

Abstract
Werner syndrome (WS) is a premature aging disorder caused by mutations in a RecQ-like DNA helicase. Mice lacking the helicase domain of the WRN homologue exhibit many phenotypic features of WS, including a prooxidant status and a shorter mean life span compared to wild-type animals. Here, we show that Wrn mutant mice also develop premature liver sinusoidal endothelial defenestration along with inflammation and metabolic syndrome. Vitamin C supplementation rescued the shorter mean life span of Wrn mutant mice and reversed several age-related abnormalities in adipose tissues and liver endothelial defenestration, genomic integrity, and inflammatory status. At the molecular level, phosphorylation of age-related stress markers like Akt kinase-specific substrates and the transcription factor NF-κB, as well as protein kinase Cδ and Hif-1α transcription factor levels, which are increased in the liver of Wrn mutants, were normalized by vitamin C. Vitamin C also increased the transcriptional regulator of lipid metabolism PPARα. Finally, microarray and gene set enrichment analyses on liver tissues revealed that vitamin C decreased genes normally up-regulated in human WS fibroblasts and cancers, and it increased genes involved in tissue injury response and adipocyte dedifferentiation in obese mice. Vitamin C did not have such effect on wild-type mice. These results indicate that vitamin C supplementation could be beneficial for patients with WS.
Keywords: ascorbate, metabolism, microarrays, liver, adipocyte, inflammation
Aging is defined as a progressive deterioration of physiological functions that impairs the ability of an organism to maintain its homeostasis and consequently increases susceptibility to disease and death. Much of the recent progress in the understanding of aging has been fueled by the study of human progeroid syndromes (1). One fascinating human aging disorder is Werner syndrome (WS). WS is a human autosomal recessive disorder characterized by genomic instability and the premature onset of a number of age-related diseases, including osteoporosis, ocular cataracts, graying and loss of hair, diabetes mellitus, arteriosclerosis, and atherosclerosis (2–5). It is, to our knowledge, the human disease model closest to normal aging. The defective enzyme responsible for WS is a helicase/exonuclease involved in DNA repair, replication, transcription, and telomere maintenance (6–10). We previously generated a mouse model with a deletion in the helicase domain of the murine WRN homologue (11) that recapitulates most of the WS phenotypes, including an abnormal hyaluronic acid excretion, higher reactive oxygen species (ROS) levels, increased genomic instability, and cancer incidence resulting in a 10 –15% decreased mean life span (12, 13). In addition, both humans with WS and WrnΔhel/Δhel mice show hallmarks of a metabolic syndrome, including premature visceral obesity, hypertriglyceridemia, insulin-resistant type 2 diabetes, and associated cardiovascular diseases (13). This is in contrast to Wrn-null mice, which do not exhibit the same phenotype as WrnΔhel/Δhel mice (14). However, Wrn-null mice fed with a high-carbohydrate diet develop hyperinsulinemia and insulin resistance (15). Notably, a cross between Wrn-null mice and mice defective in telomerase activity resulted (after several back-crosses) in the generation of progenies exhibiting metabolic abnormalities reminiscent of a severe premature aging (16). Metabolic syndrome afflicts up to one-half of Western population and is considered an age-related, proinflammatory lipidic disorder (17). Identifying compounds that ameliorate premature aging syndromes or metabolic abnormalities has thus become of major interest for both of these syndromes and for aging more generally. The WrnΔhel/Δhel mouse provides a unique opportunity to test such compounds. In this study, we assessed the effect of vitamin C on metabolic parameters and hepatic tissue status of WrnΔhel/Δhel mice.
The liver plays a pivotal role in nutrient, drug, hormone, and metabolic waste product processing, thereby maintaining body homeostasis. The liver undergoes substantial changes in structure and function in old age. There are age-related changes in liver mass, blood flow, and hepatocyte as well as sinusoidal cell morphology (18). These changes are associated with significant impairment of many hepatic metabolic and detoxification activities, with implications for systemic aging and age-related disease. For example, the age-related impairment of the hepatic metabolism of lipoproteins predisposes to cardiovascular disease (19). We therefore hypothesized that the metabolic syndrome developed by WrnΔhel/Δhel mice is due to an abnormal regulation of the redox environment and a rise in reactive oxygen species (ROS) production leading to a premature liver dysfunction normally associated with age. Here, we show for the first time that, in addition to dyslipidemia, WrnΔhel/Δhel mice exhibit a severe premature liver sinusoidal endothelial defenestration, a phenotype normally associated with old age (18). Notably, long-term vitamin C treatment restored healthy aging in WrnΔhel/Δhel mice by reversing the activity and levels of several stress and inflammatory markers, while vitamin C had no effect on wild-type (WT) mice. Since, unlike mice, humans can only rely on external sources of ascorbic acid (they are not able to synthesize it de novo), our results suggest that a continuous long-term treatment of WS with high doses of vitamin C is a promising therapeutic approach for this syndrome.
MATERIALS AND METHODS
Animal model
Mice lacking part of the helicase domain of the Wrn gene were generated by homologous recombination, as described previously (11). In the process, 121 amino acid residues of the Wrn protein were deleted (aa 710 to 831). This study was performed on WT and WrnΔhel/Δhel homozygous animals on the pure C57BL/6 genetic background. Care of mice was in accordance with the guidelines of the Centre de Recherche en Cancérologie de l’Université Laval. Mice were housed in cages (containing a top filter) on static racks in a conventional animal facility at 22 ± 2°C with 40–50% humidity and a 12:12-h light-dark cycle (light cycle: 6:00 AM to 6:00 PM). All mice were fed ad libitum with Teklad Global (Madison, WI, USA) 18% protein rodent diet (5% fat). Animals were checked every day for any external mass, infection, bleeding, gasping, and overall decrease or change in activity or behavior. Mice that became immobile or moribund were sacrificed for histological examination of their organs, as described previously (12). A cohort of WrnΔhel/Δhel homozygous animals was given 0.4% L-ascorbate (w/v) in drinking water (20) for the indicated time.
Blood analysis
Blood was harvested by puncturing the leg saphenous vein of mice immobilized under contention. No anesthesia was employed. Serum triglyceride levels were measured by an enzymatic method using a reagent kit from Roche Diagnostics (Indianapolis, IN, USA) that allows correction for free glycerol. Blood glucose concentration was measured with a FreeStyle Mini glucose meter (TheraSense, Kent, UK). Serum insulin was measured with the ultrasensitive mouse insulin ELISA assay kit from Alpco Diagnostics (Windham, NH, USA). An index of fasting insulin resistance, consisting of the product of fasting glucose and insulin, was calculated accordingly to the homeostasis model assessment of insulin resistance (HOMA-IR) described previously (21). Ascorbic acid in blood or tissue extract was measured with the ferric reducing ascorbate assay kit from BioVision Research Products (Mountain View, CA, USA).
Detection of ROS and oxidative DNA damage in tissues
ROS and oxidative DNA damage measurements were performed as described previously (13). Briefly, liver tissues were excised, rinsed with PBS to remove blood, and homogenized with a Dremel tissue homogenizer (BioSpec Products, Bartlesville, OK) in 1 ml of RIPA buffer (50 mM Tris HCl, pH 7.5; 150 mM NaCl; 1% Nonidet P-40; 0.1% SDS; and 0.5% deoxycholate). The homogenate (200 μl) was incubated with 10 μg/ml of the dye 2′-7′ DCF diacetate (Sigma-Aldrich Canada, Oakville, ON, Canada) for 1 h at 37°C. This dye is highly fluorescent on oxidation. The oxidized dye was measured as described previously (13). Protein concentrations were measured using the Bradford assay. The remaining tissues were embedded in paraffin for histology. Oxidative DNA damage was measured as described previously (13).
Sequencing of mitochondrial DNA
Mitochondrial DNA liver tissues were extracted from 10 mice as described previously (22). The D-loop control region of mitochondrial DNA was amplified and cloned into the PCR2.1 vector (Invitrogen, Carlsbad, CA, USA) for sequencing. Eight to 10 clones of each genotype and tissue were sequenced. The primers used for the PCR were MTC1 GCCAACTAGCCTCCATCTCATACTT, nt 15196–15220; and MTC2 GGGCGGGTTGTTGGTTTCAC, nt 15701–15720.
Measurements of reduced glutathione
Blood glutathione (GSH) levels were quantified with the ApoGSH Glutathione Detection Kit from BioVision Research Products. The assay utilizes monochlorobimane (MCB), a dye that appears to form adduct exclusively with GSH. The GSH-bound MCB dye fluoresces blue. Fluorescence was measured with a Fluoroskan Ascent fluorescence spectrophotometer (Thermo Electron, Milford, MA, USA). The excitation and emission wavelengths used were 355 and 460 nm, respectively. For hepatic GSH tissue levels, livers were excised, rinsed with PBS to remove blood, and homogenized with a Dremel tissue homogenizer (BioSpec Products) in 1 ml of RIPA buffer. GSH was measured from the homogenate. Protein concentrations were measured using the Bradford assay.
Measurements of ATP
Liver ATP levels were quantified with the ApoSensor ADP/ATP ratio assay kit from BioVision Research Products. The assay uses the enzyme luciferase to catalyze the formation of light from ATP and luciferin. Luminescence was measured with a Luminoskan Ascent luminometer (Thermo Electron). Livers were excised, rinsed with PBS to remove blood, and homogenized with a Dremel tissue homogenizer (BioSpec Products) in 1 ml of RIPA buffer. ATP was measured from the homogenate. Protein concentrations were measured using the Bradford assay.
Electron microscopy of the liver sinusoid
Livers were harvested from 5 WrnΔhel/Δhel control mice and 5 WrnΔhel/Δhel mice that had been treated with ascorbate, all aged 6–9 months. Liver specimens were fixed for electron microscopy, as described previously (23). The specimens were perfused with fixative by injecting an aliquot of fixative (2% glutaraldehyde/3% paraformaldehyde in 0.1 M sodium cacodylate buffer, 0.1 M sucrose, and 2 mM CaCl2) into the specimen with a 25-gauge needle until the tissue hardened. Tissue was postfixed for 2 h. Fixed liver tissue was treated with 1% (w/v) osmium, then dehydrated in an ethanol gradient to 100%, and incubated for 10 min in hexamethyl-disilazane. Samples were mounted and sputter-coated with gold. Samples for scanning microscopy were examined using a Jeol JSM 6380LV scanning electron microscope (Jeol Ltd., Akishima, Japan). Ten random sinusoids were photographed from each liver. Analysis of fenestral diameter and frequency was made using the ImageJ image analysis program (U.S. National Institutes of Health, Bethesda, MD, USA) and excluding gaps (>300 nm diameter). Porosity was determined as the percentage of the endothelial surface perforated by fenestrations and is a function of both diameter and frequency of fenestrations. The number of fenestrations counted was 768 in the WrnΔhel/Δhel controls and 3337 in the ascorbate-treated WrnΔhel/Δhel mice.
Protein analysis
Protein extraction and Western blotting were performed as described previously (24). Antibodies against phospho-Akt substrates, phospho-NF-κB, NF-κB, phospho-p38, p38, phospho-SAP/JNK, SAP/JNK, PKCδ, and phospho-PKCδ were purchased from Cell Signaling Technology (Danvers, MA, USA). The anti-TNF-α antibody was purchased from R&D Systems (Minneapolis, MN, USA). Antibodies against senescence marker protein 30 (SMP30), CTGF, and PPARα were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-Hif-1α polyclonal antibody was a kind gift from Dr. D. Richard (Centre de Recherche en Cancérologie, Quebec, QC, Canada). Finally, all horseradish peroxidase-conjugated secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ, USA). Proteins on the Western blots were visualized using an ECL kit (Amersham Biosciences). All antibodies were used as indicated by the manufacturers.
Microarray analysis
Total RNA from the liver of 9-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice was extracted on a CsCl cushion. RNAs were processed at the functional genomic platform of the Institut de Recherche Clinique de Montreal (Montreal, QC, Canada) and were hybridized onto mouse Agilent 60-mer Oligo Microarrays (44,000 genes/microarray; Agilent Technologies, Santa Clara, CA, USA) in quadruplicates with dye swap. Arrays were scanned on an Axon GenePix 4000B (Axon Instruments, Foster City, CA, USA). Data were extracted from images using GenePix 6.0 (Axon) with local background estimation. Lists of differentially expressed genes were generated using limma in BioConductor (http://www.bioconductor.org). The data were background subtracted using normexp and also normalized using the loess method. Correction for multiple hypothesis testing was performed using Benjamini-Hochberg. We have deposited all the raw data (accession number GSE14549) in the U.S. National Center for Biotechnology Information public database Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). Gene set enrichment analysis (GSEA) was applied using default parameters on a fold-change-sorted list of genes on the C2 curated gene sets database (25). Such databases contain gene sets from known pathways, expert-created pathways, and also gene sets extracted from PubMed publications. Real-time RT-PCR was performed with the FastStart SYBR Green Master kit (Roche Diagnostics) according to the manufacturer’s protocol.
Lactonase (SMP30) activity
SMP30 activity was measured by the change in absorbance of the pH indicator p-nitrophenol caused by free acid formation from the D-glucono-δ-lactone, as described previously (26). Briefly, the reaction buffer contained 10 mM D-glucono-δ-lactone, 10 mM Pipes (pH 6.4), 0.25 mM p-nitrophenol, 75 μM ZnCl2, and 2 μl of liver extract in a total volume of 200 μl. Livers were removed from mice and homogenized with ice-cold homogenization buffer (10 mM Tris·HCl, pH 8.0/1 mM phenyl methanesulfonyl fluoride) for 30 s at high speed with a Polytron homogenizer (Kinematica AG, Lucerne, Switzerland). The homogenate was centrifuged at 10,000 g for 10 min. The protein concentration of the sample was determined by the standard Bradford assay. The substrate solution was freshly prepared immediately before the assay. The reaction was followed by monitoring a decrease in absorbance at 405 nm. The rate of spontaneous hydrolysis of the lactone was subtracted from the total rate.
Thioredoxin reductase activity
The thioredoxin reductase activity was measured with a thioredoxin reductase assay kit (BioVision Research Products) following the instructions of the manufacturer. Briefly, in this assay thioredoxin reductase catalyzes the reduction of 5,5′-dithio-bis-(2-nitrobenzoic) acid (DTNB) with NADPH to 5-thio-2-nitrobenzoic acid (TNB2−), which generates a strong yellow color (λmax = 405 nm). Since in crude biological samples, other enzymes, such as glutathione reductase and glutathione peroxidase, can also reduce DTNB, a thioredoxin reductase-specific inhibitor was used to determine thioredoxin reductase specific activity. Two assays were performed: the first measured the total DTNB reduction by the sample, and the second measured the DTNB reduction by the sample in the presence of the thioredoxin reductase-specific inhibitor. The difference between the two results is the DTNB reduction by thioredoxin reductase.
Cytochrome-b5-reductase activity
The cytochrome-b5-reductase activity was measured as described previously (27, 28). Briefly, liver tissues were homogenized in ice-cold 0.25 M sucrose and 10 mM HEPES-NaOH, pH 7.8 (iso-osmotic buffer, 4 ml/g of liver) with a cocktail of protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA). The homogenate was centrifuged at 1000 g for 20 min at 4°C. The supernatant was then centrifuged at 12,000 g for 20 min at 4°C. The latter supernatant contained membranes (endoplasmic reticulum, mitochondrial, and cellular). The protein concentration of the sample was determined by the standard Bradford assay. The enzymatic reaction was carried out in a 50 mM phosphate buffer, pH 7.5 (27, 28), in the presence of 0.3 mM KCN (to inhibit oxidases) and 2 μM rotenone (to inhibit mitochondrial NADH cytochrome C reductases). The reaction was started by the addition of 0.5 mM NADH. Reduction of cytochromes was followed by monitoring the increase in absorbance at 550 nm with time.
Statistical analysis
Data are presented as means ± SE. The unpaired Student’s, Kolmogorov-Smirnov’s test for normality and the log-rank tests were all performed using an α level of 0.05 and a 2-sided hypothesis. Mitochondrial mutation rates were analyzed by the Wilcoxon test using an alpha level of 0.05 and a one-sided hypothesis. Differences between cohorts were considered significant at values of P < 0.05 in all statistical analyses. All statistical analyses were performed with R 2.6.0 (http://www.r-project.org).
RESULTS
WrnΔhel/Δhel mice exhibit decreased blood GSH levels and increased liver oxidative stress
Patients with WS exhibit an imbalance in plasma GSH levels (29), suggesting a prooxidant status in such individuals. We thus examined GSH plasma levels in 4-and 9-mo-old WrnΔhel/Δhel and WT mice. As indicated in Fig. 1A, WT and WrnΔhel/Δhel mice increased plasma GSH levels with age (2.5-fold increase from 4- to 9-mo-old animals). However, GSH levels were always significantly lower in WrnΔhel/Δhel mice (~20% lower). GSH is an important endogenous antioxidant produced by the liver. Thus, we examined ROS and oxidative DNA damage in liver tissues of these animals. There was no significant increase in ROS and oxidative DNA damage at 4 mo of age between WT and WrnΔhel/Δhel mice (data not shown). At 9 mo of age, however, significantly higher amounts of ROS (Fig. 1B) and oxidative DNA damage (Fig. 1C) were detected in the livers of WrnΔhel/Δhel mice compared to WT animals. We also examined the mitochondrial DNA mutation rate in liver tissues. Sequencing of the mitochondrial D-loop region cloned from mouse tissues showed a significant 1.7-fold mutation rate increase in WrnΔhel/Δhel livers (Wilcoxon test; P=0.036) compared to WT animals (Table 1). The higher mutation rate in liver mitochondrial DNA from WrnΔhel/Δhel mice could have an effect on energy production in such mice. Thus, we examined the ATP levels in liver tissues of each cohort. Overall ATP level was 40% lower in WrnΔhel/Δhel mice than WT animals (Fig. 1D). We finally examined lipid peroxidation and did not detect a significant difference between WT and WrnΔhel/Δhel mice (data not shown).
Figure 1.
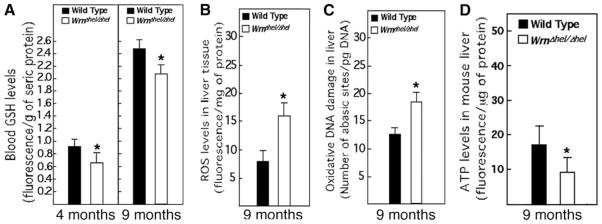
Blood GSH levels and oxidative stress in WT and WrnΔhel/Δhel mice. A) Blood ascorbate level in 4- and 9-mo-old mice; n = 6 for each cohort and measurement. B) Liver ROS levels in 9-mo-old mice; n = 10 (5 females, 5 males) for each cohort and measurement. C) Oxidative DNA damage levels in liver of 9-mo-old mice; n = 10 (5 females, 5 males) for each cohort and measurement. D) ATP levels in liver tissues of 9-mo-old WT and WrnΔhel/Δhel mice; n = 6 for each cohort and measurement. Bars represent means ± SE. *P < 0.05 vs. WT; unpaired Student’s t test.
TABLE 1.
Effect of ascorbate on mitochondrial DNA mutation rate in WrnΔhel/Δhel mice
| Genotype, liver mitochondria | Mutation/sequenced base pairsa | % | P value |
|---|---|---|---|
| WT | 10/5230 | 0.19 | |
| WrnΔhel/Δhel | 17/5214 | 0.33 | 0.036b |
| WrnΔhel/Δhel + ascorbate | 8/4607 | 0.17 | 0.068c |
From cloned PCR fragments of mitochondrial D-loop control region. Primers are MTC1 GCCAACTAGCCTCCATCTCATACTT, nt 15196–15220, and MTC2 GGGCGGGTTGTTGGTTTCAC, nt 15701–15720.
Wilcoxon test; WT vs. WrnΔhel/Δhel.
Wilcoxon test; WrnΔhel/Δhel vs. WrnΔhel/Δhel + ascorbate.
WrnΔhel/Δhel mice exhibit a severe sinusoidal endothelium defenestration
Morphological changes in the liver with old age are increasingly recognized and include defenestration of the sinusoidal endothelium (Fig. 2A, B) (18). As this change also occurs with diabetes and may contribute to dyslipidemia (19), we examined the morphology of hepatic sinusoids in WrnΔhel/Δhel mice. A severe defenestration was observed in mutants from 7 mo onward (Fig. 2C, D), which coincides with the appearance of significant hyperlipidemia and the onset of diabetes in WrnΔhel/Δhel animals (13). The overall porosity (determined as the percentage of the endothelial surface perforated by fenestrations) was very low in the WrnΔhel/Δhel mice (0.91± 0.69%) compared to age-matched WT mice (4.1± 0.3%) (23).
Figure 2.

Effect of ascorbate on liver sinusoidal fenestrations. Scanning electron microscopy showing lumenal surface of the liver sinusoidal endothelium with fenestrations (diameter ~50–100 nm) clustered into sieve plates in WT and ascorbate-treated WrnΔhel/Δhel mice. A) WT mouse at 7 mo of age. B) WT mouse at 24 mo of age. C) WrnΔhel/Δhel mouse at 7 mo of age. D) WrnΔhel/Δhel mouse at 9 mo of age. E) WrnΔhel/Δhel mouse at 9 mo of age treated with ascorbate since weaning. F) WrnΔhel/Δhel mouse at 9 mo of age treated with ascorbate since 7 mo of age. All views ×25,000.
Vitamin C restores sinusoidal endothelium fenestration and normal metabolism in WrnΔhel/Δhel animals
Most vertebrates synthesize ascorbic acid, or vitamin C, de novo, but humans and a few other species have lost this faculty, rendering this molecule a necessary nutrient or vitamin. Ascorbic acid is a major physiological antioxidant that replenishes GSH levels, prevents high-caloric-diet-induced dyslipidemia (30) and also influences the outcome of several age-related disorders, such as diabetes mellitus, obesity, and atherosclerosis (31). It is known that ascorbate delays replicative senescence of WS fibroblasts in vitro (32). Therefore, we investigated the role of ascorbate in the disease progression of our mouse model. At weaning, the drinking water of WrnΔhel/Δhel mice was supplemented daily with 0.4% sodium L-ascorbate (w/v) as described before (20). The overall porosity of the sinusoidal endothelium was increased to normal levels in ascorbate-treated WrnΔhel/Δhel mice (Fig. 2E) from less than 1% of the area perforated (as reported above) to 6.53 ± 4.97% (t test; P<0.000001, n=5 mice for each cohort). In addition, the diameter of the fenestrations was also increased by ascorbate (from 93.8±44.7 to 98.57±42.51 nm, P<0.005).
Diabetes, hyperlipidemia, and enhanced visceral fat accretion were reported in patients with WS (33) and WrnΔhel/Δhel mice (13). The unfolding of these phenotypes was monitored in ascorbate-treated and untreated WrnΔhel/Δhel mice. Overall, visceral and subcutaneous fat pads of 9-mo-old WrnΔhel/Δhel mice were at least twice the weight of WT animals (Fig. 3A, B). Ascorbate significantly limited this change by reducing the mean weight of treated WrnΔhel/Δhel mice to similar values as those observed in WT animals. The same effect was observed for blood triglyceride (Fig. 3C) and fasting glucose (Fig. 3D) levels. Although fasting insulin levels were not significantly different between treated and untreated mutant mice (Fig. 3E), a clear normalization of the insulin resistance was noted in treated WrnΔhel/Δhel mice, as revealed by the HOMA-IR index (Fig. 3F).
Figure 3.

Ascorbate treatment improves metabolism of WrnΔhel/Δhel mice. Ascorbate was added to drinking water (0.4% w/v) of WrnΔhel/Δhel mice at weaning (30 wk treatment) or at the age of 7 mo (8-wk treatment). A) Visceral fat weight. B) Subcutaneous fat weight. C) Fasting blood triglyceride levels. D) Fasting blood glucose levels. E) Fasting insulin levels. F) HOMA-IR index for fasting WT, WrnΔhel/Δhel, and ascorbate-treated WrnΔhel/Δhel mice. Bars represent means ± SE. *P < 0.05 vs. WrnΔhel/Δhel; unpaired t test. Nine-month-old males (n=5) and females (n=5) of each cohort were used for all measurements. Data from males and females were pooled for glucose, insulin, and HOMA-IR histograms.
We next examined ROS and DNA damage levels in the liver of ascorbate-treated WrnΔhel/Δhel mice. We measured ROS and oxidative DNA damage in 5 males and 5 females of each cohort at 9 mo of age. As indicated before, WrnΔhel/Δhel males and females exhibited higher amounts of ROS (Fig. 4A) and oxidative DNA damage (Fig. 4B) in their liver compared to WT animals. Ascorbate treatment of WrnΔhel/Δhel mice reduced these amounts to equal or even inferior values than those observed in the WT cohort. We also examined the mitochondrial DNA mutation rate in liver tissues. The mutation rate tended to be reduced near to the basal WT levels by ascorbate treatment (Table 1). Nonetheless, ascorbate significantly rescued the ATP levels in WrnΔhel/Δhel liver tissues (Fig. 4C). Since ATP level has an effect on AMPKα activity (34), we also examined the level of phosphorylation of this kinase in livers of each cohort. As expected, WrnΔhel/Δhel mice showed an increase in AMPKα phosphorylation (Fig. 4D). However, ascorbate treatment did not significantly decrease AMPKα phosphorylation in the liver of WrnΔhel/Δhel mice. These results indicate that ascorbate treatment of WrnΔhel/Δhel mice since weaning rescued the abnormal metabolic phenotypes associated with this mouse model and normalized the endothelial morphology in liver tissues.
Figure 4.

Ascorbate treatment improves ROS, oxidative DNA damage, and ATP balance in the liver of WrnΔhel/Δhel mice. A) Liver ROS levels in 9-mo-old males and females. B) Oxidative DNA damage in the liver of 9-mo-old animals. Ascorbate was added to drinking water (0.4% w/v) of WrnΔhel/Δhel mice containing cages at weaning (30-wk treatment) or at the age of 7 mo (8-wk treatment). C) ATP levels in liver tissues of 9-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice. Bars represent means ± SE; n = 5 for each cohort (including males and females when indicated) and measurement. *P < 0.05 vs. WrnΔhel/Δhel; unpaired t test. D) Western blot showing expression of AMPKα and phosphorylated AMPKα in liver tissues of 9-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel. β-Tubulin is used as loading control.
Finally, we measured the daily water and food intake in WT, WrnΔhel/Δhel, and ascorbate-treated WrnΔhel/Δhel animals. The daily water and food intakes (5.2±1.4 ml/d and 3.5±0.6 g/d, respectively) were not significantly different between our three cohorts (data not shown).
Short-term vitamin C treatment restores sinusoidal endothelium fenestration and overall fat tissue weight in WrnΔhel/Δhel mice
To determine whether a shorter treatment time has similar effects on WrnΔhel/Δhel mice, we supplemented 7-mo-old WrnΔhel/Δhel mice with ascorbate for 8 wk and repeated our measurements. Seven-month-old WrnΔhel/Δhel animals already displayed metabolic abnormalities and a severe liver sinusoidal endothelial defenestration (Fig. 2E). A normal sinusoidal refenestration was obtained in WrnΔhel/Δhel mice on this short-term ascorbate treatment (Fig. 2F). Concomitant with these observations, an overall reduction of fat tissue weight to WT basal levels was observed with WrnΔhel/Δhel males (Fig. 3A, B). Visceral fat tissues from WrnΔhel/Δhel females were more refractory to the short-term ascorbate treatment (Fig. 3A), but subcutaneous fat tissue weight was significantly reduced (Fig. 3B). Hypertriglyceridemia was not significantly reduced in short-term ascorbate-treated WrnΔhel/Δhel males and females (Fig. 3C). Although hyperinsulinemia was not significantly reduced in WrnΔhel/Δhel mice, a trend to normalization was observed in HOMA-IR index of WrnΔhel/Δhel animals after 8 wk of vitamin C treatment (Fig. 3D–F). Interestingly, this short-term ascorbate treatment did not relieve ROS or oxidative DNA damage in liver tissues of treated WrnΔhel/Δhel mice (Fig. 4A, B).
GSEA of WrnΔhel/Δhel mice treated with ascorbate
To gain insight into the rescuing effect of vitamin C in WrnΔhel/Δhel mice, the global liver expression profile of WT, WrnΔhel/Δhel-treated and untreated mice at 9 mo of age was analyzed with the Agilent microarray technology. Lists of genes exhibiting a 1.5-fold change difference with a Benjamini-Hochberg false discovery rate (FDR) < 0.1 are shown in Supplemental Table 1 for WrnΔhel/Δhel mice vs. WT animals and in Supplemental Table 2 for ascorbate-treated WrnΔhel/Δhel mice vs. untreated WT animals. Real time RT-PCR confirmed the microarray data by analyzing the expression of 6 randomly picked genes listed from our WT vs. ascorbate-treated WrnΔhel/Δhel mice data (see results in Supplemental Table 3). A list of the primers for the genes analyzed is shown in Supplemental Table 4.
Three hundred eighty-four genes exhibited a 1.5-fold alteration in expression in the liver of 9-mo-old WrnΔhel/Δhel mice compared to WT animals (194 down-regulated and 190 up-regulated, respectively) (Supplemental Table 1). Two hundred ninety-six genes showed a 1.5-fold change in expression in ascorbate-treated WrnΔhel/Δhel mice compared to WT animals (139 down-regulated and 157 up-regulated, respectively) (Supplemental Table 2). Sixty-eight genes were altered in both untreated and ascorbate-treated WrnΔhel/Δhel mice (Fig. 5A and Supplemental Table 5). However, 8 of these genes showed opposite expression change in ascorbate-treated and untreated WrnΔhel/Δhel mice. Such genes included the immunoglobulin heavy chain (J558 family), a C-type lectin domain family 2 encoding gene, the ethanolamine kinase 2, the squalene epoxidase, the sulfotransferase family 2A encoding gene, the PHD finger protein 3, the major urinary protein 3, and one expression sequence tag (see Supplemental Table 5 for more details).
Figure 5.

Distinct biological pathways associated with WrnΔhel/Δhel and ascorbate-treated WrnΔhel/Δhel mice. A) Venn diagram showing the number of genes overlapping between WrnΔhel/Δhel and ascorbate-treated WrnΔhel/Δhel mice. B) All significant gene set enrichments from liver tissues of WrnΔhel/Δhel and ascorbate-treated WrnΔhel/Δhel mice compared to WT liver control were grouped into biological and functional categories. Categories with family-wise error rate (FWER) < 0.05 are presented in the histogram.
To obtain an unbiased analysis of gene sets or biological pathways up- or down-regulated in untreated and ascorbate-treated WrnΔhel/Δhel mice, we analyzed the complete list of genes using the GSEA tool. GSEA is a way to assess whether a predefined list of genes in a specific pathway is significantly up- or down-regulated (25). The analysis generated a list of eight significant gene sets with a family-wise error rate (FWER) P value <0.05 for WrnΔhel/Δhel mice compared to WT animals and 11 significant gene sets for ascorbate-treated WrnΔhel/Δhel mice. Supplemental Table 6 shows the gene enrichment sets down- or up-regulated in treated and untreated WrnΔhel/Δhel mice compared to WT animals. The complete GSEA reports are provided online (http://www.crhdq.ulaval.ca/LiverWrn-Vitc). As summarized in Fig. 5B, sets from WrnΔhel/Δhel mice included genes shown to be altered in Acox1-knockout and Myc/E2F1 transgenic mice developing hepatomas (35). Liver tissue from WrnΔhel/Δhel mice also exhibited alteration in genes involved in caloric restriction (36), glutathione, and xenobiotic metabolism by cytochrome P450 pathways. These results suggest that WrnΔhel/Δhel mice respond to the observed oxidative stress by altering the necessary pathways to survive. WrnΔhel/Δhel mice also responded by up-regulating an inflammatory response in the liver (37). Ascorbate treatment, in return, significantly inhibited genes in WrnΔhel/Δhel mice known to be up-regulated in human WS fibroblasts (38) (Fig. 5B). Ascorbate also down-regulated pathways normally up-regulated in cancer, such as Myc or Myb transcriptional activities (35, 39). Interestingly, ascorbate up-regulated genes in the pathway leading to the dedifferentiation of adipocytes in obese mice (40). It also up-regulated genes normally decreased during B-lymphomagenesis (41). Vitamin C augmented significantly several pathways involved in the response to tissue injury, including clearance of sick cells from infection or graft rejection (42–45). This is consistent with the increase in genes associated with the immune response category seen in Supplemental Table 2. (Fisher exact test; P=0.02). Concomitant with such tissue injury response, ascorbate increased pathways involved in stem cell maturation and maintenance (46, 47).
Vitamin C normalizes GSH metabolism in the liver of WrnΔhel/Δhel mice
The increase in the expression of genes involved in xenobiotic and glutathione metabolism prompted us to examine GSH and ascorbate levels in WrnΔhel/Δhel mice. Plasma ascorbate levels were not different in 4-mo-old WrnΔhel/Δhel mice, which contrasts with the 3-fold increase observed in 9-mo-old WrnΔhel/Δhel mice compared to WT animals (Fig. 6A). Despite this increase in blood ascorbate, GSH blood levels, which usually correlate with ascorbate amounts, remained significantly lower in mutant mice (Fig. 1A). We next examined ascorbate levels in the liver of our 9-mo-old cohorts. As showed in Fig. 6B, WrnΔhel/Δhel mice have higher ascorbate concentrations in the liver compared to WT animals. Finally, WrnΔhel/Δhel mice showed higher kidney ascorbate levels and lower urinary ascorbate concentration than WT animals at 9 mo of age (Fig. 6C, D). Our results corroborate the findings in patients with WS (29) and suggest that WrnΔhel/Δhel mice react to their metabolic anomalies by increasing their overall body ascorbate levels. We then quantified GSH levels in the liver of WrnΔhel/Δhel mice. Unlike the blood GSH concentration, GSH levels were higher in the liver of WrnΔhel/Δhel mice compared to WT animals (Fig. 6E) in support of the liver GSEA data (Fig. 5B). Interestingly, ascorbate treatment normalized both liver GSH and blood GSH levels in WrnΔhel/Δhel mice (Fig. 6E, F). The GSEA data of livers of ascorbate-treated WrnΔhel/Δhel mice did not show significant change in the glutathione metabolic pathway (Fig. 5B and Supplemental Table 6) and correlated with normal GSH levels in treated mice. As expected, ascorbate levels were significantly increased in the blood of young WrnΔhel/Δhel mice treated with vitamin C (Fig. 6G).
Figure 6.
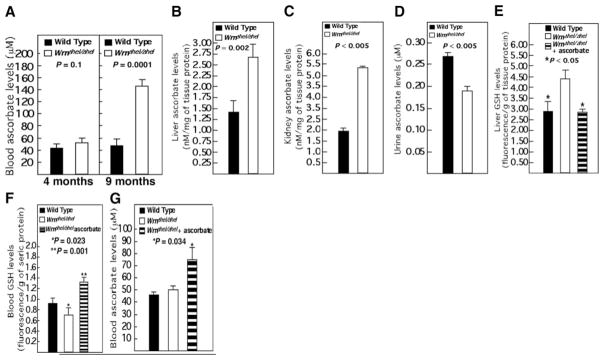
Ascorbate and GSH levels in WT and WrnΔhel/Δhel mice. A) Blood ascorbate levels in 4 and 9-mo-old mice. B) Liver ascorbate levels in 9-mo-old mice. C) Kidney ascorbate levels in 9-mo-old mice. D) Urine ascorbate levels from 9-mo-old mice. E) Liver GSH level in 9-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice. F) Blood GSH levels in 4-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice. G) Blood ascorbate levels in 4-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice. All P values are from unpaired t tests; n = 5 for each cohort and measurement. Bars represent means ± SE.
SMP30 (also known as RGN) is the penultimate enzyme in ascorbate biosynthesis and is known to decrease in liver with age (26). Examination of SMP30 protein level by Western blot analysis in 9-mo-old animals, however, failed to reveal any difference between WrnΔhel/Δhel and WT animals (Supplemental Fig. 1A). The enzymatic activity of SMP30 was also the same in liver tissues of WrnΔhel/Δhel and WT animals (Supplemental Fig. 1B).
The last enzyme in ascorbate biosynthesis is the L-gulonolactone oxidase (Gulo). Real-time RT-PCR was performed for Gulo expression in liver samples, as no commercial antibody is available against mouse Gulo gene product. No difference in Gulo mRNA expression level was detected between WT and WrnΔhel/Δhel animals (Supplemental Table 3), confirming the microarray data.
There are two other enzymes important for the intracellular metabolism of ascorbate, namely thioredoxin reductase and cytochrome-b5-reductase (48, 49). There are several isoforms of thioredoxin reductase and cytochrome-b5-reductase in mice, but there is no commercial antibody to detect all of the different isoforms of these enzymes. On the basis of microarray data, however, there is no significant change in mRNA expression levels for all of these enzymes in WrnΔhel/Δhel compared to WT animals (Supplemental Fig. 2A). We next examined the overall enzymatic activities of thioredoxin reductase and cytochrome-b5-reductase in the liver of these mice. No difference was detected in the enzymatic activities of thioredoxin reductase and cytochrome-b5-reductase in the liver tissues of WT and WrnΔhel/Δhel animals (Supplemental Fig. 2B, C).
Finally, the amount of liver and kidney ascorbate transporter SVCT1 (50) was not changed in WrnΔhel/Δhel mice compared to WT animals (data not shown). GSEA did not revealed changes in genes sets involved in ascorbate metabolism or transport in WrnΔhel/Δhel mice confirming the above results. Thus, the reason for the ascorbate increase in 9-mo-old WrnΔhel/Δhel mice is unknown.
Vitamin C normalizes several WrnΔhel/Δhel mouse liver stress markers and limits innate inflammation
The GSEA on the expression data of livers from WrnΔhel/Δhel mice suggested the increase in oxidative stress and inflammatory responses were reversed by ascorbate treatment. We first examined p38 activation, as this marker was also shown to be involved in senescence of WS fibroblasts (51). A proportional increase in p38 expression and phosphorylation levels was noted in the liver of WrnΔhel/Δhel animals compared to WT mice, but ascorbate had no effect on these changes (Fig. 7A–C). We also examined JNK and TNF-α stress markers, but no significant change was seen in their expression and/or activation status between WT and WrnΔhel/Δhel mice (data not shown).
Figure 7.
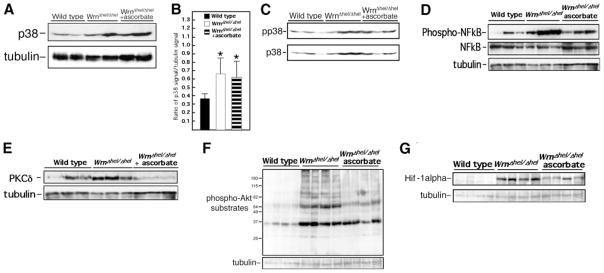
Effect of ascorbate on liver stress markers of WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice. A) Western blot showing expression of p38 in liver tissues of 9-mo-old mice. β-Tubulin is used as loading control. B) Scanning analyses of Western blots, expressed as ratio of p38 signal to β-tubulin signal. Bars represent means ± SE. *P < 0.05 vs. WT; unpaired t test. C) Phosphorylation of the p38 kinase (pp38) in liver tissues of mice at 9 mo of age. D) Western blots showing levels of phosphorylated NF-κB and total NF-κB proteins in liver tissues of 9-mo-old mice. E) Western blots showing levels of PKCδ protein in liver tissues of 9-mo-old mice. β-Tubulin is used as loading control. F) Western blots showing levels of phosphorylated Akt substrates in liver tissues of 9-mo-old mice. β-Tubulin is used as loading control. G) Western blots showing levels of Hif-1α in liver tissues of 9-mo-old mice. β-Tubulin is used as a loading control.
The NF-κB transcription factor determines cell response to a wide variety of stresses, including inflammation, and was recently shown to be one of the most strongly age-associated markers (52). As indicated in Fig. 7D, NF-κB phosphorylation levels were significantly higher in the livers of WrnΔhel/Δhel mice than in those of WT mice. This change was largely prevented by ascorbate treatment (Fig. 7D and Supplemental Fig. 3A, B).
Protein kinase B (PKB/Akt) and C family members are critical upstream regulators of cell survival, metabolism, or inflammation. They regulate NF-κB and are altered in a broad range of diseases (53–55). Therefore, we extended our molecular survey to these factors and examined the expression and activation status of PKCα, β, and δ. No differences were noted in PKCα and β protein or phosphorylation levels between WT and WrnΔhel/Δhel liver tissues (data not shown). In contrast, liver PKCδ protein levels were ~55% increased in WrnΔhel/Δhel mice compared to WT animals (Fig. 7E and Supplemental Fig. 3C). This result is consistent with the role of PKCδ in fatty acid-induced hepatic insulin resistance (56). Ascorbate prevented this change in WrnΔhel/Δhel animals and even reduced PKCδ levels to values below those of WT mice (Fig. 7E and Supplemental Fig. 3C). Finally, the overall Akt catalytic activity was assessed by the detection of its endogenous phosphorylated substrates. There was a net increase of Akt activity in the liver of WrnΔhel/Δhel mice compared to WT animals (Fig. 7F), and this change was prevented in WrnΔhel/Δhel mice on ascorbate supplementation (Fig. 7F and Supplemental Fig. 3D).
It is known that Hif-1α is stabilized under chronic inflammatory conditions or under chronic cellular redox alterations due to accumulation of ROS (57, 58). As shown in Fig. 7G, Hif-1α protein level was ~5.5-fold higher in WrnΔhel/Δhel animals than WT mice (Supplemental Fig. 3E). Ascorbate treatment decreased this level in WrnΔhel/Δhel animals by 2-fold (Supplemental Fig. 3E). Altogether, these results indicate that ascorbate treatment decreased the activities of several markers associated with oxidative stress and inflammation in the liver of WrnΔhel/Δhel mice.
PPARα expression changes on both long-term and short-term ascorbate treatment in WrnΔhel/Δhel mice
Peroxisome proliferator-activated receptor α (PPARα) is a major upstream regulator of lipid metabolism, and its expression is also modulated by aging (59). Microarray analyses indicated that PPARα expression was increased in ascorbate-treated WrnΔhel/Δhel mice. Western blot analyses of liver extracts revealed similar PPARα protein content in WT and WrnΔhel/Δhel mice. In contrast, ascorbate dramatically increased PPARα expression in mutant animals (Fig. 8). To determine whether a shorter treatment had similar effects on WrnΔhel/Δhel mice, we supplemented 7-mo-old animals with ascorbate for 8 wk and repeated our measurements. As indicated previously, 7-mo-old WrnΔhel/Δhel animals already displayed metabolic abnormalities and a severe liver sinusoidal endothelial defenestration (Fig. 2C). An 8-wk ascorbate treatment led to refenestration of the liver sinusoidal endothelium (Fig. 2F). Interestingly, liver PPARα protein level was also significantly increased after 8 wk of ascorbate treatment (Fig. 8). In contrast, the increased activation and expression of p38, phospho-Akt substrates, Hif-1α, phospho-NFκB, and PKCδ were not reversed by a short-term ascorbate treatment (data not shown).
Figure 8.
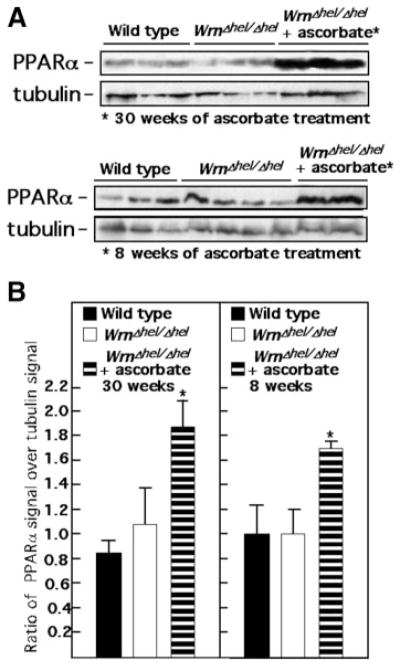
Short-term and long-term effect of ascorbate on liver PPARα in WrnΔhel/Δhel mice. A) Examples of Western blots showing levels of PPARα protein in liver tissues of 9-mo-old WT, WrnΔhel/Δhel, and ascorbate-treated WrnΔhel/Δhel mice (treated for 30 or 8 wk before protein extraction). B) Scanning analyses of Western blots, expressed as ratio of PPARα signal to β-tubulin signal in liver tissues. WrnΔhel/Δhel mice were treated with ascorbate since the age of 7 mo (8-wk treatment) or since weaning (30-wk treatment). Bars represent means ± SE. *P < 0.05 vs. WT; unpaired t test.
Vitamin C restores the mean life-span defect observed with WrnΔhel/Δhel mice
We investigated the effect of ascorbate supplementation on the mean life span of our mutant cohort. Strikingly, the ascorbate treatment (since weaning) rescued the mean life-span decrease observed in WrnΔhel/Δhel mice (log rank test; P=0.0065), allowing them to reach the same values as WT animals (Fig. 9A). Although the incidence of heart fibrosis and tumor formation was the same between untreated and treated WrnΔhel/Δhel mice, the oldest WrnΔhel/Δhel mouse died at 26 mo of age, while the oldest ascorbate-treated WrnΔhel/Δhel mouse died at 30 mo of age like the oldest WT animal.
Figure 9.

Percentage of disease-free animals with age. A) Comparison of WT, WrnΔhel/Δhel, and ascorbate-treated (since weaning) WrnΔhel/Δhel mice. B) Comparison of untreated and ascorbate-treated (since weaning) WT animals.
Vitamin C has no beneficial effect on WT animals
A cohort of WT animals ase also provided with vitamin C, drinking water supplemented daily with 0.4% sodium L-ascorbate (w/v) at weaning. Although ascorbate tended to decrease ROS levels (Supplemental Fig. 4A) and significantly decreased oxidative DNA damage in the liver of 9-mo-old WT animals (Supplemental Fig. 4B), it did not increase their mean life span. Ascorbate had no significant beneficial effect on fat tissues, blood triglyceride, glucose, or insulin resistance (Supplemental Fig. 4C–H). Furthermore, ascorbate did not increase PPARα expression in the liver of WT mice and had no effect on the expression of liver p38 kinase, phospho-Akt substrates, NF-κB, Hif-1α, and PKCδ (data not shown).
DISCUSSION
Mouse model
The study of the age-associated biological defects relies on animal models that closely mimic the clinical setting. One of the numerous consequences of aging is the appearance of metabolic syndrome. Our mouse model, the WrnΔhel/Δhel mutant mice, prematurely develops most of the symptoms associated with normal human aging and mimics the human WS. These mice exhibit increased blood insulin, glucose, triglycerides, cholesterol, hyaluronic acid, systemic ROS, heart failure, and different types of cancers (13). The WrnΔhel/Δhel mutant mouse contains a deletion in part of the helicase domain of the Wrn protein (11). These mice synthesize a stable mutant protein with no DNA helicase activity. This is apparently different from humans with WS, who neither synthesize a stable WRN protein nor produce a truncated WRN protein that cannot translocate to the nucleus. As such, patients with WS are considered null mutants (60). Although most WrnΔhel/Δhel mice do not die prematurely (for example, before the age of 12 mo), the mean life span of WrnΔhel/Δhel mouse cohort is significantly reduced compared to WT animals. This is in contrast to Wrn-null animals that do not have a reduced life span (14) or develop metabolic syndrome (such as hyperglycemia and hyperinsulinemia) unless given a high-carbohydrate diet (15). The reason for the difference between Wrn-null and WrnΔhel/Δhel mice is unknown. Although we cannot rule out the possibility that a DNA helicase mutant Wrn protein could act as a dominant-negative peptide, this Wrn mutant protein does not associate with the DNA replication complex like the normal protein (61). Experiments are currently under way to determine whether the helicase mutant Wrn protein can interact and affect the enzymatic activity of known Wrn protein partners.
In addition to the observed metabolic abnormalities, WrnΔhel/Δhel mice have increased circulating ascorbate levels like humans with WS (29). Every organ extract from mutant mice that we examined had an increased ascorbate level. WrnΔhel/Δhel mice also secreted less ascorbate in their urine than WT animals. Interestingly, blood ascorbate levels increased with phenotypic progression in WrnΔhel/Δhel mice. Ascorbate being protective, it is possible that the retention of vitamin C is a defense mechanism to counterbalance the age-dependent rise in oxidative stress. We cannot rule out, however, the possibility that the increased ascorbate levels observed in WrnΔhel/Δhel mice are due to a modification in ascorbate metabolism only specific to these mice. Interestingly, blood ascorbate levels are 2-fold higher in patients with WS compared to other patients with segmental progeroid syndrome, such as patients with Bloom syndrome, ataxia telangectasia, Down syndrome, Fanconi anemia, and xeroderma pigmentosum (62). The molecular mechanism by which WrnΔhel/Δhel mice retain ascorbate in their liver, for example, is unknown. Microarray analyses of liver tissues between WT and WrnΔhel/Δhel mice did not indicate significant expression changes in pathways involved in ascorbate metabolism or transportation. Accordingly, expression of enzymes directly involved in ascorbate synthesis (SMP30 and Gulo gene products) and the activity of enzymes involved in ascorbate metabolism (thioredoxin reductase and cytochrome-b5-reductase) were not changed in WrnΔhel/Δhel mice compared to WT animals. Finally, the protein levels of the ascorbate transporter SVCT1 in the liver and kidneys were similar in WrnΔhel/Δhel and WT mice. It is possible, however, that post-translational modification of key components of ascorbate transport are affecting their activities in WrnΔhel/Δhel mice. Thorough proteomic analyses will be required to resolve this issue.
GSEA on liver tissues indicate increased oxidative stress and inflammatory response in WrnΔhel/Δhel mice
GSEA on the liver of WrnΔhel/Δhel mice revealed changes in the expression of genes involved in xenobiotic and glutathione metabolisms, caloric restriction, and inflammation. Although blood GSH levels were lower in WrnΔhel/Δhel mice than WT animals, the increased glutathione metabolism pathway in liver helped these mice to maintain hepatic GSH levels above normal. Again, such findings indicate that WrnΔhel/Δhel mice may be up-regulating the necessary pathways to survive. Correspondingly, it was reported that these so-called shared longevity assurance pathways are similarly altered in both delayed and accelerated aging mouse models (63, 64). However, the alteration in these pathways and the greater retention of ascorbate were insufficient for WrnΔhel/Δhel mice to maintain a healthy life. As already mentioned, blood GSH levels in WrnΔhel/Δhel mutant mice were still lower than in normal mice, and mutant mice developed all of the metabolic traits associated with human WS (such as dyslipidemia, hyperglycemia, hyperinsulinemia). Furthermore, liver tissues from WrnΔhel/Δhel mice exhibited increased expression and phosphorylation of the stress markers phospho-p38 kinase, phospho-Akt substrates, phospho-NFκB, phospho-PKCδ, and Hif-1α. Finally, the mean life span of WrnΔhel/Δhel mice were reduced compared to WT animals. Interestingly, WrnΔhel/Δhel mice exhibited a severe premature liver sinusoidal endothelial defenestration at 7 mo of age (Fig. 2). Such defenestration is normally observed in much older animals (18). The defenestration corresponded to the onset of diabetes in WrnΔhel/Δhel mice (13). Finally, several older WrnΔhel/Δhel mice developed liver tumors, which is consistent with the gene expression changes observed in the microarray studies.
Vitamin C has a beneficial effect on WrnΔhel/Δhel mice but not on WT animals
Because increased ROS was observed prior to any of the metabolic changes measured in WrnΔhel/Δhel mice (13), we decided to treat mutant mice with suprapharmacological levels of vitamin C based on a previous study on the beneficial effect of ascorbate on mice unable to synthesize vitamin C (20). Note that the decision of using vitamin C was taken before we performed blood ascorbate measurements. To our knowledge, this is the first report showing that vitamin C is beneficial in a mouse model for WS. A daily ascorbate supplementation allowed WrnΔhel/Δhel mice to recover a normal mean life span and healthy aging. Although the number of animals used in each cohort was not big enough for a statistical testing on maximum life span (65), ascorbate treatment did prevent the appearance of WS characteristic redox imbalance and related genomic damage. It also prevented the liver proinflammatory status observed in WrnΔhel/Δhel mice. A normalization of phospho-Akt substrates, phospho- NF-κB, phospho-PKCδ, and Hif-1α levels was obtained with the treatment. In addition, ascorbate supplementation dramatically improved the metabolic profile of WrnΔhel/Δhel animals, although the lack of change in liver steatosis (as defined by total lipid levels) was a curious exception (data not shown). These metabolic improvements were paralleled by an increased expression of the homeostasis regulator PPARα and by a complete refenestration of the sinusoidal endothelium in treated WrnΔhel/Δhel -mouse livers. PPARα activation is known to cause lipid clearance via β-oxidation enhancement and exerts anti-inflammatory effects on both the vascular wall and the liver (66). Finally, the increase in ascorbate levels in WrnΔhel/Δhel mice was insufficient to stabilize GSH levels. Implementation of suprapharmacological amounts of ascorbate was required to normalize GSH levels in blood and tissues of WrnΔhel/Δhel mice.
Vitamin C only induced a trend in reducing the number of mitochondrial DNA mutations in liver tissues of WrnΔhel/Δhel mice. This is not surprising, as mice were treated with ascorbate at weaning. Mitochondrial DNA mutations could have accumulated during embryogenesis and before weaning in such tissues of mutant mice. Vitamin C cannot reverse mutations once fixed in mitochondrial DNA.
In contrast to WrnΔhel/Δhel mice, ascorbate had no beneficial effects on the health or mean life span of WT animals. Even though oxidative DNA damage was reduced significantly in the liver of ascorbate-treated WT mice, the expression and activation of oxidative stress and inflammatory response markers did not change. Thus, the beneficial effect of vitamin C could only be seen with WrnΔhel/Δhel mice. These results suggest that vitamin C supplementation would be a very relevant regimen to alleviate abnormal cellular functions associated with WS only. However, additional testing will be required on Wrn-null mice lacking telomerase activity, which exhibits a more severe premature aging phenotype related to WS (16), to determine its beneficial effect. Ascorbate treatments on other progeroid mouse models will also be required to determine whether it can alleviate several age-related syndromes.
GSEA of vitamin C-treated WrnΔhel/Δhel mice reveals changes in pathways associated with the immune system, stem cell maturation, and adipocyte differentiation
The first remarkable set of liver genes affected by ascorbate is the one known to be up-regulated in human WS fibroblast. Ascorbate significantly inhibited genes that have been shown to be up-regulated in human WS fibroblasts (38) (Fig. 5B). Ascorbate also down-regulated pathways normally up-regulated in cancers exhibiting increased Myc or Myb transcriptional activities (35, 39). In addition, pathways related to several types of tissue injury response were up-regulated by ascorbate. Concomitantly, ascorbate altered biological pathways related to stem-cell maturation. It is unknown at this point whether this maturation is entirely related to the immune system associated with the liver tissue, or the replacement of abnormal sinusoidal endothelial cells and/or to the renewal of hepatocytes. Although refenestration of the sinusoidal endothelium points to endothelial maturation, thorough examination of each cellular compartment in the liver with the appropriate techniques will indicate which cell type is rejuvenated with vitamin C in this mouse model. Regardless of future results, our observations indicate that vitamin C stimulated a positive response for liver tissue renewal (e.g., refenestration) and slowed WS senescence-associated gene expression (38). Finally, ascorbate up-regulated genes normally involved in dedifferentiation of adipocytes in obese mice (40). This GSEA result is also consistent with the observation that WrnΔhel/Δhel mice lose weight and visceral fat tissue on exposure to vitamin C. It is noteworthy that ascorbate-treated WrnΔhel/Δhel mice drank and ate as much water and food as untreated or WT animals.
Short-term ascorbate treatment reversed liver endothelium defenestration and normalized several metabolic abnormalities, even in the presence of a significant oxidative stress
Although ROS scavenging would be a mechanism by which vitamin C could indirectly affect gene expression (ROS can also be used as a second messenger of several signal transduction pathways), the short-term treatment indicated another potential mechanism for gene regulation. Metabolic normalization, PPARα induction, and liver sinusoidal endothelial refenestration in WrnΔhel/Δhel mice were readily observable on a short 8-wk treatment with vitamin C. This occurred even in the presence of significant amounts of ROS and oxidative DNA damage in liver tissues. It is worthy to note that activation and overexpression of stress markers such as p38 kinase, phospho-Akt, phospho- NF-κB, and PKCδ were still up-regulated after an 8-wk treatment. These results suggest that PPARα could be a target molecule to revert liver sinusoidal endothelial defenestration in WrnΔhel/Δhel mice and ameliorate lipid as well as glucose metabolism. Interestingly, a recent report has indicated that an agonist of another member of this family of transcription factor, PPARγ, ameliorated insulin resistance, glucose, and lipid metabolism and decreased the inflammatory marker IL-6 in several patients with WS (67, 68). The agonist also increased the median life span of several patients with WS (69). Treatment of WrnΔhel/Δhel mice with PPARα agonists is under way to assess their effect on liver sinusoid endothelial fenestration.
The exact mechanism by which vitamin C alters gene expression is currently unknown. It is less clear how PPARα expression is increased in WrnΔhel/Δhel mice but not in WT animals. Notably, ascorbate can be used by cells as a cofactor for a family of dioxygenase enzymes. An important class of enzymes that directly affects the activity of several transcription factors is the prolyl hydroxylase. Hydroxylation of prolyl residues on Hif-1α will induce its polyubiquitination and its proteosomal degradation (57, 58). On one hand, ROS will stabilize Hif-1α, as seen in the liver of WrnΔhel/Δhel mice. On the other hand, large implementation of ascorbate may increase cellular prolyl hydroxylase activities and hence Hif-1α degradation, as seen in vitamin C-treated mutant mice. Recently, several reports indicated that the same hydroxylases that alter the Hif pathway may also regulate important components of the NF-κB pathway (58). Furthermore, there are several other transcription factors that can be directly or indirectly activated by prolyl hydroxylases or by the cellular redox status. Such transcription factors include Hif-1α, NF-κB, AP-1 (Fos/Jun), CREB, GATA-2, p53, Sp-1, C/EBPβ, certain Ets family members, Gadd153, and NF-IL-6 (58). The next step will be to examine the role of prolyl hydroxylases in our mouse model treated with or without vitamin C and to identify the transcription factors that affect the pathways leading to the WS phenotypes.
CONCLUSIONS
The present study is the first to demonstrate that treatment of a mouse model phenocopying many aspects of the human WS with vitamin C reverses most of the phenotypes associated with this syndrome. We also show that such Wrn-mutant mice exhibit a severe premature defenestration of their liver sinusoidal endothelial cells, a phenotype usually associated with old age in different animal models (18). Long-term (since weaning) and short-term (8 wk) treatments of vitamin C reversed the liver sinusoidal endothelial defenestration phenotype. Notably, the major upstream regulator of lipid metabolism PPARα was increased by both long-and short-term treatments. Finally, vitamin C (or ascorbate) increased the mean life span of our Wrn mutant mice. In contrast, vitamin C had no beneficial effect on the health of WT animals. Our results suggest that long-term vitamin C supplementation could have beneficial effects for patients with WS.
Supplementary Material
Acknowledgments
We thank Dr. O. Neyret-Djossou of the functional genomic platform of the Institut de Recherche Clinique de Montreal (Montreal, QC, Canada) for the microarray services. We are also grateful to M. Sild and A. Labbé for real-time RT-PCRs (Centre de Recherche en Cancérologie de l’Université Laval). This work was supported in part by grants from the Canadian Institutes of Health Research and from the National Sciences and Engineering Research Council of Canada to M.L. and from The National Health and Medical Research Council of Australia and from the Aging and Alzheimers Research Foundation (a Division of the Medical Foundation of the University of Sydney) to D.G.L. M.L. is a senior scholar of the Fonds de la Recherche en Santé du Québec.
References
- 1.Kipling D, Davis T, Ostler EL, Faragher RGA. What can progeroid syndromes tell us about human aging? Science. 2004;305:1426–1431. doi: 10.1126/science.1102587. [DOI] [PubMed] [Google Scholar]
- 2.Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner’s syndrome: a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine. 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Salk D. Werner’s syndrome: a review of recent research with an analysis of connective tissue metabolism, growth control of cultured cells, and chromosomal aberrations. Hum Genet. 1982;62:1–5. doi: 10.1007/BF00295598. [DOI] [PubMed] [Google Scholar]
- 4.Yu C, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 5.Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Balajee AS, Machwe A, May A, Gray MD, Oshima J, Martin GM, Nehlin JO, Brosh R, Orren DK, Bohr VA. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell. 1999;10:2655–2668. doi: 10.1091/mbc.10.8.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;4:907–912. [PMC free article] [PubMed] [Google Scholar]
- 8.Shen JC, Loeb LA. The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet. 2000;16:213–220. doi: 10.1016/s0168-9525(99)01970-8. [DOI] [PubMed] [Google Scholar]
- 9.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in Werner Syndrome. Mol Cell Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 11.Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci U S A. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebel M, Lavoie J, Gaudreault I, Bronsard M, Drouin R. Genetic cooperation between the Werner syndrome protein and poly(ADPribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol. 2003;162:1559–1569. doi: 10.1016/S0002-9440(10)64290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massip L, Garand C, Turaga RV, Deschênes F, Thorin E, Lebel M. Increased insulin, triglycerides, reactive oxygen species, and cardiac fibrosis in mice with a mutation in the helicase domain of the Werner syndrome gene homologue. Exp Gerontol. 2006;41:157–168. doi: 10.1016/j.exger.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 14.Lombard DB, Beard C, Johnson B, Marciniak RA, Dausman J, Bronson R, Buhlmann JE, Lipman R, Curry R, Sharpe A, Jaenisch R, Guarente L. Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol Cell Biol. 2000;20:3286–3291. doi: 10.1128/mcb.20.9.3286-3291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore G, Knoblaugh S, Gollahon K, Rabinovitch P, Ladiges W. Hyperinsulinemia and insulin resistance in Wrn null mice fed a diabetogenic diet. Mech Aging Dev. 2008;129:201–206. doi: 10.1016/j.mad.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 17.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 18.Le Couteur DG, Warren A, Cogger VC, Smedsrød B, Sørensen KK, De Cabo R, Fraser R, McCuskey RS. Old age and the hepatic sinusoid. Anat Rec (Hoboken) 2008;291:672–683. doi: 10.1002/ar.20661. [DOI] [PubMed] [Google Scholar]
- 19.Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology. 2005;42:1349–1354. doi: 10.1002/hep.20937. [DOI] [PubMed] [Google Scholar]
- 20.Maeda N, Hagihara H, Nakata Y, Hiller S, Wilder J, Reddick R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci U S A. 2000;97:841–846. doi: 10.1073/pnas.97.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 22.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 23.Warren A, Bertolino P, Cogger VC, McLean AJ, Fraser R, Le Couteur DG. Hepatic pseudocapillarization in aged mice. Exp Gerontol. 2005;40:807–812. doi: 10.1016/j.exger.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Deschênes F, Massip L, Garand C, Lebel M. In vivo misregulation of genes involved in apoptosis, development and oxidative stress in mice lacking both functional Werner syndrome protein and poly(ADP-ribose) polymerase-1. Hum Mol Genet. 2005;14:3293–3308. doi: 10.1093/hmg/ddi362. [DOI] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondo Y, Inai Y, Sato Y, Handa S, Kubo S, Shimokado K, Goto S, Nishikimi M, Maruyama N, Ishigami A. Senescence marker protein 30 functions as gluconolactonase in L-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc Natl Acad Sci U S A. 2006;103:5723–5728. doi: 10.1073/pnas.0511225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sottocasa GC, Kuylenstierna B, Ernster L, Bergstrand A. An electron-transport system associated with the outer membrane of liver mitochondria. J Cell Biol. 1967;32:415–438. doi: 10.1083/jcb.32.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgese N, Pietrini G. Distribution of the integral membrane protein NADH-cytochrome b5 reductase in rat liver cells, studied with a quantitative radioimmunoblotting assay. Biochem J. 1986;239:393–403. doi: 10.1042/bj2390393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagano G, Zatterale A, Degan P, d’Ischia M, Kelly FJ, Pallardó FV, Calzone R, Castello G, Dunster C, Giudice A, Kilinç Y, Lloret A, Manini P, Masella R, Vuttariello E, Warnau M. In vivo prooxidant state in Werner syndrome (WS): results from three WS patients and two WS heterozygotes. Free Radic Res. 2005;39:529–533. doi: 10.1080/10715760500092683. [DOI] [PubMed] [Google Scholar]
- 30.Otsuka M, Matsuzawa M, Ha TY, Arakawa N. Contribution of a high dose of L-ascorbic acid to carnitine synthesis in guinea pigs fed high-fat diets. J Nutr Sci Vitaminol (Tokyo) 1999;45:163–171. doi: 10.3177/jnsv.45.163. [DOI] [PubMed] [Google Scholar]
- 31.Massie HR, Aiello VR, Doherty TJ. Dietary vitamin C improves the survival of mice. Gerontology. 1984;30:371–375. doi: 10.1159/000212659. [DOI] [PubMed] [Google Scholar]
- 32.Kashino G, Kodama S, Nakayama Y, Suzuki K, Fukase K, Goto M, Watanabe M. Relief of oxidative stress by ascorbic acid delays cellular senescence of normal human and Werner syndrome fibroblast cells. Free Radic Biol Med. 2003;35:438–443. doi: 10.1016/s0891-5849(03)00326-5. [DOI] [PubMed] [Google Scholar]
- 33.Mori S, Murano S, Yokote K, Takemoto M, Asaumi S, Take A, Saito Y. Enhanced intra-abdominal visceral fat accumulation in patients with Werner’s syndrome. Int J Obes Relat Metab Disord. 2001;25:29229–29225. doi: 10.1038/sj.ijo.0801529. [DOI] [PubMed] [Google Scholar]
- 34.Bokko PB, Francione L, Bandala-Sanchez E, Ahmed AU, Annesley SJ, Huang X, Khurana T, Kimmel AR, Fisher PR. Diverse cytopathologies in mitochondrial disease are caused by AMP-activated protein kinase signaling. Mol Biol Cell. 2007;18:1874–1886. doi: 10.1091/mbc.E06-09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JS, Chu IS, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–1311. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 36.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 37.Zhu H, Cong JP, Mamtora G, Gingeras T, Shenk T. Cellular gene expression altered by human cytomegalo-virus: global monitoring with oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:14470–14475. doi: 10.1073/pnas.95.24.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kyng KJ, May A, Kølvraa S, Bohr VA. Gene expression profiling in Werner syndrome closely resembles that of normal aging. Proc Natl Acad Sci U S A. 2003;100:12259–12264. doi: 10.1073/pnas.2130723100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shepard JL, Amatruda JF, Stern HM, Subramanian A, Finkelstein D, Ziai J, Finley KR, Pfaff KL, Hersey C, Zhou Y, Barut B, Freedman M, Lee C, Spitsbergen J, Neuberg D, Weber G, Golub TR, Glickman JN, Kutok JL, Aster JC, Zon LI. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc Natl Acad Sci U S A. 2005;102:13194–13199. doi: 10.1073/pnas.0506583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS, Attie AD. The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci U S A. 2000;97:11371–11376. doi: 10.1073/pnas.97.21.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu D, Cozma D, Park A, Thomas-Tikhonenko A. Functional validation of genes implicated in lymphomagenesis: an in vivo selection assay using a Myc-induced B-cell tumor. Ann N Y Acad Sci. 2005;1059:145–159. doi: 10.1196/annals.1339.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ichiba T, Teshima T, Kuick R, Misek DE, Liu C, Takada Y, Maeda Y, Reddy P, Williams DL, Hanash SM, Ferrara JL. Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood. 2003;102:763–771. doi: 10.1182/blood-2002-09-2748. [DOI] [PubMed] [Google Scholar]
- 43.Flechner SM, Kurian SM, Head SR, Sharp SM, Whisenant TC, Zhang J, Chismar JD, Horvath S, Mondala T, Gilmartin T, Cook DJ, Kay SA, Walker JR, Salomon DR. Kidney transplant rejection and tissue injury by gene profiling of biopsies and peripheral blood lymphocytes. Am J Transplant. 2004;4:1475–1489. doi: 10.1111/j.1600-6143.2004.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A. 2004;101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLachlan JL, Smith AJ, Bujalska IJ, Cooper PR. Gene expression profiling of pulpal tissue reveals the molecular complexity of dental caries. Biochim Biophys Acta. 2005;1741:271–281. doi: 10.1016/j.bbadis.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 46.Hoffmann R, Seidl T, Neeb M, Rolink A, Melchers F. Changes in gene expression profiles in developing B cells of murine bone marrow. Genome Res. 2002;12:98–111. doi: 10.1101/gr.201501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 48.Villalba JM, Navas P. Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal. 2000;2:213–230. doi: 10.1089/ars.2000.2.2-213. [DOI] [PubMed] [Google Scholar]
- 49.Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 50.Kuo SM, MacLean ME, McCormick K, Wilson JX. Gender and sodium-ascorbate transporter isoforms determine ascorbate. J Nutr. 2004;134:2216–2221. doi: 10.1093/jn/134.9.2216. [DOI] [PubMed] [Google Scholar]
- 51.Davis T, Baird DM, Haughton MF, Jones CJ, Kipling D. Prevention of accelerated cell aging in Werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J Gerontol A Biol Sci Med Sci. 2005;60:1386–1393. doi: 10.1093/gerona/60.11.1386. [DOI] [PubMed] [Google Scholar]
- 52.Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsieh CC, Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 55.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lam TK, Yoshii H, Haber CA, Bogdanovic E, Lam L, Fantus IG, Giacca A. Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab. 2002;283:E682–E691. doi: 10.1152/ajpendo.00038.2002. [DOI] [PubMed] [Google Scholar]
- 57.Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflügers Arch. 2005;450:363–371. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 58.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J Physiol. 2008;586:4055–4059. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Masternak MM, Bartke A. PPARs in calorie restricted and genetically long-lived mice [Online] PPAR Res. 2007;28436 doi: 10.1155/2007/28436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, Lucci-Cordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, von Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27:558–567. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lebel M, Spillare EA, Harris CC, Leder P. Werner syndrome gene product co-purifies with the DNA replication complex and interact with PCNA and topoisomerase I. J Biol Chem. 1999;274:37795–37799. doi: 10.1074/jbc.274.53.37795. [DOI] [PubMed] [Google Scholar]
- 62.Lloret A, Calzone R, Dunster C, Manini P, d’Ischia M, Degan P, Kelly FJ, Pallardo FV, Zatterale A, Pagano G. Different patterns of in vivo pro-oxidant states in a set of cancer- or aging-related genetic diseases. Free Rad Biol Med. 2008;44:495–503. doi: 10.1016/j.freeradbiomed.2007.10.046. [DOI] [PubMed] [Google Scholar]
- 63.Swindell WR. Gene expression profiling of long-lived dwarf mice: longevity-associated genes and relationships with diet, gender and aging. BMC Genomics. 2007;8:353–374. doi: 10.1186/1471-2164-8-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schumacher B, van der Pluijm I, Moorhouse MJ, Kosteas T, Robinson AR, Suh Y, Breit TM, van Steeg H, Niedernhofer LJ, van Ijcken W, Bartke A, Spindler SR, Hoeijmakers JH, van der Horst GT, Garinis GA. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008;4:e1000161. doi: 10.1371/journal.pgen.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang C, Li Q, Redden DT, Weindruch R, Allison DB. Statistical methods for testing effects on “maximum lifespan. Mech Ageing Dev. 2004;125:629–632. doi: 10.1016/j.mad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 66.Zandbergen F, Plutzky J. PPARα in atherosclerosis and inflammation. Biochim Biophys Acta. 2007;1771:972–982. doi: 10.1016/j.bbalip.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto H, Kurebayashi S, Kouhara H, Yoshiuchi K, Matsuhisa M, Yamasaki Y, Kasayama S. Impacts of long-term treatments with testosterone replacement and pioglitazone on glucose and lipid metabolism in male patients with Werner’s syndrome. Clin Chim Acta. 2007;379:167–170. doi: 10.1016/j.cca.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 68.Honjo S, Yokote K, Fujishiro T, Maezawa Y, Sato S, Koshizaka M, Saito Y. Early amelioration of insulin resistance and reduction of interleukin-6 in Werner syndrome using pioglitazone. J Am Geriatr Soc. 2008;56:173–174. doi: 10.1111/j.1532-5415.2007.01484.x. [DOI] [PubMed] [Google Scholar]
- 69.Yokote K, Saito Y. Extension of the life span in patients with Werner syndrome. J Am Geriatr Soc. 2008;56:1770–1771. doi: 10.1111/j.1532-5415.2008.01817.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.