

Abstract
S-Nitrosylation is a redox-mediated posttranslational modification that regulates protein function via covalent reaction of nitric oxide (NO)-related species with a cysteine thiol group on the target protein. Under physiological conditions, S-nitrosylation can be an important modulator of signal transduction pathways, akin to phosphorylation. However, with aging or environmental toxins that generate excessive NO, aberrant S-nitrosylation reactions can occur and affect protein misfolding, mitochondrial fragmentation, synaptic function, apoptosis or autophagy. Here, we discuss how aberrantly S-nitrosylated proteins (SNO-proteins) play a crucial role in the pathogenesis of neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases. Insight into the pathophysiological role of aberrant S-nitrosylation pathways will enhance our understanding of molecular mechanisms leading to neurodegenerative diseases and point to potential therapeutic interventions.
Keywords: S-Nitrosylation, Nitric oxide, Alzheimer’s disease, Parkinson’s disease, Neurodegeneration, Oxidative stress
Introduction
The global prevalence of neurodegenerative disorders such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS), is increasing with extended life expectancy. Although precise cellular and molecular mechanisms underlying neurodegeneration still remain enigmatic, key features of these devastating disorders have been identified, including elevated oxidative/nitrosative stress, mitochondrial dysfunction, protein misfolding/aggregation, synapse loss, and decreased neuronal survival. Notably, oxidative/nitrosative stress appears to influence the manifestation of other pathological features, including synaptic loss and neuronal cell death, suggesting that these pathways may be a common determinant of disease pathogenesis and progression (Nakamura and Lipton, 2007).
In most mammalian cells, reactive oxygen/nitrogen species (ROS/RNS) are normally produced at low levels and act as important physiological messengers of intracellular signaling pathways (Finkel, 2011). However, exposure to environmental toxins or even the normal process of brain aging can trigger an imbalance between the production of ROS/RNS and the availability of cell defense systems, including antioxidant enzymes, glutathione and molecular chaperones, resulting in an overabundance of ROS/RNS that causes oxidative and nitrosative stress. Neurons are particularly vulnerable to oxidative/nitrosative stress due to their high demand for energy from ROS/RNS-generating mitochondrial metabolism and the fact that they contain lower levels of certain antioxidants compared to other cells (Mattson et al., 2002). Additionally, in some cases genetic mutations that are pathogenic for inherited neurodegenerative diseases can lead to increases in basal ROS/RNS production, thus rendering neurons more vulnerable to additional oxidative/nitrosative stress.
ROS/RNS include reactive free radical groups that exert their biological effects, at least in part, via reaction with cellular macromolecules. NO is a small, highly diffusible molecule generated by a family of NO synthases (NOS) that convert L-arginine to L-citrulline using molecular oxygen and NADPH (Bredt et al., 1991). This family includes three members: neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3) (Forstermann et al., 1991). nNOS and eNOS have been named after cell types in which they are constitutively and predominantly expressed, while expression of iNOS is typically induced by acute inflammatory stimuli. All three NOS subtypes are expressed in the mammalian brain. We and our colleagues have shown that production of NO-related species or related compounds, possibly including dinitrogen trioxide (N2O3) or the reaction intermediate nitrosonium cation (NO+), can lead to S-nitrosylation, a reversible, covalent chemical reaction involving the addition of an NO moiety to a critical cysteine thiol (-SH) group (or more properly thiolate anion, -S−) on a target protein to regulate its function. This nitrosation reaction forms an S-nitrosothiol (-SNO), and an S-nitrosylated protein is thus referred to as a SNO-protein (Lei et al., 1992; Stamler et al., 1992; Lipton et al., 1993; Stamler et al., 2001). It is important to note that S-nitrosylation of cysteine thiol is a chemically distinct redox reaction from nitration of tyrosine residues, representing another NO-dependent posttranslational modification generated, for example, via reaction of tyrosine with peroxynitrite (ONOO−) (Ischiropoulos et al., 1992).
Under normal physiological conditions, S-nitrosylation modulates the function of substrate proteins, thus playing a dynamic role in a variety of biological processes (Figure 1). Like other posttranslational modifications, S-nitrosylation can trigger conformational changes, activate or inhibit protein activity, alter protein-protein interactions, affect protein aggregation, or influence protein localization. These alterations affect cell signal transduction pathways and neuronal function (Choi et al., 2000; Qu et al., 2011; Shi et al., 2013; Uehara et al., 2006). Under pathological conditions, aberrant S-nitrosylation of specific proteins stimulates cell destructive processes, contributing to neurodegeneration. Insults mediated by S-nitrosylation include protein misfolding, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, synaptic degeneration, and apoptosis (Nakamura and Lipton, 2007). Nitrosative stress-associated excitotoxicity is implicated in a number of neurological disorders, ranging from acute hypoxia-ischemia to chronic neurodegenerative diseases. Manipulating S-nitrosylation can affect pathology; for example, pharmacological inhibition of nNOS or knockout of the nNOS gene provides neuroprotection against ischemia (Huang et al., 1994).
Figure 1. NO and SNO Signaling in the CNS.

(A) S-Nitrosylation plays a dynamic role in both normal and aberrant neuronal signal transduction pathways. Initially, activation of soluble guanylate cyclase, with consequent increase in production of cGMP, was identified as an NO-mediated signal transduction pathway. Additionally, peroxynitrite (ONOO−), derived from reaction of NO· and superoxide anion (O2−·), can mediate neurotoxicity in part via a protein posttranslational modification of tyrosine residues termed nitration. However, emerging evidence suggests that NO species mediate signal transduction predominantly via protein S-nitrosylation, a posttranslational redox modification of critical cysteine residues that affects protein activity and function. Under physiological conditions, NO is produced in neurons predominantly by nNOS, which is activated by calcium influx through NMDAR-associated ion channels. Extrasynaptic in addition to synaptic NMDARs can lead to NO production, particularly during excitotoxic injury when excessive NO is generated. This can lead to aberrant protein S-nitrosylation whereby cysteine residues, which would not ordinarily be S-nitrosylated by physiological levels of NO, undergo S-nitrosylation because of the high levels of NO generated in disease states.
(B) S-Nitrosylation as a determinant of CNS disease progression. Physiological (normal) levels of NO can mediate neuroprotective effects, at least in part, by S-nitrosylating caspase and HDAC2. Additionally, during periods of moderate stress, NO can still facilitate protection of neurons, for instance, via S-nitrosylation of NMDARs to downregulate excessive activity, representing a negative feedback mechanism. However, persistent hyperactivation of neuronal NMDARs (predominantly extrasynaptic receptors) induces excessive stimulation of nNOS and thus increased production of NO that contributes to synaptic injury and cell death. Glial cells (astrocytes and microglia) can also generate high levels of NO via iNOS activity. We and others have reported evidence that overproduction of NO can be neurotoxic via aberrant S-nitrosylation of parkin, PDI, GAPDH, MMP-2/9, Cdk5, Drp1 and other proteins that contribute to accumulation of misfolded proteins, mitochondrial dysfunction, synaptic damage, and neuronal cell death.
In this review, we summarize progress made in the identification and characterization of key S-nitrosylated proteins that have been found to play a role in the pathogenesis of AD, PD and other neurodegenerative diseases. SNO-proteins discussed in this review are summarized in Table 1. We will also discuss how S-nitrosylation affects the function of specific target proteins and influences the onset or development of neurodegeneration. Further, we develop a concept for disease pathogenesis concerning the nearly ubiquitous nature of aberrant S-nitrosylation reactions in sporadic neurodegenerative disorders. This contrasts with the rarity of familial forms of these diseases due to genetic mutations. We propose that excessive nitrosative/oxidative stress might cause the more common “sporadic” form of neurodegenerative diseases by lowering the threshold for, or even mimicking, the effect of rare genetic mutations. In support of this hypothesis, a number of studies have found aberrantly S-nitrosylated proteins contributing to disease pathogenesis, and these proteins are encoded by genes that manifest rare mutations causing the same disease phenotype.
Table 1.
Summary of SNO-proteins discussed in this review.
| Category | Target of S-Nitrosylation | Effect of S-Nitrosylation |
|---|---|---|
| Synapse/membrane -related proteins | NMDA receptor | Downregulation of excessive receptor activity |
| PSD-95 | Decrease in its synaptic expression | |
| NSF | Increase in surface expression of AMPA receptors | |
| Stargazin | Increase in surface expression of AMPA receptors | |
| AMPAR (GluA1) | Increase in its phosphorylation, and enhanced endocytosis | |
| Serine racemase | Decrease in generation of D-serine | |
| Syntaxin 1 | Possibly prevention of excessive release of glutamate | |
| SNAP-25 | Decrease in evoked neurotransmitter release, while increasing spontaneous release | |
| RyR | Increased channel opening | |
| GRK2 | Prevention of receptor internalization | |
| β-arrestin | Acceleration of receptor internalization | |
| Quality control machineries | Parkin | Dysregulation of its ubiquitin E3 ligase activity |
| PDI | Inhibition of its chaperone and isomerase activities | |
| JNK1/IKKβ |
|
|
| HSP90 | Inhibition of it schaperone activity | |
| Proteasome | Inhibition of 26S proteasome activity | |
| N-end-rule | Regulation of UPS-dependent N-end rule | |
| Cell death/survival or other signaling pathways | Caspases | Inhibition of its protease activity |
| XIAP |
|
|
| FLIP | Inhibition of its proteasomal degradation | |
| Bcl-2 | Inhibition of its proteasomal degradation | |
| GAPDH |
|
|
| GOSPEL | Inhibition of SNO-GAPDH-mediated cell death | |
| MMPs | Activation of its metalloproteinase activity | |
| DJ-1 | Possible regulation of its anti-cell death activity | |
| Akt | Inhibition of its kinase activity | |
| PTEN | Inhibition of its phosphatase activity | |
| SHP-2 | Inhibition of its phosphatase activity | |
| MAP1B | Increase of its microtubule binding affinity | |
| Cdk5 |
|
|
| Prx2 | Inhibition of its antioxidant activity | |
| COX-2 | Activation of its prostaglandin synthesis activity | |
| IDE | Inhibition of its metalloprotease activity | |
| ApoE | Possibly lowering its binding affinity to LDL receptors | |
| Mitochondrial function | Drp1 | Upregulation of its GTPase activity, excessive mitochondrial fission, bioenergetic failure, synaptic dysfunction and loss |
| Complex I | Inhibition of mitochondrial respiration activity | |
| Complex IV | Inhibition of mitochondrial respiration activity | |
| F1 ATPase | Inhibition of its ATPase activity | |
| Aconitase | Suppression of the citric acid cycle | |
| ALDH2 | Suppression of alcohol metabolism | |
| Iron homeostasis | IRP2 | Increase in its proteasome degradation |
| Dexras | Augmentation of iron uptake |
Generation of NO in the CNS
A well-established route for NO production in several neurodegenerative disorders involves activation of NMDA receptors (NMDARs). In brain, nNOS is predominantly expressed in neurons and physically tethered to NMDARs due to mutual interactions with PSD-95 at the postsynaptic density (Brenman et al., 1996). Activated NMDARs are permeable to Ca2+, which in turn activates nNOS to produce NO (Bredt et al., 1991). Additionally, activation of NMDARs also generates ROS (Lafon-Cazal et al., 1993).
Neuroinflammatory stimuli or other toxins (e.g., Aβ oligomers or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP]) can induce the expression of iNOS in the brain, predominantly in astrocytes, macrophages, and microglia cells. iNOS can produce high concentrations of NO to contribute to neurotoxicity, and knockdown or knockout of iNOS has been shown to confer resistance against MPTP-induced neurotoxicity in an animal model of PD (Liberatore et al., 1999). Additionally, genetic ablation of iNOS in an animal model of AD (APP-PS1 double transgenic) ameliorated AD-like symptoms such as premature mortality, cerebral plaque formation, increased β-amyloid (Aβ) levels, and astrocytosis/microgliosis (Nathan et al., 2005). However, in a study utilizing a different mouse AD model (Tg2576 APP transgenic), deletion of iNOS worsened spatial memory, learning, and tau pathology, suggesting that NO manifested a neuroprotective effect in this case (Wilcock et al., 2008).
Protein S-Nitrosylation, Denitrosylation, and Transnitrosylation
Hundreds, if not thousands, of proteins with potential S-nitrosylation sites have been identified (Seth and Stamler, 2011). Although the majority of cellular proteins possess multiple cysteine residues, only specific cysteine residues are S-nitrosylated. One well-characterized determinant of S-nitrosylation depends on proximity. For example, formation of a complex with nNOS regulates the S-nitrosylation of NMDARs and PSD-95 (Lei et al., 1992; Lipton et al., 1993; Ho et al., 2011). Similarly, the brain-enriched small GTPase Dexras1 is linked to nNOS via an adaptor protein CAPON, and Dexras1 is activated by S-nitrosylation (Fang et al., 2000). Another mechanism for the specificity of S-nitrosylation entails the presence of a signature SNO motif of amino acid residues adjacent to the target cysteine. Acidic and/or basic amino acids typically exist within 6 to 8 Å of the S-nitrosylated thiol group, affecting the deprotonation of the sulfhydryl to facilitate S-nitrosylation (Stamler et al., 1997; Hess et al., 2005; Doulias et al., 2010; Marino and Gladyshev, 2010). In addition, local hydrophobicity may also promote the specificity of S-nitrosylation via increased stability of the S-nitrosothiol. Accordingly, S-nitrosylation can occur in a facile manner within or near biological membranes.
The process of SNO-protein formation is counterbalanced by denitrosylation enzymes, such as S-nitrosoglutathione reductase, the thioredoxin system, and PDI (see Benhar et al., 2009 for a detailed review of protein denitrosylation). These denitrosylases are involved in the removal of NO from S-nitrosylated cysteine residues and thus can potentially ameliorate nitrosative stress under disease conditions.
Transnitrosylation may be the major enzymatic mechanism to generate S-nitrosylated proteins in biological systems. Recent studies have identified S-nitrosylating enzymes, acting via protein-protein transnitrosylation, as a primary source of S-nitrosylase activity (Kornberg et al., 2010; Mitchell and Marletta, 2005; Nakamura et al., 2010). Transnitrosylation reactions catalyze the transfer of an NO group from a donor protein to the reactive cysteine residue on an acceptor protein, producing both a denitrosylated protein and a SNO-protein. Thus, the protein accepting the NO group in a transnitrosylation reaction may also serve as a denitrosylating enzyme because the NO group is transferred to its protein thiol from another S-nitrosylated protein. Examples of transnitrosylases (representing the NO donor protein) demonstrated to date include SNO-hemoglobin, SNO-thioredoxin (Trx), SNO-GAPDH, SNO-Caspase-3, and SNO-cyclin-dependent kinase 5 (Cdk5) (Figure 2).
Figure 2. Protein-Protein Transnitrosylation as an Enzymatic Nitrosation Mechanism to Produce SNO-Proteins.
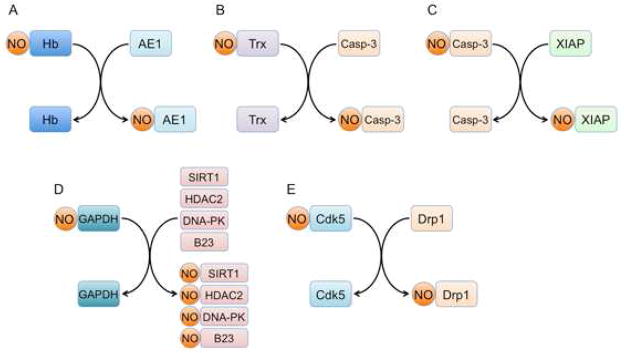
Increasing evidence suggests that a prominent S-nitrosylating reaction mechanism involves protein-protein transnitrosylation reactions (representing transfer of an NO group from one protein thiol to another). Transnitrosylation reactions reported to date include:
(A) hemoglobin-to-anion exchanger 1 (AE1).
(B) Thioredoxin 1 (Trx1)-to-Caspase-3 (Casp-3).
(C) Caspase-3 (Casp-3)-to-X-linked inhibitor of apoptosis (XIAP).
(D) GAPDH-to-HDAC2/SIRT1/DNA-PK.
(E) Cdk5-to-dynamin related protein 1 (Drp1).
Oxidation Reactions Downstream of S-Nitrosylation
S-Nitrosylation can result in conformational changes in protein structure, which may facilitate further oxidation reactions with less active ROS, resulting in sulfenic acid (-SOH), sulfinic acid (-SO2H), or sulfonic acid (-SO3H) derivatization of the cysteine thiol group. Sulfonation (-SO3H) reactions cannot be reversed by known enzymes, and can therefore result in permanent/pathological changes to protein structure and activity (Gu et al., 2002). Interestingly, sulfination (-SO2H), unlike sulfonation (-SO3H), can be reduced back towards free thiol if the enzyme sulfiredoxin is transcriptionally induced and that this can occur in neurons via increased synaptic NMDAR activity (Papadia et al., 2008).
In addition, S-nitrosylation has been reported to influence additional posttranslational modifications of cysteine residues. For instance, when there are two neighboring cysteine residues, S-nitrosylation of one of them can facilitate disulfide bond formation between them (Lipton et al., 2002; Stamler and Toone, 2002; Cho et al., 2009). In contrast, if both cysteine residues are S-nitrosylated, e.g., under severe nitrosative conditions, S-nitrosylation inhibits disulfide formation (Hess et al., 2005; Uehara et al., 2006). Moreover, S-nitrosylation of a cysteine residue can precede and thus inhibit palmitoylation (Ho et al., 2011). Since palmitoylation increases the association of a protein to the cell membrane, S-nitrosylation can decrease membrane targeting. Furthermore, S-nitrosylated as well as sulfenated cysteine residues may react with glutathione (Martinez-Ruiz and Lamas, 2007), but it is unclear whether SNO-proteins are intermediates for protein glutathionylation in vivo. Future studies are likely to reveal novel relationships between S-nitrosylation and other types of posttranslational modifications.
Oxidative and Nitrosative Stress in Alzheimer’s and Parkinson’s Diseases
The increase in free radicals and decrease in antioxidant potential that occur during aging may contribute to the development of neurodegenerative conditions, such as AD and PD. Consistent with this idea, postmortem human AD brains exhibit increased oxidative and nitrosative stress with elevated levels of free radicals (Sayre et al., 2008). While relatively rare genetic mutations, for example to the genes encoding amyloid precursor protein (APP) or presenilin 1/2 (PS1/PS2) can cause AD, it appears that this same phenotype can be mimicked by much more common nitrosylation/oxidation reactions. A number of proteins critical to synaptic function and neuronal survival are aberrantly S-nitrosylated in AD, contributing to synaptic loss and neurodegeneration (Figure 3).
Figure 3. Representative Protein Substrates for Aberrant S-Nitrosylation.
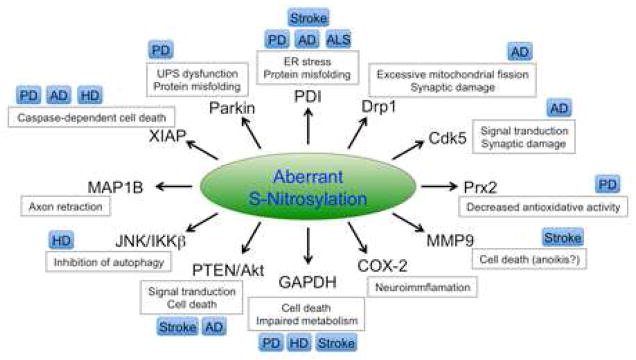
Aberrant protein S-nitrosylation plays a pathological role in many neurodegenerative conditions. Production of excessive NO can S-nitrosylate parkin, PDI, Drp1, Cdk5, Prx2, MMP-9, COX-2, GAPDH, PTEN/Akt, JNK/IKKβ, MAP1B, and XIAP. Subsequently, these aberrant S-nitrosylation reactions trigger neurotoxic signaling pathways leading to ER stress, protein misfolding, mitochondrial fragmentation, bioenergetic compromise, and consequent synaptic/neuronal damage. These processes can contribute to the pathogenesis of PD, AD, HD, ALS, stroke, and potentially other neurodegenerative disorders.
PD is the second most common neurodegenerative disease and most common motor disorder. It is characterized by specific loss of dopaminergic neurons in the substantia nigra pars compacta and is often accompanied by protein inclusions known as Lewy bodies (LBs). The major proteinaceous component of LBs is α-synuclein (α-syn), whose encoding gene is mutated in an autosomal dominant familial form of PD with LB dementia (Polymeropoulos et al., 1997). Mutations in additional genes, for example, encoding parkin, PINK1 and DJ-1, have also been identified as causal factors for autosomal recessive forms of familial PD in humans (Bonifati, 2012). In animal models, mutation of these genes can cause abnormal responses to oxidative stress, resulting in protein misfolding, mitochondrial dysfunction, ER stress, synaptic injury, and ultimately cell death (Dawson et al., 2010). However, similar to AD, the majority of PD cases (>90–95%) are sporadic (although mutations in the LRRK2 gene may in fact be present in a number of these cases). Studies suggest that sporadic PD can be affected by both genetic factors, usually causing early-onset disease, and environmental factors, possibly including agricultural pesticides, herbicides, fungicides, or other neurotoxins that are known to act as mitochondrial toxins to generate oxidative and nitrosative stress (Betarbet et al., 2000; Yao et al., 2004; Chung et al., 2004; Uehara et al., 2006). Thus, PD may represent another example of a disease in which a rare genetic mutation can be phenocopied by more common nitrosative/oxidative posttranslational modifications of critical proteins.
Dopaminergic neurons are especially susceptible to oxidative damage (Miller et al., 2009). Compelling evidence suggests that S-nitrosylation induced by nitrosative stress is a major contributing factor in the development of PD. As discussed below, S-nitrosylation of proteins, including parkin, DJ-1, X-linked inhibitor of apoptosis protein (XIAP or IAP3), peroxiredoxin (Prx) 2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), is involved in the pathological process, and can contribute to ubiquitin-proteasome impairment, ER stress, mitochondrial dysfunction, and apoptosis (Figure 3)
S-Nitrosylation of Parkin
Parkin is an E3 ubiquitin ligase that participates in the ubiquitin-proteasome system (UPS) responsible for targeting specific proteins for degradation (Shimura et al., 2000). Parkin is also involved in protein degradation during ER stress via its interaction with the chaperones, heat shock protein (Hsp)70 and carboxyl-terminus of Hsp70 interacting protein (CHIP), and thus participates in ER-associated degradation (ERAD) (Wang and Takahashi, 2007). Disruption of parkin activity causes dysfunction in protein degradation, leading to accumulation/aggregation of neurotoxic proteins and resulting ER stress (Dawson and Dawson, 2003; Lindholm et al., 2006).
In addition to its E3 ligase activity, parkin also suppresses the transcription of the oncogene p53 and serves a neuroprotective function against PD-associated apoptosis of dopaminergic neurons (da Costa et al., 2009). Mutations in parkin (PARK2) have been identified as causal for autosomal recessive juvenile PD and early onset PD (Kitada et al., 1998). Emerging evidence points to the possibility that parkin, together with PINK1, whose gene can also be mutated in familial PD, participates in mitophagy whereby damaged mitochondria are removed by quality control machinery such as autophagy (Youle and van der Bliek, 2012). In this proposed model, PINK1 is initially translocated to impaired mitochondria, which in turn recruit parkin from the cytosol to the damaged mitochondrial membrane. Recent evidence suggests that PINK1-phosphorylated mitofusin 2 is a parkin receptor on the mitochondrial membrane (Chen and Dorn, 2013). Parkin then ubiquitinates mitochondrial outer membrane proteins to enhance autophagic removal of the unhealthy mitochondria. Parkin’s neuroprotective activity and role in protein ubiquitination/degradation can also be compromised by environmental neurotoxic factors, such as pesticides and herbicides, probably because these toxins generate excessive amounts of NO and ROS. Several studies have shown that high levels of NO and ROS can precipitate S-nitrosylation or further oxidation/sulfonation of parkin (Chung et al., 2004; Yao et al., 2004; Meng et al., 2011). Parkin has multiple cysteine residues that can react with NO to form SNO-parkin. For instance, exposure to the pesticide rotenone or the neurotoxin MPTP leads to S-nitrosylation of parkin (Chung et al., 2004; Yao et al., 2004). Intriguingly, upon S-nitrosylation, parkin’s E3 ligase activity initially increases but is subsequently inhibited, possibly because of autoubiquitination. Consequently, the downregulated E3 ligase activity impairs ubiquitination and degradation of substrate proteins, potentially contributing to LB formation and neuronal cell injury or death (Chung et al., 2004; Yao et al., 2004; Lipton et al., 2005) (Figure 4).
Figure 4. Neurodegenerative Signaling Pathways Triggered by Aberrant S-Nitrosylation Reactions.

Genetic mutations associated with neurodegenerative diseases, environmental toxins such as certain pesticides, and misfolded proteins including Aβ oligomers can all lead to excessive nitrosative stress. The resulting aberrant S-nitrosylation of a number of proteins has been implicated in the pathogenesis of several neurodegenerative disorders. For example, S-nitrosylation of specific proteins can cause ‘loss-of-function’ by impairing E3 ligase ubiquitin ligase activity (parkin and XIAP) or molecular chaperone activity (PDI). These processes can contribute to accumulation of neurotoxic proteins and activation of apoptotic pathways. Additionally, oligomeric Aβ peptide can result in increased generation of NO and S-nitrosylation of Drp1, resulting in excessive mitochondrial fragmentation, bioenergetic compromise, and consequent synaptic damage. Additionally, Cdk5 is activated when Aβ increases calpain activity to cleave the Cdk5 regulatory subunit p35 to p25. The resulting neurotoxic kinase activity of Cdk5 is further enhanced by S-nitrosylation. Formation of SNO-Cdk5 may also contribute to Aβ-induced spine loss by transnitrosylating Drp1, with the resultant SNO-Drp1 participating in mitochondrial fragmentation and synaptic loss.
Additionally, levels of SNO-parkin are significantly upregulated in the brains of human cases of sporadic PD as well as in animal models of PD, further supporting a role for S-nitrosylated parkin in disease pathogenesis (Chung et al., 2004; Yao et al., 2004). These findings are consistent with our hypothesis that aberrant S-nitrosylation can contribute to more common forms of neurodegenerative diseases by mimicking the effects of rare genetic mutations.
S-Nitrosylation of PDI
In neurodegenerative conditions, accumulation of unfolded or misfolded proteins can induce ER stress. In response, cellular defense proteins, such as PDI, can be upregulated to increase chaperone and isomerase activity (Conn et al., 2004; Tanaka et al., 2000). However, this neuroprotective effect can be inhibited by S-nitrosylation, as formation of SNO-PDI compromises its ability to correct protein misfolding (Uehara et al., 2006) (Figure 4). Mitochondrial complex I inhibitors, including pesticides thought to possibly contribute to the pathogenesis of PD, also increase SNO-PDI formation in cell-based experiments (Uehara et al., 2006). Additionally, in animal models of ALS and stroke, iNOS-dependent formation of SNO-PDI increased the aggregation of ubiquitinated proteins (Chen et al., 2012; Chen et al., 2013). Consistent with a pathological role, substantial levels of SNO-PDI are present in human brains manifesting sporadic PD, AD, and ALS (Uehara et al., 2006; Walker et al., 2010). These results suggest that PDI is aberrantly S-nitrosylated during neurodegeneration and that this posttranslational modification may contribute to the progression of the disease, as it compromises chaperone/isomerase activity of PDI and thus aggravates protein misfolding and ER stress. In addition to PDI, S-nitrosylation of another ER chaperone folding protein, glucose-related protein (GRP), may also occur in these diseases (Dall’Agnol et al., 2006). Moreover, rare mutations in genes encoding PDI-family proteins have been suggested to contribute to neurodegenerative conditions, consistent with the hypothesis that rare mutations may be mimicked by environmental factors that induce S-nitrosylation.
S-Nitrosylation of Cdk5
Cdk5 is a serine/threonine kinase that is important in brain development, regulating neuronal differentiation and migration, axon guidance, and synaptic plasticity (Kim et al., 2006; Ohshima et al., 1996). In addition, excessive Cdk5 activity has been implicated in the development of several neurological disorders, including AD (Cheung and Ip, 2012). In the adult brain, Cdk5 is in close proximity to a membrane-bound protein complex containing nNOS/PSD-95/NMDAR. Cdk5 can form a complex with nNOS and become a target of S-nitrosylation under pathological conditions (Zhang et al., 2010; Qu et al., 2011; Qu et al., 2012). Indeed, SNO-Cdk5 is highly expressed in human AD postmortem brains but not in normal control brains (Qu et al., 2011).
NO was found to S-nitrosylate Cdk5 on residues Cys83 and Cys157, and thus activate Cdk5 (Figure 4). This activation leads to increased phosphorylation of substrates such as ataxia telangiectasia mutated (ATM), a proapoptotic protein kinase (Qu et al., 2011). However, S-nitrosylation induced by high (nonphysiological) concentrations of NO can exert the opposite effect on Cdk5 kinase activity, i.e., inhibiting the enzyme (Zhang et al., 2010). Nonetheless, when adding high exogenous levels of NO donors, the pH of the solution can be dramatically lowered, and buffering the change in pH can prevent inhibition of Cdk5 activity after adding high concentrations of exogenous NO donors (Qu et al., 2011). Thus, lowered pH may be at least one reason for the observed discordant effects of NO on Cdk5 activity. SNO-Cdk5 has also been shown to contribute to Aβ-induced synaptic degeneration; Aβ-induced dendritic spine loss in cultured cortical neurons was at least partially inhibited by non-nitrosylatable mutant-Cdk5, and the NOS inhibitor, N-nitro-L-arginine (NNA) also blocked synaptic spine loss (Qu et al., 2011). These results suggest that NO production by NOS and subsequent S-nitrosylation of Cdk5 are important steps in Aβ-induced synaptic loss. Interestingly, Aβ-induced synaptic loss is also in part dependent on NMDAR activity (Shankar et al., 2007; Snyder et al., 2005). NMDAR activity activates nNOS, and NMDARs are aberrantly phosphorylated and activated by Cdk5 under pathological conditions (Wang et al., 2003), apparently representing a positive-feedback loop. Thus, NMDAR-mediated activity may play an important role in SNO-Cdk5-mediated synaptic loss. Consistent with this postulate, NMDA-induced spine loss in cultured cortical neurons was blocked by pretreatment with NNA and partially blocked by the Cdk5 inhibitor, Roscovitine (Qu et al., 2011). Taken together, these results describe a unique regulatory mechanism whereby SNO-Cdk5 mediates, at least in part, Aβ-induced spine loss in AD.
S-Nitrosylation of Drp1
Mitochondria provide energy and play a crucial role in normal neuronal synaptic activity and maintenance of synapses (Li et al., 2004). Aβ-induced oxidative stress can contribute to mitochondrial dysfunction, including excessive fragmentation, which in turn leads to synaptic dysfunction, neuronal injury, and ultimately cell death (Knott et al., 2008; Reddy and Beal, 2008). Drp1 is a GTPase involved in normal mitochondrial fission. In cultured primary cortical neurons, exposure to oligomeric Aβ results in S-nitrosylation of Drp1 at Cys644. Such formation of SNO-Drp1 results in excessive mitochondrial fission, energy compromise, and eventually dendritic spine loss (Cho et al., 2009) (Figure 4). S-Nitrosylation also increases the GTPase activity of both dynamin 1 and 2, close homologues of Drp1 (Kang-Decker et al., 2007; Wang et al., 2006). Importantly, preventing SNO-Drp1 formation with the non-nitrosylatable mutant Drp1(C644A) abrogated Aβ-induced synaptic damage (Cho et al., 2009). In support of a pathophysiological role for SNO-Drp1, Drp1 is apparently S-nitrosylated aberrantly because it is found at high levels in postmortem human AD brains but not in control brains (Cho et al., 2009; Wang et al., 2009), as well as in peripheral blood lymphocytes of AD but not control patients (Wang et al., 2012). Taken together, these findings also suggest that SNO-Drp1 may represent a potential therapeutic target for protecting neurons and their synapses in AD.
Interestingly, S-nitrosylation of Drp1 is likely mediated at least in part by transnitrosylation from SNO-Cdk5, which is present at high levels in human postmortem AD brains. SNO-Cdk5 can function as a nitrosylase enzyme and NO donor by transferring its NO group to Drp1 (Qu et al., 2011). Importantly, these findings indicate that Cdk5 can regulate Aβ-induced spine loss by a mechanism independent of its kinase activity, suggesting that both phosphorylase and nitrosylase activity can be important for the pathological consequences of this dual-function enzyme (Qu et al., 2011) (Figures 2 and 4).
S-Nitrosylation of XIAP and Caspases
Another transnitrosylase system is represented by the XIAP/caspase pair of enzymes. Caspases belong to a cysteine protease family that is important for the execution of apoptotic cell death. NO has been reported to S-nitrosylate caspases-3, -8 and -9 at the active site cysteine, thus inhibiting enzymatic activity and affording neuroprotection (Dimmeler et al., 1997; Melino et al., 1997; Tenneti et al., 1997; Mannick et al., 1999). Interestingly, after exposure to cell death stimuli, mitochondrial Trx2 selectively de-nitrosylates SNO-caspase-3 (Benhar et al., 2008), relieving inhibition of the active site cysteine from -SNO. This form of denitrosylation of caspase-3 requires the oxidoreductase activity of Trx. In contrast, however, after Trx1 is S-nitrosylated at Cys73 (one of its non-active site cysteines), the resulting SNO-Trx1 has been reported to transnitrosylate caspase-3 (Mitchell and Marletta, 2005) (Figure 2). Since S-nitrosylation of Cys73 occurs only when the active site cysteines of Trx1 have been oxidized, Trx1 acts as a nitrosylase only under conditions of oxidative/nitrosative stress (Barglow et al., 2011).
Additionally, S-nitrosylation of the E3 ubiquitin ligase XIAP can occur under pathological conditions (Nakamura et al., 2010; Tsang et al., 2009). Normally, XIAP targets active caspases-3, -7, and -9 for ubiquitination and degradation, thus suppressing caspase-mediated apoptosis and promoting cell survival (Deveraux et al., 1999). XIAP is the most potent endogenous caspase inhibitor among the family of inhibitors of apoptosis proteins (IAPs) (Eckelman et al., 2006). However, this neuroprotective activity can be abrogated during nitrosative stress via S-nitrosylation of XIAP (Nakamura et al., 2010; Tsang et al., 2009). This nitrosylation inhibits the E3 ubiquitin ligase activity of XIAP, which would otherwise lead to the degradation and hence inactivation of caspases (Figure 4). The level of SNO-XIAP is significantly increased in human brains from AD, PD, and Huntington’s disease (HD) patients compared to controls (Nakamura et al., 2010; Tsang et al., 2009), consistent with the notion that SNO-XIAP plays a pathological role in these diseases. A significant increase in SNO-XIAP is also found in both cell-based and animal models of PD (Nakamura et al., 2010; Tsang et al., 2009). Additionally, XIAP can be S-nitrosylated via transnitrosylation from SNO-caspase-3. Since S-nitrosylation of caspases inhibit their apoptotic function, transnitrosylation from SNO-caspase to XIAP acts as an apoptotic switch, both relieving direct inhibition of caspases by S-nitrosylation and inhibiting caspase degradation by XIAP via SNO-XIAP formation (Nakamura et al., 2010).
In addition, NO can indirectly influence the apoptotic caspase cascade via S-nitrosylation of FLICE inhibitory protein (FLIP) and Bcl-2 (Azad et al., 2006; Chanvorachote et al., 2005). FLIP is an antiapoptotic protein that inhibits binding of caspase-8 to the Fas-associated death domain (FADD). Bcl-2 can inhibit mitochondrial-mediated apoptosis via binding to and inhibiting pro-apoptotic proteins such as Bax and Bak. S-Nitrosylation of FLIP and Bcl-2 inhibits their proteasomal degradation, stabilizing both of these anti-apoptotic proteins to enhance their pro-survival function. Thus, under physiological conditions or during the pre-symptomatic stage of neurodegenerative diseases, NO may act as an anti-apoptotic messenger via formation of SNO-FLIP and SNO—Bcl-2.
S-Nitrosylation of GAPDH
GAPDH is an important glycolytic enzyme, but S-nitrosylation of GAPDH at Cys150 initiates an apoptotic cell death cascade, for example, in the MPTP mouse model of PD (Hara et al., 2005; Hara et al., 2006). S-Nitrosylation enhances the binding of GAPDH to Siah1, an E3 ubiquitin ligase, and the SNO-GAPDH/Siah1 protein complex is translocated to the nucleus, where it mediates apoptosis (Figure 5) (Hara et al., 2005). The GAPDH-Siah1 pathway also participates in nuclear translocation of mutant huntingtin protein (mtHtt), mediating, at least in part, neurotoxicity in HD (Bae et al., 2006). In the nucleus, GAPDH stimulates the activity of p300/CBP and activates downstream targets including p53 to precipitate neuronal cell death (Sen et al., 2008). Additionally, Siah1, translocated to the nucleus along with SNO-GAPDH, promotes degradation of SUV39H1, facilitating acetylation of histone H3 and enhanced CREB-mediated neurite outgrowth (Sen and Snyder, 2011).
Figure 5. Schematic Representation of SNO-GAPDH Pathways.

Formation of SNO-GAPDH can trigger multiple signaling pathways leading to neurodegeneration.
Pathway 1: NO generated from nNOS, iNOS, or eNOS can S-nitrosylate GAPDH in the cytosol.
Pathway 2: S-Nitrosylation of GAPDH enhances its interaction with Siah1, a ubiquitin E3 ligase bearing a nuclear localization signal, allowing SNO-GAPDH to translocate into the nucleus.
Pathway 3: In the nucleus, Siah promotes degradation of nuclear proteins such as nuclear corepressor (NcoR).
Pathway 4: SNO-GAPDH also increases p300/CBP activity and induces downstream gene expression.
Pathway 5: SNO-GAPDH serves as a nuclear nitrosylase, producing SNO-SIRT1 and SNO—DNA-PK.
Pathway 6: The SNO-GAPDH/Siah1 complex can also facilitate nuclear translocation of mutant huntingtin (mtHtt) protein.
Pathway 7: Physiological levels of NO can S-nitrosylate GOSPEL, to form a SNO-GOSPEL/GAPDH complex, preventing the association of GAPDH and Siah 1.
Pathway 8: In addition to its nuclear function, SNO-GAPDH regulates iNOS activity as well as ribosomal protein L13a degradation.
Intriguingly, SNO-GAPDH can also transnitrosylate other nuclear proteins, including the deacetylating enzyme sirtuin-1 (SIRT1), histone deacetylase-2 (HDAC2), and DNA-activated protein kinase (DNA-PK) (Kornberg et al., 2010). A similar pathway involving S-nitrosylation of GAPDH and Siah1-mediated nuclear translocation has been reported to occur in a rat model of cerebral ischemia during the early stages of reperfusion injury (Li et al., 2012). A negative regulator of the SNO-GAPDH pathway involves S-nitrosylation of the protein GOSPEL, which has been shown to protect neurons from NMDAR-mediated excitotoxicity. SNO-GOSPEL binds to GAPDH, thereby inhibiting the SNO-GAPDH-mediated apoptosis cascade (Sen et al., 2009). Another inhibitory mechanism of the pro-apoptotic SNO-GAPDH pathway involves transnitrosylation of B23/nucleophosmin by SNO-GAPDH. This reaction results in decreased GAPDH-Siah binding and enhanced B23-Siah interaction, abrogating the ubiquitin E3 ligase activity of Siah1 (Lee et al., 2012).
In addition to its effect in the nucleus, recent studies have suggested that SNO-GAPDH also influences cell function in the cytosol. For instance, GAPDH binds to iNOS in a SNO-dependent manner: GAPDH enhances heme insertion into iNOS to form active iNOS (Chakravarti et al., 2010); S-nitrosylation of GAPDH inhibits heme insertion into iNOS since SNO-GAPDH fails to bind to iNOS. Thus, SNO-GAPDH may mediate a negative feedback loop to suppress iNOS-dependent NO production in glial cells under neurodegenerative conditions. Moreover, in the cytosol, S-nitrosylation of GAPDH inhibits its ability to protect free ribosomal protein L13a from ubiquitin-proteasome degradation. The resulting degradation of L13a causes defective translational control (Jia et al., 2012).
Taken together, these studies support the existence of multiple pathways to cell injury and dysfunction mediated by SNO-GAPDH. In the canonical pathway, formation of SNO-GAPDH results in Siah1-dependent nuclear translocation and neuronal apoptosis, and this cascade may contribute to the pathogenesis of PD, HD, stroke, and other disorders. However, SNO-GAPDH also appears to mediate physiological functions of NO, e.g., via degradation of SUV39H1 or transnitrosylation of HDAC2 (Kornberg et al., 2010; Sen and Snyder, 2011). Thus, additional studies are warranted to elucidate the mechanism determining whether SNO-GAPDH contributes to physiological or pathological pathways involving NO.
S-Nitrosylation of Prx2
The Prx family of antioxidant proteins belongs to a class of enzymes that catalyze reduction of intracellular peroxides by redox reactions (Rhee et al., 2005). Prx2 is the most abundant in mammalian neurons (Sarafian et al., 1999), and the level of Prx2 is increased in a number of neurodegenerative diseases, probably in an attempt to counteract oxidative stress (Krapfenbauer et al., 2003). The antioxidant activity of Prx2 is inhibited by S-nitrosylation of the two critical cysteine residues that are involved in its antioxidant activity (Cys51 and Cys172) (Fang et al., 2007; Romero-Puertas et al., 2007). The result is compromise of the normal redox cycle to detoxify peroxides, involving coordinated action of Prx with the thioredoxin pathway (thioredoxin reductase, thioredoxin and NADPH to reduce -SOH to -SH) and with the sulfiredoxin pathway (reducing -SO2H to -SH).
Consistent with the pathophysiological relevance of this nitrosylation reaction, the level of SNO-Prx2 is significantly elevated in both animal models and human postmortem PD brains (Fang et al., 2007). Since formation of SNO-Prx2 prevents reduction and thus detoxification of peroxides, the neuroprotective activity of Prx2 is compromised by S-nitrosylation, apparently contributing to the pathogenesis of PD.
S-Nitrosylation of DJ-1
The DJ-1 (PARK7) gene has been shown to be deleted or mutated in patients with early-onset autosomal recessive PD and also in rare cases of adult PD (Bonifati et al., 2003). The DJ-1 protein can be S-nitrosylated under pathological conditions, in some sense mimicking the effect of these rare mutations. DJ-1 co-localizes with Hsp70 and CHIP, and may thus function as a redox-sensitive chaperone involved in the oxidative stress response (Batelli et al., 2008). In fact, consistent with an anti-oxidant mechanism of defense, DJ-1 expression increases in response to the nitrosative/oxidative stress induced by the herbicide paraquat (Mitsumoto et al., 2001). Moreover, exogenous expression of DJ-1 protects neurons from the toxicity induced by A53T mutant or overexpression of WT α-synuclein in cell-based models of PD (Batelli et al., 2008). Additionally, downregulation of DJ-1 enhances cell death induced by oxidative stress not only in cell-based models but also in vivo in flies and mice (Kim et al., 2005a; Meulener et al., 2005; Taira et al., 2004). Taken together, these results suggest that DJ-1 plays an important neuroprotective role in response to oxidative stress. DJ-1 possesses three potentially redox-active cysteine residues (Cys46, Cys53, and Cys106). Cys106 is sulfinated (forming a cysteine sulfinic acid, -SO2H) in response to oxidative stress and may thus detoxify reactive oxygen species to offer neuroprotection (Blackinton et al., 2009), while Cys46 and Cys53 have been reported to be S-nitrosylated (Ito et al., 2006). However, these sites of nitrosylation remain contentious and further work will be needed to determine the exact site(s) and effect of S-nitrosylation of DJ-1. Importantly, whether S-nitrosylation regulates the neuroprotective activity of DJ-1 remains an open question.
S-Nitrosylation of Matrix Metalloproteinases
In focal cerebral ischemia, specific subtypes of metalloproteinases (MMPs), including MMP-2 and -9, are acutely activated. These MMPs degrade components of the extracellular matrix, leading to deleterious consequences in the affected region of the brain. As a mechanism for MMP involvement, NO, generated by the excitotoxic and neuroinflammatory consequences of a stroke, was shown to directly activate MMP-9 by S-nitrosylation of the so-called ‘cysteine switch,’ whose oxidation leads to enzyme activation. The resulting acute, excessive activity of MMPs early on in the evolution of ischemia/reperfusion contributes to cell damage and death, probably via anoikis in which separation of cells from the extracellular matrix triggers an apoptotic form of cell death (Gu et al., 2002; Manabe et al., 2005).
Mechanistically, the catalytic site of the latent (or proform) of MMP-9 contains a Zn2+ atom that is coordinated by three His and one Cys residues (constituting the cysteine switch), which prevent substrates in the extracellular matrix from reaching the active site. Gu et al. (2002) discovered that one mechanism of activation of MMPs involves an NO group reacting with the critical cysteine residue in this region to disrupt the Cys-Zn2+ interaction, exposing the catalytic Zn2+ in the active site to substrate. Consistent with a role for NO in MMP activation, MMP-9 colocalizes with nNOS during cerebral ischemia and with iNOS in migrating cells (Gu et al., 2002; Harris et al., 2008). Following S-nitrosylation, the critical Cys residue in the cysteine switch of MMPs can also undergo further oxidation by ROS to form sulfinic (-SO2H) or sulfonic (-SO3H) acid derivatives, the latter resulting in apparently irreversible activation of the enzyme. Thus, S-nitrosylation and subsequent oxidation can result in pathological activation of MMPs, contributing to neuronal injury and death during stroke and possibly other neurological disorders.
S-Nitrosylation of Akt, PTEN and Other Protein Tyrosine Phosphatases
Phosphatase and tensin homolog (PTEN) was initially identified as a tumor suppressor that antagonizes the oncogenic phosphatidylinositol-3′-kinase (PI3K)/protein kinase B (PKB or Akt) pathway by dephosphorylating phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Accordingly, in the brain, inactivation or deletion of PTEN may contribute to the pathogenesis of glioblastoma. However, recent evidence suggests that downregulation of PTEN can also play a neuroprotective role in neurodevelopmental and neurodegenerative disorders. For example, cells heterozygous for PTEN manifest resistance to oxidative stress compared to WT cells (Li et al., 2002). Additionally, S-nitrosylation of PTEN at Cys83 inhibits PTEN activity and hence exerts a neuroprotective effect via the Akt pathway (Kwak et al., 2010; Numajiri et al., 2011). Moreover, S-nitrosylated PTEN can be selectively ubiquitinated by NEDD4-1 and thus degraded via the ubiquitin-proteasome pathway (Kwak et al., 2010). Suggesting that these findings may be of pathophysiological significance, the formation of SNO-PTEN was discovered in postmortem human AD brain as well as in ischemic mouse brain (Kwak et al., 2010; Numajiri et al., 2011; Pei et al., 2009). These nitrosylation events may represent a negative feedback mechanism during nitrosative stress whereby NO can curb excessive cell death under specific circumstances by activating the Akt neuroprotective pathway. However, S-nitrosylation of Akt can directly inhibit its kinase activity to effectively “trump” the effect of SNO-PTEN formation (Yasukawa et al., 2005; Numajiri et al., 2011; Banerjee et al., 2012). Recently, these effects of S-nitrosylation, which seemingly produce opposite effects on the PTEN/PI3K/Akt pathway, were clarified by the demonstration that low (and thus neuroprotective) concentrations of NO preferentially induce SNO-PTEN formation, while higher (neurotoxic) concentrations of NO also S-nitrosylate Akt to directly inhibit its neuroprotective activity (Numajiri et al., 2011).
In addition to PTEN, another member of the protein tyrosine phosphatase (PTP) family, Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) was identified as an important substrate for S-nitrosylation during ischemic stroke (Shi et al., 2013). S-Nitrosylation of SHP-2 occurs at the catalytic cysteine and thus inhibits its phosphatase activity. Formation of SNO—SHP-2 therefore blocks downstream activation of the neuroprotective extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, thereby enhancing susceptibility to excitotoxicity. This finding is in agreement with recent studies demonstrating an inhibitory effect of S-nitrosylation on other PTPs in non-neuronal cells (Barrett et al., 2005; Chen et al., 2008; Mikkelsen and Wardman, 2003). Additionally, ROS can induce sulfination (-SO2H) or sulfonation (-SO3H) of the active site cysteine of PTPs, thus inhibiting their activity (Tonks, 2006). Since S-nitrosylation of other proteins can facilitate further oxidation of their redox-active cysteine residues to sulfinic or sulfonic acid derivatives (Gu et al., 2002; Hara et al., 2005; Uehara et al., 2006), it is conceivable that the S-nitrosylated cysteine residue of SHP-2 or other PTPs can also initiate further oxidation in this manner. Nonetheless, for PTEN, S-nitrosylation by NO, unlike oxidation by H2O2, occurs at an allosteric cysteine residue rather than at the active site cysteine (Lee et al., 2002; Numajiri et al., 2011).
S-Nitrosylation of Microtubule Associated Protein 1B (MAP1B)
Axonal retraction and degeneration frequently occur in neurodegenerative diseases, including AD, PD, and multiple sclerosis. NO has been suggested to participate in this process by S-nitrosylating microtubule-associated protein 1B (MAP1B) (Stroissnigg et al., 2007). MAP1B is highly expressed in neurons and binds to microtubules to actively extend axon length. Stroissnigg et al. (2007) demonstrated that MAP1B interacts with nNOS, facilitating S-nitrosylation of human MAP1B at Cys2464 in the protein’s light chain. S-Nitrosylation of MAP1B causes a conformational change in the protein that increases its microtubule binding affinity. Enhanced binding affinity results in inhibition of the molecular motor dynein, thus contributing to axonal retraction in response to NO. Recently, S-nitrosylation of MAP1B in mitochondria was also shown to promote its own degradation, mediated by the mitochondrial ubiquitin E3 ligase MITOL (Yonashiro et al., 2012). In that study, MITOL-dependent degradation of mitochondrial SNO-MAP1B protected neurons from NO-induced mitochondrial dysfunction and subsequent cell death. Taken together, these results suggest that SNO-MAP1B can mediate axonal retraction, but, as a negative-feedback mechanism, may also serve as a ubiquitination signal for its degradation by MITOL; the latter effect results in protection of neurons from mitochondrially-mediated cell death induced by NO. Further work will be needed to determine how these effects may be involved in specific neurodegenerative disorders.
S-Nitrosylation of COX-2
Cyclooxygenase (COX-2) converts arachidonic acid (AA) to prostaglandin (PG)H2, a precursor of many other biologically active PGs. During inflammation, iNOS has been shown to interact with, S-nitrosylate, and thus enhance the activity of COX-2 (Kim et al., 2005b). Similarly, iNOS appears to bind to, S-nitrosylate, and increase the activity of phospholipase A2 (PLA2), an AA-generating enzyme (Xu et al., 2008). Thus, NO can promote PG generation via S-nitrosylation of the two key enzymes (PLA2 and COX-2) involved in PG biosynthesis. Moreover, COX-2 also binds to nNOS, and S-nitrosylation via nNOS activates COX-2-mediated NMDAR neurotoxicity (Tian et al., 2008). Thus, S-nitrosylation of COX-2 and PLA2 may contribute to the neuroinflammatory component of neurodegeneration.
S-Nitrosylation of Insulin-Degrading Enzyme (IDE)
IDE is a zinc metalloprotease that cleaves a number of biologically active peptides, including both insulin and Aβ. Exposure to NO donors such as S-nitrosoglutathione (GSNO) can decrease IDE activity via S-nitrosylation of multiple cysteine residues (Cordes et al., 2009; Malito et al., 2008). Hence, S-nitrosylation of IDE may result in increased levels of both insulin and Aβ, as observed in Metabolic Syndrome/Type 2 Diabetes mellitus and AD, respectively. Mechanistically, the NO group initially reacts with IDE at Cys819, located near the catalytic site of the enzyme. This reaction perturbs local structure, facilitating S-nitrosylation of a second cysteine residue (Cys110), which is found near the zinc-binding center (Ralat et al., 2009). The resulting S-nitrosylation of both Cys819 and Cys110 results in complete inactivation of IDE, at least in vitro. Alternatively, S-nitrosylation at Cys789 and Cys966 has been reported to trigger not only inhibition of IDE but also its oligomerization. Finally, S-nitrosylation of Cys178 reverses the inhibition of IDE caused by S-nitrosylation of the aforementioned four cysteine residues (Ralat et al., 2009). Although further work is needed to elucidate the effects of these reactions in vivo, it appears that S-nitrosylation of IDE could contribute to AD pathophysiology by decreasing degradation of Aβ.
S-Nitrosylation of JNK1 and IKKβ in Autophagy
Autophagic/lysosomal degradation of misfolded proteins or damaged organelles is an important cellular response to stress that affects the pathogenesis of neurodegenerative diseases. Well-characterized cell signaling pathways regulating the autophagic process include the JNK/Bcl-2/Beclin 1 and IKKβ/AMPK/mTORC 1 cascades. Aberrant S-nitrosylation of JNK1 and IKKβ inhibits autophagy in models of HD (Sarkar et al., 2011). Specifically, S-nitrosylation of JNK1 inhibits its activity, disrupting Beclin 1 complex formation, which is required for the development of the autophagosome. Additionally, inhibition of IKKβ activity by S-nitrosylation leads to decreased phosphorylation of AMPK, resulting in activation of the autophagy inhibitor, mTORC1. Conversely, and consistent with these findings, downregulation of endogenous NO production enhances autophagy flux. Additional SNO-proteins have also been implicated in the control of autophagy, and further work will be necessary to determine these pathways.
S-Nitrosylation of ApoE
The apolipoprotein E (ApoE) gene encodes a class of lipoproteins that plays a critical role in CNS cholesterol homeostasis. ApoE4 represents a major genetic risk factor for developing late-onset AD (Bertram et al., 2010). The ApoE isoforms can bind to nNOS and both ApoE2/E3 can be S-nitrosylated in human hippocampal lysates (Abrams et al., 2011). In silico analysis of the ApoE 3D structure further suggested that S-nitrosylation may result in conformational alteration of the protein, lowering its binding affinity for low-density lipoprotein (LDL) receptors (Abrams et al., 2011), raising the interesting question of whether S-nitrosylation of ApoE might contribute to the onset or progression of AD by disrupting normal lipid metabolism.
Protein S-Nitrosylation Regulates Diverse Aspects of Neuronal Function
Although less well characterized, a large number of additional SNO-proteins have also been linked to modulation of normal or pathological brain function. During the neurodegenerative process, these SNO-proteins may impact a wide variety of cellular mechanisms (Figure 6). Below, we discuss possible roles for several of these SNO-proteins under both physiological and pathophysiological conditions in the brain.
Figure 6. Physiological and Pathophysiological Protein S-Nitrosylation Plays an Important Role in Diverse Aspects of Cell Function in the CNS.
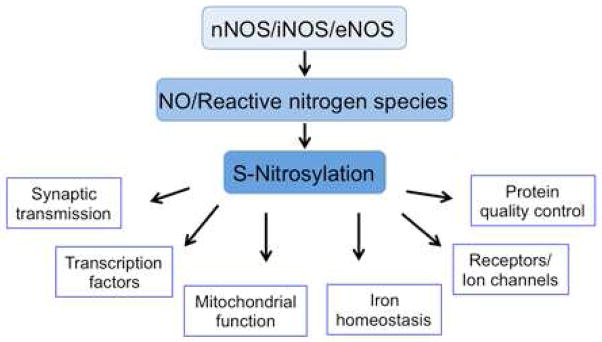
Production of both normal and pathological levels of NO results in S-nitrosylation of a number of proteins implicated in synaptic transmission, receptors/ion channels, transcription factors, iron homeostasis, protein quality control, and mitochondrial function.
S-Nitrosylation of Synaptic Proteins
In neurons, NO is typically generated by nNOS coupled to NMDARs (Bredt et al., 1991; Sattler et al., 1999). This process in turn leads to S-nitrosylation of various proteins in the cell. One such protein is N-ethylmaleimide sensitive factor (NSF); its S-nitrosylation leads to increased surface expression of AMPA receptors (AMPARs) (Huang et al., 2005; Matsushita et al., 2003). Surface expression of AMPARs can also be upregulated by S-nitrosylation of the regulatory subunit stargazin (Selvakumar et al., 2009). In addition, S-nitrosylation of the AMPAR subunit GluA1 facilitates its phosphorylation, producing an increase in single-channel conductance, and enhancing endocytosis (Selvakumar et al., 2013). Additionally, PSD-95 can be S-nitrosylated, which decreases its synaptic expression as well as the expression of proteins that are associated with it, including NMDARs and AMPARs (Ho et al., 2011). Under pathological conditions, NMDAR-mediated excitotoxicity (which appears to be mediated predominantly by extrasynaptic NMDARs) can cause massive Ca2+ influx that subsequently leads to the production of excessive amounts of NO (Dawson et al., 1991; Lipton et al., 1993; Bonfoco et al., 1995; Hardingham and Bading, 2010). As a negative-feedback mechanism, NMDARs are S-nitrosylated by NO, and this S-nitrosylation can downregulate excessive receptor activity (Lei et al., 1992; Lipton et al., 1993; Choi et al., 2000).
Additionally, NO may act as a retrograde messenger to presynaptic sites, leading, for example, to S-nitrosylation of serine racemase (SR); this nitrosylation reaction decreases the generation of D-serine, a co-agonist at NMDARs (Mustafa et al., 2007). Furthermore, S-nitrosylation of syntaxin 1 can possibly prevent excessive release of glutamate (Palmer et al., 2008); this coupled with the finding that SNAP-25 can also be S-nitrosylated (Hess et al., 1993) may account for the observation that NO can decrease evoked neurotransmitter release while increasing spontaneous release (Pan et al., 1996). Effects on these or other members of the core complex of synaptic proteins involved in docking and fusion could contribute to a phenotype of decreased evoked and increased spontaneous release. Therefore, S-nitrosylation of synaptic proteins potentially mediates both physiological and neuroprotective aspects of NO signaling.
S-Nitrosylation of Other Membrane Proteins
NO can affect many signaling pathways by S-nitrosylating membrane receptors and ion channels. In addition to NMDARs, a number of SNO-regulated membrane proteins that potentially affect brain function have been discovered, including the ryanodine receptor (RyR) (Eu et al., 2000), transient receptor potential (TRP) channels (Yoshida et al., 2006), Na+ channels (Renganathan et al., 2002), K+ channels (Nunez et al., 2006; Asada et al., 2009), and voltage-gated Ca2+ channels (Chen et al., 2002). The effects of S-nitrosylation on NMDARs and RyRs have been particularly well characterized. There are five sites of nitrosylation on NMDARs (Choi et al., 2000; Choi et al., 2001; Lipton et al., 2003). The relative hypoxia of the brain enhances S-nitrosylation of the NMDAR by a unique mechanism involving an “NO-reactive oxygen sensor motif” whose determinants include Cys744 and Cys798 of the GluN1 (NR1) subunit (Takahashi et al., 2007). Free thiols at these two cysteine residues are involved in redox reactions that sensitize other NMDAR cysteines to S-nitrosylation and result in receptor inhibition. Interestingly, however, S-nitrosylation of Cys744/Cys798 themselves has little effect on NMDAR activity (Figure 7). Solving the crystal structure of the ligand-binding domain of NR1 under oxidizing conditions revealed a flexible disulfide bond (Cys744-Cys798), which may account for its susceptibility to reduction and subsequent reaction with NO that is observed with biochemical techniques in the presence of NO. In accord with these findings, when crystals of NR1 were formed under relatively hypoxic conditions in the presence of NO, electron density consistent with S-nitrosylation in the region of Cys744/Cys798 was observed (Takahashi et al., 2007). These thiols appear to be nitrosylated preferentially during increasing (pathological) hypoxia, thus preventing excessive activity associated with cytotoxicity while avoiding blockade of physiologically active NMDARs.
Figure 7. Hypoxia Sensitizes Thiol to S-Nitrosylation-Mediated Inhibition of the NMDAR.
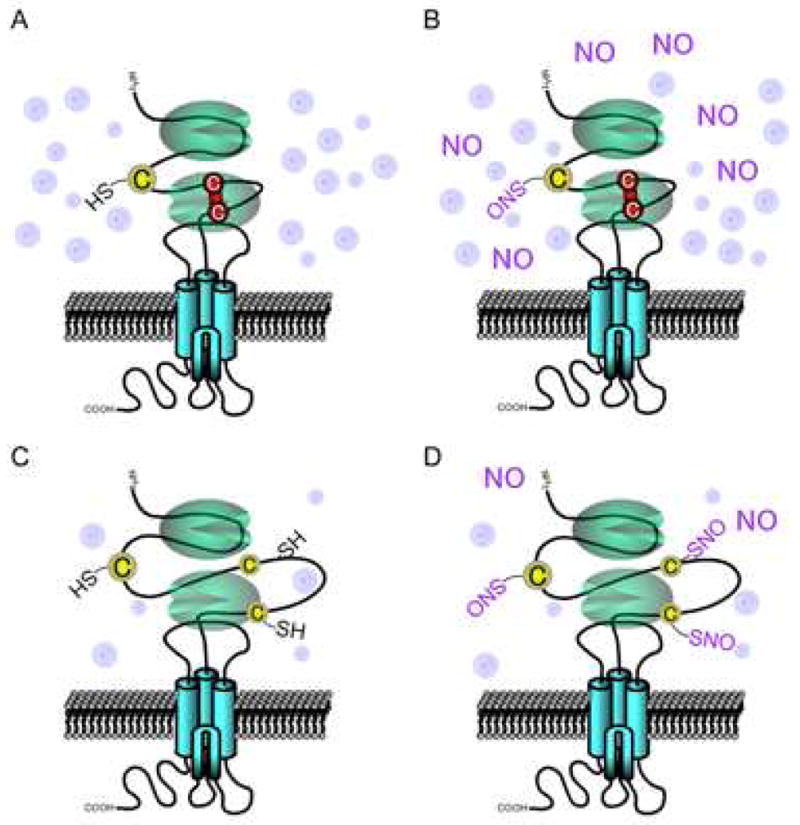
(A) Ambient O2 levels are significantly higher than levels in living brain tissue or during hypoxic insults, favoring disulfide formation between cysteine residues Cys744 and Cys798 on the GluN1 (NR1) subunit of the NMDAR (red line).
(B) S-Nitrosylation can occur on various NMDAR subunits (designated GluNs) at two or three different sites on each subunit. However, in the presence of disulfide, NO cannot be transferred to cysteine residues since no free thiol exists for reaction. S-Nitrosylation can occur at free thiols on the GluN2A (NR2A) subunit at Cys399 (cysteine residue at left) as well as on NR2A Cys87 and Cys320 (and homologous cysteine residues on GluN2B (NR2B, not shown)), but only in the presence of high concentrations of NO donor.
(C) Under physiological or even more hypoxic (pathological) conditions, the relatively reducing state favors free thiol groups on the NMDAR over disulfide bond formation.
(D) Under relatively hypoxic conditions, the free thiol groups are more readily available to react with NO to form S-nitrosothiol, and even low concentrations of an NO donor can effect this reaction. The exact reaction route that leads from S-nitrosylation to disulfide formation of vicinal thiols [NR1(C744,C798)] remains unknown, although it is likely that in this scenario, only one of these thiols is S-nitrosylated. During relative hypoxia, our hypothesis is that NO is more likely to react with both Cys744 and Cys798 to S-nitrosylate their thiol groups, and under these conditions, disulfide formation would be blocked. Such dual S-nitrosylation of Cys744/Cys798 would then lead to the increased NO sensitivity that we have observed experimentally at other NMDAR sites, such as Cys399, Cys87 and Cys320 on NR2A. Therefore, NO itself may not be regulatory at NR1(C744,C798), but the redox status of these cysteine residues would exert an allosteric influence on S-nitrosylation of other thiol groups on the NMDAR (i.e., Cys399 in the loose linker region of NR2A, and Cys87 and Cys320 on the amino terminal domain of NR2A (or the homologous cysteine residues of NR2B, not shown). Adapted from Takahashi et al. (2007).
The RyR1-type ryanodine receptor mediates Ca2+ release from the ER (or sarcoplasmic reticulum in skeletal muscle cells) into the cytosol, and S-nitrosylation of RyR1 at Cys3635 has been shown to increase channel opening in muscle cells. Although RyR1 is expressed in brain cells, the role of SNO-RyR1 in neuronal cells has remained enigmatic until recently when, in cortical neurons, S-nitrosylation of RyR1 was shown to contribute to NO-induced Ca2+ release and neuronal cell death (Kakizawa et al., 2012).
Additionally, NO appears to regulate G protein-coupled receptor (GPCR) signaling machinery via S-nitrosylation of GPCR-associated proteins such as G-protein receptor kinase 2 (GRK2) (Whalen et al., 2007) and β-arrestin (Ozawa et al., 2008). As exemplified for β-adrenergic receptors, S-nitrosylation of GRK2 prevents receptor internalization, while SNO—β-arrestin displays increased binding to clathrin heavy chain/β-adaptin to accelerate receptor internalization. As GPCR signaling is implicated in many aspects of neuronal function, these studies suggest that SNO-dependent regulation of these proteins may play a role in neurodegenerative conditions, although its exact involvement is as yet to be determined.
S-Nitrosylation of Transcriptional Regulators
S-Nitrosylation can also regulate gene transcription by targeting transcription factors and their regulatory proteins, including nuclear factor-κB (NF-κB)/IKKβ (Marshall and Stamler, 2001), hypoxia-inducible factor (HIF) (Li et al., 2007; Sumbayev et al., 2003; Yasinska and Sumbayev, 2003), c-Jun N-terminal kinase (JNK)/c-Jun (Park et al., 2000), human homolog of mouse double minute-2 (HDM2)/p53 (Schonhoff et al., 2002), Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor erythroid 2-related factor 2 (Nrf2) (Fourquet et al., 2010; Um et al., 2011), nuclear hormone receptors such as the estrogen receptor (Garban et al., 2005), and histone deacetylase 2 (HDAC2) (Nott et al., 2008). Additionally, NO can indirectly regulate the activity of transcriptional factors via S-nitrosylation of a number of upstream effectors, including Ras, Src, and ASK1 (Akhand et al., 1999; Lander et al., 1995; Park et al., 2004; Hess et al., 2005). Despite the well-characterized effects of S-nitrosylation on many transcriptional activities, the pathophysiological significance of these redox modifications on neurodegenerative diseases is only now being elucidated.
S-Nitrosylation of Iron Homeostasis Regulatory Proteins
Increasing evidence suggests that progressive accumulation of iron in the aged brain may underlie a number of neurodegenerative disorders due to induction of oxidative stress (Zecca et al., 2004). Iron homeostasis is tightly regulated by a series of carriers, enzymes, and associated proteins, including transferrin, transferrin receptor (TfR), ferritin, and iron-regulatory proteins (IRPs). IRPs interact with the mRNA of the TfR (which is involved in iron uptake) and ferritin (an iron storage protein), stabilizing TfR mRNA and blocking ferritin mRNA translation. Accordingly, IRP2 knockout mice manifest significant accumulation of iron deposits as well as profound movement disorders and neurodegeneration (LaVaute et al., 2001). Interestingly, S-nitrosylation of IRP2 at Cys178 reportedly promotes UPS-dependent degradation of IRP2, leading to significant accumulation of iron with the iron storage protein, ferritin (Kim et al., 2004). These findings suggest that SNO-IRP2 formation can result in malfunction of the iron homeostasis pathway, and is implicated in nitrosative stress-mediated neurotoxicity.
Additionally, Dexras1, a small GTPase that is activated by S-nitrosylation, affects NO-regulated iron homeostasis. NMDAR stimulation of nNOS can produce S-nitrosylation of Dexras on Cys11 (Fang et al., 2000; Jaffrey et al., 2002). Dexras1 forms a complex with the divalent metal transporter 1 (DMT1), and SNO activation of Dexras1 augments iron uptake via DMT1 (Cheah et al., 2006). Importantly, selective iron chelation ameliorates NMDA-nNOS neurotoxicity, implying that iron uptake in response to NMDAR-nNOS activation can play an important pathophysiological role.
S-Nitrosylation Protein Quality Control Machinery
Abnormal accumulation of toxic misfolded proteins, such as oligomeric Aβ and α-synuclein, represents a characteristic feature of many neurodegenerative diseases. It is generally believed that malfunction in protein quality control can contribute to the appearance of misfolded proteins. The quality control system encompasses multiple cellular elements, including molecular chaperones, the UPS, and the autophagy/lysosomal pathway. As discussed above, S-nitrosylation disrupts both autophagic degradation of misfolded proteins and the protein refolding/chaperone activity of PDI and GRP in the ER. In addition, S-nitrosylation of HSP90 inhibits its ATPase activity and chaperone function in the cytoplasm (Martinez-Ruiz et al., 2005).
Concerning components of the UPS, S-nitrosylation has been shown to cause dysfunctional ubiquitin E3 ligase activity in parkin, XIAP, and pVHL (Yao et al., 2004; Chung et al., 2004; Nakamura et al., 2010; Tsui et al., 2011). Additionally, S-nitrosylation can directly inhibit 26S proteasome activity via targeting 10 cysteine residues in the 20S catalytic core (Kapadia et al., 2009). Moreover, S-nitrosylation and further oxidation of an N-terminally located cysteine residue appear to control the UPS-dependent N-end rule (Hu et al., 2005). The N-end rule determines the in vivo stability of a protein depending on the identity of its N-terminal amino acid residue(s); in this manner, Asn, Gln, or Cys at the N-terminus destabilizes the protein in mammalian cells (Sriram et al., 2011). Hu et al. (2005) demonstrated that NO targets the N-terminal cysteine when a basic residue is located next to the cysteine (reminiscent of the prior report of an S-nitrosylation motif in peptides (Stamler et al., 1997)), and subsequent oxidation of the cysteine residue to a sulfonic acid derivative can trigger its arginylation, which represents the destabilizing signal. The UPS then recognizes this signal and degrades the protein. Thus, nitrosative/oxidative stress can cause specific degradation of proteins bearing an N-terminal “cysteine-basic residue motif” via S-nitrosylation and further oxidation.
S-Nitrosylation and Mitochondrial Dysfunction
Aberrant redox reactions triggered by excessive amounts of NO can result in mitochondrial dysfunction, although basal levels of NO serve as a physiological regulator of mitochondrial activity. As described above, arguably the best-characterized SNO-protein that affects mitochondrial function during neurodegeneration is SNO-Drp1 (Cho et al., 2009). Additionally, ubiquitination of mitochondrial proteins by parkin is implicated in mitophagy (Youle and van der Bliek, 2012). As S-nitrosylation of parkin regulates its ubiquitin E3 ligase activity, it is interesting to hypothesize that SNO-parkin might affect mitophagy during the degenerative process.
The main function of mitochondria entails ATP synthesis via oxidative phosphorylation (OX/PHOS). NO has been reported to negatively affect mitochondrial respiratory activity in part through cysteine S-nitrosylation of complexes I and IV (Cleeter et al., 1994; Clementi et al., 1998; Zhang et al., 2005; Dahm et al., 2006; Burwell et al., 2006). It should also be noted that NO directly reacts with iron in the iron-sulfur center of mitochondrial complexes I and II, decreasing both the transfer of electrons and ATP production (Drapier and Hibbs, 1988). Additionally, S-nitrosylation may decrease F1 ATPase activity to further disrupt OX/PHOS, as demonstrated in cardiomyocytes (Sun et al., 2007). However, direct evidence demonstrating the link between SNO-mediated inhibition of mitochondrial complexes and neurodegeneration is still lacking, although this mechanism has been suggested for diseases such as PD (Brown and Borutaite, 2004). In fact, in animal models of PD, administration of complex I inhibitors, such as MPTP or rotenone, recapitulates many features of sporadic PD, including degeneration of dopaminergic neurons, overproduction and aggregation of α-synuclein, accumulation of Lewy body-like intraneuronal inclusions, and impairment of behavioral function (Beal, 2001; Betarbet et al., 2000). Inhibition of mitochondrial complexes by S-nitrosylation may potentially generate excessive ROS/RNS, providing a possible positive feedback loop to accelerate neuronal injury (Beal, 2001; Betarbet et al., 2000; Chung et al., 2004; Uehara et al., 2006; Yao et al., 2004). Further studies are needed to determine whether regulation of other mitochondrial metabolic pathways by S-nitrosylation contributes to neurodegenerative conditions.
Conclusions and Perspectives
Protein S-nitrosylation plays an important role in the pathogenesis of a number of neurodegenerative disorders, including AD, PD, HD, and ALS. Aberrant protein S-nitrosylation reactions can occur in response to excessive nitrosative/oxidative stress and may contribute to neurodegeneration via disruption of a number of pathways. To date, aberrant protein S-nitrosylation has been identified on an increasing number of targets, and we expect that many more S-nitrosylated proteins will be found to play a role in neurodegenerative diseases. There is the potential for hundreds of such proteins to be identified by improved SNO-proteome technology coupled with bioinformatics (Forrester et al., 2009; Hao et al., 2006; Paige et al., 2008; Xue et al., 2010). Elucidation of the aberrant SNO-proteome in the brain should expedite our understanding of the pathological role of S-nitrosylation in these neurodegenerative diseases. Better characterization of the aberrant SNO-proteome will also facilitate identification of potential therapeutic targets for drug discovery. Interestingly, although multiple targets for S-nitrosylation appear to be present in each neurodegenerative condition, intervention that affects a single nitrosylation event can ameliorate the condition, at least in animal models (Gu et al., 2002; Uehara et al., 2006; Hara et al., 2006; Cho et al., 2009). This finding suggests that there may be redundancy in redox-mediated pathways to injury, but interfering with even a single pathway can offer therapeutic benefit – raising hope for the development of future therapies.
Acknowledgments
The authors’ work related to this research area was supported in part by funding from the Alzheimer’s Association (to T.N.), grants from the Michael J. Fox Foundation (to T.N. and S.A.L.), and NIH grants R21 NS080799, P01 HD29587, P01 ES01673, and P30 NS076411 (to S.A.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams AJ, Farooq A, Wang G. S-Nitrosylation of ApoE in Alzheimer’s disease. Biochemistry. 2011;50:3405–3407. doi: 10.1021/bi200266v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhand AA, Pu M, Senga T, Kato M, Suzuki H, Miyata T, Hamaguchi M, Nakashima I. Nitric oxide controls src kinase activity through a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J Biol Chem. 1999;274:25821–25826. doi: 10.1074/jbc.274.36.25821. [DOI] [PubMed] [Google Scholar]
- Asada A, Yamamoto N, Gohda M, Saito T, Hayashi N, Hisanaga S-i. Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cycline-dependent kinase 5 complexes. J Neurochem. 2008;106:1325–1336. doi: 10.1111/j.1471-4159.2008.05500.x. [DOI] [PubMed] [Google Scholar]
- Asada K, Kurokawa J, Furukawa T. Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J Biol Chem. 2009;284:6014–6020. doi: 10.1074/jbc.M807158200. [DOI] [PubMed] [Google Scholar]
- Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J Biol Chem. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- Bae BI, Hara MR, Cascio MB, Wellington CL, Hayden MR, Ross CA, Ha HC, Li XJ, Snyder SH, Sawa A. Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc Natl Acad Sci USA. 2006;103:3405–3409. doi: 10.1073/pnas.0511316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Liao L, Russo R, Nakamura T, McKercher SR, Okamoto S, Haun F, Nikzad R, Zaidi R, Holland E, Eroshkin A, Yates JR, 3rd, Lipton SA. Isobaric tagging-based quantification by mass spectrometry of differentially regulated proteins in synaptosomes of HIV/gp120 transgenic mice: implications for HIV-associated neurodegeneration. Exp Neurol. 2012;236:298–306. doi: 10.1016/j.expneurol.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barglow KT, Knutson CG, Wishnok JS, Tannenbaum SR, Marletta MA. Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc Natl Acad Sci USA. 2011;108:E600–606. doi: 10.1073/pnas.1110736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Black SM, Todor H, Schmidt-Ullrich RK, Dawson KS, Mikkelsen RB. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005;280:14453–14461. doi: 10.1074/jbc.M411523200. [DOI] [PubMed] [Google Scholar]
- Batelli S, Albani D, Rametta R, Polito L, Prato F, Pesaresi M, Negro A, Forloni G. DJ-1 modulates α-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson’s disease and involvement of HSP70. PLoS ONE. 2008;3:e1884. doi: 10.1371/journal.pone.0001884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Beal MF. Experimental models of Parkinson’s disease. Nat Rev Neurosci. 2001;2:325–334. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, Cookson MR, Wilson MA. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–6485. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V. Autosomal recessive parkinsonism. Parkinsonism Relat Disord. 2012;18(Supplement 1):S4–S6. doi: 10.1016/S1353-8020(11)70004-9. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MCJ, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ Domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide peroxynitrite and S-nitrosothiols, Biochim. Biophys, Acta. 2004;1658:44–49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti R, Aulak KS, Fox PL, Stuehr DJ. GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc Natl Acad Sci USA. 2010;107:18004–18009. doi: 10.1073/pnas.1008133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanvorachote P, Nimmannit U, Wang L, Stehlik C, Lu B, Azad N, Rojanasakul Y. Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degradation of FLICE inhibitory protein. J Biol Chem. 2005;280:42044–42050. doi: 10.1074/jbc.M510080200. [DOI] [PubMed] [Google Scholar]
- Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE, 3rd, Papadopoulos V, Snyder SH. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51:431–440. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Daggett H, De Waard M, Heinemann SH, Hoshi T. Nitric oxide augments voltage-gated P/Q-type Ca2+ channels constituting a putative positive feedback loop. Free Radic Biol Med. 2002;32:638–649. doi: 10.1016/s0891-5849(02)00748-7. [DOI] [PubMed] [Google Scholar]
- Chen X, Guan T, Li C, Shang H, Cui L, Li XM, Kong J. SOD1 aggregation in astrocytes following ischemia/reperfusion injury: a role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI) J Neuroinflammation. 2012;9:237. doi: 10.1186/1742-2094-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li C, Guan T, Shang H, Cui L, Li XM, Kong J. S-Nitrosylated protein disulphide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J Neurochem. 2013;124:45–58. doi: 10.1111/jnc.12046. [DOI] [PubMed] [Google Scholar]
- Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, Khoo KH, Meng TC. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem. 2008;283:35265–35272. doi: 10.1074/jbc.M805287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., II PINK1-phosphorylated mitofusin 2 Is a parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22:169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-Nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Chen HSV, Lipton SA. Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the NMDA receptor. J Neurosci. 2001;21:392–400. doi: 10.1523/JNEUROSCI.21-02-00392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn KJ, Gao W, McKee A, Lan MS, Ullman MD, Eisenhauer PB. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004;1022:164–172. doi: 10.1016/j.brainres.2004.07.026. [DOI] [PubMed] [Google Scholar]
- Cordes CM, Bennett RG, Siford GL, Hamel FG. Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation. Biochem Pharmacol. 2009;77:1064–1073. doi: 10.1016/j.bcp.2008.12.006. [DOI] [PubMed] [Google Scholar]
- da Costa CA, Sunyach C, Giaime E, West A, Corti O, Brice A, Safe S, Abou-Sleiman PM, Wood NW, Takahashi H, Goldberg MS, Shen J, Checler F. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat Cell Biol. 2009;11:1370–1375. doi: 10.1038/ncb1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- Dall’Agnol M, Bernstein C, Bernstein H, Garewal H, Payne CM. Identification of S-nitrosylated proteins after chronic exposure of colon epithelial cells to deoxycholate. Proteomics. 2006;6:1654–1662. doi: 10.1002/pmic.200500240. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1β-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci USA. 2010;107:16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapier JC, Hibbs JB., Jr Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in l-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macrophage effector cells. J Immunol. 1988;140:2829–2838. [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Fang J, Nakamura T, Cho DH, Gu Z, Lipton SA. S-Nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104:18742–18747. doi: 10.1073/pnas.0705904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Schmidt HH, Pollock JS, Sheng H, Mitchell JA, Warner TD, Nakane M, Murad F. Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochem Pharmacol. 1991;42:1849–1857. doi: 10.1016/0006-2952(91)90581-o. [DOI] [PubMed] [Google Scholar]
- Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem. 2010;285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garban HJ, Marquez-Garban DC, Pietras RJ, Ignarro LJ. Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc Natl Acad Sci USA. 2005;102:2632–2636. doi: 10.1073/pnas.0409854102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-Nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y. S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci USA. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-Nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314:1765–1776. doi: 10.1016/j.yexcr.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hess DT, Patterson SI, Smith DS, Skene JH. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature. 1993;366:562–265. doi: 10.1038/366562a0. [DOI] [PubMed] [Google Scholar]
- Ho GP, Selvakumar B, Mukai J, Hester LD, Wang Y, Gogos JA, Snyder SH. S-Nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron. 2011;71:131–141. doi: 10.1016/j.neuron.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu RG, Sheng J, Qi X, Xu Z, Takahashi TT, Varshavsky A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature. 2005;437:981–986. doi: 10.1038/nature04027. [DOI] [PubMed] [Google Scholar]
- Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-Nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Ito G, Ariga H, Nakagawa Y, Iwatsubo T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem Biophys Res Commun. 2006;339:667–672. doi: 10.1016/j.bbrc.2005.11.058. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Fang M, Snyder SH. Nitrosopeptide mapping: a novel methodology reveals S-nitrosylation of dexras1 on a single cysteine residue. Chem Biol. 2002;9:1329–1335. doi: 10.1016/s1074-5521(02)00293-4. [DOI] [PubMed] [Google Scholar]
- Jia J, Arif A, Willard B, Smith JD, Stuehr DJ, Hazen SL, Fox PL. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol Cell. 2012;47:656–663. doi: 10.1016/j.molcel.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa S, Yamazawa T, Chen Y, Ito A, Murayama T, Oyamada H, Kurebayashi N, Sato O, Watanabe M, Mori N, Oguchi K, Sakurai T, Takeshima H, Saito N, Iino M. Nitric oxide-induced calcium release via ryanodine receptors regulates neuronal function. EMBO J. 2012;31:417–428. doi: 10.1038/emboj.2011.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- Kapadia MR, Eng JW, Jiang Q, Stoyanovsky DA, Kibbe MR. Nitric oxide regulates the 26S proteasome in vascular smooth muscle cells. Nitric Oxide. 2009;20:279–288. doi: 10.1016/j.niox.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci USA. 2005a;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Wing SS, Ponka P. S-Nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol Cell Biol. 2004;24:330–337. doi: 10.1128/MCB.24.1.330-337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005b;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, Greengard P. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–817. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapfenbauer K, Engidawork E, Cairns N, Fountoulakis M, Lubec G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003;967:152–160. doi: 10.1016/s0006-8993(02)04243-9. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, Lipton SA, Masliah E, Xu H, Liao FF. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener. 2010;5:49. doi: 10.1186/1750-1326-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, 3rd, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim CK, Lee KH, Ahn JY. S-Nitrosylation of B23/nucleophosmin by GAPDH protects cells from the SIAH1-GAPDH death cascade. J Cell Biol. 2012;199:65–76. doi: 10.1083/jcb.201205015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HSV, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Li C, Feng JJ, Wu YP, Zhang GY. Cerebral ischemia-reperfusion induces GAPDH S-nitrosylation and nuclear translocation. Biochemistry (Mosc) 2012;77:671–678. doi: 10.1134/S0006297912060156. [DOI] [PubMed] [Google Scholar]
- Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q, Vujaskovic Z, Dewhirst, Mark W, Li CY. Regulation of HIF-1α stability through S-nitrosylation. Mol Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu F, Salmonsen RA, Turner TK, Litofsky NS, Di Cristofano A, Pandolfi PP, Jones SN, Recht LD, Ross AH. PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol Cell Neurosci. 2002;20:21–29. doi: 10.1006/mcne.2002.1115. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Takahashi T, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function — as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Nakamura T, Uehara T, Shi ZQ, Gu Z. Comment on S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2005;308:1870. doi: 10.1126/science.1110353. [DOI] [PubMed] [Google Scholar]
- Malito E, Ralat LA, Manolopoulou M, Tsay JL, Wadlington NL, Tang WJ. Molecular bases for the recognition of short peptide substrates and cysteine-directed modifications of human insulin-degrading enzyme. Biochemistry. 2008;47:12822–12834. doi: 10.1021/bi801192h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe S, Gu Z, Lipton SA. Activation of matrix metalloproteinase-9 via neuronal nitric oxide synthase contributes to NMDA-induced retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2005;46:4747–4753. doi: 10.1167/iovs.05-0128. [DOI] [PubMed] [Google Scholar]
- Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol. 2010;395:844–859. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry (Mosc) 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- Martinez-Ruiz A, Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc Res. 2007;75:220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-Nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O’Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL, Duan W. Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol Rev. 2002;82:637–672. doi: 10.1152/physrev.00004.2002. [DOI] [PubMed] [Google Scholar]
- Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G, Finazzi-Agro A. S-Nitrosylation regulates apoptosis. Nature. 1997;388:432–433. doi: 10.1038/41237. [DOI] [PubMed] [Google Scholar]
- Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, Ma Y, Moosmann B, Masliah E, Lipton SA, Gu Z. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener. 2011;6:34. doi: 10.1186/1750-1326-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol. 2005;15:1572–1577. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- Miller RL, James-Kracke M, Sun GY, Sun AY. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem Res. 2009;34:55–65. doi: 10.1007/s11064-008-9656-2. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- Mitsumoto A, Nakagawa Y, Takeuchi A, Okawa K, Iwamatsu A, Takanezawa Y. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic Res. 2001;35:301–310. doi: 10.1080/10715760100300831. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Kumar M, Selvakumar B, Ho GP, Ehmsen JT, Barrow RK, Amzel LM, Snyder SH. Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of D-serine formation. Proc Natl Acad Sci USA. 2007;104:2950–2955. doi: 10.1073/pnas.0611620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. S-Nitrosylation and uncompetitive/fast off-rate (UFO) drug therapy in neurodegenerative disorders of protein misfolding. Cell Death Differ. 2007;14:1305–1314. doi: 10.1038/sj.cdd.4402138. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, Tzitzilonis C, Meng F, Gu Z, Holland EA, Clemente AT, Okamoto S-i, Salvesen GS, Riek R, Yates JR, 3rd, Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, Uehara T. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN) Proc Nati Acad Sci USA. 2011;108:10349–10354. doi: 10.1073/pnas.1103503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez L, Vaquero M, Gomez R, Caballero R, Mateos-Caceres P, Macaya C, Iriepa I, Galvez E, Lopez-Farre A, Tamargo J, Delpon E. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc Res. 2006;72:80–89. doi: 10.1016/j.cardiores.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-Nitrosylation of β-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Xu G, Stancevic B, Jaffrey SR. Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem Biol. 2008;15:1307–1316. doi: 10.1016/j.chembiol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer ZJ, Duncan RR, Johnson JR, Lian LY, Mello LV, Booth D, Barclay JW, Graham ME, Burgoyne RD, Prior IA, Morgan A. S-Nitrosylation of syntaxin 1 at Cys(145) is a regulatory switch controlling Munc18–1 binding. Biochem J. 2008;413:479–491. doi: 10.1042/BJ20080069. [DOI] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Huh SH, Kim MS, Lee SH, Choi EJ. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc Natl Acad Sci USA. 2000;97:14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Yu JW, Cho JH, Kim MS, Huh SH, Ryoo K, Choi EJ. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J Biol Chem. 2004;279:7584–7590. doi: 10.1074/jbc.M304183200. [DOI] [PubMed] [Google Scholar]
- Pei DS, Sun YF, Song YJ. S-Nitrosylation of PTEN Invovled in ischemic brain injury in rat hippocampal CA1 region. Neurochem Res. 2009;34:1507–1512. doi: 10.1007/s11064-009-9938-3. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation Activates Cdk5 and contributes to synaptic spine loss induced by β-Amyloid peptide. Proc Natl Acad Sci USA. 2011:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Nakamura T, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation of Cdk5: Potential implications in amyloid-beta-related neurotoxicity in Alzheimer disease. Prion. 2012;6:364–370. doi: 10.4161/pri.21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralat LA, Ren M, Schilling AB, Tang WJ. Protective role of Cys-178 against the inactivation and oligomerization of human insulin-degrading enzyme by oxidation and nitrosylation. J Biol Chem. 2009;284:34005–34018. doi: 10.1074/jbc.M109.030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Beal MF. Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Nitric oxide blocks fast, slow, and persistent Na+ channels in C-type DRG neurons by S-nitrosylation. J Neurophysiol. 2002;87:761–775. doi: 10.1152/jn.00369.2001. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Romero-Puertas MC, Laxa M, Matte A, Zaninotto F, Finkemeier I, Jones AM, Perazzolli M, Vandelle E, Dietz KJ, Delledonne M. S-Nitrosylation of peroxiredoxin II E promotes peroxynitrite-mediated tyrosine nitration. Plant Cell. 2007;19:4120–4130. doi: 10.1105/tpc.107.055061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafian TA, Verity MA, Vinters HV, Shih CCY, Shi L, Ji XD, Dong L, Shau H. Differential expression of peroxiredoxin subtypes in human brain cell types. J Neurosci Res. 1999;56:206–212. [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O’Kane CJ, Rubinsztein DC. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–188. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Schonhoff CM, Daou MC, Jones SN, Schiffer CA, Ross AH. Nitric oxide-mediated inhibition of Hdm2-p53 binding. Biochemistry. 2002;41:13570–13574. doi: 10.1021/bi026262q. [DOI] [PubMed] [Google Scholar]
- Selvakumar B, Huganir RL, Snyder SH. S-Nitrosylation of stargazin regulates surface expression of AMPA-glutamate neurotransmitter receptors. Proc Natl Acad Sci USA. 2009;106:16440–16445. doi: 10.1073/pnas.0908949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumar B, Jenkins MA, Hussain NK, Huganir RL, Traynelis SF, Snyder SH. S-Nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc Natl Acad Sci USA. 2013;110:1077–1082. doi: 10.1073/pnas.1221295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Hara MR, Ahmad AS, Cascio MB, Kamiya A, Ehmsen JT. GOSPEL: a neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron. 2009;63:81–91. doi: 10.1016/j.neuron.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, Sawa A. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Snyder SH. Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc Natl Acad Sci USA. 2011;108:20178–20183. doi: 10.1073/pnas.1117820108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D, Stamler JS. The SNO-proteome: causation and classifications. Curr Opin Chem Biol. 2011;15:129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi ZQ, Sunico CR, McKercher SR, Cui J, Feng GS, Nakamura T, Lipton SA. S-Nitrosylated SHP-2 contributes to NMDA receptor-mediated excitotoxicity in acute ischemic stroke. Proc Natl Acad Sci USA. 2013;110:3137–3142. doi: 10.1073/pnas.1215501110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sriram SM, Kim BY, Kwon YT. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol. 2011;12:735–747. doi: 10.1038/nrm3217. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-Nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ. The decomposition of thionitrites. Curr Opin Chem Biol. 2002;6:779–785. doi: 10.1016/s1367-5931(02)00383-6. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Stroissnigg H, Trancikova A, Descovich L, Fuhrmann J, Kutschera W, Kostan J, Meixner A, Nothias F, Propst F. S-nitrosylation of microtubule-associated protein 1B mediates nitric-oxide-induced axon retraction. Nat Cell Biol. 2007;9:1035–1045. doi: 10.1038/ncb1625. [DOI] [PubMed] [Google Scholar]
- Sumbayev VV, Budde A, Zhou J, Brune B. HIF-1α protein as a target for S-nitrosation. FEBS Lett. 2003;535:106–112. doi: 10.1016/s0014-5793(02)03887-5. [DOI] [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Taira T, Saito Y, Niki T, Iguchi-Ariga SMM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Shin Y, Cho SJ, Zago WM, Nakamura T, Gu Z, Ma Y, Furukawa H, Liddington R, Zhang D, Tong G, Chen H-SV, Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron. 2007;53:53–64. doi: 10.1016/j.neuron.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Uehara T, Nomura Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J Biol Chem. 2000;275:10388–10393. doi: 10.1074/jbc.275.14.10388. [DOI] [PubMed] [Google Scholar]
- Tenneti L, D’Emilia DM, Lipton SA. Suppression of neuronal apoptosis by S-nitrosylation of caspases. Neurosci Lett. 1997;236:139–142. doi: 10.1016/s0304-3940(97)00780-5. [DOI] [PubMed] [Google Scholar]
- Tian J, Kim SF, Hester L, Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci USA. 2008;105:10537–10540. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Tsang AHK, Lee YI, Ko HS, Savitt JM, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, Chung KKK. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proc Natl Acad Sci USA. 2009;106:4900–4905. doi: 10.1073/pnas.0810595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui AK, Marsden PA, Mazer CD, Adamson SL, Henkelman RM, Ho JJ, Wilson DF, Heximer SP, Connelly KA, Bolz SS, Lidington D, El-Beheiry MH, Dattani ND, Chen KM, Hare GM. Priming of hypoxia-inducible factor by neuronal nitric oxide synthase is essential for adaptive responses to severe anemia. Proc Natl Acad Sci USA. 2011;108:17544–17549. doi: 10.1073/pnas.1114026108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Um HC, Jang JH, Kim DH, Lee C, Surh YJ. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide. 2011;25:161–168. doi: 10.1016/j.niox.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Walker AK, Farg MA, Bye CR, McLean CA, Horne MK, Atkin JD. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain. 2010;133:105–116. doi: 10.1093/brain/awp267. [DOI] [PubMed] [Google Scholar]
- Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci USA. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HQ, Takahashi R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid Redox Signal. 2007;9:553–561. doi: 10.1089/ars.2006.1524. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003;6:1039–1047. doi: 10.1038/nn1119. [DOI] [PubMed] [Google Scholar]
- Wang S, Song J, Tan M, Albers KM, Jia J. Mitochondrial fission proteins in peripheral blood lymphocytes are potential biomarkers for Alzheimer’s disease. Eur J Neurol. 2012;19:1015–1022. doi: 10.1111/j.1468-1331.2012.03670.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Han C, Lim K, Wu T. Activation of cytosolic phospholipase A2alpha through nitric oxide-induced S-nitrosylation. Involvement of inducible nitric-oxide synthase and cyclooxygenase-2. J Biol Chem. 2008;283:3077–3087. doi: 10.1074/jbc.M705709200. [DOI] [PubMed] [Google Scholar]
- Xue Y, Liu Z, Gao X, Jin C, Wen L, Yao X, Ren J. GPS-SNO: computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PLoS One. 2010;5:e11290. doi: 10.1371/journal.pone.0011290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, Lipton SA. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasinska IM, Sumbayev VV. S-Nitrosation of Cys-800 of HIF-1α protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549:105–109. doi: 10.1016/s0014-5793(03)00807-x. [DOI] [PubMed] [Google Scholar]
- Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-Nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- Yonashiro R, Kimijima Y, Shimura T, Kawaguchi K, Fukuda T, Inatome R, Yanagi S. Mitochondrial ubiquitin ligase MITOL blocks S-nitrosylated MAP1B-light chain 1-mediated mitochondrial dysfunction and neuronal cell death. Proc Natl Acad Sci USA. 2012;109:2382–2387. doi: 10.1073/pnas.1114985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jin B, Li L, Block ER, Patel JM. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288:C840–849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- Zhang P, Yu PC, Tsang AH, Chen Y, Fu AK, Fu WY, Chung KK, Ip NY. S-Nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J Neurosci. 2010;30:14366–14370. doi: 10.1523/JNEUROSCI.3899-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]