

Abstract
Replication stress (RS) is a source of DNA damage that has been linked to cancer and aging, which is suppressed by the ATR kinase. In mice, reduced ATR levels in a model of the ATR-Seckel syndrome lead to RS and accelerated aging. Similarly, ATR-Seckel embryonic fibroblasts (MEF) accumulate RS and undergo cellular senescence. We previously showed that senescence of ATR-Seckel MEF cannot be rescued by p53-deletion. Here, we show that the genetic ablation of the INK4a/Arf locus fully rescues senescence on ATR mutant MEF, but also that induced by other conditions that generate RS such as low doses of hydroxyurea or ATR inhibitors. In addition, we show that a persistent exposure to RS leads to increased levels of INK4a/Arf products, revealing that INK4a/ARF behaves as a bona fide RS checkpoint. Our data reveal an unknown role for INK4a/ARF in limiting the expansion of cells suffering from persistent replication stress, linking this well-known tumor suppressor to the maintenance of genomic integrity.
Keywords: H2AX, ATR, replicative stress, INK4a/ARF, DNA damage
Introduction
DNA damage leads to mutations and cellular responses that ultimately compromise mammalian health. To minimize its effects, the DNA damage response (DDR) is a kinase-initiated signaling cascade that detects, signals and promotes the repair of chromosomal breaks.1,2 One of the cellular outcomes to DNA damage is senescence, which is defined as a permanent state of cell cycle arrest and which has been linked to cancer and aging. In vitro, all the conditions that induce senescence are known triggers of the DDR, such as telomere attrition,3 oncogene activation4,5 or reagents that induce DNA double-strand breaks (DSB).6 In all these cases, deletion of the tumor suppressor p53 rescues senescence, allowing cells to grow in the presence of damage. In addition to p53, the products of the INK4a/ARF locus (p16Ink4A and p19ARF, in mice) are also important mediators of cellular senescence.7,8 The relevance of these gene products is evidenced by the frequent loss of the INK4a/ARF locus in human cancer. However, and in contrast to p53, several studies have failed to show a role for INK4a/ARF in the acute response to DNA breaks, such as those induced by ionizing radiation,9-11 which has frequently lead to unlinking this tumor suppressor from the response to DNA damage.
Among the different sources of endogenous DNA damage that cells can suffer, replication stress (RS) has gained significant attention in the recent years. This is in part due to the finding that oncogene-induced DNA damage starts with the generation of RS, which has important implications to explain how genomic aberrations evolve during tumor development.12 Specifically, RS derives from the accumulation of single-stranded DNA, which challenges genome integrity due to its propensity to recombine and to initiate genomic rearrangements. In mammalian cells, an RS response (RSR) initiated by ATR and Chk1 kinases limits the accumulation of ssDNA during DNA replication and delays cell cycle progression,13,14 likely to prevent the entry into mitosis with unreplicated loci, which would then derive into DNA breaks.15 Similarly to the DDR, a persistent activation of the ATR-dependent RSR is sufficient to initiate p53-dependent senescence.16
Not only ATR activation, but also reduced levels of ATR, promote cellular senescence. This was first seen in MEF from a mouse model of the ATR-Seckel Syndrome, which accumulate RS and enter into premature senescence.17 Interestingly, spontaneous immortalization of ATR-Seckel MEF does not occur, and subsequent efforts to transform these MEF with various oncogenes also failed,18 raising doubts as to whether cellular immortalization was at all possible in the context of low ATR activity. Moreover, and in contrast to the key role of p53 in the senescence induced by DNA breaks, senescence of ATR mutant MEF was not rescued by p53 deletion.17 On the contrary, p53 deficiency aggravated the symptoms of ATR hypomorphic and deficient mice.17,19 These and other observations led to the proposal that ATR inhibitors could be of particular use for the treatment of tumors with high levels of RS, particularly for those lacking p53.18,20-22 We here show that, in contrast to p53, deletion of the INK4a/ARF locus rescues senescence of ATR-Seckel MEF, as well as that induced by other conditions that generate RS such as nucleotide deprivation. In the context of INK4a/ARF deletion, cells can sustain growth in the presence of abundant levels of RS. Consistent with these findings in MEF, we also find low levels of p16INK4a expression in human cancer cell lines containing high levels of copy number variants (CNV), which are the outcome to a persistent exposure to RS.23 Collectively, these data reveal an important role of INK4a/ARF in limiting the growth of RS-harboring cells, and help to illustrate an active role for INK4a/ARF in genome maintenance.
Results
Depletion of INK4a/ARF rescues senescence on ATR-Seckel MEF
Early studies in MEF showed that the bypass of spontaneous or oncogene-induced senescence invariably occurs through the mutually exclusive loss of p53 or INK4a/ARF.24,25 Given that loss of p53 not only does not rescue but rather aggravates the phenotypes of ATR-Seckel MEF,17 we explored the impact of losing INK4a/ARF. First, we infected ATR mutant MEF with retroviruses expressing short hairpin RNAs targeting both products of the INK4a/ARF locus, and which have been previously validated in several studies.26,27 In contrast to MEF that had been infected with a control retrovirus, INK4a/ARF depletion rescued proliferation on ATR-Seckel (ATRS/S) cells (Fig. 1A). We then tested whether INK4a/ARF depletion had an impact on ATR levels. However, growing cultures of ATRS/S MEF depleted for INK4a/ARF presented the same reduction of ATR as the one seen in ATR mutant cells (Fig. 1B). These data suggested that ATR-Seckel MEF depleted for INK4a/ARF could be growing in the presence of RS. In mammalian cells, RS leads to a pan-nuclear phosphorylation of the histone H2A variant H2AX, which we have shown in the past can be rigorously quantified through high-throughput microscopy (HTM).18,20 As shown in Figure 1C, growing cultures of INK4a/ARF-depleted ATRS/S MEF presented similar levels of RS as INK4a/ARF-proficient Seckel MEF. In summary, depletion of INK4a/ARF allows the growth of ATR-Seckel MEF in the presence of substantial amounts of RS.
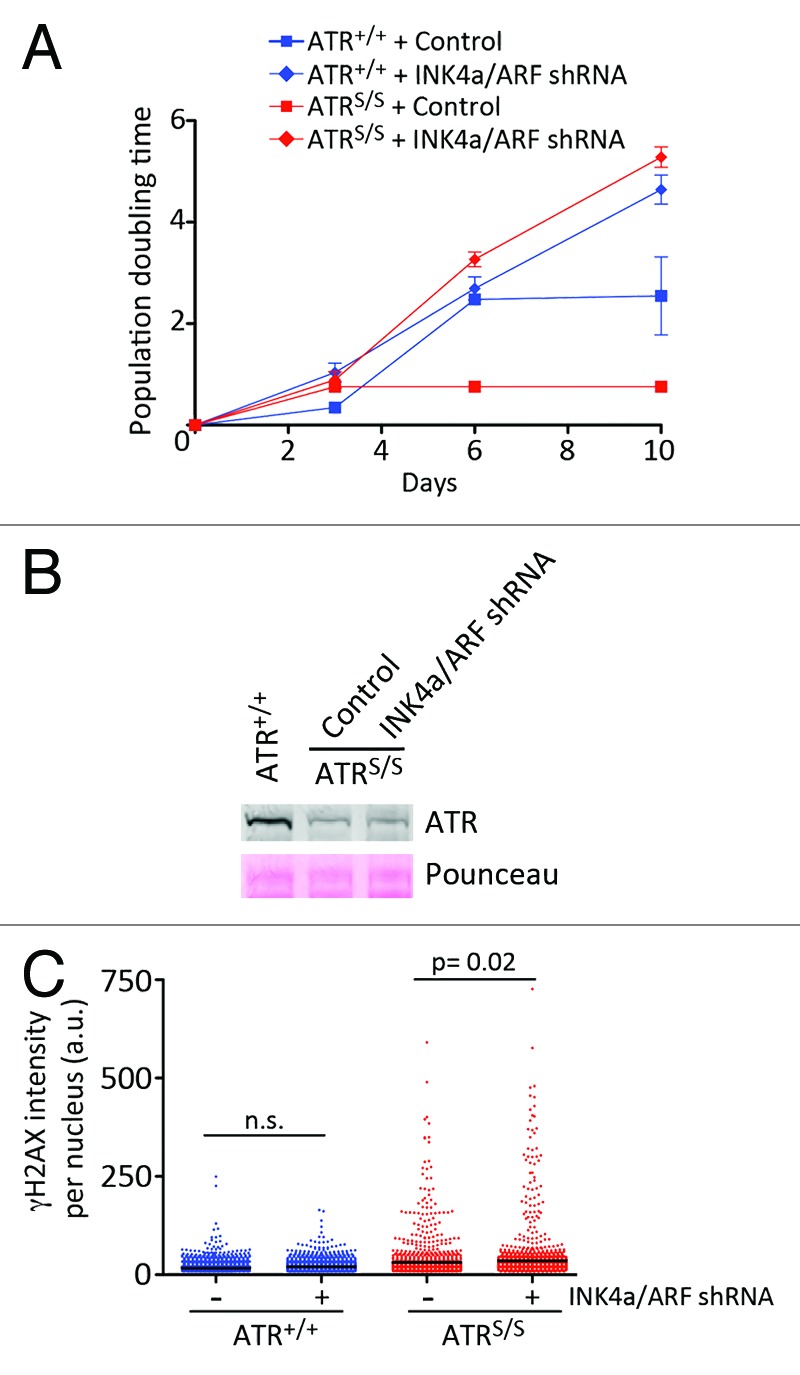
Figure 1. INK4a/ARF limits the growth of ATR-Seckel MEF. (A) Growth curves of ATR+/+ and ATRS/S MEF that had been infected with a control retrovirus or with a retrovirus expressing an shRNA against both products of the INK4a/ARF locus. Data represent mean and s.d. (n = 3). (B) Western blot analysis of ATR levels in wt MEF, as well as in ATR-Seckel MEF that had been infected with a control retrovirus, or with a retrovirus expressing a shRNA against both products of the INK4a/ARF locus. (C) High-throughput microscopy (HTM)-mediated quantification of the levels of γH2AX per nucleus found on ATR+/+ and ATRS/S MEF that had been infected with a control retrovirus or with a retrovirus expressing a shRNA against both products of the INK4a/ARF locus. The levels on the INK4a/ARF-depleted cells were measured 10 d after infection, to illustrate the levels of RS on growing cultures of ATR mutant cells. Data are representative of three independent analyses; n.s., non-significant.
INK4a/ARF regulate senescence in response to RS
To explore the impact of RS on p16Ink4A and p19ARF expression, wild type (wt) MEF were exposed to an inhibitor of the ribonucleotide reductase (hydroxyurea, HU), which leads to nucleotide depletion and RS, as well to an ATR inhibitor (ATRi) recently developed in our group.20 Given their low size and expression, which frequently complicates their biochemical detection, and in order to facilitate a rigorous quantification of p16Ink4A and p19ARF levels, we implemented a HTM assay that could measure the expression of both proteins in each individual cell. The specificity of this assay was validated with the inclusion of INK4a/ARF-deficient MEF as a negative control for p16Ink4A and p19ARF signals. As shown in Figure 2A, a persistent exposure to low doses of HU or ATRi led to the accumulation of cells with increased levels of p16Ink4A and p19ARF, showing that a persistent exposure to RS promotes the expression of this locus. Similarly, the levels of INK4a/ARF gene products were also constitutively higher in cultures of ATRS/S MEF, which, as mentioned, present high levels of RS (Fig. 2B). Moreover, p16Ink4A and γH2AX levels correlated on a cell-by-cell basis on ATR mutant cells, further supporting that RS can induce the expression of the INK4a/ARF locus (Fig. 2C). Finally, and consistent with HTM data, protein and mRNA levels of p16Ink4A and p19ARF were also induced by RS (Fig. 2D and E). In summary, these data support that a persistent exposure to RS leads to the activation of INK4a/ARF-dependent senescence.

Figure 2. Activation of INK4a/ARF in response to RS. (A) HTM-mediated quantification of the nuclear levels of γH2AX, p16INK4a and p19ARF in wt MEF that had been exposed for 5 d with HU (0.5 mM) and ATRi (1 μM). INK4a/ARF−/− MEF were included as a negative control. (B) HTM-mediated quantification of the nuclear levels of γH2AX, p16INK4a and p19ARF in ATR+/+ and ATRS/S MEF. INK4a/ARF−/− MEF were included as a negative control. (C) 2D plot illustrating the relationship between nuclear γH2AX and p16INK4a levels found on ATR+/+ and ATRS/S MEF. (D) Western blot analysis of p16INK4a and p19ARF in wt MEF that had been exposed to ATRi for 5 d (0.05, 0.1 and 0.5 μM). MEF that had been infected with a retrovirus expressing RasV12 and E1A oncogenes were used as a positive control of INK4a/ARF activation. INK4a/ARF−/− MEF were included as a negative control. (E) qRT-PCR analysis of the mRNA levels of p16INK4a and p19ARF in wt MEF that were grown in the presence of ATRi (2 μM) or HU (0.5 mM) for six passages. mRNA levels were normalized to the expression of GAPDH in each case. Data are representative of three independent analyses. In (A–C) at least 2,000 nuclei were quantified per condition. ***p < 0.001.
A limited role for INK4a/ARF on ATR-Seckel phenotypes
Chemical inhibitors of ATR kinases are actively being explored in cancer therapy, particularly for their use in tumors with high levels of RS (reviewed in ref. 21). Given that loss of INK4a/ARF is a frequent event in human cancer, our previous results raised the question of whether those tumors might not be sensitive to ATR inhibition. Consistent with this, INK4a/ARF-deficient MEF were able to sustain growth in the presence of low doses of ATRi or HU that induced senescence on wt MEF (Fig. S1). To explore the impact of INK4a/ARF loss on limited ATR activity in vivo, we generated INK4a/ARF-deficient ATR-Seckel mice. Similar to our previous findings with shRNAs, deletion of INK4a/ARF fully rescued growth on ATRS/S MEF (Fig. 3A). Again, INK4a/ARF deletion rescued the growth of ATR mutant cells without restoring ATR levels or the presence of RS (Fig. 3B–D). Surprisingly, and despite the rescue of MEF senescence, loss of INK4a/ARF did not have an impact on the viability and lifespan of ATR-Seckel mice (Fig. 3E). Accordingly, INK4a/ARF-deletion did not significantly alter the frequency of ATR-Seckel mice that reached birth, nor the apparent progeroid phenotypes that are found on ATR-Seckel mice.
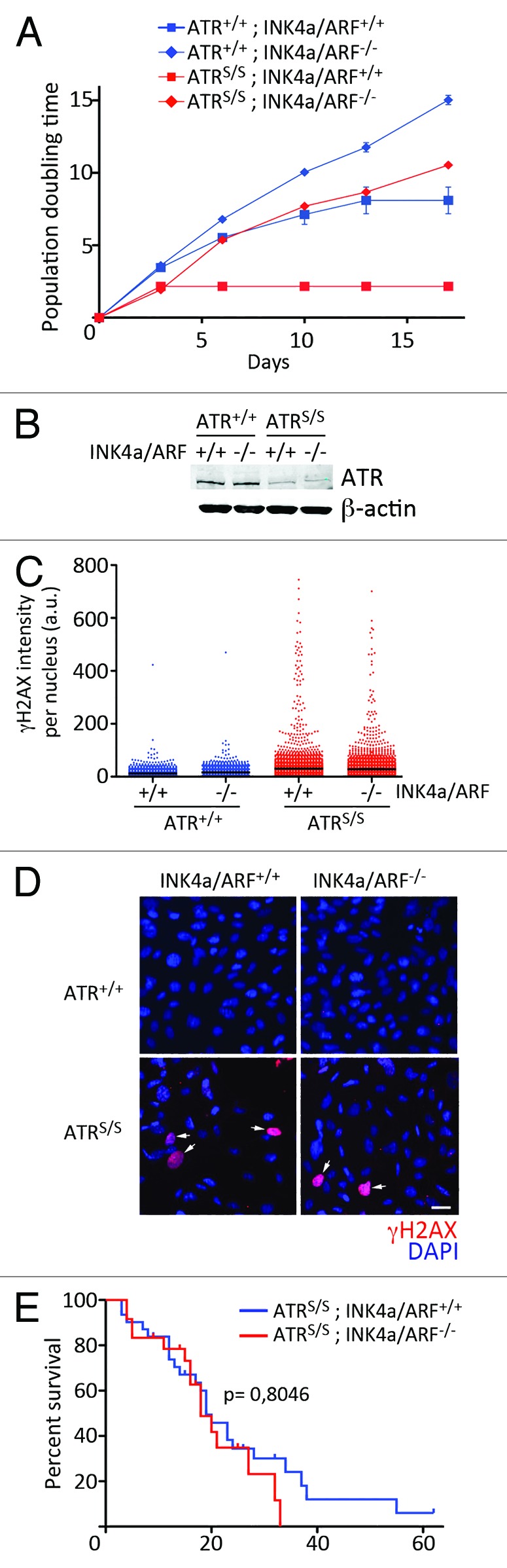
Figure 3. Rescuing senescence does not alter the lifespan of ATR-Seckel mice. (A) Growth curves of ATR+/+INK4a/ARF+/+, ATR+/+INK4a/ARF−/−, ATRS/SINK4a/ARF+/+ and ATRS/SINK4a/ARF−/− MEF. Data represent mean and s.d. (n = 3). (B) Western blot analysis of ATR levels in ATR+/+INK4a/ARF+/+, ATR+/+INK4a/ARF−/−, ATRS/SINK4a/ARF+/+ and ATRS/SINK4a/ARF−/− MEF. (C) HTM-mediated quantification of the levels of γH2AX per nucleus found on ATR+/+INK4a/ARF+/+, ATR+/+INK4a/ARF−/−, ATRS/SINK4a/ARF+/+ and ATRS/SINK4a/ARF−/− MEF. (D) Representative examples from the HTM images for γH2AX (red) used in (C). DAPI was used (blue) to stain DNA. Scale bar (white) indicates 10 μm. All MEF data are representative of three independent analyses. (E) Kaplan-Meyer analysis of the lifespan of ATRS/SINK4a/ARF+/+ and ATRS/SINK4a/ARF−/− mice. The p value indicates the statistical significance obtained on the log-rank Mantel-Cox test.
One possibility to explain the lack of an impact of INK4a/ARF in the phenotypes of ATR-Seckel mice is that, in vivo, the main outcome to a severe reduction in ATR levels is cell death and not senescence. Several facts support that this is the case. First, we cannot find evidence of senescence in ATR-Seckel embryos, which is, in contrast, abundant in a mouse model with impaired Brca1 function (Fig. S2A).28 Second, and in contrast to senescence, ATRS/S embryos present substantial amounts of cell death, which correlates with the severity of the progeroid phenotype in mice (Fig. S2B).17,29 Third, genetic deletion of the Suv39h1 histone H3 methyltransferase, which has been shown to abrogate senescence in vivo,30 also fails to rescue the viability and overall phenotype of ATR-Seckel animals. Fourth, in cell types that are more prone to cell death than senescence, such as splenocytes, INK4a/ARF deletion does not modify the cytotoxic effects of ATRi (Fig. S3A). Similarly, an in vivo treatment of mice with the Chk1 inhibitor UCN-01 leads to RS and apoptosis in the thymus regardless of INK4a/ARF status (Fig. S3B). Altogether, these observations suggest that cell death rather than senescence is the main outcome to limited ATR-Chk1 function in vivo.
Contribution of INK4a/ARF to the RS-response in the context of cancer
The limited impact of INK4a/ARF deletion on ATR-Seckel mice contrasts with its role in regulating RS-induced senescence in MEF. To explain this discrepancy, we propose that INK4a/ARF-dependent senescence would be the outcome of a persistent exposure to moderate amounts of RS, such as in the case of MEF chronically exposed to low doses of HU or ATRi. In contrast, high amounts of RS would inevitably lead to cytotoxicity, given that cells would not be able to divide in the presence of unreplicated regions. This would be particularly true in the case of ATR inhibitors, where the compounds would not only perturb DNA replication, but also abrogate the checkpoint activity that limits entry into mitosis with unreplicated DNA. Consistent with this model, whereas INK4a/ARF deletion facilitates the growth of MEF exposed to low doses of ATRi, it does not alter the cytotoxic effects of ATRi at higher doses of the inhibitor (Fig. S3C). This clarification is important, since it indicates that the cytotoxicity of ATR inhibitors in cancer would also not be affected by INK4a/ARF status. Supporting this view, the toxicity of ATRi for pancreatic ductal adenocarcinoma (PDAC) cell lines established from a mouse model of carcinogenesis driven by the K-ras oncogene31 was not dependent on INK4a/ARF status (Fig. S4).
Regardless of its limited impact on ATRi-mediated cytotoxicity, the RS-induced senescence pathway discovered here might still play a relevant role in cancer, where cells could be exposed to low but continuous levels or RS. One context where this might happen is in response to oncogenes. Oncogene-induced senescence was first observed in 1997, which already identified increased levels of p16INK4a expression associated to this process.25 Later works suggested that oncogene-induced senescence could be the outcome to RS,32,33 which would be the origin of the activated DDR that is observed in cancer.4,5,12 In this context, the findings we report in this manuscript could indicate that one of the roles of INK4a/ARF in cancer would be to limit the expansion of cells suffering from oncogene-induced RS. In agreement with this notion, INK4a/ARF deletion facilitates transformation with oncogenes in MEF.25,34
To explore this hypothesis on a large data set of human tumors, we benefited from the recently developed “Cancer Cell Line Encyclopedia” (CCLE) project (www.broadinstitute.org/ccle/home).35 The CCLE contains genome-wide gene expression and copy number variant (CNV) data for 947 human cancer cell lines of different origin. Given that CNVs are the outcome to a persistent exposure to RS,23 we evaluated the relationship between p16INK4a expression and total number of CNVs present in each of the cell lines from the CCLE. This analysis shows that cells with high numbers of CNV tend to present low levels of p16INK4a in human cancer cell lines (Fig. S5). The absence of unique probes for p19ARF in the CCLE data set precluded a similar analysis for this transcript. Nevertheless, p16Ink4A is thought to be the main contributor from the INK4a/ARF locus to tumor suppression in human cancers.7 The tumor suppressor activity of p16INK4a is linked to the activation of the retinoblastoma (Rb) pathway. Remarkably, the only other condition besides INK4a/ARF deletion which we have found rescues senescence on ATR-Seckel MEF is the expression of a fragment of the large T antigen from SV40 virus (T121), which selectively inactivates Rb and not p53 (Fig. S6). Altogether, the data presented in this manuscript suggest that an important function of the p16INK4a-Rb pathway in tumor suppression would be to limit the expansion of cells suffering from RS, revealing a new role for this pathway in safeguarding genome integrity during tumor evolution.
Discussion
p16INK4a was originally described as a CDK4-interacting protein,36 capable of inhibiting CDK4 and CDK6.37 Soon afterwards, the locus encoding p16INK4a was found to be mutated in cancer.38,39 However, the locus also encoded a different transcript named p19ARF, which shares the last two exons with p16INK4a, although in a different reading frame.40 Interestingly, p19ARF is not a CDK inhibitor but rather an activator of p53. This makes the INK4a/ARF a key player in cancer, since it can activate the two main tumor suppressor pathways in human cells (p16INK4a/Rb and p19ARF/p53). In human cells, however, p16INK4a seems to be the main contributor to tumor suppression, and cancer-associated mutations involve either the entire locus, or p16INK4a alone.40 Nevertheless, given that p19ARF works through activating p53, which is a bona fide DNA damage-responsive protein, much debate has been placed over the years upon a potential role for the INK4a/ARF locus in genome maintenance.
The initial studies unlinked INK4a/ARF from the response to DNA damage.9,11 In contrast, the locus was shown to be activated by “oncogenic stress,” suggesting that it was only in this context where p53 activation would depend on p19ARF 25. In agreement with this, p19ARF deficiency does not compromise radiation-induced apoptosis and p53-activation in vivo, but it is essential for the activation of p53 in response to oncogenic stress.10 However, several reports also indicated a potential involvement for the locus in the response to DNA damage, particularly upon the exposure to chronic doses of the genotoxic agent.6,41-45 One possibility to reconcile these observations emerges in the light that “oncogenic stress” could relate to the generation of chronic and low amounts of RS.4,5,32,33 Thus, it is possible that whereas INK4a/ARF does not play an active role in the acute response to DNA breaks, it might otherwise be relevant in the context of a persistent exposure to RS.
In agreement with this view, we show here that low amounts of RS can induce INK4a/ARF expression and INK4a/ARF-dependent senescence. From the two products of the locus, several observations suggest that p16INK4a is the main contributor to this phenomenon. First, whereas INK4a/ARF deletion rescues the senescence of ATR-Seckel MEF, p53 deletion aggravates the phenotype of Seckel MEF and mice.17 Second, a fragment of the large T antigen that inactivates Rb but not p53 also rescues senescence of ATR-Seckel MEF. Consistent with this, we previously reported that Rb was activated by a continuous exposure to doxorubicin, which damages DNA during replication, and this activation was necessary to maintain a persistent cell cycle arrest of the damaged cells.46 Finally, our meta-analysis of the CCLE data shows that p16INK4a levels are particularly low on cancer cells with a high number of CNV. Altogether, we propose that the p16INK4a/Rb pathway plays an active role in genome maintenance by restricting the growth of cells suffering from a persistent exposure to low amounts of RS.
At this point, it remains to be understood how a chronic exposure to low doses of RS leads to the activation of the INK4a/ARF locus. Nevertheless, several reasons suggest that this activation is not dependent on the ATR-dependent RSR. First, the activation of INK4a/ARF in response to RS demands several days, which contrasts with the immediate activation of the RSR. Second, ATR inhibition or hypomorphism lead to increased levels of p16INK4a and p19ARF. Finally, we previously showed that a persistent activation of ATR is able to induce senescence in an INK4a/ARF-independent manner, which formally demonstrates that ATR-induced senescence does not require INK4a/ARF.16 If not ATR, how a chronic exposure to RS ends up activating the locus remains an open question. We should note, however, that understanding how oncogenes trigger the activation of the INK4a/ARF locus remains unsolved after many years since its discovery. Whichever the mechanism, our work reveals a new role for the INK4a/ARF locus in limiting the expansion of cells suffering from RS, which places this tumor suppressor locus as a key player in the maintenance of genomic integrity during tumor evolution.
Materials and Methods
Mice
ATR-Seckel17 and INK4a/ARF−/− 34 mice have already been described before. Mice were kept under standard conditions at serum-pathogen free facility of the Spanish National Cancer Centre in a mixed C57BL/6-129/Sv background. All mouse work was performed in accordance with the Guidelines for Humane Endpoints for Animals Used in Biomedical Research and under the supervision of the Ethics Committee for Animal Research of the Instituto de Salud Carlos III.
Cell culture
MEFs were isolated from 13.5 dpc embryos. All cells were grown in Dulbecco’s minimum essential media (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (Sigma) and antibiotics. INK4a/Arf targeting shRNAs were previously validated in several studies.26,27 Retroviral infections were done using standard procedures, and infected MEFs were selected with puromycin (2 μg/ml) for 3 d. For senescence assays, MEF and embryos were fixed with 2% formaldehyde and subsequently stained with a senescence-associated β-Galactosidase Staining Kit (Cell Signaling), following manufacturer’s instructions. HU (SIGMA) and ATRi were added at the indicated concentrations and times. The ATRi has been recently described by our group.20
Immunobloting
Cells were lysed in RIPA buffer (50 mM TRIS-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl and 1 mM EDTA) containing protease and phosphatase inhibitors (Sigma-Aldrich). Samples were resolved by SDS-PAGE and analyzed by standard western blotting techniques. Antibodies against ATR (Serotec), p16INK4a (Santa Cruz p16 M-156), p19ARF (Santa Cruz sc-32748) and β-actin (Sigma-Aldrich), were used. Protein blot analyses were performed on the LICOR platform (eBioscience).
High-throughput microscopy
The procedure for our HTM analyses of RS has been previously documented.29 Briefly, MEF were grown on μCLEAR bottom 96-well plates (Greiner Bio-One), and immunofluorescence was performed using standard procedures. Images were automatically acquired from each well by an Opera High-Content Screening System (Perkin Elmer). A 20× or 40× magnification lens was used, and pictures were taken at nonsaturating conditions. Images were segmented using the DAPI staining to generate masks matching cell nuclei from which the mean fluorescence intensity signals were calculated. γH2AX (Upstate Biotechnology), p16INK4a (Santa Cruz p16 M-156), p19ARF (Santa Cruz sc-32748) as well as secondary antibodies conjugated with Alexa 488, Alexa 594 or Alexa 647 (Molecular probes) were used.
Immunohistochemistry
Embryos and organs were fixed in formalin and embedded in paraffin for subsequent processing. Consecutive 2.5 μm sections were treated with citrate for antigenic recovery and processed for immunohistochemistry with γH2AX (Upstate) and activated caspase-3 (R&D Systems) antibodies. Hematoxiline was used to counterstain.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
A.M. is funded by a PhD fellowship from the Spanish Government (MINECO). A.J.L. is the recipient of a postdoctoral fellowship from the Spanish Association Against Cancer (AECC). Work in O.F. laboratory is supported by grants from the Spanish Ministry of Science (SAF2011-23753), the Association for International Cancer Research (12-0229), the Howard Hughes Medical Institute and the European Research Council (ERC-210520).
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/25017
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25017
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satyanarayana A, Wiemann SU, Buer J, Lauber J, Dittmar KEJ, Wüstefeld T, et al. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J. 2003;22:4003–13. doi: 10.1093/emboj/cdg367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorgoulis VG, Vassiliou L-VF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 5.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 6.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP, Poele te RH. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–83. [PubMed] [Google Scholar]
- 7.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–77. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 9.Kamijo T, van de Kamp E, Chong MJ, Zindy F, Diehl JA, Sherr CJ, et al. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 1999;59:2464–9. [PubMed] [Google Scholar]
- 10.Efeyan A, Garcia-Cao I, Herranz D, Velasco-Miguel S, Serrano M. Tumour biology: Policing of oncogene activity by p53. Nature. 2006;443:159. doi: 10.1038/443159a. [DOI] [PubMed] [Google Scholar]
- 11.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–14. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 13.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.López-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair (Amst) 2010;9:1249–55. doi: 10.1016/j.dnarep.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–53. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- 16.Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302. doi: 10.1101/gad.452308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009;41:891–8. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–5. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruzankina Y, Schoppy DW, Asare A, Clark CE, Vonderheide RH, Brown EJ. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat Genet. 2009;41:1144–9. doi: 10.1038/ng.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–7. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol Oncol. 2011;5:368–73. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, et al. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest. 2012;122:241–52. doi: 10.1172/JCI58928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arlt MF, Wilson TE, Glover TW. Replication stress and mechanisms of CNV formation. Curr Opin Genet Dev. 2012;22:204–10. doi: 10.1016/j.gde.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59. doi: 10.1016/S0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 25.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 26.Dickins RA, McJunkin K, Hernando E, Premsrirut PK, Krizhanovsky V, Burgess DJ, et al. Tissue-specific and reversible RNA interference in transgenic mice. Nat Genet. 2007;39:914–21. doi: 10.1038/ng2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–9. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao L, Li W, Kim S, Brodie SG, Deng C-X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–13. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.López-Contreras AJ, Gutierrez-Martinez P, Specks J, Rodrigo-Perez S, Fernández-Capetillo O. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J Exp Med. 2012;209:455–61. doi: 10.1084/jem.20112147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 31.Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–20. doi: 10.1016/S1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 32.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 33.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 34.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/S0092-8674(00)81079-X. [DOI] [PubMed] [Google Scholar]
- 35.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 1993;7:1572–83. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 37.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 38.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–40. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 39.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–6. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 40.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 41.Khan SH, Moritsugu J, Wahl GM. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci USA. 2000;97:3266–71. doi: 10.1073/pnas.97.7.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Mohanna MA, Al-Khalaf HH, Al-Yousef N, Aboussekhra A. The p16INK4a tumor suppressor controls p21WAF1 induction in response to ultraviolet light. Nucleic Acids Res. 2007;35:223–33. doi: 10.1093/nar/gkl1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Mohanna MA, Manogaran PS, Al-Mukhalafi Z, A Al-Hussein K, Aboussekhra A. The tumor suppressor p16(INK4a) gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene. 2004;23:201–12. doi: 10.1038/sj.onc.1206927. [DOI] [PubMed] [Google Scholar]
- 44.Sarkar-Agrawal P, Vergilis I, Sharpless NE, DePinho RA, Rünger TM. Impaired processing of DNA photoproducts and ultraviolet hypermutability with loss of p16INK4a or p19ARF. J Natl Cancer Inst. 2004;96:1790–3. doi: 10.1093/jnci/djh307. [DOI] [PubMed] [Google Scholar]
- 45.Lau WM, Ho TH, Hui KM. p16INK4A-silencing augments DNA damage-induced apoptosis in cervical cancer cells. Oncogene. 2007;26:6050–60. doi: 10.1038/sj.onc.1210405. [DOI] [PubMed] [Google Scholar]
- 46.Cuadrado M, Gutierrez-Martinez P, Swat A, Nebreda AR, Fernandez-Capetillo O. p27Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 2009;69:8726–32. doi: 10.1158/0008-5472.CAN-09-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.