden KG, Obst-Sander U, Hilpert K, Kühne H, Banner DW, Böhm HJ, Stahl M, Ackermann J, Alig L, Weber L, Wessel HP, Riederer MA, Tschopp TB, Lavé T. Bioorg Med Chem Lett. 2005;15:5344. doi: 10.1016/j.bmcl.2005.04.079. [DOI] [PubMed] [Google Scholar]; (b) Zbinden KG, Banner DW, Ackermann J, D'Arcy A, Kirchhofer D, Ji YH, Tschopp TB, Wallbaum S, Weber L. Bioorg Med Chem Lett. 2005;15:817. doi: 10.1016/j.bmcl.2004.10.092. [DOI] [PubMed] [Google Scholar]
- 67.Zbinden KG, Banner DW, Hilpert K, Himber J, Lavé T, Riederer MA, Stahl M, Tschopp TB, Obst-Sander U. Bioorg Med Chem. 2006;14:5357. doi: 10.1016/j.bmc.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 68.Xu P, Lin W, Zou X. Synthesis. 2002;2002:1017. [Google Scholar]
- 69.Gilley CB, Buller MJ, Kobayashi Y. Org Lett. 2007;9:3631. doi: 10.1021/ol701446y. [DOI] [PubMed] [Google Scholar]
- 70.(a) Lage S, Villaluenga I, Sotomayor N, Lete E. Synlett. 2008;2008:3188. [Google Scholar]; (b) Kreye O, Westermann B, Wessjohann LA. Synlett. 2007;2007:3188. [Google Scholar]; (c) Isaacson J, Gilley CB, Kobayashi Y. J Org Chem. 2007;72:3913. doi: 10.1021/jo0700225. [DOI] [PubMed] [Google Scholar]
- 71.Oefner C, D'Arcy A, Mac Sweeney A, Pierau S, Gardiner R, Dale GE. Acta Crystallogr, Sect D: Biol Crystallogr. 2003;59:1206. doi: 10.1107/s0907444903010059. [DOI] [PubMed] [Google Scholar]
- 72.Behnke D, Taube R, Illgen K, Nerdinger S, Herdtweck E. Synlett. 2004;2004:688. [Google Scholar]
- 73.Elders N, Ruijter E, de Kanter Frans JJ, Janssen E, Lutz M, Spek Anthony L, Orru RVA. Chem -Eur J. 2009;15:6096. doi: 10.1002/chem.200900785. [DOI] [PubMed] [Google Scholar]
- 74.Dunn BM. Chem Rev. 2002;102:4431. doi: 10.1021/cr010167q. [DOI] [PubMed] [Google Scholar]
- 75.Zaman MA, Oparil S, Calhoun DA. Nat Rev Drug Discovery. 2002;1:621. doi: 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]
- 76.(a) Jensen C, Herold P, Brunner HR. Nat Rev Drug Discovery. 2008;7:399. doi: 10.1038/nrd2550. [DOI] [PubMed] [Google Scholar]; (b) Boogers JAF, Felfer U, Kotthaus M, Lefort L, Steinbauer G, de Vries AHM, de Vries JG. Org Process Res Dev. 2007;11:585. [Google Scholar]
- 77.Oefner C, Binggeli A, Breu V, Bur D, Clozel JP, D'Arcy A, Dorn A, Fischli W, Grüninger F, Güller R, Hirth G, Märki HP, Mathews S, Müller M, Ridley RG, Stadier H, Vieira E, Wilhelm M, Winkler FK, Wostl W. Chem Biol. 1999;6:127. doi: 10.1016/s1074-5521(99)89004-8. [DOI] [PubMed] [Google Scholar]
- 78.Dömling A, Beck B, Herdtweck E, Antuch W, Oefner C, Yehia N, Gracia-Marques A. Arkivoc. 2007;2007:99. [Google Scholar]
- 79.Wood JM, Maibaum J, Rahuel J, Grütter MG, Cohen NC, Rasetti V, Rüger H, Göschke R, Stutz S, Fuhrer W, Schilling W, Rigollier P, Yamaguchi Y, Cumin F, Baum HP, Schnell CR, Herold P, Mah R, Jensen C, O'Brien E, Stanton A, Bedigian MP. Biochem Biophys Res Commun. 2003;308:698. doi: 10.1016/s0006-291x(03)01451-7. [DOI] [PubMed] [Google Scholar]
- 80.(a) van Leusen D, van Leusen AM. In: Organic Reactions. Overman LE, editor. Vol. 57. John Wiley & Sons; New York: 2001. p. 417. [Google Scholar]; (b) van Leusen AM, Wildeman J, Oldenziel OH. J Org Chem. 2002;42:1153. [Google Scholar]
- 81.(a) Wright CM, Seguin SP, Fewell SW, Zhang H, Ishwad C, Vats A, Lingwood CA, Wipf P, Fanning E, Pipas JM, Brodsky JL. Virus Res. 2009;141:71. doi: 10.1016/j.virusres.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhdanko AG, Gulevich AV, Nenajdenko VG. Tetrahedron. 2009;65:4692. [Google Scholar]; (c) Sisko J, Kassick AJ, Mellinger M, Filan JJ, Allen A, Olsen MA. J Org Chem. 2000;65:1516. doi: 10.1021/jo991782l. [DOI] [PubMed] [Google Scholar]
- 82.Flight MH. Nat Rev Drug Discovery. 2008;7:475. doi: 10.1038/nrd3484. [DOI] [PubMed] [Google Scholar]
- 83.Wolfe MS. Chem Rev. 2009;109:1599. doi: 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barrow JC, Stauffer SR, Rittle KE, Ngo PL, Yang Z, Selnick HG, Graham SL, Munshi S, McGaughey GB, Holloway MK, Simon AJ, Price EA, Sankaranarayanan S, Colussi D, Tugusheva K, Lai MT, Espeseth AS, Xu M, Huang Q, Wolfe A, Pietrak B, Zuck P, Levorse DA, Hazuda D, Vacca JP. J Med Chem. 2008;51:6259. doi: 10.1021/jm800914n. [DOI] [PubMed] [Google Scholar]
- 85.Umbreen S, Brockhaus M, Ehrenberg H, Schmidt B. Eur J Org Chem. 2006;2006:4585. [Google Scholar]
- 86.Rossen K, Pye PJ, DiMichele LM, Volante RP, Reider PJ. Tetrahedron Lett. 1998;39:6823. [Google Scholar]
- 87.Yehia NAM, Antuch W, Beck B, Hess S, Schauer-Vukasinovic V, Almstetter M, Furer P, Herdtweck E, Dömling A. Bioorg Med Chem Lett. 2004;14:3121. doi: 10.1016/j.bmcl.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 88.Kazantsev AG, Thompson LM. Nat Rev Drug Discovery. 2008;7:854. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 89.Hubbs JL, Zhou H, Kral AM, Fleming JC, Dahlberg WK, Hughes BL, Middleton RE, Szewczak AA, Secrist JP, Miller TA. Bioorg Med Chem Lett. 2008;18:34. doi: 10.1016/j.bmcl.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 90.Grolla AA, Podestà V, Chini MG, Di Micco S, Vallario A, Genazzani AA, Canonico PL, Bifulco G, Tron GC, Sorba G, Pirali T. J Med Chem. 2009;52:2776. doi: 10.1021/jm801529c. [DOI] [PubMed] [Google Scholar]
- 91.(a) Otto HH, Schirmeister T. Chem Rev. 1997;97:133. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]; (b) Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 92.Lescop C, Herzner H, Siendt H, Bolliger R, Henneböhle M, Weyermann P, Briguet A, Courdier-Fruh I, Erb M, Foster M, Meier T, Magyar JP, Sprecher Av. Bioorg Med Chem Lett. 2005;15:5176. doi: 10.1016/j.bmcl.2005.08.064. [DOI] [PubMed] [Google Scholar]
- 93.Weyermann P, Herzner H, Lescop C, Siendt H, Bolliger R, Hennebohle M, Rummey C, Briguet A, Courdier-Fruh I, Erb M, Foster M, Magyar JP, Sprecher Av, Meier T. Lett Drug Des Discovery. 2006;3:152. [Google Scholar]
- 94.(a) Denault JB, Salvesen GS. Chem Rev. 2002;102:4489. doi: 10.1021/cr010183n. [DOI] [PubMed] [Google Scholar]; (b) Kemnitzer W, Jiang S, Wang Y, Kasibhatla S, Crogan-Grundy C, Bubenik M, Labrecque D, Denis R, Lamothe S, Attardo G, Gourdeau H, Tseng B, Drewe J, Cai SX. Bioorg Med Chem Lett. 2008;18:603. doi: 10.1016/j.bmcl.2007.11.078. [DOI] [PubMed] [Google Scholar]
- 95.Kemnitzer W, Drewe J, Jiang S, Zhang H, Crogan-Grundy C, Labreque D, Bubenick M, Attardo G, Denis R, Lamothe S, Gourdeau H, Tseng B, Kasibhatla S, Cai SX. J Med Chem. 2008;51:417. doi: 10.1021/jm7010657. [DOI] [PubMed] [Google Scholar]
- 96.Shestopalov AM, Shestopalov AA, Rodinovskaya LA. Synthesis. 2008;2008:1. [Google Scholar]
- 97.Wang K, Nguyen K, Huang Y, Dömling A. J Com Chem. 2009;11:920. doi: 10.1021/cc9000778. [DOI] [PubMed] [Google Scholar]
- 98.Wang K, Kim D, Dömling A. J Com Chem. 2010;12:111. doi: 10.1021/cc9001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.(a) Lapenna S, Giordano A. Nat Rev Drug Discovery. 2009;8:547. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]; (b) Adams JA. Chem Rev. 2001;101:2271. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]; (c) Vieth M, Higgs RE, Robertson DH, Shapiro M, Gragg EA, Hemmerle H. Biochim Biophys Acta, Proteins Proteomics. 2004;1697:243. doi: 10.1016/j.bbapap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 100.Sisko J, Mellinger M. Pure Appl Chem. 2002;74:1349. [Google Scholar]
- 101.Wang Z, Canagarajah BJ, Boehm JC, Kassisà S, Cobb MH, Young PR, Abdel-Meguid S, Adams JL, Goldsmith EJ. Structure. 1998;6:1117. doi: 10.1016/s0969-2126(98)00113-0. [DOI] [PubMed] [Google Scholar]
- 102.(a) Winterfeldt E, Schumann D, Dillinger HJ. Chem Ber. 1969;102:1656. [Google Scholar]; (b) De Moliner F, Banfi L, Riva R, Basso A. Comb Chem High Throughput Screening. 2011 doi: 10.2174/1386211215996242073. [DOI] [PubMed] [Google Scholar]
- 103.Nair V, Vinod AU. Chem Comm. 2000;2000:1019. [Google Scholar]
- 104.Malek Taher M, Hazeri N, Navvabian H, Razmjoo Z, Marandi GJ. Chem Res. 2005;2005:401. [Google Scholar]
- 105.Adib M, Koloogani SA, Abbasi A, Bijanzadeh HR. Synthesis. 2007;2007:3056. [Google Scholar]
- 106.Alizadeh A, Oskueyan Q, Rostamnia S. Synthesis. 2007;2007:2637. [Google Scholar]
- 107.Teimouri MB, Bazhrang R, Eslamimanesh V, Nouri A. Tetrahedron. 2006;62:3016. [Google Scholar]
- 108.Nair V, Deepthi A. Tetrahedron Lett. 2006;47:2037. [Google Scholar]
- 109.Adib M, Sheibani E, Mostofi M, Ghanbary K, Bijanzadeh HR. Tetrahedron. 2006;62:3435. [Google Scholar]
- 110.Esmaeili AA, Darbanian M. Tetrahedron. 2003;59:5545. [Google Scholar]
- 111.Adib M, Sayahi Mohammad H, Mahmoodi N, Bijanzadeh Hamid R. Helv Chim Acta. 2006;89:1176. [Google Scholar]
- 112.Yavari I, Djahaniani H, Nasiri F. Tetrahedron. 2003;59:9409. [Google Scholar]
- 113.Adib M, Ghanbary K, Mostofi M, Reza Bijanzadeh H. Tetrahedron. 2005;61:2645. [Google Scholar]
- 114.Yavari I, Adib M, Sayahi MHJ. Chem Soc Perkin Trans 1. 2002;2002:2343. [Google Scholar]
- 115.Alizadeh A, Hosseinpour R, Rostamnia S. Synthesis. 2008;2008:2462. [Google Scholar]
- 116.Heravi MM, Baghernejad B, Oskooie HA, Hekmatshoar R. Tetrahedron Lett. 2008;49:6101. [Google Scholar]
- 117.Holagh MV, Ahmadi E, Ramazani A. Phosphorus, Sulfur Silicon Relat Elem. 2008;183:2173. [Google Scholar]
- 118.Yavari I, Mokhtarporyani-Sanandaj A, Moradi L, Mirzaei A. Tetrahedron. 2008;64:5221. [Google Scholar]
- 119.Alizadeh A, Rostamnia S, Hu ML. Synlett. 2006;2006:1592. [Google Scholar]
- 120.Adib M, Hosein Sayahi M, Ziyadi H, Bijanzadeh HR, Zhu LG. Tetrahedron. 2007;63:11135. [Google Scholar]
- 121.Pasquale EB. Nat Rev Cancer. 2010;10:165. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kolb P, Kipouros CB, Huang D, Caflisch A. Proteins: Struct, Funct, Bioinf. 2008;73:11. doi: 10.1002/prot.22028. [DOI] [PubMed] [Google Scholar]
- 123.Gewald K, Schinke E, Böttcher H. Chem Ber. 1966;99:94. [Google Scholar]
- 124.(a) Huang Y, Dömling A. Mol Divers. 2011;15:3. doi: 10.1007/s11030-010-9229-6. [DOI] [PubMed] [Google Scholar]; (b) Huang Y, Wolf S, Bista M, Meireles L, Camacho C, Holak TA, Dömling A. Chem Biol Drug Des. 2010;76:116. doi: 10.1111/j.1747-0285.2010.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang Y, Dömling A. Chem Biol Drug Des. 2010;76:130. doi: 10.1111/j.1747-0285.2010.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Puterova Z, Krutošíková A, Végh D. Arkivoc. 2010;2010:209. [Google Scholar]
- 125.(a) Barnes DM, Haight AR, Hameury T, McLaughlin MA, Mei J, Tedrow JS, Riva Toma JD. Tetrahedron. 2006;62:11311. [Google Scholar]; (b) Dai Y, Guo Y, Frey RR, Ji Z, Curtin ML, Ahmed AA, Albert DH, Arnold L, Arries SS, Barlozzari T, Bauch JL, Bouska JJ, Bousquet PF, Cunha GA, Glaser KB, Guo J, Li J, Marcotte PA, Marsh KC, Moskey MD, Pease LJ, Stewart KD, Stoll VS, Tapang P, Wishart N, Davidsen SK, Michaelides MRJ. Med Chem. 2005;48:6066. doi: 10.1021/jm050458h. [DOI] [PubMed] [Google Scholar]
- 126.Phoujdar MS, Kathiravan MK, Bariwal JB, Shah AK, Jain KS. Tetrahedron Lett. 2008;49:1269. [Google Scholar]
- 127.(a) Zavarzin IV, Smirnova NG, Chernoburova EI, Yarovenko VN, Krayushkin MM. Russ Chem Bull. 2004;53:1257. [Google Scholar]; (b) Doss SH, Mohareb RM, Elmegeed GA, Luoca NA. Pharmazie. 2003;58:607. doi: 10.1002/chin.200350216. [DOI] [PubMed] [Google Scholar]; (c) Mohanan K, Devi SN. Orient J Chem. 2003;19:89. [Google Scholar]; (d) Frohlich J, Shaifullah Chowdhury AZM, Hametner C. Arkivoc. 2001;2001:163. [Google Scholar]; (e) El-Ahl AAS, Ismail MA, Amer FA. Phosphorus, Sulfur Silicon Relat Elem. 2003;178:245. [Google Scholar]; (f) Elkholy YM. Phosphorus, Sulfur Silicon Relat Elem. 2002;177:115. [Google Scholar]
- 128.Che C, Xiang J, Wang GX, Fathi R, Quan JM, Yang ZJ. Com Chem. 2007;9:982. doi: 10.1021/cc070058a. [DOI] [PubMed] [Google Scholar]
- 129.Hardcastle IR, Arris CE, Bentley J, Boyle FT, Chen Y, Curtin NJ, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Jewsbury P, Menyerol J, Mesguiche V, Newell DR, Noble MEM, Pratt DJ, Wang LZ, Whitfield HJ. J Med Chem. 2004;47:3710. doi: 10.1021/jm0311442. [DOI] [PubMed] [Google Scholar]
- 130.Sehon CA, Wang GZ, Viet AQ, Goodman KB, Dowdell SE, Elkins PA, Semus SF, Evans C, Jolivette LJ, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Wright LL, Smith GK, Behm DJ, Bentley R, Doe CP, Hu E, Lee DJ. Med Chem. 2008;51:6631. doi: 10.1021/jm8005096. [DOI] [PubMed] [Google Scholar]
- 131.(a) Karpov AS, Merkul E, Rominger F, Müller TJJ. Angew Chem Int Ed. 2005;44:6951. doi: 10.1002/anie.200501703. [DOI] [PubMed] [Google Scholar]; (b) Franco LH, Joffe EBdK, Puricelli L, Tatian M, Seldes AM, Palermo JAJ. Nat Prod. 1998;61:1130. doi: 10.1021/np970493u. [DOI] [PubMed] [Google Scholar]
- 132.Bettayeb K, Tirado OM, Marionneau-Lambot S, Ferandin Y, Lozach O, Morris JC, Mateo-Lozano S, Drueckes P, Schachtele C, Kubbutat MHG, Liger F, Marquet B, Joseph B, Echalier A, Endicott JA, Notario V, Meijer L. Cancer Res. 2007;67:8325. doi: 10.1158/0008-5472.CAN-07-1826. [DOI] [PubMed] [Google Scholar]
- 133.Foster JD, Nordlie RC. Exp Bio Med. 2002;227:601. doi: 10.1177/153537020222700807. [DOI] [PubMed] [Google Scholar]
- 134.Brauer S, Almstetter M, Antuch W, Behnke D, Taube R, Furer P, Hess SJ. Com Chem. 2005;7:218. doi: 10.1021/cc049867+. [DOI] [PubMed] [Google Scholar]
- 135.Zographos SE, Oikonomakos NG, Tsitsanou KE, Leonidas DD, Chrysina ED, Skamnaki VT, Bischoff H, Goldmann S, Watson KA, Johnson LN. Structure. 1997;5:1413. doi: 10.1016/s0969-2126(97)00292-x. [DOI] [PubMed] [Google Scholar]
- 136.Ulaczyk-Lesanko A, Pelletier E, Lee M, Prinz H, Waldmann H, Hall DGJ. Com Chem. 2007;9:695. doi: 10.1021/cc0700344. [DOI] [PubMed] [Google Scholar]
- 137.Ren S, Wu SK, Lien EJ. Pharm Res. 1998;15:286. doi: 10.1023/a:1011978904905. [DOI] [PubMed] [Google Scholar]
- 138.Hansen M, Nours JL, Johansson E, Antal T, Ullrich A, Löffler M, Larsen S. Protein Sci. 2004;13:1031. doi: 10.1110/ps.03533004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lacerda RB, de Lima CKF, da Silva LL, Romeiro NC, Miranda ALP, Barreiro EJ, Fraga CAM. Bioorg Med Chem. 2009;17:74. doi: 10.1016/j.bmc.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 140.Hofmann B, Franke L, Proschak E, Tanrikulu Y, Schneider P, Steinhilber D, Schneider G. ChemMedChem. 2008;3:1535. doi: 10.1002/cmdc.200800153. [DOI] [PubMed] [Google Scholar]
- 141.Kranz M, Wall M, Evans B, Miah A, Ballantine S, Delves C, Dombroski B, Gross J, Schneck J, Villa JP, Neu M, Somers DO. Bioorg Med Chem. 2009;17:5336. doi: 10.1016/j.bmc.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 142.(a) Tyndall JDA, Pfeiffer B, Abbenante G, Fairlie DP. Chem Rev. 2005;105:793. doi: 10.1021/cr040689g. [DOI] [PubMed] [Google Scholar]; (b) Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. ChemMedChem. 2006;1:760. doi: 10.1002/cmdc.200600134. [DOI] [PubMed] [Google Scholar]
- 143.(a) Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]; (b) Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Topiol S, Sabio M. Bioorg Med Chem Lett. 2008;18:1598. doi: 10.1016/j.bmcl.2008.01.063. [DOI] [PubMed] [Google Scholar]; (d) Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Proc Natl Acad Sci U S A. 2008;105:877. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Topiol S, Sabio M. Biochem Pharmacol. 2009;78:11. doi: 10.1016/j.bcp.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 144.(a) Boss C, Brisbare-Roch C, Jenck F. J Med Chem. 2009;52:891. doi: 10.1021/jm801296d. [DOI] [PubMed] [Google Scholar]; (b) Gatfield J, Brisbare-Roch C, Jenck F, Boss C. ChemMedChem. 2010;5:1197. doi: 10.1002/cmdc.201000132. [DOI] [PubMed] [Google Scholar]
- 145.Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S, Mueller C, Nayler O, van Gerven J, de Haas SL, Hess P, Qiu C, Buchmann S, Scherz M, Weller T, Fischli W, Clozel M, Jenck F. Nat Med. 2007;13:150. doi: 10.1038/nm1544. [DOI] [PubMed] [Google Scholar]
- 146.Aissaoui H, Cappi M, Clozel M, Fischli W, Koberstein R. WO2001068609. Preparation of 1,2,3,4-tetrahydroisoquinolines as orexin receptor antagonists. 2001
- 147.Liddle J, Allen MJ, Borthwick AD, Brooks DP, Davies DE, Edwards RM, Exall AM, Hamlett C, Irving WR, Mason AM, McCafferty GP, Nerozzi F, Peace S, Philp J, Pollard D, Pullen MA, Shabbir SS, Sollis SL, Westfall TD, Woollard PM, Wu C, Hickey DMB. Bioorg Med Chem Lett. 2008;18:90. doi: 10.1016/j.bmcl.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 148.McCafferty GP, Pullen MA, Wu C, Edwards RM, Allen MJ, Woollard PM, Borthwick AD, Liddle J, Hickey DMB, Brooks DP, Westfall TD. Am J Physiol Regul Integr Comp Physiol. 2007;293:R299. doi: 10.1152/ajpregu.00057.2007. [DOI] [PubMed] [Google Scholar]
- 149.Wood S, Tickle I, Treharne A, Pitts J, Mascarenhas Y, Li J, Husain J, Cooper S, Blundell T, Hruby V, Buku A, Fischman AJ, Wyssbrod HR. Science. 1986;232:633. doi: 10.1126/science.3008332. [DOI] [PubMed] [Google Scholar]
- 150.(a) Wyatt PG, Allen MJ, Borthwick AD, Davies DE, Exall AM, Hatley RJD, Irving WR, Livermore DG, Miller ND, Nerozzi F, Sollis SL, Szardenings AK. Bioorg Med Chem Lett. 2005;15:2579. doi: 10.1016/j.bmcl.2005.03.045. [DOI] [PubMed] [Google Scholar]; (b) Borthwick AD, Davies DE, Exall AM, Livermore DG, Sollis SL, Nerozzi F, Allen MJ, Perren M, Shabbir SS, Woollard PM, Wyatt PG. J Med Chem. 2005;48:6956. doi: 10.1021/jm050557v. [DOI] [PubMed] [Google Scholar]
- 151.Sollis SL. J Org Chem. 2005;70:4735. doi: 10.1021/jo0501137. [DOI] [PubMed] [Google Scholar]
- 152.Fischer PM. J Pept Sci. 2003;9:9. doi: 10.1002/psc.446. [DOI] [PubMed] [Google Scholar]
- 153.Molteni V, Penzotti J, Wilson DM, Termin AP, Mao L, Crane CM, Hassman F, Wang T, Wong H, Miller KJ, Grossman S, Grootenhuis PDJ. J Med Chem. 2004;47:2426. doi: 10.1021/jm049974i. [DOI] [PubMed] [Google Scholar]
- 154.Petasis NA, Zavialov IA. J Am Chem Soc. 1997;119:445. [Google Scholar]
- 155.Pan SC, List B. Angew Chem Int Ed. 2008;47:3622. doi: 10.1002/anie.200800494. [DOI] [PubMed] [Google Scholar]
- 156.(a) Sato N, Jitsuoka M, Shibata T, Hirohashi T, Nonoshita K, Moriya M, Haga Y, Sakuraba A, Ando M, Ohe T, Iwaasa H, Gomori A, Ishihara A, Kanatani A, Fukami T. J Med Chem. 2008;51:4765. doi: 10.1021/jm8003587. [DOI] [PubMed] [Google Scholar]; (b) Mashiko S, Ishihara A, Iwaasa H, Sano H, Ito J, Gomori A, Oda Z, Moriya R, Matsushita H, Jitsuoka M, Okamoto O, MacNeil DJ, Van der Ploeg LHT, Fukami T, Kanatani A. Mol Pharmacol. 2007;71:602. doi: 10.1124/mol.106.029991. [DOI] [PubMed] [Google Scholar]; (c) Ishihara A, Kanatani A, Mashiko S, Tanaka T, Hidaka M, Gomori A, Iwaasa H, Murai N, Egashira Si, Murai T, Mitobe Y, Matsushita H, Okamoto O, Sato N, Jitsuoka M, Fukuroda T, Ohe T, Guan X, MacNeil DJ, Van der Ploeg LHT, Nishikibe M, Ishii Y, Ihara M, Fukami T. Proc Natl Acad Sci U S A. 2006;103:7154. doi: 10.1073/pnas.0510320103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chakravarti GC, Chattopadhyaya H, Ghosh PC. J Indian Inst Sci. 1932;A14:141. [Google Scholar]
- 158.(a) Bon RS, Hong C, Bouma MJ, Schmitz RF, de Kanter FJJ, Lutz M, Spek AL, Orru RVA. Org Lett. 2003;5:3759. doi: 10.1021/ol035521g. [DOI] [PubMed] [Google Scholar]; (b) Bon RS, van Vliet B, Sprenkels NE, Schmitz RF, de Kanter FJJ, Stevens CV, Swart M, Bickelhaupt FM, Groen MB, Orru RVA. J Org Chem. 2005;70:3542. doi: 10.1021/jo050132g. [DOI] [PubMed] [Google Scholar]; (c) Elders N, Schmitz RF, de Kanter FJJ, Ruijter E, Groen MB, Orru RVA. J Org Chem. 2007;72:6135. doi: 10.1021/jo070840x. [DOI] [PubMed] [Google Scholar]; (d) Bon RS, Sprenkels NE, Koningstein MM, Schmitz RF, de Kanter FJJ, Dömling A, Groen MB, Orru RVA. Org Biomol Chem. 2008;6:130. doi: 10.1039/b713065a. [DOI] [PubMed] [Google Scholar]
- 159.Merla B, Oberboersch S, Hennies HH, Maier M, Khartulyari A. DE102005061430. Preparation of 4,5-dihydro-1H-imidazole-4-carboxylic acids as micro -opioid receptor inhibitors. 2007
- 160.Goss JM, Schaus SE. J Org Chem. 2008;73:7651. doi: 10.1021/jo801463j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sanganee HJ, Baxter A, Barber S, Brown AJH, Grice D, Hunt F, King S, Laughton D, Pairaudeau G, Thong B, Weaver R, Unitt J. Bioorg Med Chem Lett. 2009;19:1143. doi: 10.1016/j.bmcl.2008.12.104. [DOI] [PubMed] [Google Scholar]
- 162.(a) Humphries PS, Balan G, Bechle BM, Conn EL, Dirico KJ, Hui Y, Oliver RM, Southers JA, Yang X. Tetrahedron Lett. 2009;50:2140. [Google Scholar]; (b) Humphries PS, Benbow JW, Bonin PD, Boyer D, Doran SD, Frisbie RK, Piotrowski DW, Balan G, Bechle BM, Conn EL, Dirico KJ, Oliver RM, Soeller WC, Southers JA, Yang X. Bioorg Med Chem Lett. 2009;19:2400. doi: 10.1016/j.bmcl.2009.03.082. [DOI] [PubMed] [Google Scholar]
- 163.Dow RL, Hadcock JR, Scott DO, Schneider SR, Paight ES, Iredale PA, Carpino PA, Griffith DA, Hammond M, DaSilva-Jardine P. Bioorg Med Chem Lett. 2009;19:5351. doi: 10.1016/j.bmcl.2009.07.130. [DOI] [PubMed] [Google Scholar]
- 164.Arabanian A, Mohammadnejad M, Balalaie S, Gross JH. Bioorg Med Chem Lett. 2009;19:887. doi: 10.1016/j.bmcl.2008.11.111. [DOI] [PubMed] [Google Scholar]
- 165.(a) Urban R, Marquarding D, Seidel P, Ugi I, Weinelt A. Chem Ber. 1977;110:2012. [Google Scholar]; (b) Urban R, Ugi I. Angew Chem Int Ed Engl. 1975;14:61. doi: 10.1002/anie.197500611. [DOI] [PubMed] [Google Scholar]
- 166.(a) Raymond V, Sattelle DB. Nat Rev Drug Discovery. 2002;1:427. doi: 10.1038/nrd821. [DOI] [PubMed] [Google Scholar]; (b) Kawate T, Michel JC, Birdsong WT, Gouaux E. Nature. 2009;460:592. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Krishnamurthy H, Piscitelli CL, Gouaux E. Nature. 2009;459:347. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Verkman AS, Galietta LJV. Nat Rev Drug Discovery. 2009;8:153. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, Robins LI, Dicus CW, Willenbring D, Nantz MH, Kurth MJ, Galietta LJV, Verkman AS. Mol Pharmacol. 2005;67:1797. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- 170.De La Fuente R, Namkung W, Mills A, Verkman AS. Mol Pharmacol. 2008;73:758. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- 171.Goldmann S, Stoltefuss J. Angew Chem Int Ed Engl. 1991;30:1559. [Google Scholar]
- 172.Zonouz AM, Hosseini SB. Synth Commun. 2008;38:290. [Google Scholar]
- 173.Chai L, Zhao Y, Sheng Q, Liu ZQ. Tetrahedron Lett. 2006;47:9283. [Google Scholar]
- 174.Hopes PA, Parker AJ, Patel I. Org Process Res Dev. 2006;10:808. [Google Scholar]
- 175.Dondoni A, Massi A, Minghini E, Bertolasi V. Helv Chim Acta. 2002;85:3331. [Google Scholar]
- 176.Ugi I, Steinbrueckner C. DE1103337. Aminocarboxylic acid amides and derivatives. 1961
- 177.Melchiorre C, Bolognesi ML, Minarini A, Rosini M, Tumiatti V. J Med Chem. 2010;53:5906. doi: 10.1021/jm100293f. [DOI] [PubMed] [Google Scholar]
- 178.Liu N, Cao S, Wu J, Yu J, Shen L, Feng X, Qian X. Tetrahedron. 2008;64:3966. [Google Scholar]
- 179.Pirali T, Callipari G, Ercolano E, Genazzani AA, Giovenzana GB, Tron GC. Org Lett. 2008;10:4199. doi: 10.1021/ol801612r. [DOI] [PubMed] [Google Scholar]
- 180.(a) Kim YB, Choi EH, Keum G, Kang SB, Lee DH, Koh HY, Kim Y. Org Lett. 2001;3:4149. doi: 10.1021/ol016716w. [DOI] [PubMed] [Google Scholar]; (b) Ku IW, Cho S, Doddareddy MR, Jang MS, Keum G, Lee JH, Chung BY, Kim Y, Rhim H, Kang SB. Bioorg Med Chem Lett. 2006;16:5244. doi: 10.1016/j.bmcl.2006.05.031. [DOI] [PubMed] [Google Scholar]; (c) Doddareddy MR, Jung HK, Lee JY, Lee YS, Cho YS, Koh HY, Pae AN. Bioorg Med Chem. 2004;12:1605. doi: 10.1016/j.bmc.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 181.Vaccaro W, Huynh T, Lloyd J, Atwal K, Finlay HJ, Levesque P, Conder ML, Jenkins-West T, Shi H, Sun L. Bioorg Med Chem Lett. 2008;18:6381. doi: 10.1016/j.bmcl.2008.10.099. [DOI] [PubMed] [Google Scholar]
- 182.Lloyd J, Finlay HJ, Atwal K, Kover A, Prol J, Yan L, Bhandaru R, Vaccaro W, Huynh T, Huang CS, Conder M, Jenkins-West T, Sun H, Li D, Levesque P. Bioorg Med Chem Lett. 2009;19:5469. doi: 10.1016/j.bmcl.2009.07.083. [DOI] [PubMed] [Google Scholar]
- 183.Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CBA, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Pain. 2005;114:386. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 184.Gleave RJ, Livermore DGH, Walter DS. WO2008116814. Preparation of pyrrole and isoindole carboxamide derivatives as modulators of P2×7 receptor. 2008
- 185.Harriman GCB. Tetrahedron Lett. 1997;38:5591. [Google Scholar]
- 186.Jensen AA, Erichsen MN, Nielsen CW, Stensbøl TB, Kehler J, Bunch L. J Med Chem. 2009;52:912. doi: 10.1021/jm8013458. [DOI] [PubMed] [Google Scholar]
- 187.Watanabe T, Arisawa M, Narusuye K, Alam MS, Yamamoto K, Mitomi M, Ozoe Y, Nishida A. Bioorg Med Chem. 2009;17:94. doi: 10.1016/j.bmc.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 188.(a) Jacoby E, Boettcher A, Mayr LM, Brown N, Jenkins JL, Kallen J, Engeloch C, Schopfer U, Furet P, Masuya K, Lisztwan J. In: Chemogenomics: Methods and Applications. Jacoby E, editor. Vol. 575. Springer; Heidelberg: 2009. p. 173. Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]; (b) Reynès C, Host H, Camproux AC, Laconde G, Leroux F, Mazars A, Deprez B, Fahraeus R, Villoutreix BO, Sperandio O. PLoS Comput Biol. 2010;6:e1000695. doi: 10.1371/journal.pcbi.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fry D, Vassilev L. J Mol Med. 2005;83:955. doi: 10.1007/s00109-005-0705-x. [DOI] [PubMed] [Google Scholar]
- 190.(a) Habashita H, Kokubo M, Hamano Si, Hamanaka N, Toda M, Shibayama S, Tada H, Sagawa K, Fukushima D, Maeda K, Mitsuya H. J Med Chem. 2006;49:4140. doi: 10.1021/jm060051s. [DOI] [PubMed] [Google Scholar]; (b) Nishizawa R, Nishiyama T, Hisaichi K, Matsunaga N, Minamoto C, Habashita H, Takaoka Y, Toda M, Shibayama S, Tada H, Sagawa K, Fukushima D, Maeda K, Mitsuya H. Bioorg Med Chem Lett. 2007;17:727. doi: 10.1016/j.bmcl.2006.10.084. [DOI] [PubMed] [Google Scholar]
- 191.Wade M, Wahl GM. Mol Cancer Res. 2009;7:1. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 193.(a) Fry DC, Emerson SD, Palme S, Vu BT, Liu CM, Podlaski F. J Biomol NMR. 2004;30:163. doi: 10.1023/B:JNMR.0000048856.84603.9b. [DOI] [PubMed] [Google Scholar]; (b) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 194.Vassilev LT. Trends Mol Med. 2007;13:23. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 195.Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, Cummings MD, LaFrance LV, Milkiewicz KL, Calvo RR, Maguire D, Lattanze J, Franks CF, Zhao S, Ramachandren K, Bylebyl GR, Zhang M, Manthey CL, Petrella EC, Pantoliano MW, Deckman IC, Spurlino JC, Maroney AC, Tomczuk BE, Molloy CJ, Bone RF. J Med Chem. 2005;48:909. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 196.(a) Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Gupta V, Lattanze J, Ramachandren K, Carver TE, Petrella EC, Cummings MD, Maguire D, Grasberger BL, Lu T. Bioorg Med Chem Lett. 2005;15:765. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]; (b) Marugan JJ, Leonard K, Raboisson P, Gushue JM, Calvo R, Koblish HK, Lattanze J, Zhao S, Cummings MD, Player MR, Schubert C, Maroney AC, Lu T. Bioorg Med Chem Lett. 2006;16:3115. doi: 10.1016/j.bmcl.2006.03.067. [DOI] [PubMed] [Google Scholar]; (c) Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, José Marugán J, Raboisson P, Schubert C, Koblish HK, Zhao S, Franks CF, Lattanze J, Carver TE, Cummings MD, Maguire D, Grasberger BL, Maroney AC, Lu T. Bioorg Med Chem Lett. 2006;16:3310. doi: 10.1016/j.bmcl.2006.03.055. [DOI] [PubMed] [Google Scholar]; (d) Leonard K, Marugan JJ, Raboisson P, Calvo R, Gushue JM, Koblish HK, Lattanze J, Zhao S, Cummings MD, Player MR, Maroney AC, Lu T. Bioorg Med Chem Lett. 2006;16:3463. doi: 10.1016/j.bmcl.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 197.Rothweiler U, Czarna A, Krajewski M, Ciombor J, Kalinski C, Khazak V, Ross G, Skobeleva N, Weber L, Holak TadA. ChemMedChem. 2008;3:1118. doi: 10.1002/cmdc.200800025. [DOI] [PubMed] [Google Scholar]
- 198.Krajewski M, Rothweiler U, D'Silva L, Majumdar S, Klein C, Holak TA. J Med Chem. 2007;50:4382. doi: 10.1021/jm070365v. [DOI] [PubMed] [Google Scholar]
- 199.Czarna A, Beck B, Srivastava S, Popowicz Grzegorz M, Wolf S, Huang Y, Bista M, Holak Tad A, Dömling A. Angew Chem Int Ed. 2010;49:5352. doi: 10.1002/anie.201001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Murray CW, Rees DC. Nat Chem. 2009;1:187. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- 201.Popowicz G, Czarna A, Wolf S, Wang K, Wang W, Dömling A, Holak T. Cell Cycle. 2010;9:1104. doi: 10.4161/cc.9.6.10956. [DOI] [PubMed] [Google Scholar]
- 202.Srivastava S, Beck B, Wang W, Czarna A, Holak TA, Domling A. J Com Chem. 2009;11:631. doi: 10.1021/cc9000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.(a) Dömling A, Beck B. WO2001025213. Preparation of pyrrolylimidazoles as antibiotics and antitumor agents. 2001; (b) Boettcher A, Buschmann N, Furet P, Groell JM, Kallen J, Hergovich Lisztwan J, Masuya K, Mayr L, Vaupel A. WO2008119741. 3-Imidazolylindoles for treatment of proliferative diseases and their preparation. 2008
- 204.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. Oncogene. 2008;27:6252. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 205.Eckelman BP, Salvesen GS, Scott FL. EMBO Rep. 2006;7:988. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Monfardini I, Huang JW, Beck B, Cellitti JF, Pellecchia M, Dömling A. J Med Chem. 2011;54:890. doi: 10.1021/jm101341m. [DOI] [PubMed] [Google Scholar]
- 207.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 208.Dömling A, Antuch W, Beck B, Schauer-Vukasinovic V. Bioorg Med Chem Lett. 2008;18:4115. doi: 10.1016/j.bmcl.2008.05.096. [DOI] [PubMed] [Google Scholar]
- 209.(a) Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. J Biol Chem. 2004;279:51131. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]; (b) Wright CM, Chovatiya RJ, Jameson NE, Turner DM, Zhu G, Werner S, Huryn DM, Pipas JM, Day BW, Wipf P, Brodsky JL. Bioorg Med Chem. 2008;16:3291. doi: 10.1016/j.bmc.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shonhai A, Boshoff A, Blatch GL. Protein Sci. 2007;16:1803. doi: 10.1110/ps.072918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Haney CM, Schneider C, Beck B, Brodsky JL, Dömling A. Bioorg Med Chem Lett. 2009;19:3828. doi: 10.1016/j.bmcl.2009.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bedjeguelal K, Bienaymé H, Dumoulin A, Poigny S, Schmitt P, Tam E. Bioorg Med Chem Lett. 2006;16:3998. doi: 10.1016/j.bmcl.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 213.(a) Ruoslahti E, Pierschbacher M. Science. 1987;238:491. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]; (b) Heckmann D, Meyer A, Marinelli L, Zahn G, Stragies R, Kessler H. Angew Chem Int Ed. 2007;46:3571. doi: 10.1002/anie.200700008. [DOI] [PubMed] [Google Scholar]
- 214.(a) Banfi L, Basso A, Damonte G, Pellegrini FD, Galatini A, Guanti G, Monfardini I, Riva R, Scapolla C. Bioorg Med Chem Lett. 2007;17:1341. doi: 10.1016/j.bmcl.2006.11.085. [DOI] [PubMed] [Google Scholar]; (b) Vercillo OE, Andrade CKZ, Wessjohann LA. Org Lett. 2007;10:205. doi: 10.1021/ol702521g. [DOI] [PubMed] [Google Scholar]
- 215.Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RNA, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. Science. 2003;299:893. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 216.Haigh JM, Hussain A, Mimmack ML, Lowe CR. J Chromatogr B. 2009;877:1440. doi: 10.1016/j.jchromb.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 217.Faber C, Shan L, Fan Zc, Guddat LW, Furebring C, Ohlin M, Borrebaeck CAK, Edmundson AB. Immunotechnology. 1998;3:253. doi: 10.1016/s1380-2933(97)10003-3. [DOI] [PubMed] [Google Scholar]
- 218.(a) Richter HGF, Benson GM, Blum D, Chaput E, Feng S, Gardes C, Grether U, Hartman P, Kuhn B, Martin RE, Plancher JM, Rudolph MG, Schuler F, Taylor S, Bleicher KH. Bioorg Med Chem Lett. 2011;21:191. doi: 10.1016/j.bmcl.2010.12.123. [DOI] [PubMed] [Google Scholar]; (b) Richter HGF, Benson GM, Bleicher KH, Blum D, Chaput E, Clemann N, Feng S, Gardes C, Grether U, Hartman P, Kuhn B, Martin RE, Plancher JM, Rudolph MG, Schuler F, Taylor S. Bioorg Med Chem Lett. 2011;21:1134. doi: 10.1016/j.bmcl.2010.12.123. [DOI] [PubMed] [Google Scholar]
- 219.Cavalli A, Bolognesi ML. J Med Chem. 2009;52:739. doi: 10.1021/jm9004835. [DOI] [PubMed] [Google Scholar]
- 220.WHO Model Lists of Essential Medicines. http://www.who.int/medicines/publications/essentialmedicines/en/
- 221.(a) Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. The Lancet Infectious Diseases. 2006;6:411. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]; (b) Caffrey CR. Curr Opin Chem Biol. 2007;11:433. doi: 10.1016/j.cbpa.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 222.Hotez PJ, Fenwick A, Kjetland EF. PLoS Negl Trop Dis. 2009;3:e430. doi: 10.1371/journal.pntd.0000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Khoury K, Doemling A. ChemMedChem. 2010;5:1420. doi: 10.1002/cmdc.201000202. [DOI] [PubMed] [Google Scholar]
- 224.Cao H, Liu H, Dömling A. Chem -Eur J. 2010;16:12296. doi: 10.1002/chem.201002046. [DOI] [PubMed] [Google Scholar]
- 225.Liu H, Dömling A. Submitted [Google Scholar]
- 226.(a) Musonda CC, Taylor D, Lehman J, Gut J, Rosenthal PJ, Chibale K. Bioorg Med Chem Lett. 2004;14:3901. doi: 10.1016/j.bmcl.2004.05.063. [DOI] [PubMed] [Google Scholar]; (b) Pontillo J, Tran JA, Fleck BA, Marinkovic D, Arellano M, Tucci FC, Lanier M, Nelson J, Parker J, Saunders J, Murphy B, Foster AC, Chen C. Bioorg Med Chem Lett. 2004;14:5605. doi: 10.1016/j.bmcl.2004.08.055. [DOI] [PubMed] [Google Scholar]; (c) Chipeleme A, Gut J, Rosenthal PJ, Chibale K. Bioorg Med Chem. 2007;15:273. doi: 10.1016/j.bmc.2006.09.055. [DOI] [PubMed] [Google Scholar]; (d) Musonda CC, Little S, Yardley V, Chibale K. Bioorg Med Chem Lett. 2007;17:4733. doi: 10.1016/j.bmcl.2007.06.070. [DOI] [PubMed] [Google Scholar]; (e) Musonda CC, Yardley V, Souza RCCd, Ncokazi K, Egan TJ, Chibale K. Org Biomol Chem. 2008;6:4446. doi: 10.1039/b813007h. [DOI] [PubMed] [Google Scholar]
- 227.Srinivas N, Palne S, Nishi, Gupta S, Bhandari K. Bioorg Med Chem Lett. 2009;19:324. doi: 10.1016/j.bmcl.2008.11.094. [DOI] [PubMed] [Google Scholar]
- 228.Fan X, Zhang X, Bories C, Loiseau PM, Torrence PF. Bioorg Chem. 2007;35:121. doi: 10.1016/j.bioorg.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 229.Odell LR, Nilsson MT, Gising J, Lagerlund O, Muthas D, Nordqvist A, Karlén A, Larhed M. Bioorg Med Chem Lett. 2009;19:4790. doi: 10.1016/j.bmcl.2009.06.045. [DOI] [PubMed] [Google Scholar]
- 230.Ishikawa H, Suzuki T, Hayashi Y. Angew Chem Int Ed. 2009;48:1304. doi: 10.1002/anie.200804883. [DOI] [PubMed] [Google Scholar]
- 231.Magano J. Chem Rev. 2009;109:4398. doi: 10.1021/cr800449m. [DOI] [PubMed] [Google Scholar]
- 232.Sawin KE, Mitchison TJ. Proc Natl Acad Sci U S A. 1995;92:4289. doi: 10.1073/pnas.92.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.(a) Yan Y, Sardana V, Xu B, Homnick C, Halczenko W, Buser CA, Schaber M, Hartman GD, Huber HE, Kuo LC. J Mol Biol. 2004;335:547. doi: 10.1016/j.jmb.2003.10.074. [DOI] [PubMed] [Google Scholar]; (b) Garcia-Saez I, DeBonis S, Lopez R, Trucco F, Rousseau B, Thuery P, Kozielski F. J Biol Chem. 2007;282:9740. doi: 10.1074/jbc.M608883200. [DOI] [PubMed] [Google Scholar]
- 234.Pinkerton AB, Lee TT, Hoffman TZ, Wang Y, Kahraman M, Cook TG, Severance D, Gahman TC, Noble SA, Shiau AK, Davis RL. Bioorg Med Chem Lett. 2007;17:3562. doi: 10.1016/j.bmcl.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 235.Krzysiak TC, Wendt T, Sproul LR, Tittmann P, Gross H, Gilbert SP, Hoenger A. EMBO J. 2006;25:2263. doi: 10.1038/sj.emboj.7601108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Schiemann K, Finsinger D, Zenke F, Amendt C, Knöchel T, Bruge D, Buchstaller HP, Emde U, Stähle W, Anzali S. Bioorg Med Chem Lett. 2010;20:1491. doi: 10.1016/j.bmcl.2010.01.110. [DOI] [PubMed] [Google Scholar]
- 237.Oefner C, Schulz H, D'Arcy A, Dale GE. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:613. doi: 10.1107/S0907444906009474. [DOI] [PubMed] [Google Scholar]
- 238.Sugiyama S, Arai S, Kiriyama M, Ishii K. Chem Pharm Bull. 2005;53:100. doi: 10.1248/cpb.53.100. [DOI] [PubMed] [Google Scholar]
- 239.Rosenblum SB, Huynh T, Afonso A, Davis HR, Yumibe N, Clader JW, Burnett DA. J Med Chem. 1998;41:973. doi: 10.1021/jm970701f. [DOI] [PubMed] [Google Scholar]
- 240.Jiao L, Liang Y, Xu J. J Am Chem Soc. 2006;128:6060. doi: 10.1021/ja056711k. [DOI] [PubMed] [Google Scholar]
- 241.(a) Hubschwerlen C, Specklin JL. Org Synth. 1995;72:14. [Google Scholar]; (b) Hubschwerlen C, Specklin JL. Organic Syntheses Collection. 1998;9:13. [Google Scholar]
- 242.Alonso F, Acebedo SL, Bruttomesso AC, Ramírez JA. Steroids. 2008;73:1270. doi: 10.1016/j.steroids.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 243.Siddiqui IR, Siddique SA, Srivastava V, Singh PK, Singh J. Arkivoc. 2008;2008:277. [Google Scholar]
- 244.Hammond M, Patterson JR, Manns S, Hoang TH, Washburn DG, Trizna W, Glace L, Grygielko ET, Nagilla R, Nord M, Fries HE, Minick DJ, Laping NJ, Bray JD, Thompson SK. Bioorg Med Chem Lett. 2009;19:2637. doi: 10.1016/j.bmcl.2009.03.146. [DOI] [PubMed] [Google Scholar]
- 245.Shaabani A, Soleimani E, Sarvary A, Rezayan AH. Bioorg Med Chem Lett. 2008;18:3968. doi: 10.1016/j.bmcl.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 246.Teimouri MB, Mansouri F. J Com Chem. 2008;10:507. doi: 10.1021/cc8000103. [DOI] [PubMed] [Google Scholar]
- 247.Lamberth C, Jeanguenat A, Cederbaum F, De Mesmaeker A, Zeller M, Kempf HJ, Zeun R. Bioorg Med Chem. 2008;16:1531. doi: 10.1016/j.bmc.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 248.Callens E, Hüter O, Lamy E, Lüthi P, Winkler T, Jung PMJ. Tetrahedron Lett. 2007;48:707. [Google Scholar]
- 249.Ancel JE, El Kaïm L, Gadras A, Grimaud L, Jana NK. Tetrahedron Lett. 2002;43:8319. [Google Scholar]
- 250.(a) Mori K, Rikimaru K, Kan T, Fukuyama T. Org Lett. 2004;6:3095. doi: 10.1021/ol048857e. [DOI] [PubMed] [Google Scholar]; (b) Rikimaru K, Mori K, Kan T, Fukuyama T. Chem Comm. 2005;2005:394. doi: 10.1039/b415030a. [DOI] [PubMed] [Google Scholar]; (c) Endo A, Yanagisawa A, Abe M, Tohma S, Kan T, Fukuyama T. J Am Chem Soc. 2002;124:6552. doi: 10.1021/ja026216d. [DOI] [PubMed] [Google Scholar]
- 251.(a) Heal W, Thompson MJ, Mutter R, Cope H, Louth JC, Chen B. J Med Chem. 2007;50:1347. doi: 10.1021/jm0612719. [DOI] [PubMed] [Google Scholar]; (b) Thompson MJ, Chen B. Tetrahedron Lett. 2008;49:5324. [Google Scholar]; (b) Walborsky H, Niznik G. J Org Chem. 1972;37:187. [Google Scholar]
- 252.Elders N, van der Born D, Hendrickx Loes JD, Timmer Brian JJ, Krause A, Janssen E, de Kanter Frans JJ, Ruijter E, Orru Romano VA. Angew Chem Int Ed. 2009;48:5856. doi: 10.1002/anie.200902683. [DOI] [PubMed] [Google Scholar]