

Summary
Symptomatic infection with Neisseria gonorrhoeae (Gc) promotes inflammation driven by polymorphonuclear leukocytes (PMNs, neutrophils), yet some Gc survive PMN exposure during infection. Here we report a novel mechanism of gonococcal resistance to PMNs: Gc phagosomes avoid maturation into phagolysosomes by delayed fusion with primary (azurophilic) granules, which contain antimicrobial components including serine proteases. Reduced phagosome-primary granule fusion was observed in gonorrheal exudates and human PMNs infected ex vivo. Delayed phagosome-granule fusion could be overcome by opsonizing Gc with immunoglobulin. Using bacterial viability dyes along with antibodies to primary granules revealed that Gc survival in PMNs correlated with early residence in primary granule-negative phagosomes. However, when Gc was killed prior to PMN exposure, dead bacteria were also found in primary granule-negative phagosomes. These results suggest that Gc surface characteristics, rather than active bacterial processes, influence phagosome maturation and that Gc death inside PMNs occurs after phagosome-granule fusion. Ectopically increasing primary granule-phagosome fusion, by immunoglobulin opsonization or PMN treatment with lysophosphatidylcholine, reduced intracellular Gc viability, which was attributed in part to serine protease activity. We conclude that one method for Gc to avoid PMN clearance in acute gonorrhea is by delaying primary granule-phagosome fusion, thus preventing formation of a degradative phagolysosome.
Keywords: Neisseria gonorrhoeae, neutrophil, polymorphonuclear leukocyte, granule, antimicrobial peptide, serine protease
Introduction
Neisseria gonorrhoeae (the gonococcus or Gc) is an obligate human pathogen and the sole cause of the sexually transmitted disease gonorrhea, a major global health problem. The World Health Organization estimates there are 106 million new cases of gonorrhea worldwide each year (Ndowa et al., 2012). 330,000 cases are reported annually in the United States, although it is estimated that the actual number is at least twice as high (Workowski et al., 2010). Gonorrhea typically presents as acute urethritis in men and cervicitis in women as a consequence of close sexual contact with an infected individual. Infection can lead to serious morbidity and complications if left untreated, including pelvic inflammatory disease, sterility, arthritis-dermatitis syndrome, endocarditis, and meningitis (Wiesner et al., 1980). Gonorrhea can also be transmitted during childbirth, a leading cause of neonatal blindness. Due to the sustained prevalence of gonorrhea throughout the world (Workowski et al., 2010), Gc acquisition of resistance to multiple antibiotics (Unemo et al., 2011), and the inability to date to generate a protective vaccine (Zhu et al., 2011), gonorrhea remains a significant public health issue.
During acute gonorrheal disease, the infected mucosa releases chemokines that initiate a potent inflammatory response and recruit abundant PMNs to the site of infection (Ramsey et al., 1995; Hedges et al., 1998). PMNs possess oxygen-dependent and oxygen-independent antimicrobial components contained within three subsets of cytoplasmic granules, primary, secondary, and tertiary (Faurschou et al., 2003; Segal, 2005). In response to infection, PMNs first release tertiary (gelatinase) granules, which contain enzymes including gelatinase to degrade the extracellular matrix and allow for migration across the tissue, as well as gp91phox and p22phox subunits of the NADPH oxidase complex that generates reactive oxygen species. At the site of infection, PMNs release secondary and primary granules. Secondary (specific) granules contain complement receptor 3 (CD11b/CD18), the gp91phox and p22phox NADPH oxidase subunits, and oxygen-independent antimicrobial products such as lactoferrin and hCAP18, the precursor to the cathelicidin LL-37. Primary (azurophilic) granules contain the majority of PMN antimicrobial products, including myeloperoxidase (MPO) to generate hypochlorous acid, α-defensin antimicrobial peptides, bactericidal-permeability-increasing protein (BPI), and the serine proteases cathepsin G, neutrophil elastase, and proteinase 3. In contrast to phagosomes in macrophages and nonphagocytic cells, neutrophil phagosomes are not especially acidic and even undergo an initial period of alkalinization (Lee et al., 2003). Therefore, clearance of microbes in neutrophils is primarily due to the coordinate release of PMN granules which enhances PMN phagocytosis, generates a potent oxidative burst, and releases antimicrobial peptides and proteases (Segal, 2005).
While numerous PMNs are present at the site of Gc infection, PMNs cannot resolve gonorrheal disease, and viable Gc can be cultured from the purulent exudates of gonorrhea patients (Wiesner et al., 1980). Moreover, in primary human PMNs infected in vitro with Gc, a significant percentage of internalized bacteria remain viable (Simons et al., 2005; Criss et al., 2009). These findings indicate that Gc has evolved mechanisms to resist PMN killing (Johnson et al., 2011). We and others have shown that Gc suppresses the ability of PMNs to produce ROS, and virulence factors that defend the bacteria against oxidative damage do not contribute to Gc survival in PMNs (Criss et al., 2008; Britigan et al., 1988; Lorenzen et al., 2000; Bjerknes et al., 1995; Seib et al., 2005; Wu et al., 2009). Thus Gc survival after exposure to PMNs is primarily due to the bacteria resisting oxygen-independent PMN antimicrobial activities. A variety of Gc gene products contribute to bacterial resistance to oxygen-independent antimicrobial peptides and proteins, resulting in modifications to Gc lipooligosaccharide and expression of efflux pumps (reviewed in Johnson et al., 2011). While purified cathepsin G, BPI, and LL-37 have antigonococcal activity in vitro (Rest, 1979; Rock et al., 1988; Shafer et al., 1986a; Shafer et al., 1998; Casey et al., 1985), their effects on Gc viability have not been examined in the context of bacterial interactions with PMNs.
In this study we investigated cellular mechanisms that contribute to Gc intracellular survival in adherent, IL-8 treated, primary human PMNs. To our surprise, the majority of Gc phagosomes exhibited delayed fusion with PMN primary granules. In the absence of immediate fusion with primary granules, phagosomes were more likely to contain viable Gc. Increasing primary granule fusion with Gc phagosomes reduced the viability of the bacteria inside PMNs. We present a model in which surface characteristics of Gc drive bacteria into phagosomes which initially lack primary granule proteins, which have antigonococcal activity. The delay in primary granule fusion with Gc phagosomes supports the survival of Gc inside these normally antimicrobial cells, thereby contributing to the persistence of Gc in its obligate human host.
Results
Gc phagosomes in primary human PMNs undergo limited fusion with primary granules
To assess the maturity of PMN phagosomes containing Gc, adherent, IL-8 primed, primary human PMNs were infected with Gc of strain FA1090, and the enrichment and fusion of granule subsets with bacterial phagosomes was observed by immunofluorescence and immuno-transmission electron microscopy (TEM). Piliated, Opa-negative Gc were used to remain consistent with our previous studies (Criss et al., 2009; Stohl et al., 2005). Maturation of Gc phagosomes was compared to S. aureus-containing phagosomes, which fuse with all subsets of granules (Voyich et al., 2005). For immunofluorescence, bacterial phagosomes were considered enriched for a PMN granule subset if antibody reactivity against a protein found in that granule subset surrounded ≥ 50% of the bacterial circumference.
We initially compared S. aureus and Gc phagosomes for the presence of proteins found in secondary and tertiary granules, which are mobilized early in PMN activation (Faurschou et al., 2003). These granule subsets were detected with antibodies against the membrane proteins gp91phox and p22phox. As expected, the majority of S. aureus phagosomes were enriched for gp91phox and p22phox after 1 h infection (Figures 1A and B). We observed similar enrichment of lactoferrin, a protein found within secondary granules, in S. aureus phagosomes (Figures 1C and D). After 1 h infection, Gc phagosomes were enriched for secondary and tertiary granule proteins to the same extent as S. aureus phagosomes (Figures 1B and 1D). We then conducted immuno-TEM using an anti-lactoferrin antibody to assess whether or not secondary granules fuse with S. aureus and Gc phagosomes. All bacteria were found to reside in membrane-bound compartments inside PMNs. Both S. aureus and Gc infected PMNs exhibited lactoferrin staining within phagosomes, as well as lactoferrin-positive granules surrounding the phagosomes (Figure 1E). From these data we conclude that secondary granules, and presumably tertiary granules, fuse with Gc phagosomes in PMNs.
Figure 1. S. aureus and Gc phagosomes fuse with secondary and tertiary granules in primary human PMNs.

(A–D) Adherent, IL-8 treated, primary human PMNs were infected for 1 h with S. aureus or Gc. Extracellular S. aureus and Gc appear red/blue, while intracellular S. aureus and Gc appear blue only. Both S. aureus and Gc infected PMNs were stained with antibodies against the secondary and tertiary granule proteins gp91phox and p22phox (A) or the secondary granule protein lactoferrin (C), which appear green. Arrowheads indicate bacterial phagosomes positive for the granule proteins. The percent of S. aureus and Gc phagosomes positive for gp91phox and p22phox and for lactoferrin are reported in B and D, respectively. (E) Immuno-TEM showing phagosomes positive for lactoferrin that contain S. aureus or Gc. Arrowheads indicate clusters of gold particles accumulated within bacterial phagosomes.
As the final step in PMN activation, PMNs mobilize primary granules, which contain the majority of PMN antimicrobial peptides and proteases (Faurschou et al., 2003). We examined enrichment of primary granules to S. aureus and Gc phagosomes by immunofluorescence against the membrane protein CD63 and the content protein neutrophil elastase. As anticipated, S. aureus phagosomes were highly enriched for primary granule proteins after 1 h infection, where solid rings of CD63 or neutrophil elastase staining were seen surrounding intracellular bacteria (Figure 2A and 2C). In contrast, there was a significant decrease in the percent of Gc phagosomes enriched for CD63 or neutrophil elastase after 1 h infection (Figure 2B and 2D). While punctate CD63 and neutrophil elastase staining was detected in the vicinity of some Gc phagosomes (Figure 2A and 2C), this staining pattern did not meet the criteria for phagosomal granule enrichment. Immuno-TEM against the primary granule protein MPO confirmed that S. aureus phagosomes fused with primary granules, with MPO-positive granules also seen surrounding and docking to the phagosomal membrane (Figures 2E). Inside S. aureus phagosomes, MPO reactivity appeared to form a ring around the bacteria as it decorated the bacterial surface. Similar to observations noted for S. aureus, some Gc phagosomes exhibited fusion with MPO positive granules (arrowheads, Fig. 2E). However, Gc phagosomes which lacked the electron dense, MPO positive ring were also observed, even though MPO-positive primary granules were in their vicinity (asterisks, Fig. 2E). The immunofluorescence and immuno-TEM results indicate that most Gc phagosomes do not fuse with primary granules, such that primary granule contents, including MPO and neutrophil elastase, are not delivered to phagosomes.
Figure 2. Gc phagosomes undergo less fusion with primary granules than S. aureus phagosomes.

(A–D) PMNs were infected for 1 h with S. aureus or Gc. Extracellular S. aureus and Gc appear red/blue, while intracellular S. aureus and Gc appear blue only. PMNs were stained with antibodies against the primary granule protein CD63 (A) or neutrophil elastase (NE) (C), which appear green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of S. aureus and Gc phagosomes positive for CD63 or neutrophil elastase are reported in B and D, respectively. Asterisks indicate P < 0.05 by Student’s two-tailed t test. (E) Immuno-TEM for myeloperoxidase (MPO) in PMNs infected with S. aureus or Gc. The Gc phagosomes in H exhibit qualitiatively less MPO reactivity than the phagosome in G or either of the S. aureus phagosomes. Arrowheads indicate a MPO-positive ring surrounding the bacteria. Open white arrows indicate MPO-positive granules associated or fusing with the phagosome. Asterisks indicate phagosomes which lack the MPO-positive ring surrounding the bacteria. Black arrows indicate granules which are not fusing with the phagosome.
In order to determine if Gc phagosomes avoid fusion with primary granules in PMNs in vivo, we examined PMNs in human gonorrheal exudates for enrichment of CD63 around Gc. Similar to observations in ex vivo-infected PMNs, only 40% of Gc associated with exudatous PMNs were positive for CD63 (Figure 3). This observation supports our findings using adherent, IL-8 primed, primary human PMNs.
Figure 3. Gc phagosomes in PMNs from gonorrheal exudates have reduced enrichment of primary granule proteins.

Human gonorrheal exudate PMNs were stained with an anti-CD63 antibody (red) and DAPI (blue) to label total Gc. Arrowheads indicate bacteria positive for CD63 enrichment, while arrows indicate bacteria negative for CD63 enrichment.
To address whether the absence of primary granule enrichment on Gc phagosomes represented a block or a delay in phagosome-granule fusion, maturation of Gc phagosomes in primary human PMNs was assessed over 6 h. The presence of CD63 on Gc phagosomes steadily increased over time, from 47% after 1 h to 70% after 6 h; similar results were obtained using neutrophil elastase to mark primary granules (Figure 4). We conclude that Gc delays primary granule fusion with the phagosome, thereby initially avoiding exposure to primary granule components inside PMNs.
Figure 4. Gc phagosomes delay primary granule fusion.

(A–B) PMNs were infected for 1, 2, 4, or 6 h with Gc. Extracellular Gc appear red/blue, while intracellular Gc appear blue only. PMNs were stained with antibodies against the primary granule protein CD63, which appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The average percent of Gc phagosomes positive for CD63 or neutrophil elastase from two replicate experiments are reported in B. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
We then asked if Gc globally impacted PMN phagosome-granule fusion by assessing the maturation of phagosomes in PMNs coinfected with S. aureus and Gc as described in Experimental procedures. There was no significant difference in the percent of S. aureus phagosomes positive for the primary granule protein neutrophil elastase between PMNs coinfected with Gc and PMNs infected with S. aureus alone (Figure 5). Thus Gc infection does not alter PMN primary granule fusion with S. aureus phagosomes. Interestingly, the percent of primary granule-positive Gc phagosomes was significantly increased in coinfected PMNs compared to PMNs infected with Gc alone (Figure 5), suggesting S. aureus can stimulate the increased fusion of Gc phagosomes with primary granules.
Figure 5. The effect of Gc on PMN phagosome maturation is phagosome-autonomous.
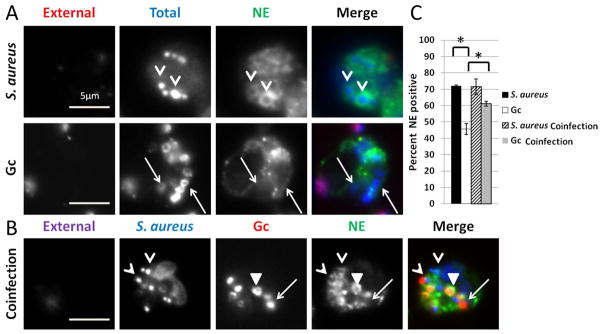
(A) PMNs were infected with S. aureus for 30 min or Gc for 1 h, then processed for immunofluorescence microscopy with the same color scheme as in Fig. 2C. (B) PMNs were infected with Gc for 30 min followed by 30 min of additional infection with S. aureus, then processed for immunofluorescence microscopy. Extracellular S. aureus appear purple/blue and extracellular Gc appear purple/red, while intracellular S. aureus appear blue only and Gc appear red only. The primary granule protein neutrophil elastase (NE) appears green. Arrowheads indicate S. aureus phagosomes positive for neutrophil elastase, triangles indicate Gc phagosomes positive for neutrophil elastase, and arrows indicate Gc phagosomes negative for neutrophil elastase. (C) The average percent of Gc and S. aureus phagosomes positive for neutrophil elastase from two replicate experiments are compared to PMNs in the same experiment infected with Gc alone for 1 h, or S. aureus alone for 30 min (not shown). Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Taken together, these results reveal that the Gc inside human PMNs reside in two subsets of phagosomes. Early in infection, the majority of Gc phagosomes remain immature, since they do not fuse with primary granules but can fuse with other granule subsets. The remaining Gc reside in phagolysosomes that have fused with all classes of PMN granules. As infection proceeds, Gc phagosomes undergo fusion with primary granules, reaching a maximal percent maturity after 6 h. The observed delay in primary granule fusion to Gc phagosomes is not due to Gc globally altering PMN primary granule fusion; instead, primary granule fusion with Gc phagosomes is determined per individual phagosome.
The delay in primary granule fusion with Gc phagosomes does not require active bacterial processes and can be overcome by IgG opsonization
We envisioned two possibilities as to how Gc delays primary granule fusion with its phagosomes in PMNs: live Gc releases factors that actively prevent early granule mobilization, or surface components on Gc influence phagosome biogenesis and granule mobilization. To test between these possibilities, PMNs were allowed to internalize nonviable, paraformaldehyde (PFA)-fixed Gc, and the composition of the Gc phagosomes was assessed by immunofluorescence microscopy. PMNs exposed to nonviable Gc exhibited reduced enrichment of neutrophil elastase and CD63 at their phagosomes, at levels statistically indistinguishable from PMNs infected with viable Gc (Figure 6). Similar results were obtained using heat-killed bacteria (data not shown). We conclude that active Gc processes are not required to prevent the initial fusion of primary granules with the Gc phagosome, suggesting instead that bacterial surface composition influences phagosome-granule fusion and delays phagosomal maturation.
Figure 6. Active Gc processes are not required to prevent primary granule fusion with Gc phagosomes.
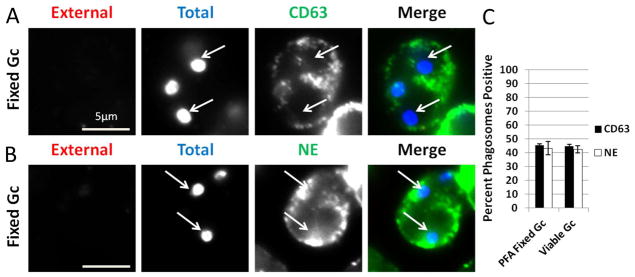
PMNs were exposed to paraformaldehyde (PFA)-fixed Gc for 1 h, and intracellular and extracellular Gc were discriminated from one another along with antibodies directed against CD63 (A) or neutrophil elastase (NE) (B). Extracellular Gc appear red/blue, while intracellular Gc appear blue only, and primary granule proteins appear green. PMNs were also infected with viable Gc; these cells are not depicted but displayed the same staining pattern for neutrophil elastase and CD63 as presented in Figure 2. Arrows indicate phagosomes negative for granule proteins. The percent of CD63-or NE-positive phagosomes containing viable and PFA-fixed Gc is reported in C.
In phagocytes, the route by which a particle enters the cell influences phagosomal maturity, and by extension, the phagosome’s ability to kill microbes (Stuart et al., 2005; Lee et al., 2003). Directing bacterial pathogens such as Mycobacterium tuberculosis and Francisella tularensis to opsonophagocytic pathways reduces the survival of these bacteria inside PMNs and macrophages, respectively (Cougoule et al., 2002; Geier et al., 2011). We hypothesized that driving Gc uptake by PMNs via complement or Fc receptors would modulate phagosome maturation. To test this hypothesis, PMNs were infected with Gc that were opsonized with normal human serum from individuals with no history of gonorrhea (serum opsonization), with polyclonal anti-Gc rabbit IgG, or left unopsonized, and phagosome maturity was assessed by immunofluorescence microscopy. Serum opsonization of Gc did not alter primary granule fusion with Gc phagosomes compared to non-opsonized Gc. However, IgG opsonization of Gc significantly increased primary granule fusion with Gc phagosomes compared to non-opsonized or complement-opsonized Gc (Figure 7). Titration of the antibody to the lowest concentration sufficient to opsonize Gc also resulted in significantly increased trafficking of the bacteria to primary granule-positive phagosomes (Supplemental Figure 1). This finding suggests that engagement of Fc receptors can drive Gc into a mature phagolysosome in PMNs, although the possibility remains that the antibody may also mask Gc surface components that prevent primary granule fusion with phagosomes. Taken together, these results indicate that the surface composition of Gc, including its opsonization state, directs interactions with PMN receptors to influence phagosome-granule fusion and consequent phagosome maturation.
Figure 7. IgG opsonization of Gc increases primary granule fusion with Gc phagosomes.

A. PMNs were infected with non-opsonized, complement opsonized, or IgG opsonized Gc for 1 h, and intracellular and extracellular Gc were discriminated from one another, along with an antibody directed against neutrophil elastase. Extracellular Gc appear red/blue, while intracellular Gc appear blue only, and neutrophil elastase staining appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of neutrophil elastase-positive phagosomes containing non-opsonized, complement opsonized, or IgG opsonized Gc is reported in B. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Gc phagosomes devoid of neutrophil primary granule proteins contain viable Gc
During ex vivo infection of primary human PMNs, up to 50% of Gc inside PMNs remain viable (Criss et al., 2009; Simons et al., 2005). As primary granules contain PMN components with known antigonococcal activity (Rest, 1979; Rock et al., 1988; Shafer et al., 1986b; Shafer et al., 1998; Casey et al., 1985), we hypothesized that primary granule fusion with Gc phagosomes reduces the viability of intracellular Gc, and the fraction of Gc that survive inside PMNs represent those bacteria residing in phagosomes that exclude primary granules. In preliminary support for this hypothesis, 86% of the phagosomes exhibiting rings of MPO staining by immuno-TEM, indicative of primary granule-phagosome fusion, contained electron-lucent, degraded bacteria (Fig. 2E).
To directly examine the relationship between phagosome maturation and bacterial viability, we combined dyes that reveal viability of individual bacteria with the detection of primary granule proteins. We modified our previously published bacterial viability assay, which used propidium iodide permeability as a surrogate for bacterial death (Criss et al., 2009). Here, nonviable Gc are permeant to Sytox Green, while the total Gc population is counterstained with DAPI. We verified that the Sytox Green/DAPI combination was able to discriminate live and dead Gc inside and attached to PMNs as effectively as the propidium iodide/SYTO9 system (Supplemental Figure 2). Detection of primary granule proteins was complicated by the fact that the bacterial viability dyes are incompatible with aldehyde fixation. However, a fluorescently coupled anti-CD63 antibody showed immunoreactivity with PMN granules and zymosan-containing phagolysosomes (Munafo et al., 2007) that colocalized with the pattern of CD63 staining in aldehyde-fixed cells (Supplemental Figure 3).
To test the hypothesis that phagosomes lacking fusion with primary granules are more likely to contain viable Gc, PMNs were infected with DAPI-labeled Gc for 1 h. PMNs were saponin-permeabilized without aldehyde fixation, then exposed to Sytox Green and fluorescently coupled anti-CD63. 25% of intracellular, Sytox Green-negative, viable Gc showed enrichment of CD63. In contrast, 60% of intracellular, nonviable, Sytox Green-positive Gc were surrounded by CD63 immunoreactivity, a significant increase (Figure 8). Thus Gc viability inside PMNs correlates with bacterial residence in immature, primary granule-negative phagosomes.
Figure 8. Increased enrichment of the primary granule protein CD63 on phagosomes containing nonviable Gc.
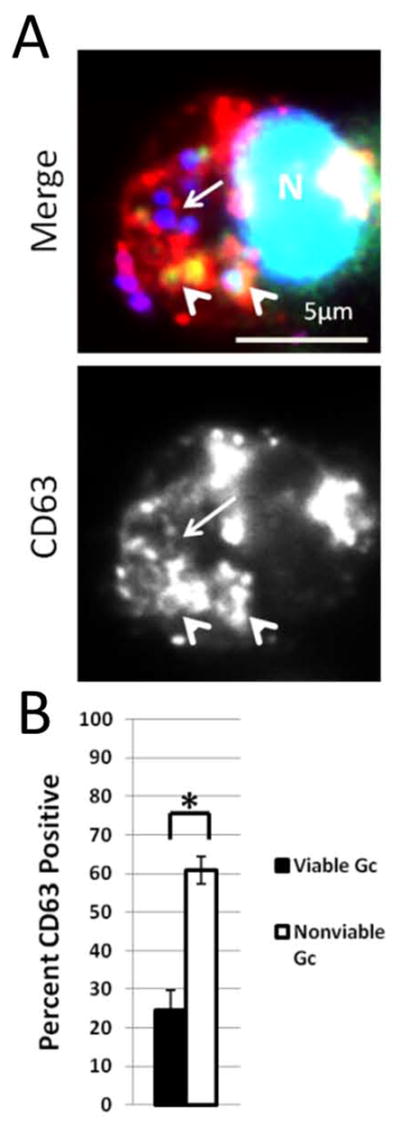
PMNs were infected with DAPI-labeled Gc (blue) for 1 h. Infected PMNs were exposed to soybean lectin (purple) to detect extracellular Gc, then permeabilized and incubated with an antibody against CD63 (red) along with Sytox Green to detect nonviable Gc (green). Viable intracellular Gc appear blue, and nonviable intracellular Gc appear green/blue. A representative infected PMN is shown in A. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of viable and nonviable Gc residing in CD63+ phagosomes is reported in B. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
The results presented thus far show that Gc has two fates within primary human PMNs. Most bacteria reside in phagosomes that undergo delayed primary granule fusion, while the remainder are found in phagosomes that fuse with primary granules and rapidly mature into phagolysosomes. This delay in phagosome maturation does not depend upon secretion or other active bacterial processes. However, when PMNs are infected with viable Gc, the fraction of bacteria that are killed with PMNs are more likely to be the ones residing in phagolysosomes, indicating that bacterial death inside PMNs occurs after phagosomes fuse with primary granules. We conclude that primary granule components have antigonococcal activity inside PMN phagosomes, and one mechanism by which Gc survives inside PMNs is by avoiding these components through a delay in phagosome maturation.
Increasing primary granule fusion with Gc phagosomes reduces internal Gc viability
Given the correlation between Gc viability and residence in primary granule-negative phagosomes, we took two approaches to examine how manipulating phagosomal composition affected Gc viability inside PMNs. First, Gc was opsonized with IgG, in order to drive Fcγ receptor-mediated uptake of the bacteria into phagolysosomes (Lee et al., 2003; Stuart et al., 2005) (see Figure 7). PMNs were infected with non-opsonized Gc or Gc opsonized with rabbit anti-Gc IgG, and bacterial viability inside PMNs was assessed using propidium iodide to detect bacteria with compromised membranes and SYTO9 for intact bacteria. IgG opsonization significantly decreased the viability of intracellular Gc, but did not affect the viability of adherent bacteria (Figure 9). Together with the results presented in Figure 7, these findings show that antibody opsonization directs Gc to a phagosome that undergoes early fusion with primary granules, which consequently decreases Gc intracellular survival.
Figure 9. IgG opsonization increases primary granule fusion with Gc phagosomes resulting in decreased Gc intracellular viability.
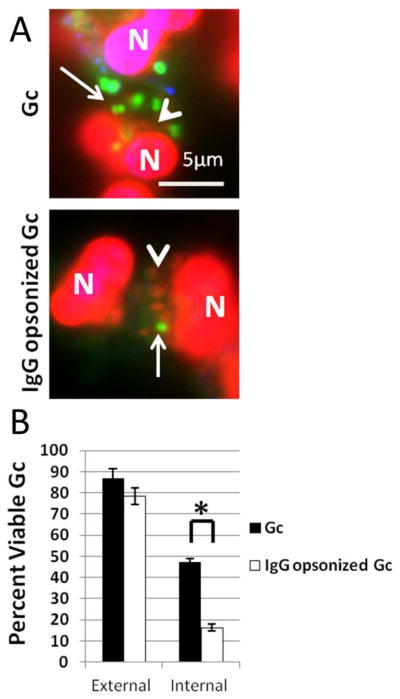
PMNs were infected with non-opsonized or IgG opsonized Gc (A). Viable Gc (green) and nonviable Gc (red) were discriminated using Baclight viability dyes SYTO9 and propidium iodide, and extracellular Gc were labeled with soybean lectin (blue). Arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. The percent of viable extracellular and intracellular bacteria is reported in B for PMNs infected with non-opsonized and IgG opsonized Gc. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
As a second approach to modulating phagosome maturation, primary granule fusion with phagosomes was ectopically induced with lysophosphatidylcholine (LPC) treatment (Hong et al., 2010). Increased primary granule fusion with neutrophil phagosomes due to LPC treatment is accompanied by calcium flux (Silliman et al., 2003). We observed LPC treatment (Figure 10A–B) and treatment with the calcium ionophore ionomycin (Supplemental Figure 4) significantly increased primary granule fusion with PMN phagosomes containing non-opsonized Gc (Figure 10A–B). As observed for IgG-opsonized Gc, intracellular Gc survival was significantly reduced in LPC-treated PMNs compared to the untreated control (Figure 10C–E). LPC treatment had no significant effect on the viability of Gc that were adherent to PMNs (Figure 10E) or were kept in media without PMNs (data not shown). From these results, we conclude that primary granules contain antimicrobial components that are capable of killing Gc inside PMN phagosomes, and driving primary granule fusion with Gc phagosomes by opsonization or LPC treatment decreases intracellular viability of the bacteria.
Figure 10. LPC treatment of primary human PMNs increases primary granule fusion with Gc phagosomes.
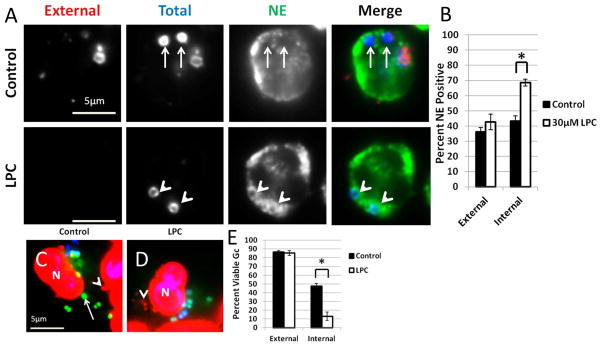
(A–B) PMNs were infected with Gc for 45 min, then given media with or without LPC. Intracellular and extracellular Gc were discriminated from one another along with an antibody raised against neutrophil elastase (NE). Extracellular Gc appear red/blue, intracellular Gc appear blue only, and neutrophil elastase staining appears green. In (A) arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of phagosomes exhibiting enrichment of neutrophil elastase with and without LPC treatment is reported in B. (C–E) PMNs were infected with Gc and left untreated (C) or treated with LPC (D). Viable Gc (green) and nonviable Gc (red) were discriminated using Baclight viability dyes SYTO9 and propidium iodide, and extracellular Gc were labeled with soybean lectin (blue). Extracellular viable Gc appear teal, intracellular nonviable Gc appear red, and intracellular viable Gc appear green. In C and D arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. The percent of viable extracellular and intracellular Gc in untreated and LPC-treated PMNs is reported in E. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Protease activity contributes to the anti-Gc activity of phagosomes enriched with primary granules
Primary granules contain the majority of oxygen-independent PMN antimicrobial components (Faurschou et al., 2003). Of these components, Gc is resistant to α-defensins (Qu et al., 1996) and suppresses the PMN oxidative burst, which provides the substrate H2O2 for MPO activity (Criss et al., 2008; Britigan et al., 1988; Lorenzen et al., 2000; Bjerknes et al., 1995; Seib et al., 2005; Wu et al., 2009). However, primary granule proteases, specifically the serine protease cathepsin G, display antigonococcal activity in vitro (Rest, 1979; Rock et al., 1988; Shafer et al., 1986a; Shafer et al., 1998; Casey et al., 1985). Cathepsin G can enzymatically cleave Gc outer-membrane proteins (Rest et al., 1981; Shafer et al., 1987) and also has protease-independent antibacterial activity (Bangalore et al., 1990; Shafer et al., 1986b). The role of these primary granule components in killing Gc has not been examined in the context of PMN infection. To test whether PMN serine proteases contribute to the antigonococcal activities of mature PMN phagolysosomes, Gc viability was measured in PMNs treated with a protease inhibitor cocktail prior to infection (see Experimental procedures), in the presence or absence of LPC. We confirmed that treatment of PMNs with the protease inhibitor cocktail inhibited the proteolytic activities of cathepsin G, neutrophil elastase, and proteinase 3 (Figure 11F). In the absence of LPC treatment, protease inhibitor treatment significantly increased Gc viability inside PMNs compared to untreated PMNs (Figures 11A, C, quantified in E). Moreover, protease inhibitor exposure rescued the survival of Gc inside LPC-treated PMNs to levels seen in protease inhibitor-exposed PMNs not given LPC (Figures 11B, D, E). Protease inhibitor treatment did not affect the fusion of primary granules with PMN phagosomes (Supplemental Figure 5). These data show for the first time that proteases are a major contributor to the antigonococcal activity associated with some PMN phagosomes. We conclude that by initially avoiding fusion with primary granules, the Gc phagosomes in PMNs exhibit reduced protease activity, which promotes Gc survival inside PMNs.
Figure 11. Ectopically increasing primary granule fusion with Gc phagosomes reduces intracellular Gc viability by a protease-dependent process.
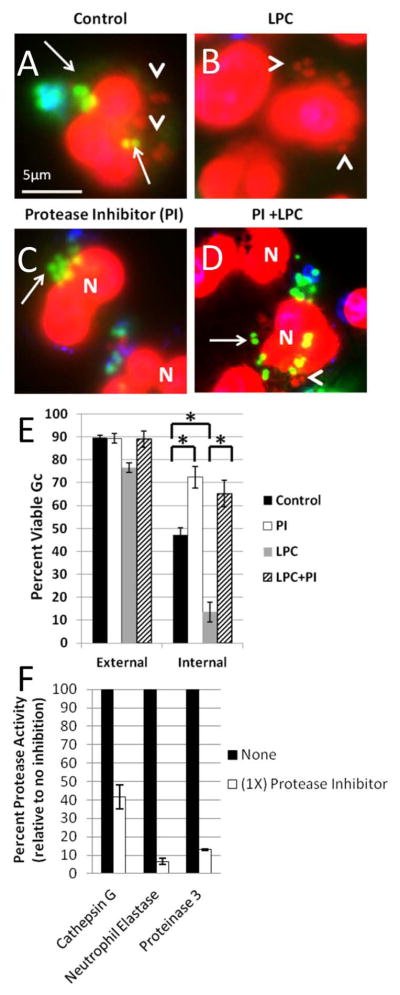
PMNs were infected with Gc and treated with LPC (B, D) or left untreated (A, C) as described in Figure 9. Prior to infection, PMNs were treated with protease inhibitor cocktail (C, D). Viable Gc (green) and nonviable Gc (red) were discriminated using Baclight viability dyes SYTO9 and propidium iodide, and extracellular Gc were labeled with soybean lectin (blue). Extracellular viable Gc appear teal, intracellular nonviable Gc appear red, and intracellular viable Gc appear green. Arrows indicate viable, intracellular Gc and arrow heads indicate nonviable, intracellular Gc. The percent of viable extracellular and intracellular Gc in control PMNs, protease inhibitor treated PMNs, LPC treated PMNs, and PMNs treated with both the protease inhibitor and LPC is reported in E. Asterisks indicate P < 0.05 by Student’s two-tailed t test. The proteolytic activity of cathepsin G, neutrophil elastase, and proteinase 3 was measured for PMNs treated with the protease inhibitor cocktail and is expressed relative to untreated PMNs (F).
Discussion
Although there is evidence for Gc survival inside PMNs during acute gonorrhea, the mechanisms that allow the bacteria to avoid killing by PMNs have remained elusive. In this work we show for the first time that most Gc inside PMNs delay phagosome fusion with primary granules. These immature phagosomes are more likely to contain viable Gc, since primary granule proteases and other components have antigonococcal activity. Our findings reveal a novel approach Gc has evolved in order to evade PMN antimicrobial activities, thereby enabling its survival in its obligate human hosts.
We identified two compartments containing Gc in primary human PMNs. Most phagosomes fuse with secondary and/or tertiary granules but not primary granules, while others fuse with all granule subsets. Primary granules contain a variety of antimicrobial components including neutrophil elastase, cathepsin G, proteinase 3, BPI, and α defensins (Faurschou et al., 2003). Previously, purified BPI and cathepsin G have been shown to have antigonococcal activity in vitro (Casey et al., 1985; Shafer et al., 1986a), which we have confirmed with strain FA1090 Gc (our unpublished observations). Thus we reasoned that delaying primary granule fusion with the Gc phagosome would confer a survival advantage on Gc found in PMNs. In support of this reasoning, nonviable Gc were more likely to be found in primary granule-positive phagosomes than viable bacteria. Moreover, ectopically increasing primary granule fusion with phagosomes by LPC treatment or antibody opsonization of Gc reduced intracellular bacterial viability. These observations strongly suggest that the components located within PMN primary granules are capable of killing Gc in the context of the PMN phagosome, which has never before been shown.
Our results point to serine proteases as important contributors to the antigonococcal activity of mature PMN phagosomes. The serine proteases of PMNs include neutrophil elastase, proteinase 3, cathepsin G, and NSP4 (Faurschou et al., 2003; Perera et al., 2012). We envision two possibilities to explain the contribution of these proteases to intraphagosomal Gc killing. First, the proteases act directly on Gc. In support of this possibility, in vitro studies have shown that cathepsin G degrades porin and Opa proteins in the Gc outer membrane (Rest et al., 1981; Shafer et al., 1987). Digestion of outer-membrane proteins could allow the proteases access to other potential targets inside Gc, although to date these are not known. It could also render the bacteria more sensitive to killing by non-proteolytic components of primary granules. These include BPI and cathepsin G itself, which contains two protease-independent peptide sequences with broad-spectrum antimicrobial activity in vitro (Bangalore et al., 1990). Second, primary granule serine proteases activate precursors of antimicrobial components that are found in other PMN granule subsets. Secondary granules contain hCAP18, the precursor of the antimicrobial peptide LL-37 (Faurschou et al., 2003), which has antigonococcal activity in vitro (Shafer et al., 1998), including against strain FA1090 Gc (our unpublished observations). Primary granule proteases including proteinase 3 are required to cleave hCAP18 to LL-37 (Sorensen et al., 2001). Therefore, even if Gc phagosomes fuse with secondary granules, no LL-37 would be generated in the phagosomes that avoid primary granules. These two possibilities are not mutually exclusive, and we propose that delaying fusion of Gc phagosomes with primary granules protects Gc against early exposure to antimicrobial components housed in both primary and secondary granules, including those known to have potent activity against Gc.
While many pathogens can avoid the phagolysosome by modulating phagolysosomal composition, this is the first time this observation has been made for Gc. One common bacterial mechanism for phagolysosome avoidance, for instance in Legionella and Salmonella, involves the manipulation of membrane traffic via the ATP-dependent secretion of bacterial proteins that target the Rab family of GTPases or Rab effectors (Urban et al., 2006). In contrast, the delay of Gc phagosome fusion with primary granules appears to be independent of active bacterial processes. This may not be surprising, as Gc does not encode secretion systems known to release proteins into host cells (van Ulsen et al., 2006). Instead, our results point to Gc surface composition as the mechanism directing phagosome maturation. Gc uses a variety of surface structures to manipulate host antimicrobial responses and confer resistance to antimicrobial components. Gc pili and porin have been implicated in altering neutrophil granule exocytosis (Bjerknes et al., 1995; Lorenzen et al., 2000; Densen et al., 1978), modifications to Gc lipooligosaccharide have been shown to increase bacterial resistance to antimicrobial peptides and proteases (Balthazar et al., 2011; Lewis et al., 2009), and the Mtr and FarAB efflux pump systems span the inner and outer membranes to export a variety of antimicrobial components out of the cell (Shafer et al., 1998; Lee et al., 1999). We speculate that as a complement to these mechanisms, Gc uses one or more surface components to direct the bacteria into a phagosomal compartment that undergoes limited, delayed fusion with primary granules. The redirection of membrane traffic could be initiated as early as during phagocytosis, since surface components on pathogens, as shown for the M and/or M-like proteins of Streptococcus pyogenes, lipophosphoglycan of Leishmania donovani, and lipoarabinomannan of Mycobacterium tuberculosis, can dictate their eventual fate inside host cells (Staali et al., 2006; Desjardins et al., 1997; Mollinedo et al., 2010; Fratti et al., 2001). Delaying of primary granule-phagosome fusion could also occur from inside the Gc phagosome, by an as-yet unappreciated mechanism. While the surface components that mediate this delay are not known, the effect is not mediated by the Gc IgA protease, which can affect the composition of Gc endosomes in non-phagocytic cells (Lin et al., 1997) (Supplemental Figure 6).
Individuals infected with Gc as well as individuals with no prior history of gonorrhea produce opsonic IgG reactive against Gc surface components. However, most of these surface components are antigenically variable, preventing individuals from developing a protective humoral response to gonorrheal disease and contributing to the difficulty in development of a protective vaccine (Zhu et al., 2011). Development of a vaccine is a long-sought after goal of the public health community and is especially pressing given the dramatic rise in multidrug-resistant Gc in the human population (Kirkcaldy et al., 2011; Unemo et al., 2011). Importantly, our results suggest that IgG opsonization of Gc is capable of enhancing primary granule fusion with the Gc phagosome and reducing Gc survival inside PMNs. This observation raises the exciting possibility that if an effective antibody-based vaccine can be raised against Gc, the abundance of PMNs at the site of infection would be sufficient for host-mediated clearance of gonorrhea, whether or not the bacteria are susceptible to antibiotics.
We propose the following working model to explain the remarkable capability of Gc to survive within human PMNs (Figure 12). In this model, Gc uses surface structures to influence its fate within PMNs. Selective membrane trafficking allows the majority of Gc phagosomes to delay fusion with PMN primary granules until 4–6 h post-infection. The Gc inside phagosomes that delay primary granule fusion are more likely to remain alive because they avoid degradation by primary granule proteases and membrane disruption by antimicrobial peptides. The early avoidance of primary granules is not complete, and early after internalization, a fraction of Gc are found in phagolysosomes that fuse with all classes of PMN granules. Yet some of the Gc found in these phagolysosomes remain viable, which is likely due to bacterial virulence factors that directly defend the bacteria from PMN antimicrobial components. Additionally, we speculate that the delay in primary granule fusion with the Gc phagosome allows Gc to adapt to the PMN intracellular environment, for instance by upregulating genes whose products confer resistance to PMN antimicrobial components. Thus Gc uses multiple, simultaneously operating mechanisms to enable its persistence and potential replication inside PMNs. Although PMNs are terminally differentiated cells, Gc has the potential to delay PMN apoptosis (Chen et al., 2011; Simons et al., 2006), which would co-opt these normally antimicrobial cells into a protected niche for Gc. We envision that the ability of Gc to persist within PMNs would facilitate long-term colonization of its obligate human host and provide increased opportunities for dissemination and transmission (Criss et al., 2012). The ability of Gc to avoid exposure to toxic PMN components reflects how effectively Gc subverts normal PMN function, which underlies the remarkable survival of Gc when confronted with the potent innate immune response of symptomatic gonorrhea.
Figure 12. Model for Gc survival inside primary human PMNs.

See text for details.
Experimental Procedures
Bacterial strains and growth conditions
The Gc of strain FA1090 used in this study constitutively expresses the pilin variant 1-81-S2 due to mutation of the guanine quartet sequence upstream of pilE (Cahoon et al., 2009) and has in-frame deletions of the genes encoding the 4 “translucent” opacity-associated (Opa) proteins, Opas B, E, G, and K (L. Ball and A. Criss, manuscript in preparation). Piliated, phenotypically Opa-negative Gc was routinely grown on Gonococcal Medium Base (Difco) plus Kellogg’s supplements (Kellogg et al., 1963) for 20 h at 37°C in 5% CO2. Gc was grown to exponential phase via successive rounds of bacterial growth in rich liquid medium as described (Criss et al., 2008). Unless otherwise stated, viable, exponential phase Gc was used to infect primary human PMNs. To kill Gc prior to PMN exposure, Gc was either heat killed by incubation at 56°C for 20 min, or fixed for 15 min with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) in PBS. Gc was labeled with 5 μg ml−1 carboxyfluorescein diacetate succinimidyl ester (CFSE) for 20 min at 37°C. Gc was complement opsonized with 50% fresh normal human serum (in Morse’s defined medium (MDM) (Morse et al., 1980)) for 20 min at 37°C. Gc was IgG opsonized with a polyclonal rabbit anti-Gc antibody (Biosource) for 30 min at 37°C. FA1090 iga::kan Gc, lacking IgA protease, were previously described (Criss et al., 2008).
S. aureus ATCC 25923 and a Δspa mutant generated in the Newman background (obtained from E. Skaar, Vanderbilt University) were grown on Luria-Bertani (LB) agar for 16 h. S. aureus was inoculated into LB liquid broth and grown with rotation at 37°C for 20 h. In immunofluorescence experiments, prior to infection, S. aureus was prelabeled with 10 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) for 20 minutes in MDM at room temperature in the dark. S. aureus was complement opsonized with 20% fresh normal human serum in MDM for 20 min at 37°C.
Zymosan
Zymosan (MP Biomedicals) was opsonized in 50% human serum in RPMI for 25 min at 37°C.
PMN isolation
Venous blood was collected from healthy human subjects after receiving written informed consent, in accordance with a protocol approved by the University of Virginia Institutional Review Board for Health Sciences Research (IRB-HSR). PMNs were isolated from heparinized blood by dextran sedimentation followed by purification on a Ficoll-Hypaque gradient as previously described (Stohl et al., 2005). PMNs were resuspended at 1 × 107 cells per ml in Dulbecco’s PBS (without calcium and magnesium; Thermo Scientific) containing 0.1% dextrose and kept on ice until use within 2 h. Preparations routinely contained > 95% PMNs, assessed morphologically by phase-contrast microscopy.
Human gonorrheal exudates
PMN-rich urethral gonorrheal exudates were obtained from men attending the Virginia Department of Health (VDH) sexually transmitted disease clinic in Charlottesville, Virginia, after receiving written informed consent, in accordance with a protocol approved by the University of Virginia IRB-HSR and the VDH. Exudates from two male subjects presenting with symptoms consistent with acute, uncomplicated gonorrhea were collected via urethral swab into 4% PFA in PBS and stored at 4 °C until use. Gram staining of the exudate prior to fixation and nucleic acid amplification tests confirmed the subjects were positive for gonorrhea and no other sexually transmitted infection.
Adherent PMN assay
Acid washed 12mm glass coverslips in 24-well plates were coated for 1 h at 37°C with 50% pooled human serum (Sigma) in PBS and washed in PBS prior to PMN addition. PMNs were diluted into RPMI (Mediatech) containing 10% fetal bovine serum (Thermo Scientific) and 10nM IL-8 (R&D Systems). 106 PMNs were added to each coverslip and incubated for 1 h at 37°C, 5% CO2 to promote PMN attachment. PMNs were infected with S. aureus, viable exponential-phase Gc, or exposed to heat killed or PFA-killed Gc at a multiplicity of infection of 1–5 bacterial colony forming units (CFU) per PMN as described in (Criss et al., 2009). PMNs were exposed to opsonized zymosan for 25 min at 37°C. For experiments extending Gc infection to 6 h, after 1 h media was replaced with Hanks’ balanced salt solution with Ca2+ and Mg2+ and supplemented with 0.15% dextrose and 1% BSA.
Gc and S. aureus coinfection
PMNs were infected with Gc as described for 30 min. The same PMNs were then infected with DAPI-labeled S. aureus as described for an additional 30 min. Coinfected PMNs were compared to PMNs infected only with Gc for 1 h or with S. aureus for 30 min.
Immunofluorescence
PMNs infected in vitro
PMNs infected for 1, 2, 4, and 6 h with Gc and/or S. aureus were fixed with 4% PFA in PBS for 15 min. External and internal Gc were distinguished using a polyclonal rabbit anti-Gc antibody before and after PMN permeabilization as previously described in (Criss et al., 2009). For experiments examining the secondary granule marker lactoferrin, Gc was labeled prior to infection with CFSE to stain the total Gc population, and external Gc were detected using the polyclonal rabbit anti-Gc antibody as described above. External S. aureus were recognized using a polyclonal biotinylated S. aureus antibody (MyBiosource) followed by exposure to streptavidin coupled to Alexa Fluor 555 (Life Technologies), prior to PMN permeabilization. Cells were then permeabilized using a 1:1 ratio of acetone and methanol. Post permeabilization, PMN secondary granules were recognized using monoclonal antibodies against p22phox (44.1) and gp91phox (54.1) (both Santa Cruz Biotechnology), or a polyclonal rabbit anti-lactoferrin antibody (MP Biomedicals). PMN primary granules were recognized using monoclonal antibodies against CD63 (H5C6-c) (Developmental Studies Hybridoma Bank and Ancell) or neutrophil elastase (AHN-10) (Millipore) followed by Alexa Fluor-coupled goat anti-rabbit or goat anti-mouse antibodies (Life Technologies). Coverslips were mounted using Flouromount G (Southern Biotech) with 2.5 mg ml−1 propyl gallate (Acros Organics).
Human gonorrheal exudates
Gonorrheal exudates were pelleted at 100 × g for 12 min. Cells were resuspended in PBS containing 10% normal goat serum (Life Technologies) and 0.2% saponin, then incubated for 1 h with Alexa Fluor 555-coupled mouse anti-CD63 antibody to label primary granules and 10 μg ml−1 DAPI to label total Gc. The anti-CD63 antibody was coupled to Alexa Fluor 555 using a Molecular Probes monoclonal antibody labeling kit (Life Technologies), following the manufacturer’s instructions. Cells were washed in PBS and mounted on slides for imaging.
Viability of bacteria associated with PMNs
Baclight viability dyes
Detection of viable and nonviable Gc associated with PMNs was conducted as described in (Criss et al., 2009).
Sytox Green in combination with DAPI
A modification of the Baclight bacterial viability assay was developed using Sytox Green and DAPI, since the propidium iodide component of Baclight emits in both the red and ultraviolet channels of the fluorescence microscope, and detection of external vs. internal bacteria, viable bacteria, nonviable bacteria, and CD63 requires four-color fluorescence microscopy. Gc was prelabeled for 20 min with 10 μg ml−1 DAPI in MDM. After 1h of PMN infection, samples were incubated with 0.4μM Sytox Green (Life Technologies) in 0.1 MOPS pH 7.2, 1mM MgCl2 (MOPS/MgCl2) for 5 min. Gc was counted as viable if labeled only with DAPI and non-viable if labeled with DAPI and Sytox Green.
Immunofluorescence for CD63 in combination with bacterial viability dyes
PMNs were infected with DAPI-labeled Gc. After 1 h, samples were washed and incubated for 10 min at RT in MOPS/MgCl2 containing 5 μg ml−1 Alexa Fluor 647-coupled soybean agglutinin (Life Technologies), which recognizes extracellular Gc. PMNs were then incubated with Alexa Fluor 555-coupled mouse anti-CD63 antibody for 20 min in MOPS/MgCl2 containing 0.2% saponin. Subsequently, PMNs were exposed to 0.4μM Sytox Green in MOPS/MgCl2 containing 0.2% saponin for 5 min to stain nonviable Gc.
Immuno-TEM
PMNs infected for 1 h with Gc or Δspa S. aureus were fixed with 2% PFA, 0.25% glutaraldehyde in phosphate buffer pH 7.2 for 45 min. PMNs were then incubated in 1% PFA in phosphate buffer and shipped to the University of Iowa Central Microscopy Research Facility. Cells were embedded in LR White and 60nm thin sections placed on Formvar carbon coated grids. Sections were stained with polyclonal antibodies against the lactoferrin (MP Biomedicals) or myeloperoxidase (Dako), followed by 10nm gold particle-labeled secondary goat anti-rabbit antibody. Sections were examined with a JEOL 1230 transmission electron microscope at an accelerating voltage of 120 kV.
PMN treatments
Lysophosphatidylcholine (LPC)
PMNs were infected with Gc as described. After 45 min, fresh RPMI +10% FBS or media containing 30μM LPC (18:0) (Sigma-Aldrich; reconstituted in PBS) was added to each well and incubated for 15 min at 37°C. PMNs were washed with fresh media and fixed for immunofluorescence or incubated an additional 30 min in fresh media and processed using Baclight viability dyes as described above. Immunofluorescence was conducted as described to discriminate extracellular vs. intracellular Gc and to detect neutrophil elastase.
Protease inhibitors
PMNs were incubated with 1X Protease Inhibitor Cocktail Set V containing AEBSF, aprotinin, E-64, and leupeptin(Calbiochem) during PMN attachment to coverslips. Protease inhibitors were present for the entire experiment. PMNs were then infected with Gc for 90 min. After 45 min, protease inhibitor-treated PMNs were exposed to LPC. Gc viability was assessed using Baclight viability dyes.
Ionomycin
PMNs were infected with Gc as described. After 30 min, fresh RPMI +10% FBS or media containing 1μM ionomycin was added to each well and incubated for 15 min at 37°C. PMNs were washed with fresh media and fixed for immunofluorescence.
Determination of serine protease activity
The proteolytic activities of cathepsin G, neutrophil elastase, and proteinase 3 were assessed using specific substrates: cathepsin G, 0.1mM Suc-Ala-Ala-Pro-Phe-pNA (Elastin Products Company) in 100 mM HEPES, 500 mM NaCl pH 7.5; neutrophil elastase, 0.85 mM MeOSuc-Ala-Ala-Pro-Val-pNA (Calbiochem) in 100 mM HEPES, 500 mM NaCl pH 7.5; proteinase 3, 0.25 mM Boc-Ala-Ala-Nva-SBzL (Elastin Products Company) in H2O with 0.1mM 5,5′-Dithiobis (2-nitro-benzoic acid) (Sigma-Aldrich). PMNs were incubated in the presence or absence of 1X Protease Inhibitor Cocktail Set V. After 1 h the media was removed. The appropriate buffer for each substrate, with 0.01% Triton X-100, was added to each well. PMNs were scraped off coverslips and kept on ice for 5 min. Debris was removed by spinning 800 × g for 8 min. An equal volume of lysed PMNs and substrate for each indicated serine protease was added in triplicate to wells of a 96 well plate. The plate was incubated in the dark for 45 min at 37°C. The OD405 of each reaction was read on a Perkin Elmer Victor 3 1420 microplate reader. Protease activity after inhibitor treatment was expressed as a percentage of the activity recovered from PMNs not treated with the inhibitor.
Image acquisition, processing, and quantification
Images were acquired on a Nikon Eclipse E800 UV/visible fluorescence microscope with Hamamatsu Orca-ER digital camera using Openlab software. Images were processed in Adobe Photoshop CS5. For consistency, all images depicting total bacteria were false colored blue, including bacteria labeled with CFSE. Images of human gonorrheal exudates were acquired on a Zeiss LSM510 confocal laser scanning microscope with a 63x, 1.4 numerical aperture objective. Images were acquired with LSM510 operating software and processed with LSM Image Browser (Zeiss) and/or Adobe Photoshop CS5 (Adobe).
Immunofluorescence
50–200 intracellular bacteria were analyzed for enrichment of PMN granule proteins. Bacterial phagosomes were classified as positive for PMN granule proteins if antibody staining surrounded ≥ 50% of the bacterial circumference.
Bacterial viability dyes
Images were acquired within 30 min of mounting coverslips and a minimum of 100 bacteria per experiment were analyzed for viability.
Immunofluorescence for CD63 in combination with bacterial viability dyes
Images were acquired within 30 min of mounting coverslips. A minimum of 50 internalized bacteria per experiment were analyzed for viability and enrichment of the neutrophil primary granule protein CD63 at the Gc phagosome. Phagosomes were classified as positive for CD63 according to guidelines described for immunofluorescence experiments.
Immuno-TEM
200 bacteria from grids stained with the anti-MPO antibody were analyzed for the presence of an electron-dense MPO-positive ring surrounding the bacterial phagosome. Bacterial viability was indirectly assessed by the electron density of their cytoplasm, where electron-lucent bacteria were considered to represent nonviable Gc at the time of fixation.
Human gonorrheal exudate
A minimum of 100 internalized bacteria were analyzed for enrichment of the neutrophil primary granule protein CD63 at the Gc phagosome. Phagosomes were classified as positive for CD63 according to guidelines described for immunofluorescence with ex vivo-infected primary human PMNs.
Statistics
All values are expressed as a mean ± standard error of the mean for three replicate experiments (unless otherwise noted), performed on different days with different donors’ PMNs for each assay. Significance was determined for each assay using a Student’s two-tailed t-test. A p value of less than 0.05 was considered statistically significant.
Supplementary Material
Supplemental Figure 1: Titration of anti-Gc polyclonal antibody results in significantly increased trafficking of opsonized Gc to primary granule-positive phagosomes. (A) Gc were left non-opsonized or opsonized with increasing concentrations of a polyclonal rabbit anti-Gc antibody (1:50, 1:100, 1:250, 1:500). Following opsonization, Gc was labeled with DAPI and then incubated for 1 h with an Alexa Fluor 555-coupled goat anti-rabbit antibody to identify the IgG-opsonized Gc. (B) PMNs were infected with non-opsonized or IgG-opsonized (1:50, 1:100, 1:250, 1:500) Gc for 1 h and processed for immunofluorescence microscopy for Gc and neutrophil elastase. Extracellular Gc appear red/blue, while intracellular Gc appear blue only, and neutrophil elastase staining appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of Gc phagosomes positive for neutrophil elastase is reported in C. The percent of Gc phagosomes positive for neutrophil elastase was significantly decreased in PMNs infected with non-opsonized Gc compared to PMNs infected with IgG opsonized Gc (1:50, 1:100, and 1:250) as determined by Student’s two-tailed t test (P < 0.05). There was no statistically significant difference in neutrophil elastase-positive phagosomes between non-opsonized Gc and Gc opsonized with IgG at 1:500 concentration. The percent of Gc phagosomes positive for PMNs infected with IgG-opsonized Gc (1:500) was significantly less than in PMNs infected with IgG-opsonized Gc (1:50, 1:100) as determined by Student’s two tailed t test (P< 0.05).
Supplemental Figure 2: Sytox Green and DAPI effectively discriminate viable and nonviable Gc during PMN infection. (A–B) PMNs were incubated with heat killed Gc for 1 h and processed using Baclight viability dyes (A) or Sytox Green in combination with DAPI (B). (C–E) PMNs were infected with exponentially-growing, viable Gc for 1 h and processed using Baclight viability dyes (C) or Sytox Green in combination with DAPI (D). In both (A) and (C) nonviable bacteria are stained with propidium iodide and appear red. (All bacteria are stained with SYTO9, but only viable bacteria appear green due to reduction of SYTO9 fluorescence in the presence of propidium iodide.) In both (B) and (D) nonviable bacteria are stained with Sytox Green and DAPI (teal) and viable bacteria are stained with DAPI only (blue). Arrowheads indicate nonviable Gc, and arrows indicate viable Gc. The percent of viable Gc discriminated using the two dye combinations is reported in E.
Supplemental Figure 3: In the absence of fixation, an anti-CD63 antibody effectively labels primary granules and zymosan-containing phagolysosomes. PMNs were incubated with serum-opsonized zymosan for 25 min. Cells were left unfixed and incubated with an Alexa Fluor 555-coupled anti-CD63 antibody (A), or fixed with PFA and incubated with a FITC-coupled anti-CD63 antibody (B). (C) Unfixed PMNs that had internalized opsonized zymosan were permeabilized and incubated with the Alexa 555-coupled anti-CD63 antibody. PMNs were then fixed and incubated with the FITC-coupled anti-CD63 antibody. Arrowheads indicate zymosan phagosomes positive for CD63.
Supplemental Figure 4: Ionomycin treatment of primary human PMNs increases primary granule fusion with Gc phagosomes. (A–B) PMNs were infected for 30 min with Gc. PMNs were then treated with 1μM ionomycin and incubated for 15 min, followed by fixation and processing for immunofluorescence microscopy. Extracellular Gc appear red/blue, while intracellular Gc appear blue only. PMNs were stained with antibodies against the primary granule protein neutrophil elastase (NE), which appears green (A). Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of Gc phagosomes positive for neutrophil elastase is reported in B. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Supplemental Figure 5: Protease inhibitor treatment does not affect the LPC-enhanced fusion of primary granules with PMN phagosomes. PMNs were infected with Gc and treated with LPC (B, D) or left untreated (A, C) as in Figure 5. Prior to infection, PMNs were treated with the protease inhibitor cocktail (C, D) as in Figure 10. Immunofluorescence for CD63 and Gc was conducted as in Figure 2. Extracellular Gc appear red/blue, intracellular Gc appear blue only, and CD63 appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins.
Supplemental Figure 6: Gc IgA protease does not affect primary granule-phagosome fusion in PMNs. (A–B) PMNs were infected for 1 h with parental or iga:kan mutant Gc and processed for immunofluorescence as in Figure 2. Extracellular Gc appear red/blue, intracellular Gc appear blue only, and neutrophil elastase (NE) appears green. Arrows indicate phagosomes negative for granule proteins. The percent of Gc and iga:kan phagosomes positive for CD63 (images not shown) or NE are reported in B.
Acknowledgments
We are grateful to Jian Shao from the Electron Microscopy Core at the University of Iowa for processing samples for immuno-TEM. We thank Joshua Eby and Cirle Warren (Dept. of Medicine, University of Virginia) for consenting human subjects at the Virginia Department of Health clinic and collecting human gonorrheal exudate samples, Eric Skaar for providing the S. aureus Δspa mutant, Brett Moreau for preliminary experiments with the S. aureus antibody, Jonathan Handing, Adam Greene, Erin Dale, and Laura Gonyar for experiments testing FA1090 susceptibility to PMN antimicrobial components and members of the Criss lab for consenting human subjects and for assistance with venipuncture. We appreciate advice from Jeff Weiser and Alistair Standish on protocols used to test neutrophil protease activity. We thank Amy Bouton and Laura Gonyar for manuscript suggestions and members of the 2012 American Society for Microbiology Scientific Writing and Publishing Institute, Jim Casanova, and Asya Smirnov for critical reading of the manuscript. The CD63 monoclonal antibody developed by J. Thomas August and James E. K. Hildreth was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute for Child Health and Human Development and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. This work was supported by NIH R00 TW008042 and R01 AI097312 (A.K.C.). M.B.J. was supported in part by the University of Virginia Infectious Diseases Training Grant NIH T32 AI007046.
Footnotes
The authors have no conflicts of interest to declare.
References
- Balthazar JT, Gusa A, Martin LE, Choudhury B, Carlson R, Shafer WM. Lipooligosaccharide Structure is an Important Determinant in the Resistance of Neisseria gonorrhoeae to Antimicrobial Agents of Innate Host Defense. Front Microbiol. 2011;2:30. doi: 10.3389/fmicb.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangalore N, Travis J, Onunka VC, Pohl J, Shafer WM. Identification of the primary antimicrobial domains in human neutrophil cathepsin G. J Biol Chem. 1990;265:13584–13588. [PubMed] [Google Scholar]
- Bjerknes R, Guttormsen HK, Solberg CO, Wetzler LM. Neisserial porins inhibit human neutrophil actin polymerization, degranulation, opsonin receptor expression, and phagocytosis but prime the neutrophils to increase their oxidative burst. Infect Immun. 1995;63:160–167. doi: 10.1128/iai.63.1.160-167.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britigan BE, Klapper D, Svendsen T, Cohen MS. Phagocyte-derived lactate stimulates oxygen consumption by Neisseria gonorrhoeae. An unrecognized aspect of the oxygen metabolism of phagocytosis. J Clin Invest. 1988;81:318–324. doi: 10.1172/JCI113323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon LA, Seifert HS. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science. 2009;325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SG, Shafer WM, Spitznagel JK. Anaerobiosis increases resistance of Neisseria gonorrhoeae to O2-independent antimicrobial proteins from human polymorphonuclear granulocytes. Infect Immun. 1985;47:401–407. doi: 10.1128/iai.47.2.401-407.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Seifert HS. Neisseria gonorrhoeae-mediated inhibition of apoptotic signalling in polymorphonuclear leukocytes. Infect Immun. 2011;79:4447–4458. doi: 10.1128/IAI.01267-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougoule C, Constant P, Etienne G, Daffe M, Maridonneau-Parini I. Lack of fusion of azurophil granules with phagosomes during phagocytosis of Mycobacterium smegmatis by human neutrophils is not actively controlled by the bacterium. Infect Immun. 2002;70:1591–1598. doi: 10.1128/IAI.70.3.1591-1598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Katz BZ, Seifert HS. Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell Microbiol. 2009;11:1074–1087. doi: 10.1111/j.1462-5822.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Seifert HS. Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol. 2008;10:2257–2270. doi: 10.1111/j.1462-5822.2008.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss AK, Seifert HS. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol. 2012;10:178–190. doi: 10.1038/nrmicro2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Densen P, Mandell GL. Gonococcal interactions with polymorphonuclear neutrophils: importance of the phagosome for bactericidal activity. J Clin Invest. 1978;62:1161–1171. doi: 10.1172/JCI109235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Descoteaux A. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J Exp Med. 1997;185:2061–2068. doi: 10.1084/jem.185.12.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol. 2001;154:631–644. doi: 10.1083/jcb.200106049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier H, Celli J. Phagocytic receptors dictate phagosomal escape and intracellular proliferation of Francisella tularensis. Infect Immun. 2011;79:2204–2214. doi: 10.1128/IAI.01382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SR, Sibley DA, Mayo MS, Hook EW, 3rd, Russell MW. Cytokine and antibody responses in women infected with Neisseria gonorrhoeae: effects of concomitant infections. J Infect Dis. 1998;178:742–751. doi: 10.1086/515372. [DOI] [PubMed] [Google Scholar]
- Hong CW, Kim TK, Ham HY, Nam JS, Kim YH, Zheng H, et al. Lysophosphatidylcholine increases neutrophil bactericidal activity by enhancement of azurophil granule-phagosome fusion via glycine.GlyR alpha 2/TRPM2/p38 MAPK signaling. J Immunol. 2010;184:4401–4413. doi: 10.4049/jimmunol.0902814. [DOI] [PubMed] [Google Scholar]
- Johnson MB, Criss AK. Resistance of Neisseria gonorrhoeae to neutrophils. Front Microbiol. 2011;2:77. doi: 10.3389/fmicb.2011.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DS, Jr, Peacock WL, Jr, Deacon WE, Brown L, Pirkle DI. Neisseria Gonorrhoeae. I. Virulence Genetically Linked to Clonal Variation. J Bacteriol. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkcaldy RD, Ballard RC, Dowell D. Gonococcal resistance: are cephalosporins next? Curr Infect Dis Rep. 2011;13:196–204. doi: 10.1007/s11908-011-0169-9. [DOI] [PubMed] [Google Scholar]
- Lee EH, Shafer WM. The farAB-encoded efflux pump mediates resistance of gonococci to long-chained antibacterial fatty acids. Mol Microbiol. 1999;33:839–845. doi: 10.1046/j.1365-2958.1999.01530.x. [DOI] [PubMed] [Google Scholar]
- Lee WL, Harrison RE, Grinstein S. Phagocytosis by neutrophils. Microbes Infect. 2003;5:1299–1306. doi: 10.1016/j.micinf.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Lewis LA, Choudhury B, Balthazar JT, Martin LE, Ram S, Rice PA, et al. Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect Immun. 2009;77:1112–1120. doi: 10.1128/IAI.01280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Ayala P, Larson J, Mulks M, Fukuda M, Carlsson SR, et al. The Neisseria type 2 IgA1 protease cleaves LAMP1 and promotes survival of bacteria within epithelial cells. Mol Microbiol. 1997;24:1083–1094. doi: 10.1046/j.1365-2958.1997.4191776.x. [DOI] [PubMed] [Google Scholar]
- Lorenzen DR, Gunther D, Pandit J, Rudel T, Brandt E, Meyer TF. Neisseria gonorrhoeae porin modifies the oxidative burst of human professional phagocytes. Infect Immun. 2000;68:6215–6222. doi: 10.1128/iai.68.11.6215-6222.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollinedo F, Janssen H, de la Iglesia-Vicente J, Villa-Pulgarin JA, Calafat J. Selective fusion of azurophilic granules with Leishmania-containing phagosomes in human neutrophils. J Biol Chem. 2010;285:34528–34536. doi: 10.1074/jbc.M110.125302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse SA, Bartenstein L. Purine metabolism in Neisseria gonorrhoeae: the requirement for hypoxanthine. Can J Microbiol. 1980;26:13–20. doi: 10.1139/m80-003. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Johnson JL, Ellis BA, Rutschmann S, Beutler B, Catz SD. Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem J. 2007;402:229–239. doi: 10.1042/BJ20060950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndowa F, Lusti-Narasimhan M. The threat of untreatable gonorrhoea: implications and consequences for reproductive and sexual morbidity. Reprod Health Matters. 2012;20:76–82. doi: 10.1016/S0968-8080(12)40653-X. [DOI] [PubMed] [Google Scholar]
- Perera NC, Schilling O, Kittel H, Back W, Kremmer E, Jenne DE. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci U S A. 2012;109:6229–6234. doi: 10.1073/pnas.1200470109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu XD, Harwig SS, Oren AM, Shafer WM, Lehrer RI. Susceptibility of Neisseria gonorrhoeae to protegrins. Infect Immun. 1996;64:1240–1245. doi: 10.1128/iai.64.4.1240-1245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KH, Schneider H, Cross AS, Boslego JW, Hoover DL, Staley TL, et al. Inflammatory cytokines produced in response to experimental human gonorrhea. J Infect Dis. 1995;172:186–191. doi: 10.1093/infdis/172.1.186. [DOI] [PubMed] [Google Scholar]
- Rest RF. Killing of Neisseria gonorrhoeae by human polymorphonuclear neutrophil granule extracts. Infect Immun. 1979;25:574–579. doi: 10.1128/iai.25.2.574-579.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rest RF, Pretzer E. Degradation of gonococcal outer membrane proteins by human neutrophil lysosomal proteases. Infect Immun. 1981;34:62–68. doi: 10.1128/iai.34.1.62-68.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JP, Rest RF. Rapid damage to membranes of Neisseria gonorrhoeae caused by human neutrophil granule extracts. J Gen Microbiol. 1988;134:509–519. doi: 10.1099/00221287-134-2-509. [DOI] [PubMed] [Google Scholar]
- Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, Simons MP, Wu HJ, McEwan AG, Nauseef WM, Apicella MA, Jennings MP. Investigation of oxidative stress defenses of Neisseria gonorrhoeae by using a human polymorphonuclear leukocyte survival assay. Infect Immun. 2005;73:5269–5272. doi: 10.1128/IAI.73.8.5269-5272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer WM, Morse SA. Cleavage of the protein III and major iron-regulated protein of Neisseria gonorrhoeae by lysosomal cathepsin G. J Gen Microbiol. 1987;133:155–162. doi: 10.1099/00221287-133-1-155. [DOI] [PubMed] [Google Scholar]
- Shafer WM, Onunka V, Hitchcock PJ. A spontaneous mutant of Neisseria gonorrhoeae with decreased resistance to neutrophil granule proteins. J Infect Dis. 1986a;153:910–917. doi: 10.1093/infdis/153.5.910. [DOI] [PubMed] [Google Scholar]
- Shafer WM, Onunka VC, Martin LE. Antigonococcal activity of human neutrophil cathepsin G. Infect Immun. 1986b;54:184–188. doi: 10.1128/iai.54.1.184-188.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer WM, Qu X, Waring AJ, Lehrer RI. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci U S A. 1998;95:1829–1833. doi: 10.1073/pnas.95.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silliman CC, Elzi DJ, Ambruso DR, Musters RJ, Hamiel C, Harbeck RJ, et al. Lysophosphatidylcholines prime the NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic calcium. J Leukoc Biol. 2003;73:511–524. doi: 10.1189/jlb.0402179. [DOI] [PubMed] [Google Scholar]
- Simons MP, Nauseef WM, Apicella MA. Interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect Immun. 2005;73:1971–1977. doi: 10.1128/IAI.73.4.1971-1977.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons MP, Nauseef WM, Griffith TS, Apicella MA. Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell Microbiol. 2006;8:1780–1790. doi: 10.1111/j.1462-5822.2006.00748.x. [DOI] [PubMed] [Google Scholar]
- Sorensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- Staali L, Bauer S, Morgelin M, Bjorck L, Tapper H. Streptococcus pyogenes bacteria modulate membrane traffic in human neutrophils and selectively inhibit azurophilic granule fusion with phagosomes. Cell Microbiol. 2006;8:690–703. doi: 10.1111/j.1462-5822.2005.00662.x. [DOI] [PubMed] [Google Scholar]
- Stohl EA, Criss AK, Seifert HS. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol. 2005;58:520–532. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity. 2005;22:539–550. doi: 10.1016/j.immuni.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Unemo M, Shafer WM. Antibiotic resistance in Neisseria gonorrhoeae: origin, evolution, and lessons learned for the future. Ann N Y Acad Sci. 2011;1230:E19–28. doi: 10.1111/j.1749-6632.2011.06215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban CF, Lourido S, Zychlinsky A. How do microbes evade neutrophil killing? Cell Microbiol. 2006;8:1687–1696. doi: 10.1111/j.1462-5822.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- van Ulsen P, Tommassen J. Protein secretion and secreted proteins in pathogenic Neisseriaceae. FEMS Microbiol Rev. 2006;30:292–319. doi: 10.1111/j.1574-6976.2006.00013.x. [DOI] [PubMed] [Google Scholar]
- Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol. 2005;175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- Wiesner PJ, Thompson SE., 3rd Gonococcal diseases. Dis Mon. 1980;26:1–44. doi: 10.1016/s0011-5029(80)80002-2. [DOI] [PubMed] [Google Scholar]
- Workowski KA, Berman S. Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59:1–110. [PubMed] [Google Scholar]
- Wu H, Soler-Garcia AA, Jerse AE. A strain-specific catalase mutation and mutation of the metal-binding transporter gene mntC attenuate Neisseria gonorrhoeae in vivo but not by increasing susceptibility to oxidative killing by phagocytes. Infect Immun. 2009;77:1091–1102. doi: 10.1128/IAI.00825-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Chen CJ, Thomas CE, Anderson JE, Jerse AE, Sparling PF. Vaccines for gonorrhea: can we rise to the challenge? Front Microbiol. 2011;2:124. doi: 10.3389/fmicb.2011.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Titration of anti-Gc polyclonal antibody results in significantly increased trafficking of opsonized Gc to primary granule-positive phagosomes. (A) Gc were left non-opsonized or opsonized with increasing concentrations of a polyclonal rabbit anti-Gc antibody (1:50, 1:100, 1:250, 1:500). Following opsonization, Gc was labeled with DAPI and then incubated for 1 h with an Alexa Fluor 555-coupled goat anti-rabbit antibody to identify the IgG-opsonized Gc. (B) PMNs were infected with non-opsonized or IgG-opsonized (1:50, 1:100, 1:250, 1:500) Gc for 1 h and processed for immunofluorescence microscopy for Gc and neutrophil elastase. Extracellular Gc appear red/blue, while intracellular Gc appear blue only, and neutrophil elastase staining appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of Gc phagosomes positive for neutrophil elastase is reported in C. The percent of Gc phagosomes positive for neutrophil elastase was significantly decreased in PMNs infected with non-opsonized Gc compared to PMNs infected with IgG opsonized Gc (1:50, 1:100, and 1:250) as determined by Student’s two-tailed t test (P < 0.05). There was no statistically significant difference in neutrophil elastase-positive phagosomes between non-opsonized Gc and Gc opsonized with IgG at 1:500 concentration. The percent of Gc phagosomes positive for PMNs infected with IgG-opsonized Gc (1:500) was significantly less than in PMNs infected with IgG-opsonized Gc (1:50, 1:100) as determined by Student’s two tailed t test (P< 0.05).
Supplemental Figure 2: Sytox Green and DAPI effectively discriminate viable and nonviable Gc during PMN infection. (A–B) PMNs were incubated with heat killed Gc for 1 h and processed using Baclight viability dyes (A) or Sytox Green in combination with DAPI (B). (C–E) PMNs were infected with exponentially-growing, viable Gc for 1 h and processed using Baclight viability dyes (C) or Sytox Green in combination with DAPI (D). In both (A) and (C) nonviable bacteria are stained with propidium iodide and appear red. (All bacteria are stained with SYTO9, but only viable bacteria appear green due to reduction of SYTO9 fluorescence in the presence of propidium iodide.) In both (B) and (D) nonviable bacteria are stained with Sytox Green and DAPI (teal) and viable bacteria are stained with DAPI only (blue). Arrowheads indicate nonviable Gc, and arrows indicate viable Gc. The percent of viable Gc discriminated using the two dye combinations is reported in E.
Supplemental Figure 3: In the absence of fixation, an anti-CD63 antibody effectively labels primary granules and zymosan-containing phagolysosomes. PMNs were incubated with serum-opsonized zymosan for 25 min. Cells were left unfixed and incubated with an Alexa Fluor 555-coupled anti-CD63 antibody (A), or fixed with PFA and incubated with a FITC-coupled anti-CD63 antibody (B). (C) Unfixed PMNs that had internalized opsonized zymosan were permeabilized and incubated with the Alexa 555-coupled anti-CD63 antibody. PMNs were then fixed and incubated with the FITC-coupled anti-CD63 antibody. Arrowheads indicate zymosan phagosomes positive for CD63.
Supplemental Figure 4: Ionomycin treatment of primary human PMNs increases primary granule fusion with Gc phagosomes. (A–B) PMNs were infected for 30 min with Gc. PMNs were then treated with 1μM ionomycin and incubated for 15 min, followed by fixation and processing for immunofluorescence microscopy. Extracellular Gc appear red/blue, while intracellular Gc appear blue only. PMNs were stained with antibodies against the primary granule protein neutrophil elastase (NE), which appears green (A). Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins. The percent of Gc phagosomes positive for neutrophil elastase is reported in B. Asterisks indicate P < 0.05 by Student’s two-tailed t test.
Supplemental Figure 5: Protease inhibitor treatment does not affect the LPC-enhanced fusion of primary granules with PMN phagosomes. PMNs were infected with Gc and treated with LPC (B, D) or left untreated (A, C) as in Figure 5. Prior to infection, PMNs were treated with the protease inhibitor cocktail (C, D) as in Figure 10. Immunofluorescence for CD63 and Gc was conducted as in Figure 2. Extracellular Gc appear red/blue, intracellular Gc appear blue only, and CD63 appears green. Arrowheads indicate bacterial phagosomes positive for granule proteins, while arrows indicate phagosomes negative for granule proteins.
Supplemental Figure 6: Gc IgA protease does not affect primary granule-phagosome fusion in PMNs. (A–B) PMNs were infected for 1 h with parental or iga:kan mutant Gc and processed for immunofluorescence as in Figure 2. Extracellular Gc appear red/blue, intracellular Gc appear blue only, and neutrophil elastase (NE) appears green. Arrows indicate phagosomes negative for granule proteins. The percent of Gc and iga:kan phagosomes positive for CD63 (images not shown) or NE are reported in B.