

Abstract
Gracilariopsis lemaneiformis has a high economic value and is one of the most important aquaculture species in China. Despite it is economic importance, it has remained largely unstudied at the genomic level. In this study, we conducted a genome survey of Gp. lemaneiformis using next-generation sequencing (NGS) technologies. In total, 18.70 Gb of high-quality sequence data with an estimated genome size of 97 Mb were obtained by HiSeq 2000 sequencing for Gp. lemaneiformis. These reads were assembled into 160,390 contigs with a N50 length of 3.64 kb, which were further assembled into 125,685 scaffolds with a total length of 81.17 Mb. Genome analysis predicted 3490 genes and a GC% content of 48%.
The identified genes have an average transcript length of 1,429 bp, an average coding sequence size of 1,369 bp, 1.36 exons per gene, exon length of 1,008 bp, and intron length of 191 bp. From the initial assembled scaffold, transposable elements constituted 54.64% (44.35 Mb) of the genome, and 7737 simple sequence repeats (SSRs) were identified. Among these SSRs, the trinucleotide repeat type was the most abundant (up to 73.20% of total SSRs), followed by the di- (17.41%), tetra- (5.49%), hexa- (2.90%), and penta- (1.00%) nucleotide repeat type. These characteristics suggest that Gp. lemaneiformis is a model organism for genetic study. This is the first report of genome-wide characterization within this taxon.
Introduction
Gracilariopsis was long indistinguishable from the genus Gracilaria in the family Gracilariaceae Nageli (Rhodophyta). In 1989, however, Fredericq and Hommersand [1] reinstated this genus based on four important differences between the two genera. Considered to be one of the most important economic macroalgae, Gp. lemaneiformis Bory is utilized for agar extraction [2], [3]. Its high agar yield accounts for more than half of total annual agar production worldwide [4]. Gp. lemaneiformis is also an ideal material for genetic research [5], and it has a potential role in the inhibition of red tides and in bioremediation [6], [7]. Single-rope floating raft cultivation of Gp. lemaneiformis has been widespread along the coastline of South China since the 1990s [5]. Currently, Gp. lemaneiformis is one of the most important aquaculture species in China [8].
Following the success of the human genome project, next-generation sequencing (NGS) technologies were launched. Compared with Roche's 454 and ABI's SOLiD, HiSeq 2000 offers the cheapest sequencing and the biggest output [9]. Illumina HiSeq 2000 also has been successfully used to characterize the genetic background of many marine species, including Saccharina japonica [10], Pyropia yezoensis [11], [12], Membranipora grandicella [13], Anguilla japonica [14], Pinctada fucata [15], Pinctada martensii [16] and Pseudomonas stutzeri AN10 [17].
Genetic studies of Gp. lemaneiformis aimed at improving its agar yield and growth rate are relatively new. Recent genetic studies have focused on the mitochondrial genome [18], functional gene cloning [19], analysis of genetic diversity [20], [21], and mutation research [22]. However, genetic studies of Gp. lemaneiformis remain underdeveloped compared with many other aquaculture species, such as S. japonica, P. yezoensis, Cynoglossus semilaevis, and Apostichopus japonicus, which might be due in part to the insufficient genetic or genomic resources available for Gp. lemaneiformis. To investigate and provide a genomic resource for further research (e.g., molecular cloning, structural and functional genomic studies, breeding, and comparative and evolutionary studies) on this species, we conducted a genome survey of Gp. lemaneiformis using NGS technology. The results of this study should be useful for crop improvement programs and better utilization of the existing genomic information in the future.
Materials and Methods
Sample preparation
A healthy female thallus of Gp. lemaneiformis, which was identified based on its morphology at maturity [23], was collected in November 2011 from the intertidal zone of Zhanshan Bay, Qingdao, China (36°02′ N, 120°20′ E). After clearing the specimen in seawater to remove epiphytes, mud, and sand, the thalli were rinsed with 1% sodium hypochlorite for 2 min and treated in antibiotic seawater containing 0.3 g L−1 penicillin, 0.2 g L−1 nystatin, 0.02 g L−1 cefotaxine, 0.1 g L−1 kanamycin, 1.0 g L−1 streptomycin sulphate, and 0.1 g ml−1 GeO2 for about 6 h to remove possible bacterial contaminants and diatoms. Part of a thallus then was used for DNA extraction, and the remaining thalli were cultured continuously in modified Provasoli's medium [24] under the following conditions: temperature of 20°C, photon flux density of 15 µmol m−2s−1, salinity of 30‰, and a 12 h light∶12 h dark photoperiod regime.
DNA extraction, library construction, and sequencing
Genomic DNA was extracted using the Plant Genomic DNA Kit (Tiangenbiotech, Beijing, China) following the manufacturer's instructions. The quantity and quality of genomic DNA were measured using 1% agarose gel electrophoresis and Gene Quant (Amersham Bioscience, San Francisco, USA). Two paired-end libraries with insert sizes of about 170 base pairs (bp) and 500 bp were constructed from fragmented random genomic DNA following the manufacturer's instructions (Illumina, Guangzhou, China). Sequence data generation, using the Illumina HiSeq 2000 sequencing platform was conducted by Beijing Genomics Institute (Shenzhen, China). All sequencing reads were deposited in the Short Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra/), which are retrievable under the accession number SRX258772.
Sequence assembly and analysis
The raw HiSeq 2000 reads were first pre-processed by trimming short tips and low quality sequences using a phred-scale quality score cut-off of 20 for the acquisition of Q20 values. Genome assembly was completed with the pre-processed reads using SOAPdenovo software [25]. All usable reads were realigned to the contig sequences, and then the paired-end relationship between reads was aligned between contigs. We constructed the scaffolds step by step using variant insert size paired-ends. Default assembly parameters of >40 bp overlap length and >90% sequence identity were used. The genome size was calculated using the total length of sequence reads and sequencing depth [26].
Analysis of G+C content
We used 10-kb non-overlapping sliding windows along the assembled sequence to calculate GC content and average sequencing depth. The x-axis was GC content percent across every 10-kb window and the y-axis was average sequencing depth, which was determined across every 10-kb window independently.
Identification of repeat sequences
Tandem repeats and interspersed repeats are two main types of repeats in the genome. The program TRF was used to search for tandem repeats. The annotated interspersed repeats were identified using RepeatMasker (version 3.3.0) and RepeatProteinMasker against the Repbase transposable element library (version 16.10). The software programs LTR-FINDER, Piler, and RepeatScout were used to construct a de novo repeat library, and then the software RepeatMasker was run to find homolog repeats in the de novo repeat library [27], [28].
Gene prediction, annotation and comparison
Homology-based and de novo methods were used to predict genes, and the predicted results were integrated by the program GLEAN 17 [29]. Protein sequences from Chondrus crispus, Cyanidioschyzon merolae, Pyropia yezoensis, Arabidopsis thaliana, Chlamydomonas reinhardtii, Chlorella variabilis, and Oryza sativa were mapped to the genome assembly using blastN with an E-value of 1e-5 [30] to perform gene prediction, using one species each time. For de novo prediction, Augustus [31] and Genscan [32] were used to predict genes with parameters trained on A. thaliana.
Each predict gene was annotated by blastP to the GenBank database. Functional assignments were mapped onto Gene Ontology (GO) using Blast2GO and then the proportions of GO categories among four red algae species were compared using WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl).
Synteny analysis of the genome
The two reference genomes (C. variabilis and A. thaliana) were downloaded from ftp://ftp.ensemblgenomes.org/pub/release-15/plants/fasta/arabidopsis_thaliana/(A. thaliana) and http://www.ncbi.nlm.nih.gov/genome/694 (C. variabilis), respectively. We used BLASTP [33] with an E-value threshold of 1e-5 to identify homologous genes between Gp. lemaneiformis and these two reference genomes. The program MCscan (version 8.0) then was used to identify syntenic blocks between the two genomes.
Identification of simple sequence repeats (SSRs)
SSRs were mined in the genome sequence using the SSRIT program with the following parameters: at least six repeats for di- and four repeats for tri-, tetra-, penta-, and hexa-nucleotides for SSRs.
Results and Discussion
Sequencing and de novo short-read assembly
The two small-insert (170 and 500 bp) libraries were sequenced to generate a total of 21.52 Gb raw reads. After filtering and correction of the sequence data, a total of about 18.70 Gb of clean reads were obtained, with a read length of 95 bp and about 113.98X coverage of the estimated 160-Mb genome [34] (Table 1).
Table 1. Summary of two paired-end libraries used for HiSeq 2000 Sequencing and paired-end sequencing datasets.
| Library | Sex of algae | Insert Size/bp | Read Length/bp | Data/Mb | Sequence Depth/X |
| L1 | female | 500 | 95 | 9,087.49 | 53.93 |
| L2 | female | 170 | 95 | 9,608.67 | 60.05 |
| Total | 18,696.16 | 113.98 | |||
The program SOAPdenovo and the 18.69 Gb clean reads were used to conduct de novo assembly [25] to produce a contig with the N50 of ∼3.64 kb, longest contig length of ∼51.34 kb, and total length of ∼77.54 Mb (Table 2). A sequence with a scaffold N50 length of ∼20 kb, total length of 81.17 Mb, and longest scaffold length of ∼159,75 kb (Table 2) also was generated. Our draft genome assembly had unclosed gaps shorter than 16.3% (∼16 Mb) based on the calculation suggested in Li et al. [35]. In addition, Li et al. [35] reported that most gaps were easy to occur in repetitive regions with high unit identity and lengths larger than the sequencing read length, which usually could not be assembled with the current data.
Table 2. Statistics of the genome assembly.
| Contig | Scaffold | |||
| Size(bp) | Number | Size(bp) | Number | |
| N90 | 127 | 94,867 | 127 | 57,716 |
| N80 | 193 | 38,389 | 560 | 7,482 |
| N70 | 648 | 15,203 | 5,754 | 2,422 |
| N60 | 1,674 | 7,523 | 12,961 | 1,511 |
| N50 | 3,638 | 4,379 | 20,007 | 1,013 |
| Longest | 51,340 | - | 159,753 | - |
| Total size | 77,537,041 | - | 81,167,384 | - |
| Total number(≥100 bp) | 160,390 | - | 125,685 | |
| Total number(≥2 kb) | 6649 | - | 3,704 | |
As sequencing depth increases, assembly quality also improves. Generally, current NGS assemblers require at least 30X coverage for a successful assembly without gaps from a standard multicell sample [36]. One of the most popular metrics to comparing assemblies is the N50 statistic [37], which is defined as a weighted median and is the smallest contig size in the set whose combined length totals 50% of the genome assembly [38], [39]. The N50 contig size of Gp. lemaneiformis (3.64 kb) in this study was larger than that of Nannochloropis gaditanathose (404 bp) [40] and P. yezoensis (1,669 bp) [11] but lower than those of prokaryotes with an average N50 contig size of 24 kb from de novo assemblies [41] or some terrestrial plants [26], [42], [43]. A large N50 contig and contig number might simply reflect a continuous and complete assembly [41]. However, it would be not valid to compare species using the contig N50 statistics from different assemblies if each N50 statistic was not calculated from the same combined length value [44].
GC content and GC-depth analysis
Generally, genomic sequences generated through NGS are not uniformly distributed across the genome, as they are wider than the Poisson distribution [45]. GC content was one of three factors found to contribute to sequence bias from Illumina's platform [46]. Compared with mid-GC content, high and low GC contents cause reduced coverage in sequencing regions [47], [48]. To measure genome-wide sequencing bias, GC content and average sequencing depth were plotted using non-overlapping 10-kb sliding windows along the assembled sequence. The density points only concentrated in the 40–60% range, with the average GC content of ∼48%. Moreover, Gp. lemaneiformis had a mid-GC content [47], [48], which did not demonstrate abnormality of the species sample (some species themselves have abnormal GC content) or sequencing bias for the present data (Figure 1).
Figure 1. GC content and average sequencing depth of the genome data used for assembly.
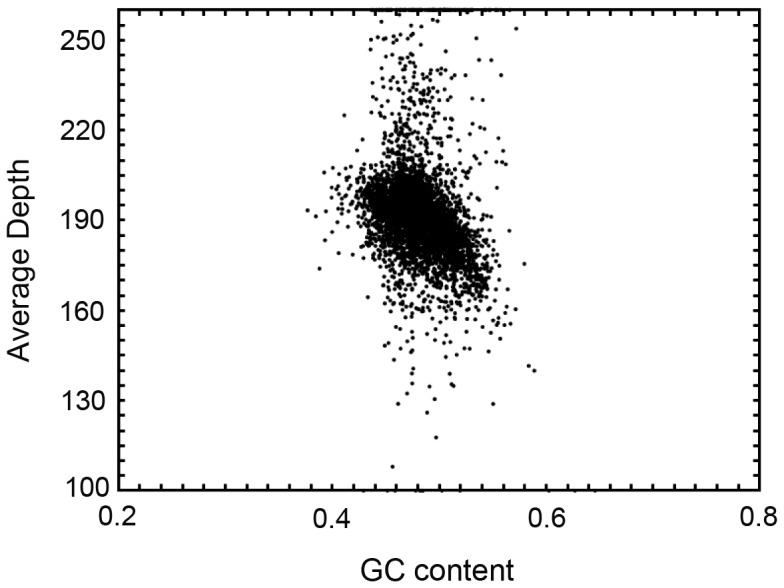
(The x-axis was GC content percent across every 10-kb non-overlapping sliding window).
The 48% GC content in our study correlated well with values previously reported by Zhang et. al. [23], who found a G+C content of 41.1–49.7% in Gp. lemaneiformis. These values were higher than that of some marine bacteria (32.8–33.2%) including Agarivorans gilvus, Aquimarina agarilytica and Vibrio tubiashii [49]–[51] generated by Illumina paired-end sequencing; similar to values for marine macroalgae, such as Solieria filiformis (48.6%) [52], Chondrus crispus (46.3%) [53], P. purpurea (44.6%) [53], and Laminaria hyperborea (42.6%) [54]; and lower than that of P. yezoensis (63.6%) [11] and Cyanidioschyzon merolae (55.0%) [55].
Estimation of genome size
Based on K-mer analysis, all sequences of about 18.70 Gb (Table 1) were used to estimate the genome size of Gp. lemaneiformis. The peak depth was at 162X, and we estimated the genome size to be 95.96 Mb. Generally, the 30X data are relatively accurate for estimating the size of the genome (based on evaluation experience of the Beijing Genomics Institute), so 3.5 Gb (18.70 Gb *30X/162X) of data were used to count and plot the distribution of 17-mer frequency. The peak depth was at 30X (Figure 2), and the number of 17-mers was 2,910,526,453 (Table 3). Their relationship could be expressed by the algorithm: Genome Size = K-mer_num/Peak_depth. The estimated genome size of 97.02 Mb (Table 3) was obtained, which was just one-quarter of the diploid size (375 Mb) for this alga based on average genetic substance content within the cells of Gracilariopsis [23]. The result also was not in accordance with the result of Kapraun and Dutcher [34], who reported a nuclear genome size of 160 Mb (haploid) in Gp. lemaneiformis using microspectrophotometry with the DNA-localizing fluorochrome hydroethidine. The same fluorescence method had been used to evaluate the genome size of Ch. crispus and obtained 150-Mb genome [53], which was about one-third larger than that of the complete genome sequencing method (105 Mb) [56]. We intend to verify the genome size using the flow cytometry method in the future. In total, haploid Gp. lemaneiformis appeared having a fairly small genome size, which would be an advantage for full sequencing of the genome.
Figure 2. Distribution of 17-mer frequency in the 3.5 Gb sequences (3.5 Gb = 18.70 Gb*30X/162X).

Table 3. Estimation of Gp. lemaneiformis based on K-mer statistics.
| K-mer value | K-mer number | Depth | Genome size (bp) | Used bases | Used reads | Depth (X) |
| 17 | 2,910,526,453 | 30 | 97,017,548 | 3,500,000,165 | 36,842,107 | 36.07 |
Note: Generally, using 30X data to estimate the size of the genome are preference because of its accuracy (based on evaluation experience of the Beijing Genomics Institute); Used bases were calculated by 18.70 Gb*30X/162X; Genome Size = K-mer_num/Peak_depth.
Arabidopsis has been documented as a model organism for genetic study, mainly because it has a small genome (120 Mb genome) that is amenable to detailed molecular analysis [57], [58]. In marine macroalgae, Pyropia was suggested as a model organism for genetic studies, partly because its haploid has a relatively small genome size (diploid: 270–530 Mb) [12]. The estimated genome size of Gp. lemaneiformis (97.02 Mb) is similar to that of A. thaliana and Ch. crispus but much smaller than that of Pyropia, which shows the potential of Gp. lemaneiformis as a model species in this regard. Additionally, Gracilariopsis species reproduce both sexually and asexually and have a typical Polysiphonia-type life history that is completed in the laboratory within 6 months. These features make Gp. lemaneiformis a good model for studying genetics and genomics. It might also prove to be a model organism for common analyses of algal genetics and molecular biology.
Repetitive sequences
Combined results from RepeatMasker and RepeatProteinMasker analyses revealed that transposable elements (TEs) constituted 54.64% (44.35 Mb) of the genome, and nearly one-third (20.46% of the genome) of them could not be classified within the TE regime (Table 4). Classification of the known TEs revealed that the majority of repeats were long-terminal repeat elements (LTRs) (26.40%), whereas 8.56% of the TEs were DNA transposons (Table 4).
Table 4. Percentage of the genome masked as each class of transposable elements.
| Type | Repbase TEs | TE protiens | De novo | Combined TEs | ||||
| Length (bp) | % in genome | Length (bp) | % in genome | Length (bp) | % in genome | Length (bp) | % in genome | |
| DNA | 228,529 | 0.28 | 2,138,557 | 2.63 | 6,682,133 | 8.23 | 6,950,958 | 8.56 |
| LINE | 63,656 | 0.08 | 117,208 | 0.14 | 1,210,955 | 1.49 | 1,342,261 | 1.65 |
| LTR | 2,255,161 | 2.78 | 7,687,608 | 9.47 | 20,671,304 | 25.47 | 21,425,837 | 26.40 |
| SINE | 3,153 | 0.004 | 0 | 0.00 | 0 | 0.00 | 3,153 | 0.004 |
| Other | 63 | 0.00 | 0 | 0.00 | 0 | 0.00 | 63 | 0.00 |
| Unknown | 0 | 0.00 | 0 | 0.00 | 16,608,826 | 20.46 | 16,608,826 | 20.46 |
| Total | 2,535,513 | 3.12 | 9,941,908 | 12.25 | 43,807,750 | 53.97 | 44,351,914 | 54.64 |
Note: RepBase TEs and TE proteins were obtained, using RepeatMasker and RepeatProteinMask respectively, based on the RepBase library; De novo repeat prediction identified repetitive DNA using RepeatMasker against the de novo repeat library of Gp. lemaneiformis, which was constructed by the programs LTR-FINDER, Piler and RepeatScout; Combined TEs were the integration and filtering redundancies of the above three methods.
The fraction of TEs in the Gp. lemaneiformis genome was similar to that of the pigeonpea (51.67%) [26]; higher than that of Theobroma cacao (24%) [59], rice (35%) [60], and animals such as Cynoglossus semilaevi (5.23%) [61], Chlamys farreri (15.84%) [62], and catfish (11.91%) [63]; and far lower than that of maize (85%) [64]. Repetitive sequences, especially TEs, are known to be prominent evolutionary factors that played a significant role in plant gene structure and genome evolution by enhancing genome plasticity, such as transposition, excision, insertion, chromosome breakage, and chromosome rearrangements [65]–[68].
In prokaryotes, DNA TEs are the major class of transposable DNAs, and RNA TEs are particularly abundant in eukaryotes [65]. Repeated sequences constituted 73% of the Ch. crispus genome, ∼55% (representing 58 Mb) of which were LTR retrotransposons [56]. As in Ch. crispus and some higher plants such as maize [69] and Ricinus communis [70], the most abundant retrotransposons in Gp. lemaneiformis are LTRs, which might be associated with potential horizontal transfer, insertional mutations, and heterochromatin near centromeres [71], [72].
Gene prediction, annotation and comparison
Both homology-based and de novo gene prediction methods were used to predict the number of genes in the Gp. lemaneiformis genome (Table 5). First, Genscan and Augustus predicted 3369 and 3363 gene loci, respectively. Next, four homologous genome sequences were mapped to the genome assembly. Predicted genes 1,858–4,393 were obtained with an average transcript length of 787.06–910.23 bp. Subsequently, all data were integrated using GLEAN 17, and 3,490 genes were identified with an average transcript length of 1,429 bp, average coding sequence size of 1,369 bp, 1.36 exons per gene, exon length of 1,008 bp, and intron length of 191 bp.
Table 5. General statistics of gene prediction for Gp. lemaneiformis.
| Method | Gene set | Number | Average transcript length (bp) | Average CDS length (bp) | Average exon per gene | Average exon length (bp) | Average intron length (bp) |
| De novo | Augustus | 3369 | 631.62 | 558.01 | 1.35 | 413.99 | 211.59 |
| Genscan | 3363 | 2330.66 | 1688.73 | 2.68 | 631.02 | 382.97 | |
| Homolog | Cy. merolae | 1860 | 766.51 | 699.30 | 1.37 | 510.34 | 181.52 |
| Ch. Crispus | 1858 | 773.80 | 702.59 | 1.36 | 515.61 | 196.36 | |
| P. yezoensis | 1878 | 764.10 | 696.32 | 1.37 | 509.31 | 184.59 | |
| A. thaliana | 4222 | 879.14 | 843.62 | 1.32 | 638.08 | 110.27 | |
| C.reinhardtii | 4039 | 808.82 | 746.46 | 1.27 | 586.79 | 229.19 | |
| O. sativa | 4393 | 910.23 | 802.41 | 1.34 | 597.55 | 314.52 | |
| C. variabilis | 3851 | 787.06 | 746.58 | 1.28 | 583.54 | 144.88 | |
| GLEAN | 3490 | 1429.57 | 1369.79 | 1.36 | 1008.36 | 191.13 |
Note: Gene length included the exon and intron regions but excluded UTRs.
The number of predicted genes in the genome of Gp. lemaneiformis was comparable to that of the ultrasmall unicellular red alga Cy. merolae,(5331) [55] but the number was much lower than that of other sequenced genomes such as Arthrospira platensis (6153) [73], Nannochloropis gaditana (9052) [40], Ch. crispus (9606) [56], Ascaris suum (18,542) [74], and Cucumis sativus (26,682) [75]. Possible reasons for these differences include insufficient sequence depth coverage, variable regulation of gene expression levels (such as an enormous wealth of alternative splicing), and low sequence homology because of shorting of gene information from closely related species.
In comparison with other red algae (Table 6), Gp. lemaneiformis genome showed similar genome characteristics, such as average CDS length, short exon/intron length (bp) and low number of exons per gene. Especially, the genes of Gp. lemaneiformis also had few introns and nearly three quarters of predicted genes are monoexonic, resulting in a low average number of introns per gene (0.36). The high proportion of monoexonic genes for Gp. lemaneiformis, which was comparable to those of other red algae, further verified Collén et al.'s conclusion that a low number of introns were a typical characteristic in red algal genomes [56].
Table 6. Comparison of general genome characteristics from four red algae.
| Species | Gp. lemaneiformis | Ch. crispus | Cy. merolae | P. yezoensis |
| Average CDS length (bp) | 1370 | - | 1552 | 1247 |
| Average exon length (bp) | 1008 | 789 | 1540 | 755 |
| Average intron length (bp) | 191 | 123 | 248 | 300 |
| Introns per gene | 0.36 | 0.32 | 0.005 | 0.7 |
| Exons per gene | 1.36 | 1.32 | 1.005 | 1.7 |
| Intron-containing genes (%) | 28 | 12 | 0.6 | ∼40 |
Of the 3,490 predicted genes in the Gp. lemaneiformis genome, 2,954 genes matched known genes in GenBank and 15.35% (536) were unknown. 2,430 genes could be assigned to one or more ontologies. Totally, 34.57% genes were grouped under biological processes 32.26% genes under cellular components, 33.17% genes under molecular functions. Furthermore, cellular process (27.86%) was the most highly represented groups under the biological process category, followed by macromolecule metabolism (19.17%); for the cellular component, intracellular (28.19%) and cytoplasm (21.56%) part were the significantly represented groups; a relatively high proportion of genes (25.43%) were involved in catalytic activity. The predicted proteins were compared with the 10,327 proteins from P. yezoensis [11], 9,606 proteins from Ch. crispus [56] and.5,331 proteins from Cy. merolae [55]. As shown in Figure 3, the vast proportion of GO categories did not show significant differences among 4 red algae except those related behavior process, electron transport process and extracellular component, which hinted that the biological processes of the above among 4 red algae were different.
Figure 3. GO category comparison among Gp. lemaneiformis, P. yezoensis, Cy. merolae and Ch. crispus.

Synteny with sequenced plant genomes
We compared about 81.17 Mb of the genomic sequence from Gp. lemaneiformis with the A. thaliana and C. variabilis genomes using the program LASTZ, and then MCscan was used to identify syntenic blocks. The genomic sequence of Gp. lemaneiformis showed minimal matching to A. thaliana (match length of 900,618 bp with only 1.11% coverage) and C. variabilis (match length of 345,067 bp with only 0.43% coverage),and the minimal matching could not be used to evaluate the general features of synteny relationships (Tables 7 and 8). Although it should be possible to analyze the evolutionary history between two species using complete genomic sequences [76], the following factors would make the process difficult or impossible: short sequence length of initial assembly, low gene distribution, and species specificity.
Table 7. Summary of match sequence between Gracilariopsis lemaneiformis and Arabidopsis thalianai.
| Species | Match length (bp) | Total length (bp) | Coverage (%) |
| A. thaliana | 1,671,544 | 119,146,348 | 1.40 |
| Gp. lemaneiformis | 900,618 | 81,167,384 | 1.11 |
Table 8. Summary of match sequence between Gracilariopsis lemaneiformis and Chlorella variabili.
| Species | Match length (bp) | Total length (bp) | Coverage (%) |
| C. variabilis | 762,232 | 46,159,512 | 1.65 |
| Gp. lemaneiformis | 345,067 | 81,167,384 | 0.43 |
SSR detection
In this study, we found 7737 derived SSRs, none of which were mononucleotide repeats and complex SSR type. Among these SSRs, the trinucleotide repeat type was the most abundant (up to 73.20% of total SSRs), followed by di- (17.41%), tetra- (5.49%), hexa- (2.90%), and penta- (1.00%) nucleotide repeat types (Figure 4).
Figure 4. Frequency of SSR types in the Genome Survey of Gp. lemaneiformis.
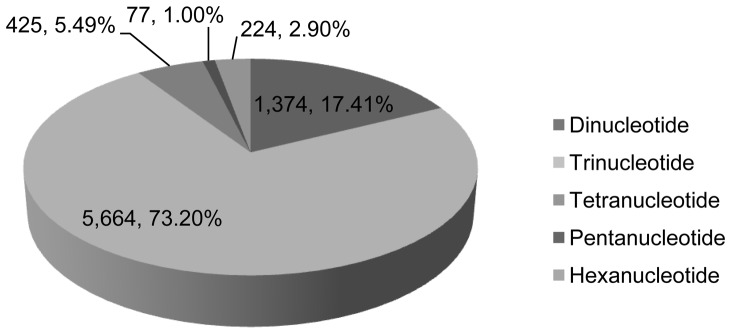
In addition, 315 motif types were identified, which consisted of di- (8), tri- (30), tetra- (68), penta- (56), and hexa- (153) nucleotide types. Table S1 shows the SSR frequency of each motif. Statistical analysis revealed that the motif AG/CT was the most abundant among the dinucleotide repeat motifs, accounting for 24.72%, followed by TG/CA at 20.86% (Figure 5). The most prominent dinucleotide motif of Gp. lemaneiformis was the same as that of the rubber tree, in which the AG/CT motif was also most abundant [41]. In embryophytes, yeast, and fungi, however, the AT/AT motif is most abundant [77]. Within the trinucleotide repeat motifs, the common motifs TGC/GCA and CAG/CTG accounted for 14.42% and 14.05%, respectively, in Gp. lemaneiformis (Figure 6).
Figure 5. Percentage of different motifs in dinucleotide repeats in Gp. lemaneiformis.

Figure 6. Percentage of different motifs in trinucleotide repeats in Gp. lemaneiformis.

In the Gp. lemaneiformis genome, the trinucleotide repeat type was predominant, as is also the case in other species such as P. yezoensis, A. thaliana, rice, maize, and tomato [12]. Yang et. al. [12] reported that the trinucleotide repeat type in the coding regions would enhance gene variation but not cause frameshift mutation. SSRs are inherently unstable, which creates and maintains quantitative genetic variation, so they must have played an important role in genome evolution [77]. It is possible that the 7737 derived SSR loci found in our study may be used as SSR markers for genetic mapping in the short term.
Supporting Information
Occurrence of SSR motifs in Genome Survey to Gracilariopsis lemaneiformis .
(DOC)
Funding Statement
Financial support for this work was provided by “The Twelfth Five-Year-Plan” in National Science and Technology for the Rural Development in China (2012AA100811-3). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fredericq S, Hommersand MH (1989) Reproductive morphology and development of the cystocarp in Curdiea flabellata Chapman (Gracilariales, Rhodophyta). New Zealand J Bot 27: 521–530. [Google Scholar]
- 2. Lapointe BE, Ryther JH (1978) Some aspects of the growth and yield of Gracilaria tikvahiae in culture. Aquaculture 15: 185–193. [Google Scholar]
- 3. Marinho-Soriano E (2001) Agar polysaccharides from Gracilaria species (Rhodophyta, Gracilariaceae). J Biotechnol 89: 81–84. [DOI] [PubMed] [Google Scholar]
- 4. McHugh DJ (1991) Worldwide distribution of commercial resources of seaweeds including Gelidium . Hydrobiologia 221: 19–29. [Google Scholar]
- 5. Zhang XC, Fei XG, Wang GC, Lin XZ, Chen WZ, et al. (2009) Genetic Studies and Large Scale Cultivation of Gracilaria lemaneiformis . Periodic Ocean Univ China 5: 029. [Google Scholar]
- 6. Wang Y, Yu Z, Song X, Tang X, Zhang S (2007) Effects of macroalgae Ulva pertusa (Chlorophyta) and Gracilaria lemaneiformis (Rhodophyta) on growth of four species of bloom-forming dinoflagellates. Aquatic botany 86: 139–147. [Google Scholar]
- 7. Zhou Y, Yang H, Hu H, Liu Y, Mao Y, et al. (2006) Bioremediation potential of the macroalga Gracilaria lemaneiformis (Rhodophyta) integrated into fed fish culture in coastal waters of north China. Aquaculture 252: 264–276. [Google Scholar]
- 8. Yang YF, Fei XG, Song JM, Hu HY, Wang GC, et al. (2006) Growth of Gracilaria lemaneiformis under different cultivation conditions and its effects on nutrient removal in Chinese coastal waters. Aquaculture 254: 248–255. [Google Scholar]
- 9.Liu L, Li Y, Li S, Hu N, He Y, et al. (2012) Comparison of Next-Generation Sequencing Systems. J Biomed Biotechnol 2012. [DOI] [PMC free article] [PubMed]
- 10. Deng Y, Yao J, Wang X, Guo H, Duan D (2012) Transcriptome Sequencing and Comparative Analysis of Saccharina japonica (Laminariales, Phaeophyceae) under Blue Light Induction. PloS ONE 7: e39704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamura Y, Sasaki N, Kobayashi M, Ojima N, Yasuike M, et al. (2013) The First Symbiont-Free Genome Sequence of Marine Red Alga, Susabi-nori (Pyropia yezoensis). PLoS ONE 8: e57122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang H, Mao YX, Kong FN, Yang GP, Ma F, et al. (2011) Profiling of the transcriptome of Porphyra yezoensis with Solexa sequencing technology. Chinese Sci. Bull 56: 2119–2130. [Google Scholar]
- 13. Shen X, Tian M, Meng X, Liu H, Cheng H, et al. (2012) Complete mitochondrial genome of Membranipora grandicella (Bryozoa: Cheilostomatida) determined with next-generation sequencing: The first representative of the suborder Malacostegina. Comp Biochem Physiol D 7: 248–253. [DOI] [PubMed] [Google Scholar]
- 14. Henkel CV, Dirks RP, de Wijze DL, Minegishi Y, Aoyama J, et al. (2012) First draft genome sequence of the Japanese eel, Anguilla japonica . Gene 511: 195–201. [DOI] [PubMed] [Google Scholar]
- 15. Huang XD, Liu WG, Guan YY, Shi Y, Wang Q, et al. (2012) Molecular cloning and characterization of class I NF-κB transcription factor from pearl oyster Pinctada fucata . Fish Shellfish Immun 33: 659–666. [DOI] [PubMed] [Google Scholar]
- 16. Zhao X, Wang Q, Jiao Y, Huang R, Deng Y, et al. (2012) Identification of Genes Potentially Related to Biomineralization and Immunity by Transcriptome Analysis of Pearl Sac in Pearl Oyster Pinctada martensii . Mar Biotechnol 14: 730–739. [DOI] [PubMed] [Google Scholar]
- 17. Brunet-Galmés I, Busquets A, Peña A, Gomila M, Nogales B, et al. (2012) Complete Genome Sequence of the Naphthalene-Degrading Bacterium Pseudomonas stutzeri AN10 (CCUG 29243). J Bacteriol 194: 6642–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Wang X, Qian H, Chi S, Liu C, et al. (2012) Complete sequences of the mitochondrial DNA of the wild Gracilariopsis lemaneiformis and two mutagenic cultivated breeds (Gracilariaceae, Rhodophyta). PloS ONE 7: e40241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bojsen K, Yu S, Marcussen J (1999) A group of α-1, 4-glucan lyase genes from the fungi Morchella costata, M. vulgaris and Peziza ostracoderma. Cloning, complete sequencing and heterologous expression. Plant mol biol 40: 445–454. [DOI] [PubMed] [Google Scholar]
- 20. Pang Q, Sui Z, Kang KH, Kong F, Zhang X (2010) Application of SSR and AFLP to the analysis of genetic diversity in Gracilariopsis lemaneiformis (Rhodophyta). J Appl Phycol 22: 607–612. [Google Scholar]
- 21. Wang W, Wang G, Gao Z, Lin X, Xu P (2007) Characterization of Gracilaria lemaneiformis Bory (Gracilariaceae, Rhodophyta) cultivars in China using the total soluble proteins and RAPD analysis. Bot Mar 50: 177–184. [Google Scholar]
- 22. Meng L, Xu D, Chen WZ, Zhang XC (2009) Selection and characterization of a new strain of Gracilaria lemaneiformis (in chinese). Periodic Ocean Univ China 39: 94–98. [Google Scholar]
- 23.Zhang XC, Qin S, Ma JH, Xu P (2005) The genetics of marine alga. Beijing: China Agriculture Press.
- 24. Pflugmacher S, Steinberg C (1997) Activity of phase I and phase II detoxification enzymes in aquatic macrophytes. Angew Bot 71: 144–146. [Google Scholar]
- 25. Li R, Fan W, Tian G, Zhu H, He L, et al. (2010) The sequence and de novo assembly of the giant panda genome. Nature 463: 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, et al. (2011) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30: 83–89. [DOI] [PubMed] [Google Scholar]
- 27. Jurka J, Kapitonov V, Pavlicek A, Klonowski P, Kohany O, et al. (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467. [DOI] [PubMed] [Google Scholar]
- 28. Price AL, Jones NC, Pevzner PA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21: i351–i358. [DOI] [PubMed] [Google Scholar]
- 29. Elsik CG, Mackey AJ, Reese JT, Milshina NV, Roos DS, et al. (2007) Creating a honey bee consensus gene set. Genome Biol 8: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kent WJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stanke M, Tzvetkova A, Morgenstern B (2006) AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol 7: S11–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salamov AA, Solovyev VV (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res 10: 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bairoch A, Apweiler R (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28: 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kapraun D, Dutcher J (1991) Cytophotometric estimation of inter-and intraspecific nuclear DNA content variation in Gracilaria and Gracilariopsis (Gracilariales, Rhodophyta). Bot Mar 34: 139–144. [Google Scholar]
- 35. Li Y, Hu Y, Bolund L, Wang J (2010) State of the art de novo assembly of human genomes from massively parallel sequencing data. Hum Genet 4: 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chitsaz H, Yee-Greenbaum JL, Tesler G, Lombardo MJ, Dupont CL, et al. (2011) Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol 29: 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salzberg SL, Yorke JA (2005) Beware of mis-assembled genomes. Bioinformatics 21: 4320–4321. [DOI] [PubMed] [Google Scholar]
- 38. Carneiro AR, Ramos RTJ, Barbosa HPM, Schneider MPC, Barh D, et al. (2012) Quality of prokaryote genomes assembly: Indispensable issues of factors affecting prokaryote genome assembly quality. Gene 505: 365–367. [DOI] [PubMed] [Google Scholar]
- 39. Earl D, Bradnam K, John JS, Darling A, Lin D, et al. (2011) Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res 21: 2224–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Radakovits R, Jinkerson RE, Fuerstenberg SI, Tae H, Settlage RE, et al. (2012) Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana . Nat Comms 3: 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li D, Deng Z, Qin B, Liu X, Men Z (2012) De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller L, et al. (2012) A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant -Microbe Interact 25: 1523–1530. [DOI] [PubMed] [Google Scholar]
- 43. D'Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, et al. (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488: 213–217. [DOI] [PubMed] [Google Scholar]
- 44. Miller JR, Koren S, Sutton G (2010) Assembly algorithms for next-generation sequencing data. Genomics 95: 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, et al. (2008) Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res 18: 1638–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheung MS, Down TA, Latorre I, Ahringer J (2011) Systematic bias in high-throughput sequencing data and its correction by BEADS. Nucleic acids Res 39: e103–e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, et al. (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, et al. (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Du ZJ, Lv GQ, Rooney AP, Miao TT, Xu QQ, et al. (2011) Agarivorans gilvus sp. nov. isolated from seaweed. Int J Syst Evol Micr 61: 493–496. [DOI] [PubMed] [Google Scholar]
- 50. Lin B, Lu G, Li S, Hu Z, Chen H (2012) Draft genome sequence of the novel agarolytic bacterium Aquimarina agarilytica ZC1. J Bacteriol 194: 2769–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Temperton B, Thomas S, Tait K, Parry H, Emery M, et al. (2011) Permanent draft genome sequence of Vibrio tubiashii strain NCIMB 1337 (ATCC19106). Stand Genomic Sci 4: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dalmon J, Loiseaux S (1981) The deoxyribonucleic acids of two brown algae: Pylaiella littoralis (L.) Kjellm. and Sphacellaria sp. Plant Sci Lett 21: 241–251. [Google Scholar]
- 53. Gall YL, Brown S, Marie D, Mejjad M, Kloareg B (1993) Quantification of nuclear DNA and GC content in marine macroalgae by flow cytometry of isolated nuclei. Protoplasma 173: 123–132. [Google Scholar]
- 54. Stam W, Bot PVM, Boele-Bos S, Van Rooij J, Van den Hoek C (1988) Single-copy DNA-DNA hybridizations among five species of Laminaria (Phaeophyceae): Phylogenetic and biogeographic implications. Helgoland Mar Res 42: 251–267. [Google Scholar]
- 55. Ohta N, Matsuzaki M, Misumi O, Miyagishima SY, Nozaki H, et al. (2003) Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae . DNA Res 10: 67–77. [DOI] [PubMed] [Google Scholar]
- 56.Collén J, Porcel B, Carré W, Ball SG, Chaparro C (2013) Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc Nat Acad Sci USA doi:10.1073/pnas.1221259110 [DOI] [PMC free article] [PubMed]
- 57. Meinke DW, Cherry JM, Dean C, Rounsley SD, Koornneef M (1998) Arabidopsis thaliana: a model plant for genome analysis. Science 282: 662–682. [DOI] [PubMed] [Google Scholar]
- 58. Meyerowitz EM, Pruitt RE (1985) Arabidopsis thaliana and plant molecular genetics. Science 229: 1214–1218. [DOI] [PubMed] [Google Scholar]
- 59. Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, et al. (2010) The genome of Theobroma cacao . Nat Genet 43: 101–108. [DOI] [PubMed] [Google Scholar]
- 60. Vij S, Gupta V, Kumar D, Vydianathan R, Raghuvanshi S, et al. (2006) Decoding the rice genome. Bioessays 28: 421–432. [DOI] [PubMed] [Google Scholar]
- 61. Wang X, Zhang Q, Sun X, Chen Y, Zhai T, et al. (2009) Fosmid library construction and initial analysis of end sequences in female half-smooth tongue sole (Cynoglossus semilaevis). Mar Biotechnol 11: 236–242. [DOI] [PubMed] [Google Scholar]
- 62. Zhang L, Bao Z, Cheng J, Li H, Huang X, et al. (2007) Fosmid library construction and initial analysis of end sequences in Zhikong scallop (Chlamys farreri). Mar Biotechnol 9: 606–612. [DOI] [PubMed] [Google Scholar]
- 63. Xu P, Wang S, Liu L, Peatman E, Somridhivej B, et al. (2006) Channel catfish BAC-end sequences for marker development and assessment of syntenic conservation with other fish species. Anim Genet 37: 321–326. [DOI] [PubMed] [Google Scholar]
- 64. Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115. [DOI] [PubMed] [Google Scholar]
- 65. Bennetzen JL (2000) Comparative sequence analysis of plant nuclear genomes: microcolinearity and its many exceptions. Plant Cell Online 12: 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grandbastien MA (1992) Retroelements in higher plants. Trends Genet 8: 103–108. [DOI] [PubMed] [Google Scholar]
- 67. Jurka J, Kapitonov VV, Kohany O, Jurka MV (2007) Repetitive sequences in complex genomes: structure and evolution. Annu Rev Genomics Hum Genet 8: 241–259. [DOI] [PubMed] [Google Scholar]
- 68. Wang S, Zhang L, Meyer E, Bao Z (2010) Discovery notes Genome-wide analysis of transposable elements and tandem repeats in the compact placozoan genome. Biol Direct 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bennetzen JL, Kellogg EA (1997) Do plants have a one-way ticket to genomic obesity? Plant Cell 9: 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, et al. (2010) Draft genome sequence of the oilseed species Ricinus communis . Nat biotechnol 28: 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, et al. (2009) The regulated retrotransposon transcriptome of mammalian cells. Nat genet 41: 563–571. [DOI] [PubMed] [Google Scholar]
- 72. Nagarajan N, Navajas-Pérez R, Pop M, Alam M, Ming R, et al. (2008) Genome-wide analysis of repetitive elements in papaya. Tropical Plant Biol 1: 191–201. [Google Scholar]
- 73. Cheevadhanarak S, Paithoonrangsarid K, Prommeenate P, Kaewngam W, Musigkain A, et al. (2012) Draft genome sequence of Arthrospira platensis C1 (PCC9438). Stand Genomic Sci 6: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jex AR, Liu S, Li B, Young ND, Hall RS, et al. (2011) Ascaris suum draft genome. Nature 479: 529–533. [DOI] [PubMed] [Google Scholar]
- 75. Huang S, Li R, Zhang Z, Li L, Gu X, et al. (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41: 1275–1281. [DOI] [PubMed] [Google Scholar]
- 76. Okuno J (1999) Izucaris masudai, new genus, new species (Decapoda: Caridea: Palaemonidae), a sea anemone associate from Japan. Journal Crustacean Biol 19: 397–407. [Google Scholar]
- 77. Tóth G, Gáspári Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10: 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Occurrence of SSR motifs in Genome Survey to Gracilariopsis lemaneiformis .
(DOC)