

Abstract
To study protection of melanocytes from stress-induced cell death by heme oxygenases during depigmentation and repigmentation in vitiligo, expression of isoforms 1 and 2 was studied in cultured control and patient melanocytes and normal skin explants exposed to UV or bleaching agent 4-TBP. Similarly, expression of heme oxygenases was followed in skin from vitiligo patients before and after PUVA treatment. Single and double immunostainings were used in combination with light and confocal microscopic analysis and Western blotting. Melanocyte expression of heme oxygenase 1 is upregulated, whereas heme oxygenase 2 is reduced in response to UV and 4-TBP. Upregulation of inducible heme oxygenase 1 was also observed in UV-treated explant cultures, in skin of successfully PUVA-treated patients and in melanocytes cultured from vitiligo non-lesional skin. Heme oxygenase encoding genes were subsequently cloned to study consequences of either gene product on cell viability, demonstrating that HO-1 but not HO-2 overexpression offers protection from stress-induced cell death in MTT assays. HO-1 expression by melanocytes may contribute to beneficial effects of UV treatment for vitiligo patients.
Keywords: heme oxygenase, melanocyte, stress, ultraviolet
Introduction
Vitiligo patients present with white skin patches because of progressive loss of melanocytes. There is support for a role of oxidative stress in precipitating the disease (1). As not every individual is susceptible to developing progressive depigmentation, the skin of vitiligo patients appears inadequately equipped to handle the precipitating events. Here, the potential role of heme oxygenase, in particular its isoform HO-1, in antioxidant defense of melanocytes is explored with reference to vitiligo.
Vitiligo is a skin disease that occurs in approximately 0.5% of the world population (2). Melanocytes are central to the disease. Hereditary factors define susceptibility to its development, and associated genes largely implement an autoimmune aetiology (3). This is further supported by the association with other autoimmune diseases and the presence of high affinity, melanocyte-reactive T cells (4,5).
Levels of hydrogen peroxide in plasma and lesional skin of vitiligo patients are elevated following oxidative stress, likely explaining initial damage to melanocytes (6). Oxygen radicals can modify cellular proteins and peptides, rendering melanocytes increasingly susceptible to an autoimmune response once such ‘neoantigens’ are presented in the context of MHC on the cell surface (7). Such consequences are best understood in the context of a limited antioxidant defense among vitiligo melanocytes, implicating antioxidant enzymes including catalase, glutathione peroxidase and glutathione reductase in vitiligo aetiology (1,8,9).
Precipitating factors include overexposure to ultraviolet light (10). Moreover, measurements of oxidative stress confirmed that levels of glutathione, heme oxygenase-1 and reactive oxygen species were elevated in human dermal fibroblasts following exposure to ultraviolet radiation (11).
Meanwhile, ultraviolet A (UVA) and ultraviolet B (UVB) radiation are used in the treatment of vitiligo, in particular for patients that do not respond to more subtle immunosuppressive measures (12). Treatment with UVA combined with psoralen photosensitizers or by NB-UVB treatment can stimulate migration of melanocyte precursor cells to the skin and increase melanocyte abudance (13).
The apparent contradiction posed by UV radiation precipitating depigmenting lesions in vitiligo while also contributing to a cure for some patients prompted the current investigations. Reduced functionality of specific components of the antioxidant defense system within melanocytes may represent stressful situations that can lead to melanocyte death (14). In this respect, heme oxygenase is an antioxidant defense enzyme representing a candidate pathway to drive UV responsiveness (15).
Heme oxygenase (HO) defines the degradation of heme. It is encoded in three isoforms, inducible HO-1 also known as heat shock protein 32 (HSP32), constitutive HO-2 and ill-defined HO-3. HO-1 and HO-2 carry approximately 80% homology over two stretches of 105 and 82 nucleotides, representing approximately 20% of the total sequence. HO-3 may instead be a pseudogene as shown in rat (16). Several functions of HO-1 define its importance to cell physiology: its role in protection from apoptosis following stress, its enzymatic function in heme degradation and its immunomodulatory effects (17). Metabolite CO has anti-inflammatory and anti-apoptotic effects on different cell types, while bilirubin has anti-oxidant and radical scavenging effects (18,19). HO-1 is a powerful immunomodulator, and elevated levels of HO-1 can eliminate inflammatory atopic dermatitis-like lesions in mice (20). In psoriasis, the levels of HO-1 expression by keratinocytes were found to be elevated in comparison with the control group (21). Finally, HO-1 promoter mutations have been reported in melanoma, linking expression of the enzyme to pigment cell physiology (22). HO-1 is one of only few heat shock proteins immediately induced by stress, and its level of expression thus offers a first line of defense, preceding the role of other anti-oxidant enzymes.
In healthy skin, UV radiation may enhance HO-1 expression, offering protection from apoptosis and inflammation. Consequently, the expression of HO-1 and HO-2 by melanocytes and its modulation in response to stress including UV exposure and bleaching compound 4-tertiary butyl phenol was studied in vitro by western blotting, using homogenates of control and patient-derived melanocytes. To follow expression of heme oxygenases within the skin, ex vivo organotypic cultures of normal human skin were exposed to UV and 4-TBP and subjected to immunohistochemistry and confocal microscopy. A functional role for heme oxygenases in the response to precipitating factors in vitiligo aetiology was studied by cloning the cDNA into expression vectors, and exposing transfectant cells to UV, measuring cell death in MTT assays. Finally, intracellular localization of heme oxygenases was followed by cell fractionation and Western blotting. These studies serve to further understand the role of HO-1 in the antioxidant defense of melanocytes.
Materials and methods
PUVA-treated patients
Biopsies were from nine patients > 12 years of age with 30–60% stable, generalized, lesional vitiligo skin before and immediately after PUVA treatment. Disease duration at time of treatment varied from 4 months to 10 years. Patients were treated with PUVA for 21–36 sessions for cumulative doses of 55–101 J/cm for 7–12 weeks. Patients remained untreated for 8 weeks and were then subjected to 4-mm skin biopsies from lesions prior to, and from successfully repigmented skin within 1 h following the last PUVA treatment. Biopsies were snap-frozen, stored at −80°C and transported on dry ice. Eight-lm cryosections were cut, fixed in cold acetone and stored at −20°C.
Melanocyte cultures
Human melanocytes were cultured in media consisting of Ham’s F-12 medium (Media Tech, Herndon, VA, USA) with 2 mM glutamine (Media Tech), 100 IU/ml penicillin, 100 μg/ml streptomycin and 100 μg/ml amphotericin (Media Tech), 0.1 mM 3-isobutyl-l-methylxanthine (IBMX) (Sigma, st Louis, MO, USA), 10 ng/ml TPA (Sigma) and 1% Ultroser G (Pall Biosepra, Cergy-Saint-Christophe, France). Non-lesional vitiligo skin biopsies were obtained with informed consent according to IRB-approved protocols at Loyola University Chicago.
Organotypic culture of skin
Neonatal skin was obtained as otherwise discarded tissue after routine circumcision according to IRB-approved protocols at the University of Chicago and Loyola University Chicago. Biopsies of 4 mm in diameter and 2 mm thickness were taken and cultured in 12-mm tissue culture inserts (Corning Incorporated, New York, NY, USA), with media added to the outer well to maintain explants at the air-liquid interface. Media used were RPMI (Mediatech) with 10% heat-inactivated normal human serum (Valley Biomedical, Winchester, VA, USA), 5 mM glutamine (Mediatech), 100 IU/ml of penicillin and 100 μg/ml streptomycin 10% (Mediatech) and 100 μg/ml of fungizone (Invitrogen, Carlsbad, CA, USA). Skin explants were treated with 250 μM of 4-TBP (Sigma) in 50 μl applied daily and incubated at 37°C for 4 days. Cryosections of snap-frozen explants were acetone fixed and stored at −20°C.
UVA exposure in vitro
Vitiligo and control melanocytes were exposed to 140 mJ/cm UVA. A phototherapy device was used, composed of 2 F6T5-350BL UVA and 2 F6T5 UVB phototherapy lamps (National Biological Corporation, Beachwood, OH, USA). PBS replaced the media during exposure, and cells were cultured in complete media for an additional 24 h.
Immunohistology
Cryosections were indirectly immunoperoxidase stained with primary polyclonal rabbit or mouse monoclonal antibody to HO-1 (HO-1-1) (Assay Designs, Ann Arbor, MI, USA) or polyclonal rabbit antiserum to HO-2 (Assay Designs). Sections were incubated with primary antibodies in 10% NHS in PBS. In single-staining procedures, biotinylated antibodies to mouse or rabbit immunoglobulins (Dakopatts, Carpinteria, CA, USA) were then used, followed by peroxidase-conjugated streptavidin. Aminoethylcarbazole was used as a substrate for peroxidase, followed by Mayer’s haematoxylin counterstaining (Dakopatts) where indicated.
Immunodouble stainings were performed using primary polyclonal rabbit anti-HO-1 antibody and mouse monoclonal anti-TRP-1 clone Ta99 (Signet, Dedham, MA, USA). Enzymatically labelled isotype-specific antibodies were used in the second step (Southern Biotechnologies, Birmingham, AL, USA). Fast Blue-BB was first added as an alkaline-phosphatase substrate, followed by detection of peroxidase by aminoethylcarbazole as above.
Cell fractionation
A volume of 1.5 × 10 (7) cells were lysed in hypotonic buffer in the presence of protease inhibitors (Boehringer Mannheim GmbH, GE) at 4°C. Cells were dounced (Pierce, Rockford, IL, USA), and sucrose was added to 8%. A 5–30% iodixanol gradient was underloaded with the homogenate in 35% iodixanol (Nycomed/GE Healthcare, Little Chalfont, UK). After 50-kg ultracentrifugation for 16 h, the resulting gradient was fractionated in 0.5-ml portions. Microsome-containing fractions were identified by Western blotting, pooled and concentrated by filtered centrifugation. Concentrated microsomal proteins were used to confirm ER localization of heme oxygenase proteins.
Western blotting
Human melanocytes from light and dark skin tones were treated with 125, 250 and 500 μM 4-TBP (Sigma). Cells were dissolved in 1 × cell lysis buffer. Protein content was estimated using Bio-Rad protein assay reagent (Hercules, CA, USA). Ten micrograms of protein was electrophoresed in an 8% polyacrylamide gel and transferred to immobilon–P (Millipore, Billerica, MA, USA). Blots were subjected to single or double staining. Antibodies used were polyclonal or monoclonal antibodies to HO-1 or polyclonal HO-2, monoclonal anti-TRP-1 (BD Biosciences, Franklin Lakes, NJ, USA) monoclonal anti- β- actin (Sigma) and polyclonal anti-TRAP-a, a kind gift from Dr C. Nicchita, Duke University, NC. The staining procedure followed was similar to immunohistochemistry described earlier, detecting alkaline phosphatase using 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt/nitro-blue tetrazoliumchloride (Sigma), and peroxidase activity using amino-ethylcarbazole.
HO-1 and HO-2 gene amplification and cloning
For HO-1 cDNA cloning, melanocytes were exposed to 140 mJ of UVA prior to RNA isolation. For HO-2, RNA was isolated from M14 melanoma cells. RNA was isolated in Tri Reagent (Invitro-gen) according to manufacturers’ instructions. The RT reaction consisted of 1.3 U/μl Rnase Out (Invitrogen), 1 × 1st-strand buffer (Invitrogen), 1 mM each of dNTPs (Invitrogen), 25 ng/μl oligo dT primer (Invitrogen), 10 μM DTT (dithiothreitol), 3.3 mM MgCL2 (Invitrogen) and 10 u/μl Superscript II enzyme (Invitrogen). The reaction was performed in an iCycler (Biorad) at 42°C for 52 min, followed by 70°C for 15 min. The PCR reaction was performed as follows: gene-specific primers used were for HO-1: ATGGAGCGTCCGCAACCCGACAG and CATGGCATAAAGCC CTACAGCAAC; for HO-2 ATGTCAGCGGAAGTGGAAACCTCA GAG and CTACATGTAGTACCAGGCCAAGAGTCC.
The PCR mix consisted of 1 × PCR buffer (Invitrogen), 2 mM MgCl2 (Invitrogen), 0.4 mM dNTPs (Invitrogen), 1 μg/μl BSA, 2 ng/μl each of forward and reverse primers (Invitrogen), (Invitrogen), 5 U/ml Taq DNA polymerase (Invitrogen) and cDNA from the first-strand reaction. PCR: the reaction was performed as 40 cycles of 95°C for 20 min, 58°C for 30 min and 72°C for 1 min, followed by 72°C for 10 min. HO-1 or HO-2 cDNA was cloned into pcDNA3.1/V5-HIS-TOPO TA (Invitrogen) and used to transform competent E.coli. Cloning of the correct PCR product in the correct orientation was verified by restriction analysis using XbaI and ESP3I enzymes (Fermentas, Glen Burnie, Maryland) for HO-1 or with ApaI enzyme for HO-2 (Fermentas). Plasmid was isolated in bulk (QIAGEN, Valencia, CA, USA) and the insert subjected to routine DNA sequencing (Retrogen, San Diego, CA, USA).
Confocal microscopy
Six-mm human biopsies were exposed to 1.5 J/cm UVA. Skin was embedded in Tissue Tek (Sakura, Torrance, CA, USA) 24 h after exposure. Eight-μm cryosections were immunostained with polyclonal rabbit anti-HO-1 (Assay Designs) and mouse monoclonal anti-TRP-1 antibody clone Ta99 (Southern Biotechnology, Birmingham, AL, USA), followed by anti-rabbit-biotin and streptavidin-PE (Dakopatts), and anti-mouse-FITC (Southern Biotechnology). Sections were counterstained with DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen) and coverslipped in fluorescent mounting media (Dakopatts). Sections were studied by confocal microscopy (Zeiss LSM-510, Thornwood, NY, USA). PE was monitored using a HeNE 633-nm laser, DAPI was monitored using a diode laser at 405 nm, and detection of FITC was performed using a multiline-argon laser exciting at 488 nm.
Viability assays
Samples were plated in triplicate. Cultured COS cells (acronym for CV-1 in origin, carrying SV40) were transfected with pHO-1 (plasmid encoding the HO-1 gene), pHO-2 (plasmid encoding the HO-2 gene) or control vector. For transfection, lipofectamine 2000 (Invitrogen) and DNA were added to melanocytes and incubated at 37°C for 24 h. Transfected melanocytes were exposed to 0.5 J/cm UVB and incubated at 37 C for 24 h. The per cent of viable cells was evaluated by an MTT assay (Bioassay system, Hay-ward, CA, USA), adding MTT reagent (tetrazole) and incubating at 37°C for 4 h for conversion to formazan in living cells. Forma-zan was quantified in an ELISA reader at 562 nm. Cell viability was calculated relative to untreated cells.
Results
HO-1 is upregulated and HO-2 is downregulated in UV-exposed skin
HO-1 expression in skin from nine vitiligo patients before and after PUVA treatment was evaluated by immunostaining, and melanocyte repopulation in response to treatment was confirmed by TRP-1 immunostaining. There was faint expression of HO-1 in lesional skin before PUVA, while HO-1 expression was apparent particularly among the basal cell layer in treated skin 1 h following UVA, and after 13 weeks of 3 × / week PUVA treatment and a cumulative dose of 100 J/cm2 as shown in a representative example in Fig. 1. The patient shown was 30% depigmented at the onset of treatment, with a disease duration of 1 year. Enhanced HO-1 expression in response to successful PUVA was observed in 8 of 9 patients, whereas expression in a single patient with 50% lesional skin prior to treatment remained unchanged following the lowest applied cumulative dose of 55 J/cm of UV-A. HO-1 expression was upregulated in human skin explants in response to 10 μl of 1 mM 4TBP for 5 days (not shown).
Figure 1.
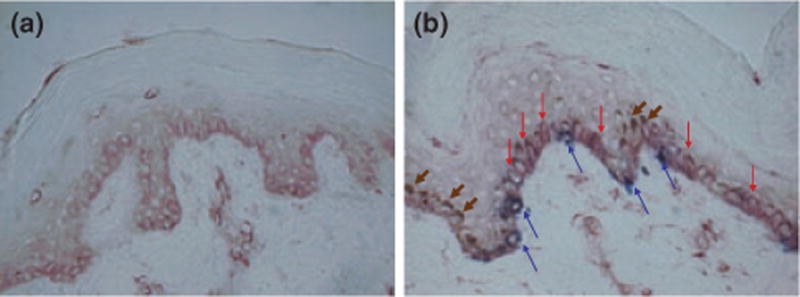
HO-1 expressed in skin repigmenting in response to PUVA treatment. HO-1 staining of skin biopsies from vitiligo (a) lesional skin before, and (b) from repigmented skin within 1 h, after 13 weeks of 3x/week PUVA treatment, total dose 100 J/cm2. Red arrows: marked HO-1 expression. Blue arrows: TRP-1 expression restricted to melanocytes. Brown arrows: nuclear capping by melanin.
HO-1 expression specifically by melanocytes in foreskin sections was estimated by immunodouble-staining to detect HO-1 and TRP1 expression. HO-1 expression was increased 24 h after a single exposure to 0.4 J/cm UVA (Fig. 2a,b). HO-2 expression was also evaluated by indirect immunoperoxidase staining with polyclonal anti-HO-2. HO-2 expression was markedly reduced 24 h after a single exposure to 0.4 J/cm UVA (Fig. 2c,d).
Figure 2.
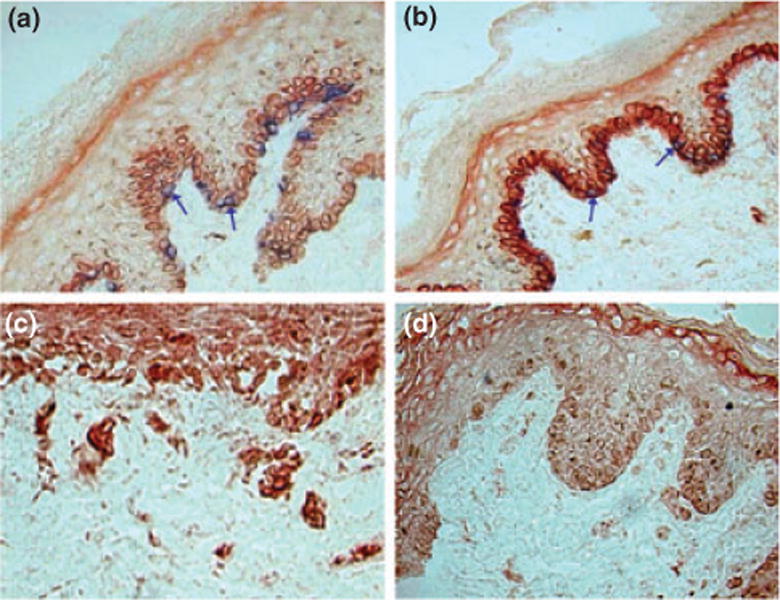
HO-1 expression is upregulated and HO-2 expression reduced in response to UVA in organotypic culture. Expression of HO-1 in the basal epidermal layer of neonatal skin (a) untreated skin and (b) after single exposure to 400 mJ/m2 UVA. Comparison between expression of HO-2 in (c) the epidermis of untreated neonatal skin and (d) 24 h after UVA. HO is shown in red and TRP-1 is shown in blue. Arrows: melanocytes.
UV affects expression of heme oxygenases in cultured melanocytes
Data shown in supplementary Fig. S1a,b demonstrate that cultured normal melanocytes displayed 1.5-fold upregulated HO-1 expression 24 h after a single exposure with and without 140 m/cm2 UVA, while a 2.4-fold upregulation was observed in vitiligo melanocytes. These data suggest a more vigorous response to UVA by non-lesional vitiligo melanocytes.
HO-1 expression increased with increasing 4-TBP exposure when melanocytes were cultured in the presence of 4-TBP in concentrations of 125 or 250 μm for 24 h. Treatment with 125 μm of 4-TBP induced a 1.1-fold HO-1 expression, while HO-1 expression was elevated 2.8-fold after 250 μm 4-TBP (Supplementary Fig. S1c).
Both heme oxygenases are found in the ER compartment of melanocytes
Blots of fractionated cells stained with antibodies to HO-1 and TRAP-α confirmed colocalization in fractions 12–15. TRP-1 staining is shown for reference (Supplementary Fig. S2a). HO-2 expression in microsomal fractions was confirmed in separate blot of purified and concentrated ER proteins stained with polyclonal antibody to HO-2 and monoclonal antibody to HO-1 (Supplementary Fig. S2b). Thus, both HO-1 and HO-2 reside in the ER.
UV-induced upregulation of HO-1 is also observed within the skin environment
Organotypic cultures of human skin (three explants per condition) were exposed to UVA at 1.5 J/cm2 and immunostained with anti-HO-1 antibody (red) and anti-TRP-1 antibody (green). Sections were DAPI (blue) counterstained to reveal nuclei and studied by confocal microscopy. Figure 3 shows a representative area of staining in treated versus untreated skin, demonstrating that HO-1 was expressed particularly by melanocytes in untreated skin and upreg-ulated epidermal expression following UV.
Figure 3.
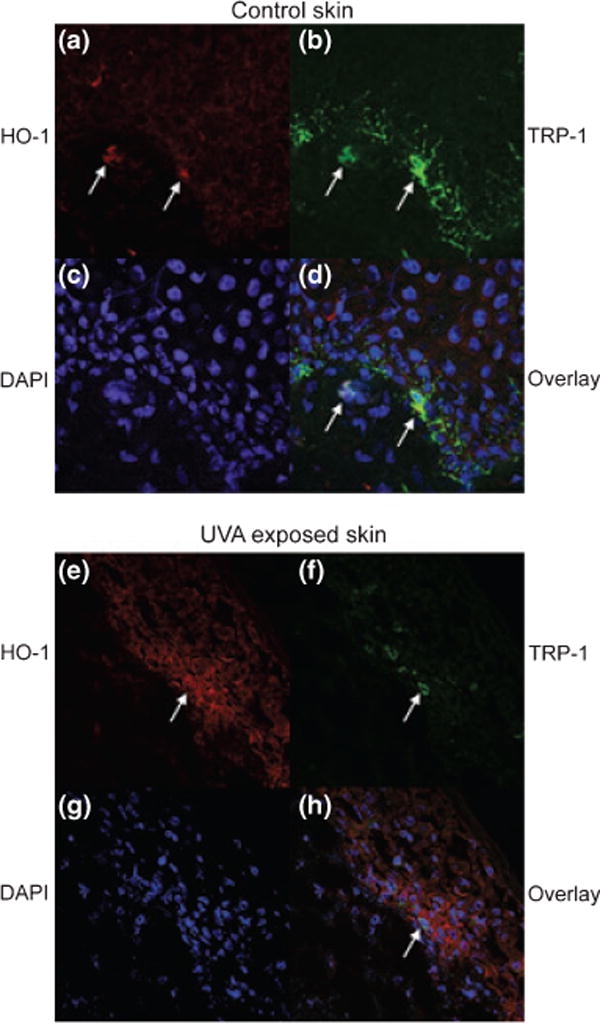
Confocal microscopy of cultured human skin treated with UVA. Skin before and after 1.5 J/cm2 UVA was immunostained with (a, e) anti-HO-1 antibody and (b, f) anti-TRP-1 antibody, and (c, g) DAPI. In (d, h) overlays are shown. (a–d) untreated skin. (e–h) 24 h after UVA treatment. Arrows point to the same cells under (a)–(d) and under (e)–(h).
Overexpression of HO-1 but not HO-2 protects cells from UV-induced cell death
As shown in Fig. 4, cell viability increased from 16 to 48% in COS cells overexpressing HO-1 (P < 0.05 in a t-test) exposed to 1 J/cm2 UV-B, whereas no significant protection from cell death was observed in cells overexpressing HO-2.
Figure 4.
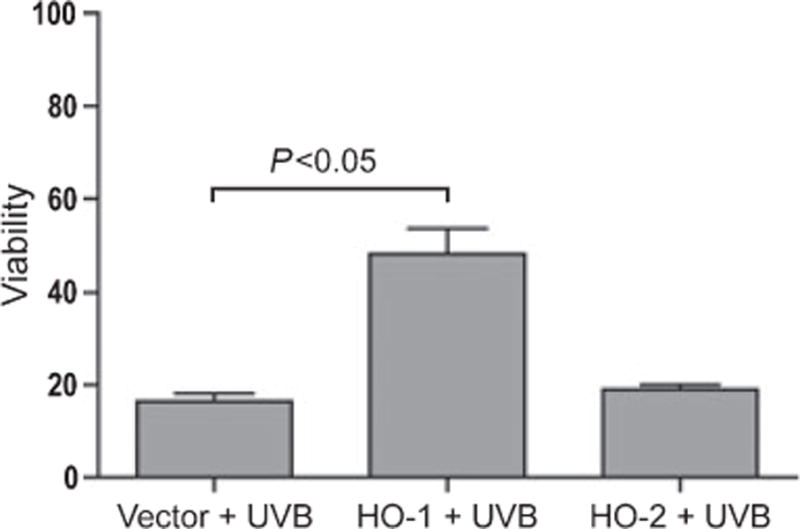
HO-1 overexpression protects cells from undergoing UV-induced apoptosis. COS cells were transfected with HO-1 and HO-2 expression plasmids and exposed to 1 J/cm2 UV-B. Viability was measured by MTT assay after 48 h.
Discussion
A cytoprotective role is well established for HO-1, yet published studies generally describe HO-1 function in the absence of assessing HO-2 (23). Given the homology among both gene products, the expression and function of both heme oxygenases was assessed in control and vitiligo skin derived melanocytes, in UV-exposed control skin and PUVA-treated patient samples.
In earlier studies, melanocytes were shown to be capable of expressing HO-1, moreso than keratinocytes (24). Moreover, HO-1 expression can increase in response to UV exposure (25). As HO-1 is important in protecting cells from oxidative stress, and oxidative stress is considered a contributing factor in depigmentation of vitiligo skin, we postulate that HO-1 expression is important in protecting vitiligo melanocytes from UV-induced cell death in response to therapeutic UV exposure.
UV-induced HO-1 expression can be of particular importance to vitiligo patients as HO-1 affects immune responses. The contribution of an autoimmune response to progressive vitiligo is well established. HO-1 expression has been associated with immunosuppressive effects (26). In fact, HO-1 can prevent activation of dendritic cells and thus indirectly affect responses by cytotoxic T cells (27). Its dual role in antioxidant defenses and immunomodulation formed the basis of the current study and points to the importance of accurate dosing when exposing vitiligo patients to UV.
Supporting the involvement of HO-1 in protecting vitiligo skin from UV, we were able to demonstrate that HO-1 is expressed after repigmentation (directly after UV) and not in lesional skin prior to PUVA treatment. We propose that the HO-1 expression observed in melanocytes contributed to their success in repopulat-ing the skin previously devoid of melanocytes. In this regard, the expression of HO-1 is detectable in melanocytes both before and in response to UV exposure in cultured normal skin explants. The relative migration and proliferation of melanocytes within vitilig-inous skin may be affected compared to controls in response to UV.
The actual cellular protection from UV by HO-1 was analysed in vitro and compared to protection by HO-2. As applying UV itself is sufficient for HO-1 induction, psoralens were not added in culture (28). Both heme oxygenases localized to the endoplasmic reticulum in melanocytes as expected (29,30). Whereas both HO-1 and HO-2 have been assigned to the endoplasmic reticulum, others have observed a mitochondrial localization of HO-1 explaining a possible involvement in protection from apoptosis (31). It is possible that the subcellular localization is cell type dependent and varies with enviromental conditions. Meanwhile, the importance of particularly HO-1 expression in the ER lies in the fact that morphological aberrancies have been demonstrated in the ER of vitiligo melanocytes. We have previously shown that ER aberrancies in the vitiligo melanocyte cell line PIG3V are associated with differential de novo protein expression (32).
In organotypic skin cultures, we were able to detect expression of HO-1 restricted to melanocytes under baseline conditions and upregulated epidermal expression of HO-1 24 h after exposure to 0.4 J/cm2 UVA. By contrast, HO-2 expression was reduced in response to the same dose, supporting the prevalent role of HO-1 versus HO-2 in protection from UV. This is comparable to earlier studies reporting a similar upregulation of HO-1, and not the constitutive form HO-2, in hairless mouse skin after UVA exposure (25). It should be noted that in the latter studies, upregulation was measured in mouse skin at higher doses (38.7 J/cm2) than used here. However, doses used here were sufficient to induce overexpression of HO-1, and not HO-2.
A 2-fold upregulation of HO-1 expression by normal human Caucasian melanocytes in culture was reported after UVA exposure corresponding to a dose of 13.95 J/cm2 (24). In the present study, the HO-1 expression increased in non-lesional vitiligo melanocytes by 2.4-fold and normal melanocytes by 1.5-fold after 24 h in comparison to unexposed melanocytes. Overall, we speculate that successful vitiligo treatment requires high induceability of HO-1 by UV to protect melanocytes from apoptosis.
Immunohistochemistry was used to confirm HO-1 expression by melanocytes in vivo in newly pigmented skin in vitiligo patients exposed to incremental weekly doses of UV for up to 11 weeks. To our knowledge, HO-1 status in repigmented skin of vitiligo patients in response to PUVA therapy has not been previously reported. This reinforced the notion that the extent to which melanocytes can upregulate expression of HO-1 may determine the efficacy of UV treatment.
The actual role of HO-1 in protection from UV is supported by chemical interference with HO-1 upregulation leaving treated cells unprotected from UV (30). Some difficulty arises in the interpretation of such data as chemical inhibitors can have limited specificity for HO-1. Thus, for the current studies, the HO-1 gene was cloned into an expression vector to induce overexpression of the gene product, and COS cells transfected to overexpress either hemoxygenase were compared with control host cells for sensitivity to UV. Assuming equal transfection efficacy for either vector, it was clearly demonstrated that HO-1 overexpression offered superior protection from UV-induced cell death.
The cytoprotective role for HO-1 is in stark contrast to the role proposed for HSP70, another heat shock protein in vitiligo pathogenesis (33). It is well possible that upregulated HO-1 expression compensates for detrimental effects of HSP70 upregulation in patients with a positive response to UV therapy.
In all, the current studies support a role for HO-1 in protecting cells from UV-induced cell death and suggest that HO-1 expression among melanocytes may be a discriminating factor that differentiates responders from non-responders to UV. Note that expression of HO-1 and its underlying transcription factor Nrf2 is impacted by levels α-MSH expression (34,35). In line with reports of reduced α-MSH levels in vitiligo melanocytes, a resulting HO-1 deficiency may result in increased UV sensitivity (36).
A role for HO-1 has long been proposed in psoriasis (15). Striking similarities among the pathogenesis of psoriasis and vitiligo have been described (37). Both involve T-cell-mediated immune responses, directed against melanocytes in vitiligo, and against keratinocytes in psoriasis. Both diseases benefit from UV therapy and application of immunosuppressants including corticosteroids and protopic. Thus, it will not be surprising if beneficial effects of UV are mediated by HO-1 in either case.
Supplementary Material
HO-1 expression in cultured melanocytes in response to stress.
Melanocytes express HO-1 in the endoplasmic reticulum.
Acknowledgments
Support by a fellowship from the Egyptian government to YEE and support by NIH RO1AR054749 to CLP are gratefully acknowledged.
Abbreviations
- HO-1
heme oxygenase-1
- HO-2
heme oxygenase-2
- HSP
heat shock protein
- 4-TBP
4-tertiary butyl phenol
- PUVA
psoralen ultraviolet A
- UVB
ultraviolet B
- ROS
reactive oxygen species
Footnotes
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Arican O, Kurutas EB. Acta Dermatovenerol Alp Panonica Adriat. 2008;17:12–16. [PubMed] [Google Scholar]
- 2.Taïeb A, Picardo M, VETF Members Pigment Cell Res. 2007;20:27–35. doi: 10.1111/j.1600-0749.2006.00355.x. [DOI] [PubMed] [Google Scholar]
- 3.Jin Y, Birlea SA, Fain PR, et al. N Engl J Med. 2010;362:1686–1697. doi: 10.1056/NEJMoa0908547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molina Garrido MJ, Enríquez R, Mora Rufete A, et al. Am J Med Sci. 2007;333:178–180. doi: 10.1097/MAJ.0b013e3180319488. [DOI] [PubMed] [Google Scholar]
- 5.Garbelli S, Mantovani S, Palermo B, et al. Pigment Cell Res. 2005;18:234–242. doi: 10.1111/j.1600-0749.2005.00244.x. [DOI] [PubMed] [Google Scholar]
- 6.Song X, Xu A, Pan W, et al. Int J Mol Med. 2008;22:9–16. [PubMed] [Google Scholar]
- 7.Westerhof W, d’Ischia M. Pigment Cell Res. 2007;5:345–359. doi: 10.1111/j.1600-0749.2007.00399.x. [DOI] [PubMed] [Google Scholar]
- 8.Schallreuter KU, Bahadoran P, Picardo M, et al. Exp Dermatol. 2008;17:139–140. doi: 10.1111/j.1600-0625.2007.00666_1.x. [DOI] [PubMed] [Google Scholar]
- 9.Shalbaf M, Gibbons NC, Wood JM, et al. Exp Dermatol. 2008;17:761–770. doi: 10.1111/j.1600-0625.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 10.Mason CP, Gawkrodger DJ. Clin Exp Dermatol. 2005;30:344–345. doi: 10.1111/j.1365-2230.2005.01779.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhong JL, Edwards GP, Raval C, et al. Photo-chem Photobiol Sci. 2010;1:18–24. doi: 10.1039/b9pp00068b. [DOI] [PubMed] [Google Scholar]
- 12.El-Mofty M, Mostafa W, Youssef R, et al. Photo-dermatol Photoimmunol Photomed. 2006;4:214–216. doi: 10.1111/j.1600-0781.2006.00229.x. [DOI] [PubMed] [Google Scholar]
- 13.Abdel-Naser MB, Hann SK, Bystryn JC. Arch Dermatol. 1997;133:1530–1533. [PubMed] [Google Scholar]
- 14.Yildirim M, Baysal V, Inaloz HS, et al. J Eur Acad Dermatol Venereol. 2004;18:683–686. doi: 10.1111/j.1468-3083.2004.01080.x. [DOI] [PubMed] [Google Scholar]
- 15.Hanselmann C, Mauch C, Werner S. Biochem J. 2001;353:459–466. doi: 10.1042/0264-6021:3530459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi S, Omata Y, Sakamoto H, et al. Gene. 2004;2:241–250. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 17.McDaid J, Yamashita K, Chora A, et al. FASEB J. 2005;19:458–460. doi: 10.1096/fj.04-2217fje. [DOI] [PubMed] [Google Scholar]
- 18.Chung SW, Liu X, Macias AA, et al. J Clin Invest. 2008;118:239–247. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mancuso C. Antioxid Redox Signal. 2004;6:878–887. doi: 10.1089/ars.2004.6.878. [DOI] [PubMed] [Google Scholar]
- 20.Kirino M, Kirino Y, Takeno M, et al. J Allergy Clin Immunol. 2008;122:290–297. doi: 10.1016/j.jaci.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 21.Kaur S, Zilmer K, Zilmer M. J Eur Acad Dermatol Venereol. 2009;1:81–82. doi: 10.1111/j.1468-3083.2008.02705.x. [DOI] [PubMed] [Google Scholar]
- 22.Was H, Cichon T, Smolarczyk R, et al. Am J Pathol. 2006;169:2181–2198. doi: 10.2353/ajpath.2006.051365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maines MD, Ewing JF. Biol Reprod. 1996;54:1070–1079. doi: 10.1095/biolreprod54.5.1070. [DOI] [PubMed] [Google Scholar]
- 24.Marrot L, Belaidi JP, Jones C, et al. Photochem Photobiol. 2005;81:367–375. doi: 10.1562/2004-10-13-RA-343. [DOI] [PubMed] [Google Scholar]
- 25.George JF, Braun A, Brusko TM, et al. Am J Pathol. 2008;173:154–160. doi: 10.2353/ajpath.2008.070963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Listopad J, Asadullah K, Sievers C, et al. Exp Der-matol. 2007;16:661–670. doi: 10.1111/j.1600-0625.2007.00581.x. [DOI] [PubMed] [Google Scholar]
- 27.Briganti S, Wlaschek M, Hinrichs C, et al. Free Radical Biol Med. 2008;45:636–644. doi: 10.1016/j.freeradbiomed.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Liu XM, Peyton KJ, Ensenat D, et al. J Biol Chem. 2005;280:872–877. doi: 10.1074/jbc.M410413200. [DOI] [PubMed] [Google Scholar]
- 29.Ma N, Ding X, Doi M, et al. J Neurocytol. 2004;33:407–415. doi: 10.1023/B:NEUR.0000046571.90786.6e. [DOI] [PubMed] [Google Scholar]
- 30.Turkseven S, Drummond G, Rezzani R, et al. J Cell Biochem. 2007;4:815–823. doi: 10.1002/jcb.21138. [DOI] [PubMed] [Google Scholar]
- 31.Allanson M, Reeve VE. J Invest Dermatol. 2004;122:1030–1036. doi: 10.1111/j.0022-202X.2004.22421.x. [DOI] [PubMed] [Google Scholar]
- 32.Le Poole IC, Boissy RE, Sarangarajan R, et al. In Vitro Cell Dev Biol Anim. 2000;5:309–319. doi: 10.1290/1071-2690(2000)036<0309:PAIHVM>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 33.Offord EA, Gautier JC, Avanti O, et al. Free Radic Biol Med. 2002;32:1293–1303. doi: 10.1016/s0891-5849(02)00831-6. [DOI] [PubMed] [Google Scholar]
- 34.Marrot L, Jones C, Perez P, et al. Pigment Cell Melanoma Res. 2008;21:79–88. doi: 10.1111/j.1755-148X.2007.00424.x. [DOI] [PubMed] [Google Scholar]
- 35.Kokot A, Metze D, Mouchet N, et al. Endocrinology. 2009;150:3197–3206. doi: 10.1210/en.2008-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham A, Westerhof W, Thody AJ. Ann N Y Acad Sci. 1999;885:470–473. doi: 10.1111/j.1749-6632.1999.tb08715.x. [DOI] [PubMed] [Google Scholar]
- 37.Asarch A, Barak O, Loo DS, et al. J Dermatolog Treat. 2008;5:259–266. doi: 10.1080/09546630802206686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HO-1 expression in cultured melanocytes in response to stress.
Melanocytes express HO-1 in the endoplasmic reticulum.