

Abstract
Protection of genome integrity depends on the coordinated activities of DNA replication, DNA repair, chromatin assembly and chromosome segregation mechanisms. DNA lesions are detected by the master checkpoint kinases ATM (Tel1) and ATR (Rad3/Mec1), which phosphorylate multiple substrates, including a C-terminal SQ motif in histone H2A or H2AX. The 6-BRCT domain protein Brc1, which is required for efficient recovery from replication fork arrest and collapse in fission yeast, binds phospho-histone H2A (γH2A)-coated chromatin at stalled and damaged replication forks. We recently found that Brc1 co-localizes with γH2A that appears in pericentromeric heterochromatin during S-phase. Our studies indicate that Brc1 contributes to the maintenance of pericentromeric heterochromatin, which is required for efficient chromosome segregation during mitosis. Here, we review these studies and present additional results that establish the functional requirements for the N-terminal BRCT domains of Brc1 in the replication stress response and resistance to the microtubule destabilizing drug thiabendazole (TBZ). We also identify the nuclear localization signal (NLS) in Brc1, which closely abuts the C-terminal pair of BRCT domains that form the γH2A-binding pocket. This compact arrangement of localization domains may be a shared feature of other γH2A-binding proteins, including Rtt107, PTIP and Mdc1.
Keywords: DNA damage response, replication stress, heterochromatin, centromere, BRCT domain, mitosis, nuclear localization signal
Introduction
The transmission of accurate copies of the genome during cell proliferation relies on the integrated functions of multiple genome protection systems. The minimal requirements for successful genome transmission are accurate DNA replication and chromosome segregation mechanisms. Overseeing these events are genome surveillance mechanisms that detect DNA replication errors, DNA damage and defects in chromosome transmission. These checkpoint systems ensure the completion of DNA replication, the repair of damaged DNA and proper distribution of duplicated chromosomes during mitosis.1-3
Of the many proteins tasked with genome surveillance and protection, perhaps none are more important than the phosphatidylinositol 3-kinase-related kinases (PIKKs) ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related).4,5 These master checkpoint kinases rapidly mobilize to double-strand breaks (DSBs) and single-strand DNA regions, respectively, where they phosphorylate multiple proteins, such as the effector kinases Chk1 and Chk2 (Cds1) and checkpoint mediator proteins Mdc1, Mrc1 and Crb2.6-12 Another notable substrate is histone H2A in yeasts and the variant histone H2AX in mammals.13,14 The phosphorylated SQ motif at the C terminus of histone H2A(X) provides a chromatin-specific docking site for genome protection factors. These proteins bind phospho-histone H2A(X), otherwise known as γH2A(X), through paired C-terminal BRCT domains, which are named after the C-terminal domain BRCA1 breast cancer susceptibility protein. Some of the γH2A(X)-binding proteins include Mdc1 in mammals and Crb2 in the fission yeast Schizosaccharomyces pombe, which mediate checkpoint responses to DSBs, as well as Brc1 in fission yeast.15-18 Brc1 is a 6-BRCT domain that has important but as yet poorly understood roles in promoting recovery from stalled or damaged replication forks.19-22
Involvement of Brc1 in Maintenance of Pericentromeric Heterochromatin
Brc1 localizes at chromosome regions that experience replication stress by binding γH2A-marked chromatin. γH2A decorates pericentromeric heterochromatin during an unperturbed DNA synthesis (S) phase in fission yeast, suggesting that replication forks stall in these regions.23 In support of this possibility, a recent study detected X-shaped DNA molecules in pericentromeric heterochromatin during S-phase.24 These DNA molecules were absent from cells lacking Clr4/Suv39, which is a subunit of the Rik1 complex that is required for dimethylation of histone H3 at lysine 9 (H3k9me2). This histone modification, which is a defining feature of pericentromeric heterochromatin, is formed via an RNAi-dependent mechanism.25-27 These data suggested a model in which collisions between the replication and transcription machineries lead to RNA interference (RNAi)-mediated release of RNA polymerase II, thereby allowing resumption of DNA replication by Rik1-associated replisomes, which is required for maintenance of pericentromeric heterochromatin.24
This model suggests that proteins involved in the responses to stalled replication forks may play roles in ensuring the efficient transmission of pericentromeric heterochromatin in fission yeast. In support of this model, we found that Brc1 is enriched in pericentromeric heterochromatin during S-phase. This localization of Brc1 largely depends on the ability to form γH2A. Specifically, mutations of the C-terminal SQ motif in the two genes encoding histone H2A (hta1-AQ and hta2-AQ mutations, also known as the htaAQ genotype) reduce the enrichment of Brc1 in pericentromeric heterochromatin.23 We recently discovered that the appearance of γH2A and Brc1 in pericentromeric heterochromatin during S-phase was substantially diminished in cells lacking Clr4.23,28 We further found that H3k9me2 was reduced in pericentromeric heterochromatin in Brc1-defective cells. Gene silencing in pericentromeric heterochromatin was also partially impaired in Brc1-null cells.28 As pericentromeric heterochromatin is required for effective cohesion of chromosome arms in pericentromeric regions and for fully efficient centromere function, we explored whether brc1Δ cells were sensitive to the antifungal drug thiabendazole (TBZ), which destabilizes microtubules. We found that brc1Δ cells are sensitive to TBZ and display increased rates of lagging chromosomes during mitosis both in the absence or presence of TBZ.19,28 Collectively, these data support a model in which Brc1-mediated stabilization of stalled replication forks in pericentromeric heterochromatin contributes to efficient maintenance of heterochromatin during DNA replication (Fig. 1).
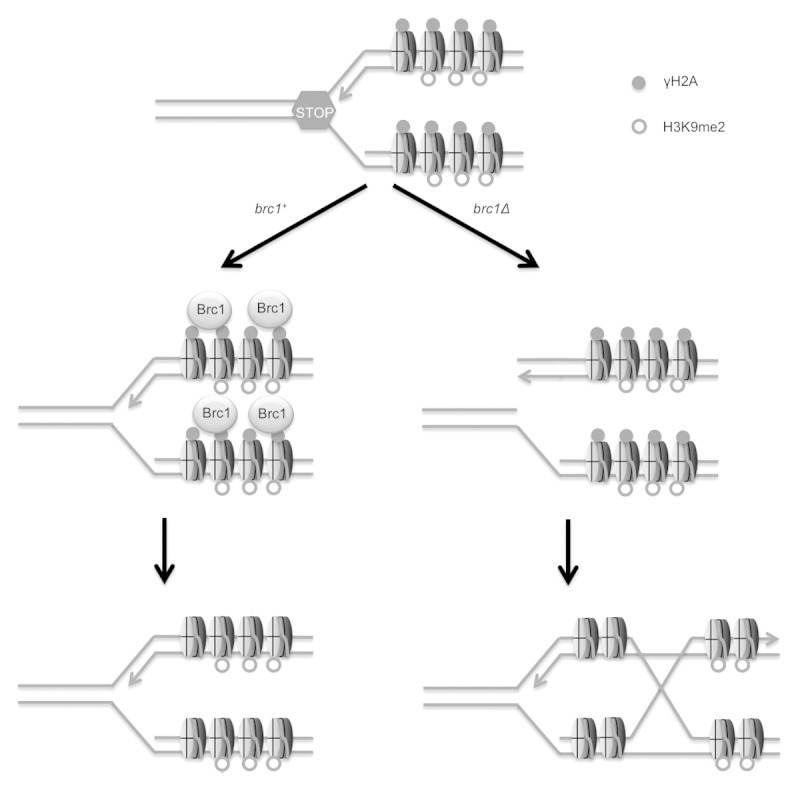
Figure 1. Model indicating that replication fork stalling in pericentromeric heterochromatin can lead to replication fork breakdown or disassociation of the Rik1-associated replisome (not shown) in brc1Δ cells, leading to defects in propagation of H3K9me2-marked chromatin.28 Note that homology directed repair of the collapsed replication fork results in formation of a Holliday junction.
Although Brc1 localization in pericentromeric heterochromatin is diminished in cells lacking γH2A, these htaAQ cells differed from brc1Δ cells in that they were insensitive to TBZ.28 This relationship is consistent with studies showing that brc1Δ mutants are more sensitive to replication stress conditions in comparison to htaAQ cells.18 Thus, while Brc1 directly binds γH2A, and the appearance of Brc1 nuclear foci in response to replication stress or DNA damage requires this physical interaction with γH2A, genetic studies reveal Brc1 retains significant activities in the absence of γH2A. If the TBZ sensitivity of brc1Δ cells is linked to a function of Brc1 at pericentromeric heterochromatin, the absence of TBZ sensitivity in htaAQ cells suggests that the remaining γH2A-independent localization of Brc1 at pericentromeric heterochromatin in htaAQ cells is sufficient to maintain gene silencing and promote proper centromere function. However, it is possible that other activities involved in insuring proper chromosome segregation may become more critical in the absence of Brc1 binding to γH2A. The most obvious candidate for such an activity is the spindle assembly checkpoint, which is partly dependent on the phosphorylation of the serine-121 residue in the C terminus of histone H2A. Phosphorylation of this residue by Bub1 kinase plays a significant role in the recruitment of shugoshin.29 We tested this model by mutating both the Rad3 and Bub1 phosphorylation sites in the C termini of both histone H2A genes. This hta1-S121,129A hta2-S121,128A strain displayed enhanced TBZ sensitivity compared with the hta1-S121A hta2-S121A strain lacking only the Bub1 phosphorylation sites, suggesting that defects in recruiting Brc1 to γH2A-marked pericentromeric heterochromatin place a burden on the spindle assembly checkpoint.28
Functional Analyses of the N-terminal BRCT Domains of Brc1 in Resistance to Replication Stress and TBZ
The 878-amino acid sequence of Brc1 indicates a protein consisting of 4 N-terminal BRCT domains connected through a linker domain to the two paired C-terminal BRCT domains that bind γH2A (Fig. 2A). This arrangement of domains is conserved in orthologous proteins throughout ascomycete fungi, including Rtt107 protein in Saccharomyces cerevisiae, which is also involved in the cellular responses to replication stress.30-32 The presence of 6-BRCT domains connected by linkers is also found in the PTIP/Swift family of proteins in metazoans, which also appear to function in DNA damage responses.33-36 As seen for Brc1, both Rtt107 and PTIP bind γH2A(X) through their C-terminal BRCT domains.18,37,38 To address the functional significance of the N-terminal BRCT domains of Brc1 for survival of replication stress and exposure to TBZ, we performed mutational analyses of conserved residues in BRCT domains 2, 3 and 4. Of the five mutants tested, brc1-TH148-9SG, brc1-R268K and brc1-HYP307-9GFG caused sensitivity to the replication stress agents camptothecin (CPT) and hydroxyurea (HU), which primarily act by causing replication fork collapse and arrest, respectively (Fig. 2B). Two other alleles, brc1-G136A and brc1-W298G-P301G, displayed no phenotype despite having mutations of conserved residues All mutant proteins properly localized in the nucleus when expressed as green fluorescent protein (GFP)-tagged constructs under the control of the nmt42 promoter, and in contrast to mutations such as brc1-T672A that ablate binding to γH2A,18 all formed both spontaneous and HU-induced foci (Figs. 2C and D and 3B). From these results, we conclude that brc1-TH148-9SG, brc1-R268K and brc1-HYP307-9GFG mutations impair Brc1 function without grossly disturbing its stability, localization or ability to bind γH2A.

Figure 2. Mutational analysis of the N-terminal BRCT domains of Brc1. (A) Diagram of Brc1 showing sites of mutations in BRCT domains. (B) HU and CPT-sensitive phenotypes of a subset of N-terminal BRCT domain mutants of brc1. Ten-fold serial dilutions of cells were exposed to the indicated DNA-damaging agent and incubated at 30°C for 3 d. (C) Photomicrographs of cells expressing wild type and mutant GFP-Brc1. In all strains, GFP-Brc1 localized in the nucleus and formed spontaneous foci. (D) Cells expressing wild type and mutant GFP-Brc1 all formed spontaneous (−HU) and HU-induced (+HU) GFP-Brc1 nuclear foci. Cells were grown for 17 h at 30°C in minimal medium before being split. Treatment was with 12 mM HU for 4 h. Data are derived from three independent experiments. (E) Combining mutations in the N-terminal BRCT domains in Brc1 with the T672A mutation that disrupts binding to γH2A does not increase sensitivity to CPT. (F) The brc1-TH148-9SG, -R268K and -HYP307-9GFG mutations cause sensitivity to TBZ. Ten-fold serial dilutions were incubated at 30°C for 2 d.

Figure 3. The nuclear localization signal (NLS) of Brc1. (A) Predicted NLS sequences in γH2A-binding proteins. The highest scoring NLS sequence calculated using cNLS Mapper is shown for each protein. The brc1-nls1 mutant allele is also indicated. (B) Live-cell microscopy of wild type, truncated and mutant GFP-Brc1. GFP-Brc1 was expressed from pREP42-GFP-brc1 plasmids. Cells were grown in EMM (Edinburgh minimal media) for 18–20 h at 30°C. Arrows indicate Brc1 foci. Wild type (WT) Brc1 and the N-terminal truncation lacking the N-terminal BRCT domains (brc1-ΔN398) showed normal Brc1 foci formation, whereas the brc1-nls1 mutant fails to localize in the nucleus. The brc1-T672A mutant that cannot bind γH2A localizes in the nucleus but fails to form foci. Wild GFP-Brc1 expressed in hta-AQ also fails to form nuclear foci.
Comparison to brc1Δ cells revealed that the brc1-TH148-9SG, brc1-R268K and brc1-HYP307-9GFG mutations did not fully ablate Brc1 activity in promoting survival of CPT exposure (Fig. 2B). The brc1-T672A mutation that impairs binding to γH2A has a similar partial effect.18,39 We found that these N-terminal BRCT domain mutations did not enhance CPT sensitivity when combined with brc1-T672A (Fig. 2E). The absence of synergy among these mutations that partially impair the function of Brc1 was unexpected and suggests that Brc1 may have at least two distinct functions in mediating CPT resistance. The mutations might fully ablate one function while leaving the other intact. The data also suggest that the N-terminal BRCT domains (specifically domains 2, 3 and 4) and C-terminal BRCT domains of Brc1 provide functional effect and localization activities, respectively.
The mutations in the N-terminal BRCT domains that caused sensitivity to genotoxins also increased sensitivity to the microtubule-destabilizing drug TBZ (Fig. 2F). These data suggest that the replication stress response functions of Brc1 that are mediated through the N-terminal BRCT domains are also required for its functions related to replication of pericentromeric heterochromatin.28
Compact Arrangement of Nuclear Localization Signal (NLS) and the C-terminal BRCT Domains in Brc1
As noted above, Brc1 is typical of several families of DNA damage response proteins that bind γH2A(X)-marked chromatin. In addition to Rtt107 and PTIP, these proteins include Mdc1 in mammals, Rad9 in S. cerevisiae and Crb2 in fission yeast. These proteins must be transported into the nucleus before they can bind γH2A(X)-marked chromatin. As the mechanism of nuclear localization for these families of proteins has not been reported, we sought to identify whether Brc1 has a functional nuclear localization signal (NLS). The most common NLSs are short stretches of basic amino acid-rich sequences that interact with the receptor importin β, either directly or through the adaptor importin α.40,41 The computer program cNLS Mapper predicted an NLS in sequences encompassing the KKRR motif starting at residue 635 in Brc1 (Fig. 3A).42 This sequence is located approximately 30 residues before the BRCT5 domain. We mutated the KKRR motif to alanine residues in the allele brc1-nls1. This mutant protein was expressed as a GFP-tagged construct under the control of the nmt42 promoter in a plasmid. In contrast to the wild type construct that was nuclear localized, the brc1-nls1 mutant protein was clearly excluded from the nucleus (Fig. 3B). These data strongly suggested that the KKRR motif starting at residue 635 is part of a functional NLS. To further test this possibility, we expressed a brc1-ΔN398 construct that contains the linker domain with the NLS and the BRCT5,6 region. In support of our model, this construct was strongly localized in the nucleus (Fig. 3B). Moreover, the brc1-ΔN398 construct formed spontaneous nuclear foci, as also seen with full-length wild type Brc1 (Fig. 3B). As seen previously, GFP-Brc1 harboring the brc1-T672A mutation localized in the nucleus but failed to form spontaneous foci, as was also observed by expressing wild type GFP-Brc1 in the hta-AQ background (Fig. 3B).18
From these data we conclude that the NLS abuts the C-terminal BRCT domains in Brc1. To investigate whether this might be a general feature of DNA damage response proteins that bind γH2A(X) through their C-terminal BRCT domains, we used cNLS mapper to predict NLSs in budding yeast Rtt107 and the human proteins Mdc1 and PTIP. In each case the strongest scoring NLS is located just upstream of the C-terminal BRCT domains (Fig. 3A). Mutational studies will be necessary to confirm if indeed these sequences are functional NLSs, but the observations raise the question of whether the targeting sequences of these proteins are closely linked to allow efficient import into the nucleus and localization at chromatin surrounding DNA lesions.
Outlook
Our recent studies support a model in which the replication stress-response protein Brc1 helps to ensure the efficient transmission of heterochromatin markers in the chromosomal regions flanking centromeres in fission yeast (Fig. 1).28 These findings are consistent with evidence that that RNAi-mediated release of RNA polymerase II is required to replicate pericentromeric heterochromatin with replisome protein complexes that maintain an interaction with the Rik1 holocomplex.24,43 In the absence of Brc1 function, it appears that partial defects in maintenance of pericentromeric heterochromatin lead to chromosome segregation defects in cells grown in the presence of TBZ.28 These findings are also consistent with other studies suggesting a requirement for replication stress-response proteins in maintaining gene silencing in pericentromeric heterochromatin.44,45 Indeed, the Smc5/6 genome protection complex localizes at pericentromeric heterochromatin during S-phase in a Clr4-dependent manner, matching the behavior of Brc1.28,45,46 These similarities strengthen the connections between Brc1 and the Smc5/6 holocomplex, of which Brc1 was initially discovered as a high-copy suppressor of the smc6-74 hypomorphic allele.19,20
Brc1 localization in pericentromeric heterochromatin largely depends on the interaction of its C-terminal BRCT domains with γH2A-marked chromatin, although, as we have discovered, the requirement for this interaction for TBZ resistance only becomes obvious when the phosphorylation of histone H2A by Bub1 kinase is also ablated.28 The new findings reported in this study indicate that the structural integrity of the N-terminal BRCT domains is also important for its functions in replication stress responses and resistance to TBZ (Fig. 2). It will be important to determine whether these mutations impair gene silencing in pericentromeric heterochromatin as observed in brc1Δ mutants.28 We speculate that the N-terminal BRCT domains mediate protein scaffolding interactions that are necessary for Brc1 function once it localizes at stalled or damaged replication forks. The nature of these interactions remains to be discovered.
We also report that the nuclear localization of Brc1 depends on a typical NLS sequence of basic amino acids that abut the N-terminal side of the BRCT5 domain. The fact that the linker region between BRCT4 and BRCT5 contains the NLS is not especially remarkable; however, it is interesting that this close arrangement of targeting sequences appears to be conserved in other Brc1-like proteins that contain 6 BRCT domains (e.g., Rtt107 and PTIP), and likely Mdc1 as well. It may be worthwhile to explore through domain shuffling whether this arrangement of targeting sequences is necessary for the optimum functions of these DNA damage response proteins.
A key question that arises from our studies is whether the functions of Brc1 in chromosome segregation and resistance to TBZ are explained by its role in survival of replication stress. An affirmative answer is suggested by the strong correlation between CPT, HU and TBZ sensitivities in the new N-terminal BRCT domain mutants (Fig. 2). However, this question cannot be definitively answered without a better understanding of the precise role of Brc1 in recovery from replication fork stalling and collapse. Earlier studies suggested that the sensitivity to DNA damaging agents in brc1Δ cells was likely caused by a DNA repair defect, which was consistent with the genetic interactions involving Brc1 and the Smc5/6 holocomplex.19-21 However, a recent study indicated that the requirement for Brc1 more likely involves a role in the resumption of DNA replication at stalled or damaged replication forks.22 In the case of collapsed replication forks, resumption of DNA replication absolutely depends on homology directed repair.47 The one-ended DSB formed by the collapsed replication fork invades the sister chromatid to re-establish the replication fork. Evidence in favor of this mode of repair comes from many studies demonstrating that homologous recombination proteins are essential for survival of CPT treatment. CPT is a topoisomerase I poison that causes replication forks to collapse when they encounter the CPT-TopI complex.48,49 The protein Ctp1 (Sae2/CtIP), which interacts with the Mre11-Rad50-Nbs1 to initiate the 5′-to-3′ resection of DSBs, is one of many HR proteins required for repair of CPT-induced DNA damage in fission yeast.50-56 Most telling is the requirement for the Mus81-Eme1 Holliday junction resolvase complex in the survival of CPT and other toxins that collapse replication forks, as the reestablishment of a broken replication fork necessitates the formation of Holliday junction that must then be resolved by a Holliday junction resolvase (Fig. 1).57-60 Both Mus81-Eme1 complex and Brc1 have important roles in survival of CPT treatment but are insensitive to ionizing radiation, which creates two-end DSBs that are repaired by homologous recombination repair without forming Holliday junctions. These observations suggest that Brc1 may be involved in the resolution of DNA junctions formed during repair of broken replication forks, or it may be involved in preventing the collapse of replication forks in the first place. For example, Brc1 might stabilize replication forks that stall because of positive supercoiling that occurs ahead of the replication fork when it encounters a CPT-TopI complex.49 Unraveling the role of Brc1 at stalled replication forks will be necessary to understand how it functions genome-wide and at pericentromeric heterochromatin.
Acknowledgments
This work was supported by NIH grants GM59447, CA77325 and CA117638 (awarded to P.R.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24900
References
- 1.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 3.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci USA. 2001;98:11289–94. doi: 10.1073/pnas.191557598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka K, Boddy MN, Chen XB, McGowan CH, Russell P. Threonine-11, phosphorylated by Rad3 and atm in vitro, is required for activation of fission yeast checkpoint kinase Cds1. Mol Cell Biol. 2001;21:3398–404. doi: 10.1128/MCB.21.10.3398-3404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–94. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melchionna R, Chen XB, Blasina A, McGowan CH. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol. 2000;2:762–5. doi: 10.1038/35036406. [DOI] [PubMed] [Google Scholar]
- 11.Zhao H, Tanaka K, Nogochi E, Nogochi C, Russell P. Replication checkpoint protein Mrc1 is regulated by Rad3 and Tel1 in fission yeast. Mol Cell Biol. 2003;23:8395–403. doi: 10.1128/MCB.23.22.8395-8403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qu M, Yang B, Tao L, Yates JR, 3rd, Russell P, Dong MQ, et al. Phosphorylation-dependent interactions between Crb2 and Chk1 are essential for DNA damage checkpoint. PLoS Genet. 2012;8:e1002817. doi: 10.1371/journal.pgen.1002817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM. H2AX: functional roles and potential applications. Chromosoma. 2009;118:683–92. doi: 10.1007/s00412-009-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura TM, Du LL, Redon C, Russell P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol. 2004;24:6215–30. doi: 10.1128/MCB.24.14.6215-6230.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 16.Du LL, Nakamura TM, Russell P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006;20:1583–96. doi: 10.1101/gad.1422606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilkenny ML, Doré AS, Roe SM, Nestoras K, Ho JC, Watts FZ, et al. Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes Dev. 2008;22:2034–47. doi: 10.1101/gad.472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 2010;29:1136–48. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O’Connell MJ. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell. 1999;10:2905–18. doi: 10.1091/mbc.10.9.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KM, Nizza S, Hayes T, Bass KL, Irmisch A, Murray JM, et al. Brc1-mediated rescue of Smc5/6 deficiency: requirement for multiple nucleases and a novel Rad18 function. Genetics. 2007;175:1585–95. doi: 10.1534/genetics.106.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheedy DM, Dimitrova D, Rankin JK, Bass KL, Lee KM, Tapia-Alveal C, et al. Brc1-mediated DNA repair and damage tolerance. Genetics. 2005;171:457–68. doi: 10.1534/genetics.105.044966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bass KL, Murray JM, O’Connell MJ. Brc1-dependent recovery from replication stress. J Cell Sci. 2012;125:2753–64. doi: 10.1242/jcs.103119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rozenzhak S, Mejía-Ramírez E, Williams JS, Schaffer L, Hammond JA, Head SR, et al. Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 2010;6:e1001032. doi: 10.1371/journal.pgen.1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, et al. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature. 2011;479:135–8. doi: 10.1038/nature10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SI. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat Genet. 2005;37:809–19. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- 26.Almeida R, Allshire RC. RNA silencing and genome regulation. Trends Cell Biol. 2005;15:251–8. doi: 10.1016/j.tcb.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Pidoux AL, Allshire RC. Kinetochore and heterochromatin domains of the fission yeast centromere. Chromosome Res. 2004;12:521–34. doi: 10.1023/B:CHRO.0000036586.81775.8b. [DOI] [PubMed] [Google Scholar]
- 28.Lee SY, Rozenzhak S, Russell P. γH2A-binding protein Brc1 affects centromere function in fission yeast. Mol Cell Biol. 2013;33:1410–6. doi: 10.1128/MCB.01654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, Watanabe Y. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science. 2010;327:172–7. doi: 10.1126/science.1180189. [DOI] [PubMed] [Google Scholar]
- 30.Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell. 2010;39:300–6. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 31.Rouse J. Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J. 2004;23:1188–97. doi: 10.1038/sj.emboj.7600129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberts TM, Kobor MS, Bastin-Shanower SA, Ii M, Horte SA, Gin JW, et al. Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol Biol Cell. 2006;17:539–48. doi: 10.1091/mbc.E05-08-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Göhler T, Munoz IM, Rouse J, Blow JJ. PTIP/Swift is required for efficient PCNA ubiquitination in response to DNA damage. DNA Repair (Amst) 2008;7:775–87. doi: 10.1016/j.dnarep.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Cho EA, Prindle MJ, Dressler GR. BRCT domain-containing protein PTIP is essential for progression through mitosis. Mol Cell Biol. 2003;23:1666–73. doi: 10.1128/MCB.23.5.1666-1673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329:917–23. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu J, Prindle MJ, Dressler GR, Yu X. PTIP regulates 53BP1 and SMC1 at the DNA damage sites. J Biol Chem. 2009;284:18078–84. doi: 10.1074/jbc.M109.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Liu K, Li F, Wang J, Huang H, Wu J, et al. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem. 2012;287:9137–46. doi: 10.1074/jbc.M111.311860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan W, Shao Z, Li F, Niu L, Shi Y, Teng M, et al. Structural basis of γH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett. 2011;585:3874–9. doi: 10.1016/j.febslet.2011.10.045. [DOI] [PubMed] [Google Scholar]
- 39.Du LL, Nakamura TM, Moser BA, Russell P. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol Cell Biol. 2003;23:6150–8. doi: 10.1128/MCB.23.17.6150-6158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bednenko J, Cingolani G, Gerace L. Nucleocytoplasmic transport: navigating the channel. Traffic. 2003;4:127–35. doi: 10.1034/j.1600-0854.2003.00109.x. [DOI] [PubMed] [Google Scholar]
- 41.Nakielny S, Dreyfuss G. Transport of proteins and RNAs in and out of the nucleus. Cell. 1999;99:677–90. doi: 10.1016/S0092-8674(00)81666-9. [DOI] [PubMed] [Google Scholar]
- 42.Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci USA. 2009;106:10171–6. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li F, Martienssen R, Cande WZ. Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature. 2011;475:244–8. doi: 10.1038/nature10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. The Cul4-Ddb1(Cdt)² ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell. 2011;144:41–54. doi: 10.1016/j.cell.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN. Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J. 2008;27:3011–23. doi: 10.1038/emboj.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pebernard S, Wohlschlegel J, McDonald WH, Yates JR, 3rd, Boddy MN. The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol Cell Biol. 2006;26:1617–30. doi: 10.1128/MCB.26.5.1617-1630.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGlynn P, Lloyd RG. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol. 2002;3:859–70. doi: 10.1038/nrm951. [DOI] [PubMed] [Google Scholar]
- 48.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 49.Koster DA, Palle K, Bot ES, Bjornsti MA, Dekker NH. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature. 2007;448:213–7. doi: 10.1038/nature05938. [DOI] [PubMed] [Google Scholar]
- 50.Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–46. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dodson GE, Limbo O, Nieto D, Russell P. Phosphorylation-regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double-strand breaks. Cell Cycle. 2010;9:1516–22. doi: 10.4161/cc.9.8.11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7:e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li J, et al. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139:100–11. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–95. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nicolette ML, Lee K, Guo Z, Rani M, Chow JM, Lee SE, et al. Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat Struct Mol Biol. 2010;17:1478–85. doi: 10.1038/nsmb.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR, 3rd, Russell P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001;107:537–48. doi: 10.1016/S0092-8674(01)00536-0. [DOI] [PubMed] [Google Scholar]
- 58.Boddy MN, Lopez-Girona A, Shanahan P, Interthal H, Heyer WD, Russell P. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol. 2000;20:8758–66. doi: 10.1128/MCB.20.23.8758-8766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaillard PH, Noguchi E, Shanahan P, Russell P. The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol Cell. 2003;12:747–59. doi: 10.1016/S1097-2765(03)00342-3. [DOI] [PubMed] [Google Scholar]
- 60.Doe CL, Ahn JS, Dixon J, Whitby MC. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem. 2002;277:32753–9. doi: 10.1074/jbc.M202120200. [DOI] [PubMed] [Google Scholar]