

Abstract
MicroRNAs (miRNAs) are a group of small non-coding RNAs that play important regulatory roles at the post-transcriptional level. Although several computational methods have been developed to compare miRNAs, it is still a challenging and a badly needed task with the availability of various biological data resources. In this study, we proposed a novel graph theoretic property based computational framework and method, called miRFunSim, for quantifying the associations between miRNAs based on miRNAs targeting propensity and proteins connectivity in the integrated protein-protein interaction network. To evaluate the performance of our method, we applied the miRFunSim method to compute functional similarity scores of miRNA pairs between 100 miRNAs whose target genes have been experimentally supported and found that the functional similarity scores of miRNAs in the same family or in the same cluster are significantly higher compared with other miRNAs which are consistent with prior knowledge. Further validation analysis on experimentally verified miRNA-disease associations suggested that miRFunSim can effectively recover the known miRNA pairs associated with the same disease and achieve a higher AUC of 83.1%. In comparison with similar methods, our miRFunSim method can achieve more effective and more reliable performance for measuring the associations of miRNAs. We also conducted the case study examining liver cancer based on our method, and succeeded in uncovering the candidate liver cancer related miRNAs such as miR-34 which also has been proven in the latest study.
Introduction
MicroRNAs (miRNAs), ∼ 22 nucleotides (nt) in length, are a major class of short endogenous non-coding RNA (ncRNA) molecules that play important regulatory roles at the post-transcriptional level by targeting mRNAs for cleavage or translational repression [1], [2]. Since the discovery of miRNA molecules lin-4 and let-7 in 1993 in Caenorhabditis elegans through forward genetic screens [3], more and more novel miRNAs have been identified in almost all metazoan genomes, including worms, flies, plants and mammals by forward genetics, direct cloning, high-throughput sequencing technology and bioinformatics approaches [4], [5], [6]. To date, 1600 miRNAs of the human genome have been annotated in the latest version of the miRBase [7].
During the past several years, many methods have been proposed to compare the functional similarities between different protein-coding genes for further better understanding of the underlying biological phenomena or discovering previously unknown gene functions [8], [9], [10], [11], [12]. With the growth of information on miRNAs, miRNAs have been shown as a group of important regulators to regulate basic cellular functions including proliferation, differentiation and death [13], [14], [15], [16]. However, the functions of most miRNAs remain unknown. Therefore, to better understand miRNAs and their roles in the underlying biological phenomena, biologists are paying more attention to compare miRNA genes and want to know the associations between them. For example, comparing similarities between miRNA with known molecular functions or associated with specific disease and that with unknown functions would allow us to infer potential functions for novel miRNAs, or help us to identify potential candidate disease-related miRNAs for guiding further biological experiments. However, until now, only several computational methods have been developed to meet the requirement [17], [18]. Therefore, comparing miRNAs is still a challenging and a badly needed task with the availability of various biological data resources.
Many studies have shown that the functions of miRNAs can be predicted or inferred by analyzing the properties of miRNA targets [19], [20], [21]. It has been reported that the targeting propensity of miRNA can be largely explained by the functional behavior of protein connectivity in the protein-protein interaction network (PPIN) [22], [23]. With the rapid advances in biotechnology, large-scale PPIN is currently available and is already rich enough to evaluate the relationship between miRNAs based on their targeting propensity in PPIN. Here, based on the above notion, we proposed a novel computational method, called miRFunSim, to quantify the associations between miRNAs in the context of protein interaction network. We evaluated and validated the performance of our miRFunSim method on miRNA family, miRNA cluster data and experimentally verified miRNA-disease associations. Further comparison analysis showed that our method is more effective and reliable as compared to other existing similar methods, and offers a significant advance in measuring the associations between miRNAs.
Materials and Methods
Construction of Integrated Human Protein Interaction Network
The high throughput protein-protein interaction data were obtained from Wang’s study [24] consisting of 69,331 interactions between 11,305 proteins, which integrated BioGRID [25], IntAct [26], MINT [27], HPRD [28] and by the Co-citation of text mining [29] databases and made further filtering to improve coverage and quality of PPIN and reduce false-positives produced by different prediction algorithms in different databases.
Human miRNA Datasets
All known human miRNAs were from miRBase Sequence Database, release 16 (http://www.mirbase.org/) [30]. We used experimentally verified miRNA targets from TarBase which houses a manually curated collection of experimentally supported miRNA targets in several animal species [31] (File S1). The predicted miRNA targets were downloaded from starBase database which provides a comprehensive integrated miRNA-target map [32]. Because of the high false positive rate of predicted miRNA targets, we only chose miRNA targets predicted by at least three prediction algorithms with readNum > = 1 and biological complexity > = 1 (File S2). The genome coordinates of miRNAs were downloaded from miRBase Sequence Database, release 16 (http://www.mirbase.org/) [30]. Those miRNAs with pair-wise distance less than 10 kb were considered as clustered miRNAs. The high-quality experimentally verified miRNA-disease association data was retrieved from Jiang’s study [33].
Statistical Analysis
The functional similarity score between miRNAs may be generated by chance. In order to take this effect into account and obtain the statistical significance of scores, we performed randomization test and repeated 1000 times. For each score, 1000 simulated miRNA pairs were generated and target genes of simulated miRNA pairs were randomly sampled from all human protein-coding genes keeping the same size as given miRNA pairs. Then the functional similarity scores between simulated miRNA pairs were recomputed for each simulated miRNA pair denoted SFSSM. M denoted the number of simulated miRNA pairs having an equal or larger SFSSM value than the true score. The estimate of the empirical statistical significance value, P-value, of true score was obtained as P = M/1001. The empirical P-value based on such randomizations represented the probability of obtaining a score greater than a given score by chance.
Results and Discussion
Overview of miRFunSim
In this study, we developed a graph theoretic property based method, miRFunSim, to quantify the associations between two miRNA in the context of targets propensity in the protein-protein interaction network. A schematic representation of the miRFunSim method is shown in Figure 1. Initially, given two interested miRNAs, miRNA A and miRNA B, we evaluate the functional relationship between them using the protein interaction network. First, we obtain the target gene lists for each miRNA, which are denoted by TA and TB respectively. There may be existing common targets between TA and TB. Second, we map the target genes from these lists onto the integrated protein interaction network. Then, the protein interaction sub-network of target genes is generated from the integrated protein interaction network. Here, the targets of miRNA A and miRNA B are marked in red and green respectively in the protein interaction sub-network of target genes. The nodes colored by both of the red and green in the protein interaction sub-network are the common targets. Thirdly, the distance between two nodes which are from TA and TB respectively is the length of the shortest path and can be calculated based on the sub-network which reflects the protein connectivity and functional associations of targets. For the overlap of targets between TA and TB, we made two hypotheses: there exists a ring on the node that represents the common target, and there exists a shortcut between two targets except they are isolated. Based on the above hypotheses, we add the hypothetical edges into the protein interaction sub-network, where the new edges are marked in red. Finally, the functional similarity score between two miRNAs is defined as the reciprocal of average pair-wise distance between TA and TB, and is computed as follows:
Figure 1. The schematic representation and overview of the miRFunSim method.

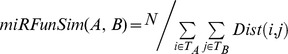 |
where N is the number of path in the protein interaction sub-network. The closer the miRNA targets are in the network, the higher the scores. The scores range from 0 to 1 and higher scores correspond to higher functional concordance between miRNAs.
Performance Evaluation of miRFunSim
The accumulating evidence revealed that miRNAs in the same family are likely to have similar functions [34], [35], [36]. Therefore, to evaluate the reliability of functional similarity scores computed by our miRFunSim method, we first downloaded miRNA family data from miRBase Database [30] and obtained 100 miRNAs whose target genes have been experimentally supported from TarBase [31]. Then we used our miRFunSim method to compute functional similarity scores of miRNA pairs between 100 miRNAs. These miRNA pairs were grouped into two classes: intrafamily miRNA pairs and interfamily miRNA pairs. We further compared the functional similarity scores of intrafamily miRNA pairs,interfamily miRNA pairs and random miRNAs pairs. As a result, the significant differences in functional similarity scores among intrafamily miRNA pairs, interfamily miRNA pairs and random miRNA pairs are observed (Figure 2A, Kruskal-Wallis test, df = 2, p-value = 0). The functional similarity scores for intrafamily miRNA pairs are significantly higher compared with interfamily miRNA pairs (p-value = 4.30e-5, Wilcoxon rank sum test) and random miRNA pairs (p-value = 1.80e-14, Wilcoxon rank sum test). Interfamily miRNA pairs also showed higher functional similarity scores than random miRNA pairs (p-value = 1.46e-20, Wilcoxon rank sum test).
Figure 2. Performance evaluation of miRFunSim using miRNA family and miRNA cluster.

(A) A comparison of functional similarity scores of intrafamily miRNA pairs, interfamily miRNA pairs and random miRNA pairs. (B) A comparison of functional similarity scores of intracluster miRNA pairs, intercluster miRNA pairs and random miRNA pairs.
It is well known that a large number of miRNAs have clustering propensity and tend to form some clusters. Previous studies have suggested that miRNA members within the same cluster are often located in a polycistron and display a homogeneous expression pattern [37], [38], which imply that these clustered miRNAs perhaps have common or similar functions. Therefore, we also computed functional similarity scores between miRNAs in the same cluster and between miRNAs not located in the same cluster using miRFunSim method (Fig. 2B). Statistical analyses showed that functional similarity scores among intracluster miRNA pairs, intercluster miRNA pairs and random miRNA pairs are also significantly different (Figure 2B, Kruskal-Wallis test, df = 2, p-value = 0). The functional similarity scores of intracluster miRNA pairs are significantly higher than those of intercluster miRNA pairs (p-value = 5.42e-6, Wilcoxon rank sum test) and random miRNA pairs (p-value = 1.40e-8, Wilcoxon rank sum test). Intercluster miRNA pairs also showed higher functional similarity scores than random miRNA pairs (p-value = 5.60e-23, Wilcoxon rank sum test). These results suggested that our method is reliable and sensible to measure the functional relationship between miRNAs. To investigate the robustness of our miRFunSim method, we first performed above analysis for the predicted miRNA target and examine whether our method is still able to measure the associations between miRNAs. The resulting scores between miRNAs using predicted targets are shown in Fig. S1A. The functional similarity scores for intrafamily and intracluster miRNA pairs are significantly higher compared with interfamily and intercluster miRNA pairs (p-value = 0 and p-value = 1.01e-6, Wilcoxon rank sum test). Then we further access our method by the removal of 5% and 10% network nodes in the protein interaction network randomly (Fig. S1B, C). As shown in Fig. S1B, C, the functional similarity scores of intrafamily and intracluster miRNA pairs are significantly higher than those of interfamily (5%:p-value = 9.47e-5 and 10%:p-value = 9.53e-5, Wilcoxon rank sum test) and intercluster miRNA pairs (p-value = 2.01e-3 and p-value = 1.6e-3, Wilcoxon rank sum test).
To further evaluate the performance of our miRFunSim method for quantifying the associations between two miRNAs, we performed a validation analysis on experimentally verified miRNA-disease associations. It has been proven that miRNAs with similar functions tend to be involved in phenotypically similar disease, and miRNAs associated with common diseases are more related in function [17], [33], [34]. Our validation analysis for performance of miRFunSim method was based on above notion. First, we obtained 270 high-quality experimentally verified miRNA-disease associations from Jiang’s study [33] and 100 miRNAs whose target genes have been experimentally supported. For each disease, the functional similarity score between every two miRNAs associated with this disease were computed using the miRFunSim method as the testing case. For each testing case, 99 simulated miRNA pairs were generated and the target genes of simulated miRNA pairs were randomly sampled from all human protein-coding genes keeping the same size as the given testing case. The functional similarity scores of 99 simulated miRNA pairs also were computed using the miRFunSim method as negative controls of the given testing case. Second, we prioritized the testing case together with 99 negative controls according to the scores derived from miRFunSim method. Therefore, for each testing case, we obtain a ranking list, that is, prioritization of 100 miRNA pairs. In total, we obtained 562 ranking lists, each with 100 prioritizations. Third, from 562 ranking lists, we calculated the sensitivity and specificity at varying thresholds. Sensitivity measures the proportion of the testing case whose ranking is higher than a given score. Specificity measures the proportion of negative controls ranked below this score. Finally, a receiver operating characteristics (ROC) curve was plotted by varying the score and the area under the curve (AUC) was calculated. We used AUC as a standard measure of the performance of miRFunSim. The maximum value of AUC is 100%, which indicates every testing case is ranked first in the ranking list. Figure 3 shows the results of performance evaluation of miRFunSim using the ROC curves obtained by calculating the sensitivity (sensitivity = TP/(TP +FN)) and 1-specificity (specificity = TN/(TN+FP)) by varying the threshold. Our miRFunSim method tested on 270 high-quality experimentally verified microRNA-disease associations achieved an AUC of 83.1%, suggesting that miRFunSim can recover the miRNA pairs associated with common disease and efficiently quantify the relationship between miRNAs.
Figure 3. Area under ROC curve (AUC) analysis on 270 high-quality experimentally verified miRNA-disease associations from Jiang’s study and 100 miRNAs whose target genes have been experimentally supported using our miRFunSim method.

Comparisons with Other Existing Similar Methods
Recently, several approaches have been proposed for comparing miRNAs. Yu et al. developed a method to determine functional similarity of miRNAs by using their target genes GO semantic similarities [18]. However, this method perhaps sometimes produces disappointing results because of some GO limitations. Another existing method, called MISIM, is to measure the similarity of their associated disease directed acyclic graph (DAG) to compare two miRNAs. However, this method relies on miRNA-disease association data, and is difficult to achieve high reliability when little miRNA-disease association data is available [17]. Here, we also performed a performance comparison analysis between miRFunSim and these two similar methods using the same datasets. First, we used the method presented by Yu et al. and MISIM to compute functional similarity scores of miRNA pairs between 100 miRNAs whose target genes have been experimentally supported from TarBase [31]. Then these miRNA pairs also were grouped into four classes: intrafamily miRNA pairs, interfamily miRNA pairs, intracluster miRNA pairs and intercluster miRNA pairs. As shown in Fig. 4A, the functional similarity scores produced by Yu’s method are significantly different among intrafamily, interfamily and random miRNA pairs (Kruskal-Wallis test, df = 2, p-value = 0), and among intracluster, intercluster and random miRNA pairs (Kruskal-Wallis test, df = 2, p-value = 0). However, there is no significant difference in functional similarity scores produced by MISIM method between intrafamily and interfamily miRNA pairs (p-value = 0.25, Wilcoxon rank sum test), and between intracluster and intercluster miRNA pairs (p-value = 0.19, Wilcoxon rank sum test) (Fig. 4B), suggesting that the functional similarity scores produced by Yu’s method and our miRFunSim method can better reflect the functional relationship of miRNAs based on miRNA families and miRNA clusters than MISIM method. Next, we also tested Yu’s method on 270 high-quality experimentally verified miRNA-disease associations to compute the functional similarity score between every two miRNAs associated with the same disease, and obtained a ROC curve as the methods described in our analysis. Finally, the method presented by Yu et al. achieved an AUC of 63.9% (Fig. 4C), but is less than an AUC of 83.1% obtained by our miRFunSim method tested on the same datasets. Taken together, these results suggested that our miRFunSim method can achieve more effective and more reliable performance for quantifying the associations between miRNAs compared with other available similar methods.
Figure 4. A performance comparison analysis between miRFunSim and other existing methods with similar functions.

(A) The distribution and comparison of functional similarity scores of intrafamily, interfamily, intracluster,intercluster and random miRNA pairs computed by Yu’s method. (B) The distribution and comparison of functional similarity scores of intrafamily, interfamily, intracluster, intercluster miRNA pairs computed by MISIM method. (C) Area under ROC curve (AUC) analysis on 270 high-quality experimentally verified miRNA-disease associations from Jiang’s study and 100 miRNAs whose target genes have been experimentally supported using Yu’s method.
Case Study of Liver Cancer
As an example, to illustrate the application of quantifying the relationship between miRNAs using miRFunSim method, we presented a case study of liver cancer, which is one of the most common cancers, and applied the miRFunSim method to identify novel candidate liver cancer-related miRNAs. First, we retrieved 15 miRNAs which have been experimentally verified to contribute to the development of liver cancer and have experimentally verified target genes in TarBase as seed miRNAs. Next, we computed the functional similarity scores between every seed miRNAs and every miRNA from the remaining 85 miRNAs using miRFunSim method. The higher the score is, the more likely the miRNAs is associated with liver cancer. Finally, we prioritized all 1275 miRNA pairs for liver cancer according to their scores. The top 15 miRNA pairs with the highest functional similarity scores (score>0.5 and p<0.005) were chosen and 12 miRNAs with the highest functional similarity scores with seed miRNAs were listed as candidate liver cancer-related miRNAs and shown in Table 1. Among top 12 miRNAs, 8 miRNAs have been recorded to be deregulated in liver cancer and possibly contribute to the development of liver cancer, and 4 miRNAs (miR-15b,miR-20, miR-15 and miR-34) have been verified to be deregulated in other cancers in miR2Disease [39], and PhenomiR [37] databases which provide comprehensive resources for miRNA deregulation in disease. When our research is in progress, a new study provided further supporting evidence for one of the remaining four candidate liver cancer-related miRNAs. Li et al. [40] found that miR-34 participate in the neoplastic transformation of liver cancer stem cells (LCSCs) during hepatocarcinogenesis.
Table 1. The top 12 miRNAs with the highest functional similarity scores with known experimentally verified liver cancer-related miRNAs.
| MiRNAname | Scores | P-value | Liver cancer-related miRNAs | References |
| miR-145 | 1.0 | 0.001 | YES | [41] |
| miR-15b | 1.0 | <0.001 | * | [42], [43] |
| miR-20 | 1.0 | <0.001 | * | [44] |
| miR-200a | 1.0 | <0.001 | YES | [45], [46] |
| miR-200c | 1.0 | 0.002 | YES | [47] |
| miR-214 | 1.0 | 0.002 | YES | [48], [49] |
| miR-222 | 1.0 | <0.001 | YES | [49], [50] |
| miR-17-5p | 0.75 | <0.001 | YES | [51] |
| miR-125a | 0.67 | <0.001 | YES | [45], [52] |
| miR-127 | 0.60 | <0.001 | YES | [53] |
| miR-15 | 0.55 | <0.001 | * | [54] |
| miR-34 | 0.51 | <0.001 | * | [55] |
Note: The miRNAs which have been recorded to be deregulated in liver cancer in previous studies were designated as “YES”. The miRNAs which have been verified to be deregulated in other cancers in miR2Disease and PhenomiR databases were designated as “*”.
Conclusions
In this study, we presented a novel computational framework and method, called miRFunSim, for quantifying the associations between miRNAs based on miRNAs targeting propensity and proteins connectivity in the integrated protein-protein interaction network. We applied the miRFunSim method to compare 100 miRNAs whose target genes have been experimentally supported from TarBase [31] and compared the distributions of functional similarity scores among intrafamily, interfamily and random miRNA pairs, and among intracluster, intercluster and random miRNA pairs. The functional similarity scores of miRNAs in the same family or in the same cluster are significantly higher compared with other miRNAs. These results suggested that the miRFunSim method can better reflect the functional similarities and differences of miRNA pairs in the different groups. We further tested miRFunSim method on 270 high-quality experimentally verified miRNA-disease associations to recover the known miRNA pairs associated with the same disease and achieved a higher AUC of 83.1%. In comparison with existing similar methods, our miRFunSim method can achieve more effective and more reliable performance for measuring the functional similarity of miRNAs. With the improvement in coverage of PPI network and in prediction accuracy of miRNA targets, the proposed miRFunSim method will perform better for quantifying the associations between miRNAs. Furthermore, this method can be extended to other species when PPIN data and targets of miRNAs are available.
Supporting Information
The robustness analysis results for measuring the relationship of miRNAs using miRFunSim. (A) A comparison of functional similarity scores between intrafamily and interfamily miRNA pairs, and between intracluster and intercluster miRNA pairs using predicted miRNA targets. (B) A comparison of functional similarity scores between intrafamily and interfamily miRNA pairs, and between intracluster and intercluster miRNA pairs by the removal of 5% network nodes in the protein interaction network randomly. (C) A comparison of functional similarity scores between intrafamily and interfamily miRNA pairs, and between intracluster and intercluster miRNA pairs by the removal of 10% network nodes in the protein interaction network randomly.
(DOC)
Information of experimentally verified miRNA targets from TarBase.
(TXT)
Information of predicted miRNA targets from starBase by at least three prediction algorithms with readNum> = 1 and biological complexity > = 1.
(TXT)
Acknowledgments
We would like to thank the reviewers for valuable comments and suggestions.
Funding Statement
This work was supported by the Foundation of Health Department of Heilongjiang Province (Grant No.2012-809). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 2. Ambros V (2004) The functions of animal microRNAs. Nature 431: 350–355. [DOI] [PubMed] [Google Scholar]
- 3. Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D (2003) MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol 13: 807–818. [DOI] [PubMed] [Google Scholar]
- 4. Cullen BR (2006) Viruses and microRNAs. Nat Genet 38 Suppl: S25–30 [DOI] [PubMed] [Google Scholar]
- 5. Zhu E, Zhao F, Xu G, Hou H, Zhou L, et al. (2010) mirTools: microRNA profiling and discovery based on high-throughput sequencing. Nucleic Acids Res 38: W392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mendes ND, Freitas AT, Sagot MF (2009) Current tools for the identification of miRNA genes and their targets. Nucleic Acids Res 37: 2419–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schlicker A, Albrecht M (2010) FunSimMat update: new features for exploring functional similarity. Nucleic Acids Res 38: D244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Du Z, Li L, Chen CF, Yu PS, Wang JZ (2009) G-SESAME: web tools for GO-term-based gene similarity analysis and knowledge discovery. Nucleic Acids Res 37: W345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schlicker A, Domingues FS, Rahnenfuhrer J, Lengauer T (2006) A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics 7: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J, Aronow BJ, Jegga AG (2009) Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinformatics 10: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharan R, Ulitsky I, Shamir R (2007) Network-based prediction of protein function. Mol Syst Biol 3: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu P, Guo M, Hay BA (2004) MicroRNAs and the regulation of cell death. Trends Genet 20: 617–624. [DOI] [PubMed] [Google Scholar]
- 14. Hwang HW, Mendell JT (2007) MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer 96 Suppl: R40–44 [PubMed] [Google Scholar]
- 15.Wang D, Qiu C, Zhang H, Wang J, Cui Q, et al. (2010) Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PLoS One 5. [DOI] [PMC free article] [PubMed]
- 16. Cui Q, Yu Z, Purisima EO, Wang E (2006) Principles of microRNA regulation of a human cellular signaling network. Mol Syst Biol 2: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang D, Wang J, Lu M, Song F, Cui Q (2010) Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26: 1644–1650. [DOI] [PubMed] [Google Scholar]
- 18. Yu G, Xiao C, Bo X, Lu C, Qin Y, et al. (2010) A new method for measuring functional similarity of microRNAs. Journal of integrated omics 1: 49–54. [Google Scholar]
- 19. Nam S, Kim B, Shin S, Lee S (2008) miRGator: an integrated system for functional annotation of microRNAs. Nucleic Acids Res 36: D159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roubelakis MG, Zotos P, Papachristoudis G, Michalopoulos I, Pappa KI, et al. (2009) Human microRNA target analysis and gene ontology clustering by GOmir, a novel stand-alone application. BMC Bioinformatics 10 Suppl 6S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ulitsky I, Laurent LC, Shamir R (2010) Towards computational prediction of microRNA function and activity. Nucleic Acids Res 38: e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang H, Li WH (2007) MicroRNA regulation of human protein protein interaction network. Rna 13: 1402–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsu CW, Juan HF, Huang HC (2008) Characterization of microRNA-regulated protein-protein interaction network. Proteomics 8: 1975–1979. [DOI] [PubMed] [Google Scholar]
- 24. Wang Q, Sun J, Zhou M, Yang H, Li Y, et al. (2011) A novel network-based method for measuring the functional relationship between gene sets. Bioinformatics 27: 1521–1528. [DOI] [PubMed] [Google Scholar]
- 25. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, et al. (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, et al. (2007) IntAct–open source resource for molecular interaction data. Nucleic Acids Res 35: D561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, et al. (2010) MINT, the molecular interaction database: 2009 update. Nucleic Acids Res 38: D532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. (2009) Human Protein Reference Database–2009 update. Nucleic Acids Res 37: D767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM (2005) Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol 6: R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36: D154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Papadopoulos GL, Reczko M, Simossis VA, Sethupathy P, Hatzigeorgiou AG (2009) The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res 37: D155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang JH, Li JH, Shao P, Zhou H, Chen YQ, et al. (2011) starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 39: D202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang Q, Hao Y, Wang G, Juan L, Zhang T, et al. (2010) Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst Biol 4 Suppl 1S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu M, Zhang Q, Deng M, Miao J, Guo Y, et al. (2008) An analysis of human microRNA and disease associations. PLoS One 3: e3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu J, Li CX, Li YS, Lv JY, Ma Y, et al. (2011) MiRNA-miRNA synergistic network: construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res 39: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaczkowski B, Torarinsson E, Reiche K, Havgaard JH, Stadler PF, et al. (2009) Structural profiles of human miRNA families from pairwise clustering. Bioinformatics 25: 291–294. [DOI] [PubMed] [Google Scholar]
- 37. Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B, et al. (2010) PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol 11: R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, Zhang R, Su B (2009) Diversity and evolution of MicroRNA gene clusters. Sci China C Life Sci 52: 261–266. [DOI] [PubMed] [Google Scholar]
- 39. Jiang Q, Wang Y, Hao Y, Juan L, Teng M, et al. (2009) miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 37: D98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li R, Qian N, Tao K, You N, Wang X, et al. (2011) MicroRNAs involved in neoplastic transformation of liver cancer stem cells. J Exp Clin Cancer Res 29: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Varnholt H, Drebber U, Schulze F, Wedemeyer I, Schirmacher P, et al. (2008) MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 47: 1223–1232. [DOI] [PubMed] [Google Scholar]
- 42. Xia H, Qi Y, Ng SS, Chen X, Chen S, et al. (2009) MicroRNA-15b regulates cell cycle progression by targeting cyclins in glioma cells. Biochem Biophys Res Commun 380: 205–210. [DOI] [PubMed] [Google Scholar]
- 43. Xia L, Zhang D, Du R, Pan Y, Zhao L, et al. (2008) miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer 123: 372–379. [DOI] [PubMed] [Google Scholar]
- 44. Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, et al. (2009) The miR-17∼92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci U S A 106: 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, et al. (2006) Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 25: 2537–2545. [DOI] [PubMed] [Google Scholar]
- 46. Alisi A, Da Sacco L, Bruscalupi G, Piemonte F, Panera N, et al. (2011) Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab Invest 91: 283–293. [DOI] [PubMed] [Google Scholar]
- 47. Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, et al. (2008) MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology 47: 1955–1963. [DOI] [PubMed] [Google Scholar]
- 48. Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, et al. (2008) Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res 14: 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Y, Lee AT, Ma JZ, Wang J, Ren J, et al. (2008) Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. J Biol Chem 283: 13205–13215. [DOI] [PubMed] [Google Scholar]
- 50. Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, et al. (2009) Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology 50: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Connolly E, Melegari M, Landgraf P, Tchaikovskaya T, Tennant BC, et al. (2008) Elevated expression of the miR-17–92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol 173: 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang XH, Wang Q, Chen JS, Fu XH, Chen XL, et al. (2009) Bead-based microarray analysis of microRNA expression in hepatocellular carcinoma: miR-338 is downregulated. Hepatol Res 39: 786–794. [DOI] [PubMed] [Google Scholar]
- 53. Tryndyak VP, Ross SA, Beland FA, Pogribny IP (2009) Down-regulation of the microRNAs miR-34a, miR-127, and miR-200b in rat liver during hepatocarcinogenesis induced by a methyl-deficient diet. Mol Carcinog 48: 479–487. [DOI] [PubMed] [Google Scholar]
- 54. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, et al. (2002) Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99: 15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gao Y, Wang C, Shan Z, Guan H, Mao J, et al. (2010) miRNA expression in a human papillary thyroid carcinoma cell line varies with invasiveness. Endocr J 57: 81–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The robustness analysis results for measuring the relationship of miRNAs using miRFunSim. (A) A comparison of functional similarity scores between intrafamily and interfamily miRNA pairs, and between intracluster and intercluster miRNA pairs using predicted miRNA targets. (B) A comparison of functional similarity scores between intrafamily and interfamily miRNA pairs, and between intracluster and intercluster miRNA pairs by the removal of 5% network nodes in the protein interaction network randomly. (C) A comparison of functional similarity scores between intrafamily and interfamily miRNA pairs, and between intracluster and intercluster miRNA pairs by the removal of 10% network nodes in the protein interaction network randomly.
(DOC)
Information of experimentally verified miRNA targets from TarBase.
(TXT)
Information of predicted miRNA targets from starBase by at least three prediction algorithms with readNum> = 1 and biological complexity > = 1.
(TXT)