

Summary
The ryanodine receptor (RyR) is a large, homotetrameric sarcoplasmic reticulum (SR) membrane protein essential for calcium cycling in both skeletal and cardiac muscle. Genetic mutations in RyR1 are associated with severe conditions including malignant hyperthermia (MH) and central core disease (CCD). One phosphorylation site (Ser 2843) has been identified in a segment of RyR1 flanked by two RyR motifs, which are found exclusively in all RyR isoforms organized as closely-associated tandem (or paired) motifs, and named after the protein itself. These motifs also contain six known MH mutations. In this study we designed, expressed, and purified the tandem RyR motifs and show that this domain contains a putative binding site for the Ca2+/calmodulin-dependent protein kinase beta isoform. We present a 2.2Å resolution crystal structure of the RyR domain revealing a two-fold, symmetric, extended four-helix bundle stabilized by a β-sheet. Docking experiments with a tetrameric EM structure of native RyR1 suggest that this domain is localized in the RyR’s clamp region, which is absent in its cousin protein inositol 1,4,5-trisphosphate receptor
Keywords: x-ray crystallography, excitation contraction, RyR
Introduction
The ryanodine receptor is a large, homotetrameric SR membrane protein that is an essential component of calcium cycling in both skeletal and cardiac muscle. In skeletal muscle, excitation-contraction (EC) begins with the depolarization of sarcolemmal and transverse tubular membranes triggering a conformational change in the dihydropyridine receptor (DHPR) and the ryanodine receptor type 1 (RyR1) leading to calcium release from the SR via RyR1 and the generation of skeletal muscle contraction. Relaxation is then initiated by the re-uptake of intracellular Ca2+ through the action of the sarco/endoplasmic reticulum Ca2+-ATPase type 1 (SERCA1a) [1,2]. The correct functioning of the contraction/relaxation cycle relies on a precise balance between Ca2+ release and re-uptake. Disruptions in this balance, such as those that result from mutations in the proteins involved in the Ca2+ uptake and release processes, lead to muscle diseases, including malignant hyperthermia (MH) and central core disease (CCD), two conditions that arise from abnormalities in RyR1 [3–5]. MH is a pharmacogenetic disorder of skeletal muscle triggered in susceptible individuals by inhalational anaesthetics and depolarizing skeletal muscle relaxants [3,5]. Susceptible individuals respond with skeletal muscle rigidity, tachycardia, unstable and rising blood pressure, and eventually dramatic hyperthermia. CCD is an autosomal dominant myopathy characterized by hypotonia during infancy, proximal muscle weakness, delayed motor development, and reduced muscle bulk [4,5].
Three isoforms of ryanodine receptor have been identified: RyR1 is predominantly expressed in skeletal muscle, RyR2 is mainly expressed in cardiac tissue, and RyR3 is expressed ubiquitously [2,6,7]. All three isoforms are composed of 4 identical ~5000 amino acid subunits. With a total molecular mass of ~2 million Daltons, the homotetrameric ryanodine receptor is the largest known calcium channel [2,8]. Monomers are composed of a large cytoplasmic region (residues 1-4300) and a transmembrane region which is predicted to contain 6–8 transmembrane helices [9].
Analysis of the primary sequence of RyR1 revealed four repeats found as two closely-associated pairs or two tandem motifs in all isoforms; the four repeats were termed RyR motifs or domains [9]. The linker between the second tandem motifs (residues 2725-2844 and 2845-2958) contains the major phosphorylation site of RyR1 (Ser2843) [10]. Mutations that give rise to MH and CCD have been clustered into three hotspots (region 1 Met1- Arg614; region 2 Arg2162-Arg2458; and region 3 Leu4800-4900), although mutations have been found throughout the RyR1 sequence including six MH mutations (E2764K, T2787S, R2840W[11,12], L2867G, E2880K, R2939K) in the region encompassing the known phosphorylation site.
The large size of this protein has made its structural analysis a challenge. Previous 3D cryo-EM structures to resolutions of up to ~9 Å revealed a very dynamic homotetramer structure of RyR1 [13–16]. Very recently, a high resolution crystal structure of the N-terminal region (amino acids 1-599) of RyR1, coupled with cryo-EM mapping studies, suggested that homotetrameric interactions play critical functional roles [17,18]. By using affinity-tag purification of the large cytosolic domain of RyR1 coupled with liquid chromatography mass spectrometry (LC-MS) we recently identified CHERP as an integral membrane protein that binds to RyR1 [19] adding to the list of several known integral membrane interacting partners of RyR1 including proteins such as DHPR, junction, triadin and soluble proteins such as such as FK506-calmodulin, protein kinases, and phosphatases [10,20,21]. Here, we employed a similar purification strategy to confirm binding of CaMKIIβ to this region, and provide a high resolution crystal structure of the central region which contains the known phosphorylation site of RyR1 and six known human Malignant Hyperthermia disease mutations.
Results
Crystal structure of domain containing phosphorylation site
Sequence analysis of the C3 construct containing the known phosphorylation site showed a highly conserved region across species with ≥75% sequence identity through all seven species and 98% sequence identity between rabbit and human RyR1 (Supplemental Figure 1), suggesting an essential role either for cellular viability and/or for protein-protein interaction [22]. To generate functional hypotheses, we sought to determine the structure of the RyR domain. We designed, cloned, and purified 25 nested loop constructs of the RyR domain of which one construct (2734-2940) produced crystal hits in sodium citrate at pH 8.5 (see methods). Upon optimization, crystals diffracted to 2.2 Å resolution (Supplement Table 1). The structure was solved using single wavelength anomalous scattering from sulphur atoms of methionine residues and a model was built and refined using standard software (Figure 1A PDB deposit I.D 3RQR [23] on 04-28-2011). Resulting from the two interwoven RyR elements, the domain was composed of a four-helix bundle stabilized by a two-stranded β-sheet. Two helices (which we refer to as the legs) extended to a symmetrical upside down U-shaped form (Figure 1A and 1B). Each RyR motif contained two α-helices (α-1 to α-4) and a β-strand; the helices were connected by a loop and a 310 helix (Figure 1B). Each RyR motif consists of a two-fold symmetrical structure in which α-1, α-2 and β1 make up one half and α-3, α-4 and β2 make up the second half. The linker connecting the two RyR motifs, which contained the known phosphorylation site (Ser2843), had limited electron density and was presumed mobile and dynamic. Observed was weak electron density for the backbone of about five residues of the 19-residue linker, and these unlabeled residues were included in the final model. The observation that the phosphorylated residue was within a 19-residue (2830-2849) flexible linker supported the notion that the residue is a physiologically relevant phosphorylatable site. A stretch of basic residues just upstream of the phosphorylation site (KKKTRKISQ) suggested that the protein kinase responsible for this activity is from the Basophilic serine/threonine kinase group, including PKA, AKT, and CAMK [24].
Figure 1. Features of RyR1 C3 construct crystal structure.

(A) Ribbon diagram of rabbit RyR1 C3 (2734-2940) construct containing two repeating RyR domains. The two structural repeats are related by an approximate 2-fold symmetry. The structure is predominantly α-helical with each repeat consisting of two α-helices (α1-4, red), a β-sheets (β1-2, yellow) and intervening loop segments (green). The electron density for residues between 2831 and 2848 was limited and the structure was approximated from the limited data available in this region (blue) (B) Schematic of sequence showing exact regions of α1-4 (red), a β 1-2 (yellow), 310 helices (pink), beta bridge (gold) amino acids and turns (purple). Indicated is the phosphorylation site Ser2843 and green squares indicate 6 MH mutations. (C) Surface charge distribution shows that the structure contains an outer positive area (red) and an inner negative are (blue) and that a 17Å central cleft is formed by the four helices (D) Comparison of our C3 crystal structure of the putative ryanodine receptor from Bacteroides thetaiotaomicron VPI-5482. Here the RyR repeats are shown individually as two RyR domain individual repeats (RyR domain 1 in gold and RyR domain 2 in blue) to aid in comparison to the putative ryanodine receptor from Bacteroides thetaiotaomicron VPI-5482 which was crystallised as a monomer.
A Dali search [25] revealed one Protein Data Bank (PDB) hit of significance, a protein of unknown function from one of the dominant intestinal microbiota, Bacteroides thetaiotaomicron VPI-5482, (PDB 3NRT). Interestingly, while the bacterial gene encoded one RyR domain, the structure revealed a homodimer. Structural comparison showed a conserved overall fold comprised of two long, right-handed α-helices which packed with an identical molecule in the asymmetric unit to create a bilobed arrangement similar to that seen in our structure (Figure 1C). This suggested that despite low primary amino acid sequence similarity (35% identity with first RyR motif) between the bacterial and mammalian proteins, the overall packing of these tandem motifs is likely quite important.
Interestingly, the feet of the bacterial structure were bound together by a magnesium atom coordinated by aspartic acids leaving a relatively large opening between the legs. A surface representation of the rabbit RyR (Figure 1D) revealed that the feet were unbound, but acidic residues did exist that might mediate similar interactions. The space between the two feet of the mammalian structure was approximately 17 Å from one side to the other and may accommodate either small molecules or peptides.
Docking of the RyR domain of in cryo-EM maps of RyR1
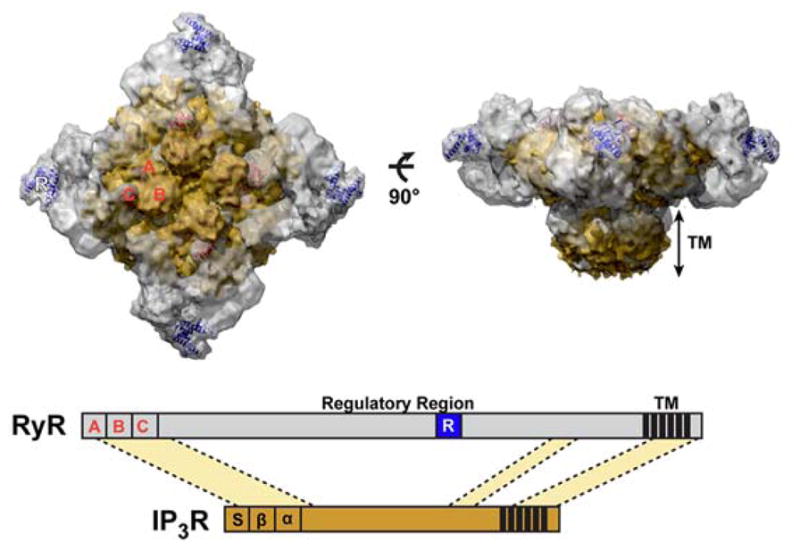
We applied a modeling approach using the SITUS program package [26]to perform rigid-body docking of our 2.2 Å C3 crystal structure into the previously determined 10.2Å cryo-EM structures of RyR1 in its open state (EMDB ID:1607) and closed state (EMDB ID: 1606). We monitored the top 10 solutions generated by the program and identified 2 possible locations for docking of the C3 crystal (Supplemental Figure 2). For the closed state the top 1-6 solutions mapped to subregion 3 whereas solutions 7-10 docked to subregion 10. For the open state the top 1-8 solutions mapped to domain 10 and solutions 9-10 mapped to domain 3 (Supplemental Figure 2). The low docking contrast of these solutions made both subregions equally plausible location for the RyR domain. Nonetheless, previous studies have provided evidence to support the structural fit of the C3 crystal into subregion 10 in the clamp region of RyR1. It has previously been determined that the epitope of the RyR1 primary antibody 34C included amino acid residues 2756 through 2803 of RyR1 [27] corresponding to residues 2722 through 2769 of RyR2 mapped to near the subregion 6 in the clamp domain in RyR2 [27,28] which is adjacent to our proposed binding domain 10 and a construct containing a GFP after Tyr-2801 localised to the bridge between domain 5 and 6 [27]. Therefore, we propose that our crystal structure docks into domain 10 of the 10.2Å cryo-EM structures of RyR1 (Figure 2A-C). Also after the deposition of our structure into the PDB database an independent study has also crystallised the phosphorylation region (2734-2940) of RyR1 which also docked the structure into domain 10 although they too emphasis ambiguity when docking this structure and provide multiple solutions of docking into the cryo-EM open structure [29]. In order to gain insight into the dynamic changes that accompany channel gating, we examined differences between the open and closed states (Figure 2D). Superposition of RyR1 in the open (blue ribbon with gray envelope) and closed (red ribbon and pink envelope) states suggested that some of the most prominent changes occur in the clamp region during gating which can be described as a downward shift of domains 10, 7, and 8a (indicated by the arrow in Figure 2D and supplemental movie 1) upon channel opening, in agreement with previous studies [16,30]. These observations suggested that this RyR domain is highly dynamic and either influences or is influenced by channel opening and closing. Interestingly, in comparison to the open state, the RyR domain fitted in the closed state is rotated ~180° about its pseudo-2-fold axis with the phosphorylation site pointing outward (Figure 2D).
Figure 2. Mapping of C3 crystal structure into Cryo-EM map of RyR1.

(A) Docking of the RyR domain into the cryo-EM map of RyR1 tetramer (EMDB-1607) (gray) in the open state. The best solution placed the RyR domain (blue) at Subregion 10 which is localized approximately 75 Å away from the N-terminal ABC region (purple) previously positioned in Subregions 1, 2a and 2b [18]. Previously determined locations of a GFP inserted after Y2801 of RyR2 and the monoclonal antibody specific for residues 2756-2803 of RyR1[27]are indicated by green and orange circles, respectively. Numbers indicate domain numbers (B) Side view of the docked molecules. (C) Close-up view of the RyR domain docked in the Subregion 10 showing the missing loop (residues 2831-2848) indicated by dashed line and phosphorylation site shown in red faces the centre of the tetramer but is located next to a cleft (D) Docking of the RyR domain in the cryo-EM maps of RyR1 in the open (blue ribbon with gray envelope) and closed (red ribbon and pink envelope) states. In comparison to the open state, the RyR domain in the closed state is rotated ~180° about its pseudo-2-fold axis with the phosphorylation site pointing outward
Disease mutation mapping
The C3 crystal structure contained six known human MH mutations E2764K [31], T2787S [32], R2840W [11,12], L2867G [31], E2880K [12], R2939K [33], the precise locations of which were analyzed (Figure 3). The R2840W mutation, being three residues upstream of the phosphorylation site (Ser2843) and localised to the missing loop, may represent a disruption of the interaction with the basophilic serine/threonine kinase and could be associated with decreased levels of phosphorylation at Ser2843. The E2764K and E2880K mutations interestingly not only have similar wild-type and mutant residues, they occurred on equivalent and symmetry-related and solvent-exposed regions of the protein and they both sat within negatively-charged patches. The E2764K mutation was located in α1 helix of the first RyR motif and the E2880K mutation was located in the opposite α3 helix in the second RyR motif. Equivalency suggested that these sites are involved in symmetric interactions and a charge reversal within negatively-charged patches could disrupt protein-protein interactions. Although the Thr2787 side-chain hydroxyl was involved in a weak hydrogen bond with the backbone carbonyl group of Asn2780, the T2787S mutation is a conserved change and is unlikely to affect possible hydrogen bonding interactions; how this mutation may affect structure was unclear. Leu2867 was part of the core residues and likely stabilized the conformational arrangement of the α3 (2869-2897) and α4 (2916-2933) helices and stabilized the conformation of an extended stretch of residues that bridged the two tandem domains. A mutation of this residue to glycine and the consequent absence of stabilizing hydrophobic interactions could affect the local and overall conformational flexibility and stability. The guanidinium group of Arg2939 formed bivalent interactions with two side-chains from α3 (Glu2870 and Gln2877), and its mutation to lysine could disrupt these interactions and destabilize the fold. Based on our fitting of this region in the molecular volume generated by electron microscopy, Arg2939 could be a solvent exposed residue forming part of an extended loop in the base of the structure, and its mutation could disrupt potential interaction with other proteins.
Figure 3. Mapping of MH mutations into C3 crystal structure.
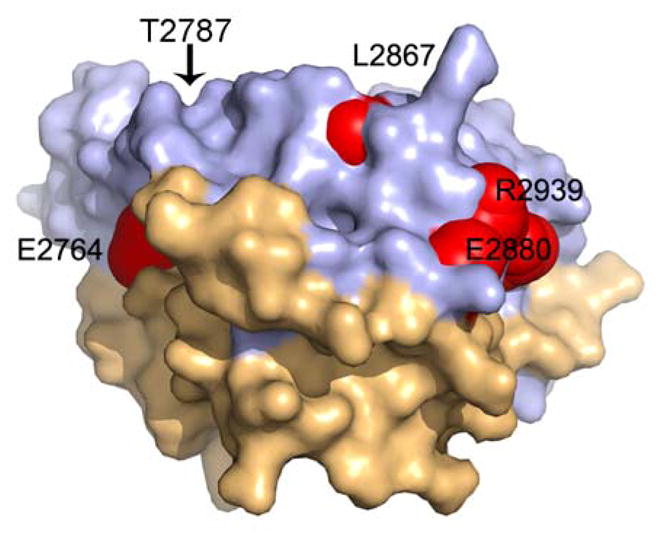
Mapping of the six known MH mutations onto our crystal structure shows that Glu2764, Glu2880, Thr2787, Arg2939 and are surface exposed residues where as Leu2867 although somewhat exposed, fits into a cleft defined by residue side-chains Q2924, L2927, K2928 and Q2931 and stabilizes the conformational arrangement of the α-3 (2869-2897) and α-4 (2916-2933) helices. The Arg2840 which is three residues upstream of the phosphorylation site localises to the missing loop in our structure.
Comparison to RyR2 and RyR3
Amino acid sequence comparison between the RyR domain of RyR1 and the equivalent domain in RyR2 (2699-2907) showed 64% sequence identity and between RyR1 and the equivalent domain in RyR3 (2599-2804) showed 62% sequence identity between the paralogs (Supplemental Figure 3). Mapping divergent residues (grey) onto a surface representation of the C3 crystal RyR domain (Figure 4A–D) indicated that the β-sheets, the surfaces of α2 and α3 helices and inner core of the structure were conserved. Divergence occurred in the α1 helix and in the loop region between α1 and α2 on one of the legs of the structure. Similarly comparison amongst seven species (Figure 4E–F and Supplemental Figure 3) also showed a conserved core; divergence occurred predominantly in the loop region between α1 and α2 on the left leg of the structure. These results were consistent with the notion that the inner curvature of the RyR domain plays a functional role, possibly by binding ligands.
Figure 4. Localisation of conserved and divergent residues mapped onto a surface representation of the C3 crystal structure.
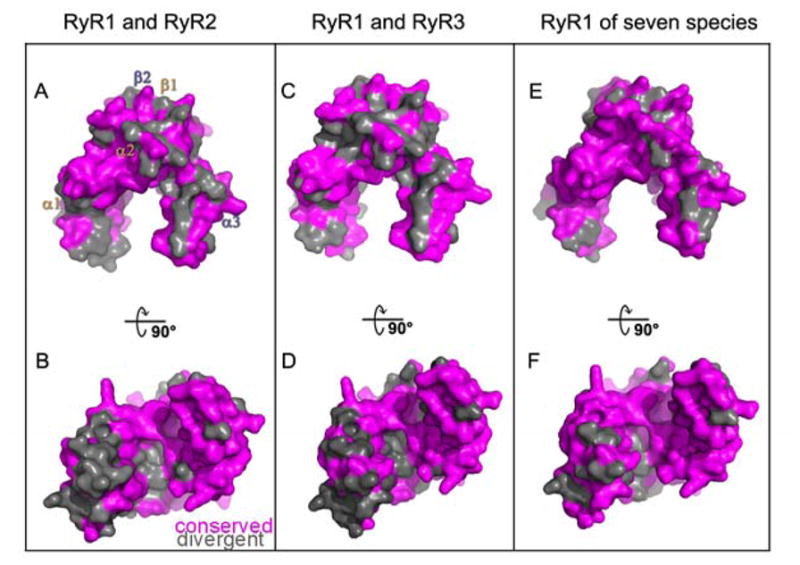
(A) Residues divergent (grey) and conserved (magenta) between RyR1 and RyR2 (B) Rotation of image in (A) by −90°. (C) Residues divergent (grey) and conserved (magenta) between RyR1 and RyR3 (D) Rotation of image in (C) by −90°(E) Residues divergent (grey) and conserved (magenta) between RyR1 isoforms across seven species listed in supplemental figure 3 (F) Rotation of image in (E) by −90°.
Comparison to the IP3 receptor
Along with the RyR receptor the Inositol 1,4,5-trisphosphate receptors (IP3R) represents the second principal channel responsible for mobilising internal calcium. IP3R are large intracellular Ca2+ release channels that like the RyR receptors form homotetramers and whose opening requires binding of two intracellular messengers IP3 and Ca2+ [34]. The two receptors share considerable structural and functional homologies and possess a similar basic architecture in the N-terminal region [17,35] and in the transmembrane domain (42). We superimposed the cryoEM structure of the IP3 receptor (EMDB-5278, yellow) and the cryoEM structure of the RyR1 in the open state (grey) aligned by their N-terminal regions (using the high similarity between the NT domain of IP3R-SD-IBC and RyR-ABC), the ABC domain of RyR [18,36] (Figure 6).Interestingly, the C3 crystal structure resides in the region which is located at a far protruded corner of the RyR structure (Figure 6) not present in the IP3 receptor, suggesting that the proposed phosphorylation-dependent modulation of the gating function at this site is unique to the RyR function.
Figure 6. CaMKII binding to RyR.
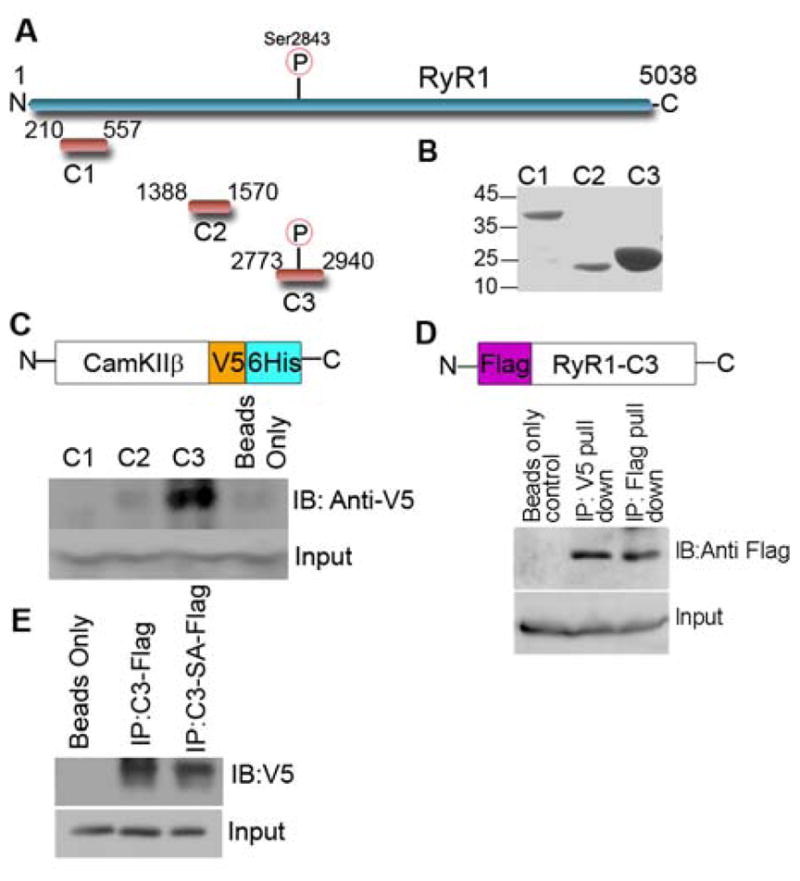
(A) Schematic showing constructs used for mass spectrometry analysis (B) Coomassie gel of purified RyR1 constructs in pET28a-LIC vector shows enrichment of each construct at the correct molecular weight (C) Schematic showing CaMKIIβ V5/6His constructs cloned into the pEF-DEST51 and western blot of C1-C3 constructs incubated with CaMKIIβ V5/6His showing binding only with C3 construct (D) Schematic showing C3 mammalian construct containing an N-terminal Flag tag used for immunoprecipitation and western blot analysis of the confirmation of binding between C3 and CaMKIIβ. Cell lysates from HEK-293 cells transfected with C3-Flag tagged and CaMKII–V5/6His tagged were subjected to immunoprecipitation experiments using the anti-V5 antibody (CaMKIIβ) (E) Western blot analysis of the binding of CaMKIIβ to wild type C3 construct and the C3S2843A mutant. Cell lysates from HEK-293 cells transfected with C3-Flag tagged, C3S2843A-Flag tagged and CaMKIIβ–V5/6His tagged were subjected to immunoprecipitation experiments using the anti-Flag antibody (RyR) and showed a 20% reduction of binding to the mutant when compared to wild type.
Binding of CaMKIIβ
CaMKII has been proposed previously to phosphorylate both RyR1 and RyR2 [2]. In order to confirm binding and determine the region-specificity of binding we cloned and amplified three constructs (C1-3) (Figure 5A) spanning various domains of the cytosolic region of RyR1 into the pET28a-LIC vector (GenBank™ EF442785). Purification of the three constructs using His-link resin yielded highly enriched, purified levels of each protein at their respective molecular weights (Figure 5B). The constructs were immobilised on His-link beads and incubated with mouse skeletal muscle lysate to allow the formation of protein interaction complexes between the RyR1 constructs and in vivo binding partners. Mass spectrometry analysis identified peptides that mapped to CaMKIIβ bound to the C3 construct but not to the C1 or C2 constructs (Table 1 and Supplemental Table 2). To validate this interaction, we immobilised the C1-C3 constructs in the bacterial pET28a-LIC vector onto His-link beads. We purified CaMKIIβ cloned into the pEF-DEST51 Gateway vector, which contained the dual tags V5 and 6-histidine tags overexpressed in HEK-293 cells and incubated this lysate with the immobilised C1-C3 constructs. The beads were then washed to remove non-specific binding and western blot analysis of boiled beads showed that CaMKIIβ was found bound only to the C3 construct when probed for using the anti-V5 antibody. Levels seen in the C1 and C2 western blots were nearly equivalent to the non-specific levels seen with empty beads (Figure 5C). This data verified the mass spectrometry findings and showed CaMKIIβ bound specifically to the C3 spanning region but not to the other constructs spanning the cytosolic region of RyR1. We further validated this interaction in a mammalian system by expressing RyR1 C3 into a mammalian vector containing an N-terminal Flag tag (Figure 5D) and incubating it with the cDNA for CaMKIIβ in the pEF-DEST51 Gateway vector. Cell lysates from HEK-293 cells transfected with Flag-tagged C3 and CaMKII–V5/6xHis were subjected to immunoprecipitation experiments using the anti-V5 antibody. Notably, CaMKII co-immunoprecipitated with C3 (Figure 5D), but neither protein was detected in the A/G resin alone or the immunoglobulin control IPs. Finally we examined the binding of CaMKIIβ to a S2843A-C3 phospho-deletion mutant. The S2843A mutant was expressed into the mammalian vector containing an N-terminal Flag tag and co-immunoprecipitated with CaMKII–V5/6xHis using the anti-Flag antibody. We found a reduction of approximately 20% in the amount of CaMKIIβ that co-immunoprecipitated the S2843A phospho-deletion mutant when compared to the wild type C3 construct (Figure 5E).
Figure 5. Comparison of RyR1 and the IP3 receptor.
(A) Overlay of the cryo-EM maps of RyR1 (EMDB-1607) (gray) and IP3R1 (EMDB-5278) (yellow) (B) Scheme of the primary sequence alignment of RyR1 and IP3R1. These proteins share 20–30% sequence identity in three different conserved regions (light yellow). The RyR domain (blue box) is present within the regulatory region of RyR, but not present in IP3R.
Table 1. Number of CaMKIIβ peptides found in each mass spectrometry run for RyR1 C1-C3 constructs.
The constructs were immobilised on His-link beads and incubated with mouse skeletal muscle lysate to allow the formation of protein interaction complexes between the RyR1 constructs and in vivo binding partners. Control spectra were obtained from beads only incubated with skeletal muscle lysate. All experiments were run in triplicate (n.d – none determined) and numbers in parentheses are total spectra for specific peptide.
| Number Of Spectra | |||||
|---|---|---|---|---|---|
|
| |||||
| Construct | Peptides | Total | Run 1 | Run 2 | Run 3 |
|
| |||||
| C1 | n.d. | 0 | 0 | 0 | 0 |
|
| |||||
| C2 | AGAYDFPSPEWDTVTPEAK (1) | 1 | 0 | 0 | 1 |
|
| |||||
| C3 | LHDSISEEGFHYLVFDLVTGGELFEDIVAR (15) | 17 | 4 | 8 | 5 |
| NLINQMLTINPAK (1) | |||||
| ESSDSTNTTIEDEDAK (1) | |||||
Discussion
The large size of the RyR protein has made its structural analysis a challenge. To overcome this challenge we generated a high-resolution crystal structure of a smaller domain containing the known phosphorylation site at Ser2843. This revealed the fold of this poorly understood domain and will provide the foundation necessary to elucidate the basis of interaction with binding partners and with other domains within the context of the full-length protein.
The clamp region of ryanodine receptors has been implicated as the likely site for: tetramerization interactions [30,37], regulatory modulator interactions, and post-translational modifications [27,30]. Phosphorylation, for example, has been hypothesized to be an allosteric modulator of channel opening (32). A comparison of RyR1 in the open and closed state shows that channel opening is associated with significant conformational changes of the cytoplasmic domain in particular of the clamp domains. Here, in keeping with the previous findings for this region in RyR2 [27], and the dynamic nature of the clamp region we mapped the domain containing the known phosphorylation site of RyR1 to clamp region 10 and propose that it is a mobile structure that contains the phosphorylation site as well as the binding site of CaMKIIβ which may in turn contribute to allosteric regulation.
In our RyR domain structure, disease-causing mutations fall within the following three categories: 1) mutations that may interfere with protein-protein interactions, including the solvent exposed E2764K, E2880K and R2939K, 2) mutations that likely disturb folding of this domain, including the L2867G mutation that is located between α3 and α4 helices, and 3) mutations that may affect the affinity with upstream protein kinases, including R2840W. The last was of particular interest because this mutation was in a dynamic loop three residues N-terminal of the known phosphorylation site [38,39]. In a systematic study using synthetic peptides to elucidate the minimal binding motif of CaMKII, a mutation at the Arg residue at position -3 from the phosphorylation motif, which is R-X-X-S/T where X can be any residue [38,39], reduced the Vmax/Km of binding of CaMKII to the substrate by ≥300 -fold [39] whereas mutations elsewhere in this motif had minimal effect on Vmax or Km. Therefore, the R2840W mutation would disrupt the essential CaMKII binding motif and it would cause reduction in phosphorylation of RyR1, the binding of regulatory modulators and ultimately in calcium dynamics of RyR1. Similarly, disease causing mutations within the recently solved crystal structure of the N-terminal domain of RyR1 (residues 1-559) were classified into three groups: 1) those that destabilized interfaces between the three amino-terminal domains, 2) those that disturbed folding of individual domains, and 3) those that affected interfaces between neighbouring domains within the receptor (21). Moreover, this study provides a framework of understanding the basis of the disease-associated mutations in this region of RyR1.
The structural and functional similarities between the RyR receptors and the IP3 receptors probably reflect a common evolutionary origin [35]. However, the lack of the clamp region in the IP3 receptor suggests that this region, where our C3 crystal fragment was docked, is likely involved in regulating either a functional or protein interaction that is unique to RyR. These unique functions may include differences recently reported in smooth muscle cells which found that RyRs underlie Ca2+ sparks, contribute to Ca2+ waves, and participate in the negative feedback regulation of myogenic tone in feed arteries but not downstream arterioles. In contrast the IP3Rs were found to contribute to Ca2+ waves and myogenic tone through a positive feedback mechanism in both feed arteries and their downstream arterioles [40].
CaMKII was shown to phosphorylate Ser2843 in RyR1 [2,41] and also Ser2814 in the equivalent region in RyR2 [42,43]. A comparison between the C3 regions in RyR1, RyR2 and RyR3 revealed that this region is highly homologous between the isoforms and that the main area of divergence is in the loop regions between helices that would thereby maintain the structural integrity of the identified α-helices and the inner core structure. These data imply that the conserved residues are critical to the structure and function of this domain throughout the two isoforms and that they may be involved in the binding of RyR and CaMKII. These findings are of critical interest since CaMKII has been shown to be upregulated and more active in congestive heart failure (HF) [44,45] a leading cause of mortality and morbidity worldwide with 50% of HF patients dying of sudden death syndrome attributed to ventricular arrhythmias [46]. Increased CaMKII-dependent phosphorylation of RyR2 appears to promote the development of ventricle arrhythmias, whereas the inhibition of CaMKII phosphorylation of RyR2 prevents ventricular arrhythmias in mice with heart failure [45,47]. The current RyR crystal structure also allows us to predict and model the interaction between RyR2 and CaMKII, however, the generation of a co-crystal structure of RyR together with CaMKII will allow us to speculate and develop pharmacological targets that may be used to modify this interaction and provide new avenues for treating ventricular arrhythmias.
The crystal structure elucidated in this study provided structural insight into understanding interaction, binding, and conformational changes that may occur in RyR1 during channel opening and closing, and how this region may be regulated by the known phosphorylation site. Interestingly, the docking of the C3 crystal in the open state showed orientation of the phosphorylation domain facing the centre of the tetramer however we showed that the C3 crystal has a 17Å space between the two legs which may accommodate either small molecules or peptides of CaMKII. It was also noted that when the C3 crystal structure is docked to the cryoEM RyR structure domain in the closed state it is rotated ~180° about its pseudo-2-fold axis with the phosphorylation site pointing outward and further investigations will be needed to determine which of these solutions is correct.
Following the release of our C3 crystal structure [23], an independent study has just crystallized the phosphorylation domain of the ryanodine receptors [29]. Their crystal structure is in agreement with ours and they too tentatively dock the crystal to domain 10 in the cryoEM structure of RyR1. Further, our study found that this region binds to CaMKII and that mutations to the known phosphorylation site (S2843A) caused a modest decrease in the ability of CaMKII binding to the region. Based on their phosphopeptide identification, they identify up to 4 additional sites in this region that are potentially phosphorylated by CaMKII which may explain why CaMKII remains associated with this domain even though the Ser2843 site is unavailable in our model. Together these studies increase our knowledge of a key regulatory domain in RyR receptors which carry human disease mutations although further informational will require an in depth analysis of neighboring domains and how they may effect or get effected by the architecture and movement of this small dynamic region in the open and closed state.
Experimental Procedures
Cloning, expression and purification of RyR1 construct
We designed, cloned, and purified 25 nested loop constructs of the RyR domain. Soluble constructs were screened in 192 sparse-matrix crystallization conditions, and one construct C3 (2734-2940) produced crystal hits. Two additional constructs located upstream of the phosphorylation region in RyR were referred to as C1 (804-1067) and C2 (1388-1570). These two constructs showed a high yield in purification but did not crystallize and thus were later used as negative controls to prove domain specific binding of CaMKIIβ. All constructs were cloned into the pET28a-LIC expression vector (GenBank, EF442785) using the In-Fusion CF Dry-Down PCR Cloning Kit (Clontech). Competent BL21 (DE3) cells (Invitrogen) were transformed and grown using the LEX system (HarbingerBiotech) at 37°C in 1L bottles containing 900 ml of TB (Sigma) supplemented with 150 mM glycerol, 100 μM kanamycin and 600 μl antifoam 204 (Sigma, A-8311). When the optical density reached a value of about 6.0, the temperature was reduced to 15°C, and one hour later the culture was induced with 1 mM IPTG (BioShop) and incubated overnight (16 hours) at 15°C. The protein was purified by the Streamline purification method using His-link resin from Promega as described previously [48]. The resin was washed 4 times using 10 mL of wash buffer (20 mM Tris-pH8.0, 500 mM NaCl and 20 mM imidazole-pH8.0). Protein was eluted with 8 ml of elution buffer (20 mM Tris-pH8.0, 500 mM NaCl and 500 mM imidazole-pH8.0) and dialyzed overnight at 4°C against 50 volumes of dialyses buffer (20 mM Tris-pH8.0, 200 mM NaCl and 5 mM DTT). The protein sample was concentrated using a 3 kDa MW cutoff concentrator (Millipore, UFC900524) at 3500 × g to a final value of 41 mg/ml. The protein yield was ~20 mg per liter of bacterial culture.
Crystallization
Crystals were grown at 18°C in sitting and hanging drop plates (Hampton, HR3-170) by mixing equal volumes of protein (41 mg/ml) and crystallization buffer (SGCB03; 1.2M Sodium citrate, 100mM Tris, pH 8.5). Crystals appeared in approximately two weeks. Suitable crystals were cryo-protected by immersion in well solution supplemented with 24% (v/v) ethylene glycol or by dipping in 100% paratone prior to flash freezing in liquid nitrogen.
Crystal structure determination and refinement
Two diffraction data sets were collected. One data set was collected on the Rigaku FR-E+ DW X-ray generator at an incident wavelength of 2.29Å, and a second, high resolution data set was collected on the 19ID beamline at Advanced Photon Source. All data sets were integrated and scaled using the HKL3000 program suite [49]. Diffraction data collection and processing statistics for the two data sets are presented in Supplemental Table 1. The crystals belong to the space group P43212, and the structure was refined with the cell parameters a=68.77, b=68.77, c=91.38, α=β=γ=90° (from high resolution data). The structure was solved by the sulfur single-wavelength anomalous dispersion (S-SAD) method using the data collected on the FR-E DW. The programs SHELX [50] and SOLVE/RESOLVE [51,52] were used to locate the sulfur sites and carry out initial model building. The high-resolution data collected on the 19ID beamline at APS was used for structure refinement. Iterative model building was carried out using the graphics program Coot [53] while maximum-likelihood refinement was carried out using Buster 2.8.0 [54]. Structure refinement statistics are provided in Supplemental Table 1.
Computational docking of the phosphorylation domain
Rigid-body docking of the phosphorylation domain structure into ~10 Å cryo-electron microscopy density maps of RyR1 (EMDB-1606, 1607 and 1275) was implemented using the six-dimensional search procedure in the Situs Program package [26]. To enhance the fitting contrast, the Laplacian filter was applied and the density corresponding to the transmembrane region was omitted from the search. Docking of the crystal structure of RyR1-ABC (PDB, 2XOA) was also performed using the above procedure. The UCSF Chimera package [55]was used to visualize the docking results with the density maps, as well as the comparison of cyro_EM maps of RyR1 and InsP3R1 (EMDB, EMDB-5278).
Mass Spectrometry
Gel-free shotgun sequencing of tryptic digests was performed essentially as described previously [19]. Briefly, individual samples were first loaded onto microcapillary fused silica columns with an internal diameter of 75 μm, packed with 7 cm of reversed phase C18 resin (Magic C18). The columns were aligned with an LTQ linear ion trap mass spectrometer, and the peptides were eluted using a 2 hour water/acetonitrile gradient and ionized via electrospray ionization. The tandem mass spectra generated were matched to peptide sequences in the human IPI protein sequence database (http://www.ebi.ac.uk/IPI) using the X!Tandem algorithm (http://gpmdb.thegpm.org/).
Co-immunoprecipitation and western blot
Standard techniques were used to carry out co-immunoprecipitation and western blot as described previously [19,56]. Antibodies used were anti-V5 (Invitrogen) and anti-His (Abcam).
Supplementary Material
Supplemental Figure 1: C3 construct sequence analysis.
Supplemental Figure 2: Docking of the RyR domain into the cryo-EM maps of RyR.
Supplemental Figure 3: C3 construct sequence analysis between RyR1 and RyR2.
Supplemental Table 1: Diffraction data collection, processing and structure refinement statistics.
Supplemental Table 2: CaMKIIβ peptides found in each mass spectrometry run for RyR1 C1-C3 constructs.
Supplemental Movie 1: Dynamic movements of the RyR domain docking site.
Acknowledgments
The authors would like to thank Vladimir Ignatchenko for excellent bioinformatic support. MI is supported by the Heart and Stroke Foundation of Ontario and the Canada Research Chair program. This work was funded by the Canadian Institutes of Health Research (CIHR) (MOP-84267 to AOG and TK), Heart and Stroke Foundation of Ontario (AOG: T-6281), an Early Research Award from the Ontario Ministry of Research and Innovation (AOG), and an unrestricted grant from Boehringer Ingelheim Canada. AOG, MI and TK are Canada Research Chairs, and AOG was a New Investigator of the Heart and Stroke Foundation of Canada. TR held a Heart and Stroke/Richard Lewar Centre of Cardiovascular Excellence Studentship and a Margaret Santalo Fellowship from the University of Toronto. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust.
The abbreviations used are
- RyR1
Ryanodine receptor 1
- CCD
Central Core Disease
- MH
Malignant Hyperthermia
- PKA
Protein Kinase A
- CaMK
Calcium/Calmodulin dependent protein kinase
References
- 1.Nakai J, Sekiguchi N, Rando TA, Allen PD, Beam KG. Two Regions of the Ryanodine Receptor Involved in Coupling withl-Type Ca2+ Channels. Journal of Biological Chemistry. 1998;273:13403–13406. doi: 10.1074/jbc.273.22.13403. [DOI] [PubMed] [Google Scholar]
- 2.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb Perspect Biol. 2010:2. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacLennan DH, Phillips MS. Malignant hyperthermia. Science. 1992;256:789–794. doi: 10.1126/science.1589759. [DOI] [PubMed] [Google Scholar]
- 4.Loke J, MacLennan DH. Malignant Hyperthermia and Central Core Disease: Disorders of Ca2 Release Channels. The American Journal of Medicine. 1998;104:470–486. doi: 10.1016/s0002-9343(98)00108-9. [DOI] [PubMed] [Google Scholar]
- 5.MacLennan DH. Ca2+ signalling and muscle disease. European Journal of Biochemistry. 2000;267:5291–5297. doi: 10.1046/j.1432-1327.2000.01566.x. [DOI] [PubMed] [Google Scholar]
- 6.Inui M, Saito A, Fleischer S. Isolation of the ryanodine receptor from cardiac sarcoplasmic reticulum and identity with the feet structures. Journal of Biological Chemistry. 1987;262:15637–15642. [PubMed] [Google Scholar]
- 7.Imagawa T, Smith JS, Coronado R, Campbell KP. Purified ryanodine receptor from skeletal muscle sarcoplasmic reticulum is the Ca2+-permeable pore of the calcium release channel. Journal of Biological Chemistry. 1987;262:16636–43. [PubMed] [Google Scholar]
- 8.Lai FA, Erickson H, Block BA, Meissner G. Evidence for a junctional feet-ryanodine receptor complex from sarcoplasmic reticulum. Biochemical and Biophysical Research Communications. 1987;143:704–709. doi: 10.1016/0006-291x(87)91411-2. [DOI] [PubMed] [Google Scholar]
- 9.Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, Meissner G, MacLennan DH. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. Journal of Biological Chemistry. 1990;265:2244–2256. [PubMed] [Google Scholar]
- 10.Suko J, Maurer-Fogy I, Plank B, Bertel O, Wyskovsky W, Hohenegger M, Hellmann G. Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1993;1175:193–206. doi: 10.1016/0167-4889(93)90023-i. [DOI] [PubMed] [Google Scholar]
- 11.Ibarra MCA, et al. Malignant hyperthermia in Japan: mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology. 2006;104:1146–54. doi: 10.1097/00000542-200606000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Human Mutation. 2006;27:977–989. doi: 10.1002/humu.20356. [DOI] [PubMed] [Google Scholar]
- 13.Serysheva II, Orlova EV, Chiu W, Sherman MB, Hamilton SL, Heel Mv. Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat Struct Mol Biol. 1995;2:18–24. doi: 10.1038/nsb0195-18. [DOI] [PubMed] [Google Scholar]
- 14.Radermacher M, Rao V, Grassucci R, Frank J, Timerman AP, Fleischer S, TW Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J Cell Biol. 1994;127:411–423. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samso M, Wagenknecht T, Allen PD. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat Struct Mol Biol. 2005;12:539–544. doi: 10.1038/nsmb938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samso M, Feng W, Pessah I, Allen PD. Coordinated Movement of Cytoplasmic and Transmembrane Domains of RyR1 upon Gating. PLoS Biol. 2009;7:e1000085. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amador FJ, Liu S, Ishiyama N, Plevin MJ, Wilson A, MacLennan DH, Ikura M. Crystal structure of type I ryanodine receptor amino-terminal trefoil domain reveals a disease-associated mutation hot spot loop. PNAS. 2009;106:11040–11044. doi: 10.1073/pnas.0905186106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tung CC, Lobo PA, Kimlicka L, Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- 19.Ryan T, Sharma P, Ignatchenko A, MacLennan DH, Kislinger T, Gramolini AO. Identification of Novel Ryanodine Receptor 1 (RyR1) Protein Interaction with Calcium Homeostasis Endoplasmic Reticulum Protein (CHERP) Journal of Biological Chemistry. 2011;286:17060–17068. doi: 10.1074/jbc.M110.197186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex Formation between Junctin, Triadin, Calsequestrin, and the Ryanodine Receptor. Journal of Biological Chemistry. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 21.Brillantes AMB, et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 22.Dolinski K, DB Orthology and Functional Conservation in Eukaryotes. Annual Review of Genetics. 2007;41:465–507. doi: 10.1146/annurev.genet.40.110405.090439. [DOI] [PubMed] [Google Scholar]
- 23.Sharma P, et al. Comprehensive Analysis of Ryanodine Receptor Through Proteomic Interaction Analysis and High Resolution Crystal Structure Determination. Circulation. 2011;124:A15352. [Google Scholar]
- 24.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holm L, Kääriäinen S, Wilton C, Plewczynski D. John Wiley & Sons, Inc, editor. Current Protocols in Bioinformatics. 2002. Using Dali for Structural Comparison of Proteins. [DOI] [PubMed] [Google Scholar]
- 26.Wriggers W, Milligan RA, McCammon JA. Situs: A Package for Docking Crystal Structures into Low-Resolution Maps from Electron Microscopy. Journal of Structural Biology. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 27.Meng X, et al. Three-Dimensional Localization of Serine 2808, a Phosphorylation Site in Cardiac Ryanodine Receptor. Journal of Biological Chemistry. 2007;282:25929–25939. doi: 10.1074/jbc.M704474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones P, Meng X, BX, SC, Bolstad J, Wagenknecht T, LZ, Chen SRW. Localization of PKA phosphorylation site, Ser2030, in the three-dimensional structure of cardiac ryanodine receptor. Biochemical Journal. 2008;410:261–270. doi: 10.1042/BJ20071257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuchi Z, Lau K, Van Petegem F. Disease Mutations in the Ryanodine Receptor Central Region: Crystal Structures of a Phosphorylation Hot Spot Domain. Structure. 2012 doi: 10.1016/j.str.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton SL, Serysheva II. Ryanodine Receptor Structure: Progress and Challenges. Journal of Biological Chemistry. 2009;284:4047–4051. doi: 10.1074/jbc.R800054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galli L, Orrico A, Lorenzini S, Censini S, Falciani M, Covacci A, Tegazzin V, Sorrentino V. Frequency and localization of mutations in the 106 exons of the RYR1 gene in 50 individuals with malignant hyperthermia. Human Mutation. 2006;27:830–830. doi: 10.1002/humu.9442. [DOI] [PubMed] [Google Scholar]
- 32.Monnier N, et al. Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Human Mutation. 2005;26:413–425. doi: 10.1002/humu.20231. [DOI] [PubMed] [Google Scholar]
- 33.Zhou H, et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain. 2007;130:2024–2036. doi: 10.1093/brain/awm096. [DOI] [PubMed] [Google Scholar]
- 34.Bosanac I, Michikawa T, Mikoshiba K, Ikura M. Structural insights into the regulatory mechanism of IP3 receptor. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2004;1742:89–102. doi: 10.1016/j.bbamcr.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 35.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 36.Seo MD, et al. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature. 2012;483:108–112. doi: 10.1038/nature10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. Journal Of Cell Biology. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson RB, Woodgett JR, Cohen P, Kemp BE. Substrate specificity of a multifunctional calmodulin-dependent protein kinase. Journal of Biological Chemistry. 1985;260:14471–14476. [PubMed] [Google Scholar]
- 39.White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM. Definition of Optimal Substrate Recognition Motifs of Ca2+-Calmodulin-dependent Protein Kinases IV and II Reveals Shared and Distinctive Features. Journal of Biological Chemistry. 1998;273:3166–3172. doi: 10.1074/jbc.273.6.3166. [DOI] [PubMed] [Google Scholar]
- 40.Westcott EB, Jackson WF. Heterogeneous function of ryanodine receptors, but not IP3 receptors, in hamster cremaster muscle feed arteries and arterioles. American Journal of Physiology - Heart and Cirulatory Physiology. 2011;300:H1616–H1630. doi: 10.1152/ajpheart.00728.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu A, Sumbilla C, Inesi G, Jay SD, KPC Specific association of calmodulin-dependent protein kinase and related substrates with the junctional sarcoplasmic reticulum of skeletal muscle. Biochemistry. 1990;29:5899–905. doi: 10.1021/bi00477a003. [DOI] [PubMed] [Google Scholar]
- 42.Wehrens XHT, Lehnart SE, Reiken SR, Marks AR. Ca2+/Calmodulin-Dependent Protein Kinase II Phosphorylation Regulates the Cardiac Ryanodine Receptor. Circulation Research. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 43.Said M, et al. Calcium-calmodulin dependent protein kinase II (CaMKII): A main signal responsible for early reperfusion arrhythmias. J Mol Cell Cardiol. 2011;51:936–944. doi: 10.1016/j.yjmcc.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pogwizd SM, Bers DM. Cellular Basis of Triggered Arrhythmias in Heart Failure. Trends in Cardiovascular Medicine. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 45.van Oort RJ, et al. Ryanodine Receptor Phosphorylation by Calcium/Calmodulin-Dependent Protein Kinase II Promotes Life-Threatening Ventricular Arrhythmias in Mice With Heart Failure/Clinical Perspective. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farr MA, Basson CT. Sparking the Failing Heart. The New England Journal of Medicine. 2004;351:185–187. doi: 10.1056/NEJMcibr041466. [DOI] [PubMed] [Google Scholar]
- 47.Dobrev D, Wehrens XHT. Calmodulin Kinase II, Sarcoplasmic Reticulum Ca2+ Leak, and Atrial Fibrillation. Trends in Cardiovascular Medicine. 2010;20:30–34. doi: 10.1016/j.tcm.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alenkin D, Yermekbayeva L, Mujib S, Vesterberg A, Newman E, Yamazaki K, Cossar D, Dhe-Paganon S. A centrifugation-free high-throughput protein purification system using in-line microfluidization. Protein Expression and Purification. 2011;79:204–209. doi: 10.1016/j.pep.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 49.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallographica Section D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 50.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;A64:112–22. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 51.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallographica Section D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terwilliger T. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallographica Section D. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Bricogne G, et al. BUSTER, version 2.8.0. Cambridge, United Kingdom: Global Phasing Ltd; 2009. [Google Scholar]
- 55.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 56.Brini M, Manni S, Pierobon N, Du GG, Sharma P, MacLennan DH, Carafoli E. Ca2+ Signaling in HEK-293 and Skeletal Muscle Cells Expressing Recombinant Ryanodine Receptors Harboring Malignant Hyperthermia and Central Core Disease Mutations. Journal of Biological Chemistry. 2005;280:15380–15389. doi: 10.1074/jbc.M410421200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: C3 construct sequence analysis.
Supplemental Figure 2: Docking of the RyR domain into the cryo-EM maps of RyR.
Supplemental Figure 3: C3 construct sequence analysis between RyR1 and RyR2.
Supplemental Table 1: Diffraction data collection, processing and structure refinement statistics.
Supplemental Table 2: CaMKIIβ peptides found in each mass spectrometry run for RyR1 C1-C3 constructs.
Supplemental Movie 1: Dynamic movements of the RyR domain docking site.