

Abstract
p53 mutations and downregulation of promyelocytic leukemia (PML) are common genetic alterations in human cancers. In healthy cells these two key tumor suppressors exist in a positive regulatory loop, promoting cell death and cellular senescence. However, the influence of their interplay on tumorigenesis has not been explored directly in vivo. The contribution of PML to mutant p53 driven cancer was evaluated in a mouse model harboring a p53 mutation (p53wild-type/R172H) that recapitulates a frequent p53 mutation (p53R175H) in human sporadic and Li-Fraumeni cancers. These mice with PML displayed perturbation of the hematopoietic compartment, manifested either as lymphoma or extramedullary hematopoiesis (EMH). EMH was associated with peripheral blood leucocytosis and macrocytic anemia, suggestive of myeloproliferative- myelodysplastic overlap. In contrast, a complete loss of PML from these mice resulted in a marked alteration in tumor profile. While the incidence of lymphomas was unaltered, EMH was not detected and the majority of mice succumbed to sarcomas. Further, males lacking PML exhibited a high incidence of soft tissue sarcomas and reduced survival, while females largely developed osteosarcomas, without impact on survival. Together, these findings demonstrate that PML is an important tumor suppressor dictating disease development in a pertinent mouse model of human cancer.
Key Points: (1) A mutant p53 allele disrupts hematopoiesis in mice, by promoting lymphomas and myeloproliferative / myelodysplastic overlap. (2) Coincidental p53 allele mutation and PML loss shifts the tumor profile toward sarcoma formation, which is paralleled in human leiomyosarcomas (indicated by immunohistochemistry; IHC).
Keywords: mutant p53, PML, p19ARF, sarcoma, lymphoma, EMH, myeloproliferative overlap, myelodysplastic overlap
Introduction
The tumor suppressor p53 is the most frequently mutated protein in cancer. p53 gene mutations may not only disable the normal, tumor-suppressive functions of p53, but also confer novel capabilities that promote tumorigenesis. Newly acquired properties promote tumor cell invasion of adjacent tissues, migration from the primary tumor bed, seeding of metastases and drug resistance.1 These “gain of function” (GOF) properties are strictly imparted by mutant p53 and do not result from p53 loss (refs. 2 and 3 and reviewed in ref. 4). Mutant p53 stabilization is essential for its GOF;5 however stabilization of mutated p53 protein is not an automatic consequence of p53 gene mutation. The fundamental observation that mutant p53 does not accumulate in normal, healthy tissues of mice bearing germline p53 mutations, but can be detected in tumors, implies that like wt p53, mutant p53 is inherently labile.2,3,5 Mutant p53 accumulation in tumors must therefore result from a breakdown of the mechanisms that normally act to keep levels low. In vitro studies6,7 and mouse models have identified that the E3 ligase Mdm2 contributes to reduced mutant p53 levels as does the p16INK4a locus;5 however, a complete delineation of the determinants of mutant p53 lability is yet to be achieved. Molecules that prevent mutant p53 stabilization are presumed tumor suppressors and represent potential candidates for cancer therapy.
Wt p53 becomes stabilized in response to stress, largely through extrication from Mdm2. Multiple pathways act in concert to execute stress-induced modifications of p53, facilitated by the promyelocytic leukemia (PML) protein.8–10 PML constitutes a family of at least nine isoforms in humans.11–14 Collectively, these isoforms are considered to be tumor suppressive, as first surmised from PML dysfunction in acute promyelocytic leukemia [due to t(15:17) translocation and PML fusion with the retinoic acid receptor α]11 and further elaborated with the identification of mutant PML isoforms in APL that inhibit proper function of PML and p53.15 PML tumor-suppressive capacity was been corroborated in a mouse model for leukemia.12 PML has been identified to impede cell proliferation through both p53-dependent and -independent mechanisms (ref. 13 and review ref. 14). PML protein downregulation or complete loss from human solid human tumors was subsequently observed in immunohistochemical (IHC) studies.16 In mice, a loss of one or two alleles of PML was sufficient to exacerbate Ras- or loss of PTEN-driven specific cancers,17,18 and PML elimination increased susceptibility to chemically induced carcinogenesis.19 Together, these observations support a role for PML as a tumor suppressor that is frequently targeted during malignancy onset. However, the contribution of PML to the suppression of tumor onset in a mutant p53 context has not been established and is the subject of this study.
We identified that PML isoform IV interacts through its C terminus with mutant p53,20 as it does with wt p53 (PML3);21 however, in contrast to the stress provocation essential for its binding to wt p53 in normal cells, in cancer cells mutant p53 interaction with PML appears constitutive.20 The contribution of PML to mutant p53 accumulation was therefore rational to interrogate. We chose to adopt the mouse model of the human germline p53R175H mutant [p53R172H mutant knock-in (KI)]3 to perform a novel, in vivo investigation of the consequence of PML loss, for mutant p53 accumulation and tumor development and metastasis. Here we demonstrate that in a heterozygous wt and mutant p53 context, the presence of PML prolonged survival, although most of these mice eventually succumbed to the consequences of disrupted hematopoiesis. When p53 mutation was compounded by the absence of PML, survival was reduced, and tumor manifestation dominated in the connective tissue, with a gender-dictated tumor spectrum.
Results
PML loss reduced survival in p53 +/R172H male mice
Survival of p53+/R172H mice modeling the human Li-Fraumeni syndrome was assessed as an indication of tumor aggression (dictated by the rate of onset and progression). Survival of p53+/R172HPML+/+ mice was measured for the combined population of both males and females to be around 500 d (Fig. 1A), which is similar to previous reports2,3 and to p53+/R172HPML+/− mice. However, a significant reduction in survival (by ~50 d) was evident with the loss of two PML alleles. Strikingly, separate analysis of male and female survival demonstrated that a complete loss of PML exerted a clear gender influence. Median survival of male mice without PML (p53+/R172HPML−/−) was diminished by over 100 d (to 414 d; Fig. 1B) compared with their male counterparts with PML. Survival in p53+/R172H females in contrast, was little affected by PML loss, and although exhibiting slightly reduced median survival to 463–488 d (Fig. S1), this was not significantly different from the male counterparts with PML. These data support haplosufficiency of PML function in a gender-independent manner, as loss of a single PML allele did not reduce the mean survival of either male or female p53+/R172H mice. In contrast, a gender-discriminating response to PML loss was demonstrated for the first time by these studies, where male survival (but not female) was significantly reduced, in these heterozygous mutant p53 mice.
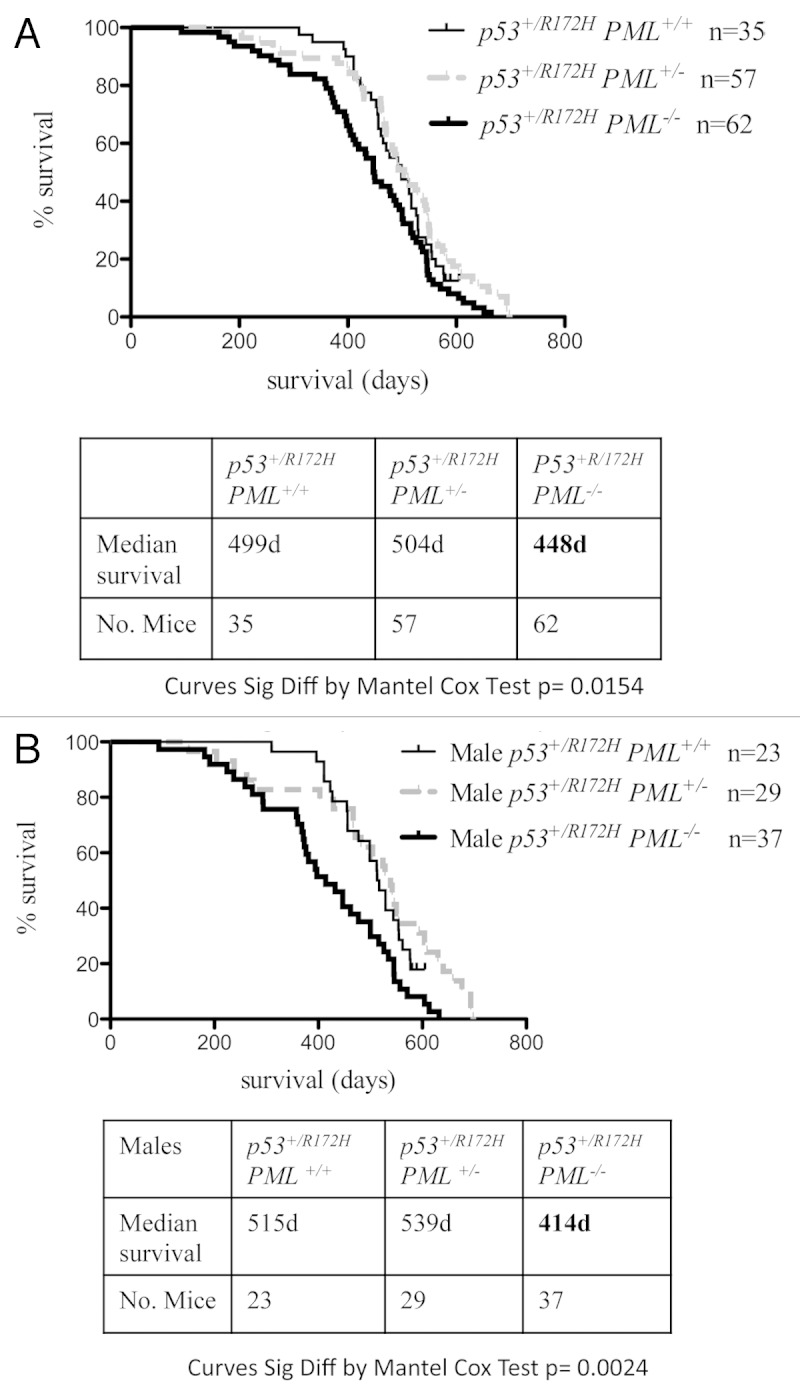
Figure 1. Kaplan-Meir survival curves for p53+/R172H mice. PML loss from p53+/R172H mice was associated with significantly lower overall survival (A); male mice survival was most profoundly reduced (B).
PML loss did not reduce p53R172H/R172H male mouse survival
In contrast to the impact of PML loss on the lifespan of male p53+/R172H mice, mean survival in p53R172H/R172H mice was ~150 d regardless of PML status (Fig. S2), which is comparable to published findings for p53R172H/R172H mice2,3,22). Together these findings suggest that PML is incapable of limiting tumor development in an exclusively mutant p53 context.
The influence of gender on survival of p53R172H/R172H mice could not be evaluated due to insufficient female births, and this was not affected by PML status (Table S1). Poor survival of female mice lacking p53 has been attributed to female-specific exencephaly23,24 (with the X-chromosome determining neural tube defects),25 and a similar phenotype appears in mutant p53 mice.2,22 This is in contrast with the near equivalent Mendelian ratios of female and male progeny of p53wt/wt and mutant p53+/R172H mice and is not influenced by PML absence (Table S1).
PML loss altered the tumor spectrum in p53+/R172H mice
Our study for the first time demonstrates that PML has a significant impact on tumor manifestation in p53+/R172H mice. A dose-dependent loss of PML (one, then two alleles) led to a reduction in the incidence of lymphomas as a percentage of the total numbers of tumors (in PML+/+ 52%; PML+/− 43% and PML−/− 36%) and an increase in sarcomas (from 43% to 52% and 59%, respectively, Table 1A), with no significant impact on carcinoma prevalence. Two tumor types were identified in some mice (Fig. 2A); however, the proportion of mice with multiple tumors did not change substantially with genotype (Table 1A). It should be added, however, that extramedullary hematopoesis (EMH; indicating altered hematopoiesis, possibly associated with a pre-leukemic myeloproliferatative neoplasm) was evident (Table 1B and Fig. 2B), without histological evidence of transformation to acute leukemia, in many of the mice containing PML, that had been ethically designated to have reached an end-point (largely due to marked hepatosplenomegaly). This EMH phenotype was associated with peripheral blood leucocytosis and macrocytic anemia, also indicative of a myeloproliferative/myelodysplastic overlap (further elaborated below).
Table 1. PML influences the disease profiles of p53+/R175 mice .
| (A) PML loss influences tumor profiles in p53+/R172H mice (% tumor type) | |||
|---|---|---|---|
| Tumor types | p53+/R172H PML+/+ | p53+/R172H PML+/− | p53+/R172H PML−/− |
| Lymphoma | 52% | 43% | 36% |
| Sarcoma | 43% | 52% | 59% |
| Carcinoma | 4% | 5% | 5% |
| No. tumors | 23 | 21 | 22 |
| No. of mice with tumors | 20 | 17 | 18 |
| (B) PML loss influences disease manifestation p53+/R172H mice (% disease/total no. mice) | |||
|---|---|---|---|
| Tumor types | P53+/R172H PML+/+ | p53+/R172H PML+/− | p53+/R172H PML−/− |
| Lymphoma | 44% | 38% | 44% |
| EMH | 41% | 33% | 0% |
| Sarcoma | 37% | 46% | 72% |
| Carcinoma | 4% | 4% | 6% |
| No. tumors | 23 | 21 | 22 |
| Total no. mice | 27 | 24 | 18 |
| Mean survival | 499d | 504d | 448d |
| (C) Tumor spectrum is influenced by gender and PML loss in p53+/R172H mice (% disease/total no. mice) | |||||
|---|---|---|---|---|---|
| p53+/R172H male mice | |||||
| Tumor types | p53+/R172H PML+/+ | p53+/R172H PML+/− | p53+/R172H PML−/− | ||
| Lymphoma | 50% | 33% | 44% | ||
| EMH | 43% | 33% | 0% | ||
| Osteosarcoma | 14% | 8% | 33% | ||
| Soft tissue sarcoma | 14% | 42% | 44% | ||
| Carcinoma | 0% | 0% | 0% | ||
| No. tumors | 10 | 11 | 11 | ||
| No. mice | 14 | 12 | 9 | ||
| p53+/R172H female mice | |||||
| Tumor types | p53+/R172H PML+/+ | p53+/R172H PML+/− | p53+/R172H PML−/− | ||
| Lymphoma | 38% | 42% | 44% | ||
| EMH | 31% | 33% | 0% | ||
| Osteosarcoma | 38% | 25% | 56% | ||
| Soft tissue sarcoma | 8% | 17% | 11% | ||
| Carcinoma | 8% | 8% | 11% | ||
| No. tumors | 13 | 10 | 11 | ||
| No. mice | 13 | 12 | 9 | ||

Figure 2. Tumor development in p53+/R172H mice. Abundance of lymphomas, sarcomas and carcinomas were dictated by PML abundance in p53+/R172H mice, as indicated by the proportion of tumors (calculated as the number of tumors of a specific tumor type/total number of tumors and expressed as a percentage) in a Venn Diagram presentation (A). Tumor prevalence as determined on an individual mouse basis indicated the extensive perturbation of the hematological niche in mice with PML; however, in mice without PML, sarcomas dominated (B).
It is pertinent to note that mutation of p53 has been suggested as a predictive marker of leukemic transformation in human myeloproliferative neoplasms,26 and, further, the prognosis of hematological malignancy in patients harboring a p53 mutation is worse than those expressing the wt p53 protein (reviewed in ref. 27). EMH has not been previously reported for this genotype; however, distinct disease manifestations between studies have been attributed to individual genetic backgrounds.2,3 Specifically, in our study on an advanced C57BL.6 genetic background, disruption of hematopoiesis in p53+/R172H mice with PML was higher than previously published.2,3 Importantly, neither male or female p53+/R172H mice lacking PML were ever identified with EMH. Since females did not exhibit a significantly curtailed lifespan, it suggests that a loss of PML offers protection from this phenotype in the context of mutant p53 mice.
Strikingly, when tumor incidence was evaluated per mouse (including those that succumbed to EMH, rather than as a % of tumors), a high incidence of sarcomas was identified to accompany the elimination of PML (increasing from PML+/+ 37%, to PML+/− 46% to PML−/− 72%, Table 1B). When tumors were segregated according to gender, and soft tissue sarcomas were distinguished from osteosarcomas, it became profoundly apparent that males compromised for PML succumbed to soft tissue sarcomas more frequently than females (Table 1C). These data therefore indicate that the proportion of males and females in a cohort influence the abundance of tumor types for a particular genotype. Most importantly, the average survival duration of male p53+/R172HPML−/− mice with soft tissue sarcomas was shorter (380 d; n = 4) than for all other mice, including male p53+/R172HPML+/− mice presenting with soft tissue sarcoma, (443 d; n = 5; which manifested with a similar frequency). Further, while both male and female p53+/R172HPML−/− mice exhibited an elevated incidence of osteosarcomas, these were proportionately more abundant in females but did not alter survival latency. These data support the finding that loss of PML in p53+/R172H resulted in an enhanced incidence of osteosarcomas in females and also males, while in males, aggressive soft tissue sarcomas were more abundant.
Hepatosplenomegaly was identified as a prominent feature of p53+/R172H mice, with diminished severity corresponding to PML loss (Fig. 3A and B; Table S2A and B, as measured by percentage body weight, and Fig. S4; C57BL.6:p53+/R172HPML+/+:p53+/R172H PML+/−:p53+/R172HPML−/−: for spleens 1:21:9:6.5; and for livers 1:2:1.5:1-fold variation). Interestingly, p53+/R172HPML+/+ mice exhibited the lowest body weights (Fig. S4) and also the greatest incidence of lymphomas (and correspondingly in humans, unintentional weight loss of > 10% body weight is defined as a “B-symptom” according to the Ann Arbor staging).28 These findings are consistent with extensive targeting of the hematopoietic system in mice with mutant p53 and PML and support the suggestion that an absence of PML results in earlier cancer development at alternative sights (apparently the connective tissue).

Figure 3. Disease manifestation in p53+/R172H mice. Significant splenomegaly (A) and hepatomegaly (B) were most pronounced in p53+/R172H mice with PML. Hematological disruption in p53+/R172H mice manifested as anemia (C), elevated MCV (D) and elevated white blood cell levels (E) were most pronounced in p53+/R172H mice with PML but only in subpopulations of p53+/R172H mice with reduced PML. Immunophenotyping of lymphocytes from terminally resected tumors of p53+/R172H mice identified B cell lymphomas to be most abundant in p53+/R172H mice with PML (F).
Anemia and elevated WBC counts in p53+/R172HPML+/+ was alleviated with PML depletion
Anemia was identified in p53+/R172H mice (Fig. 3C and Table 2; Fig. S5) and was more severe on average in the presence of two PML alleles compared with mice with a single or no PML alleles, respectively. Intriguingly, with PML reduction, two populations emerged, either with normal HGB levels or with elevated levels. Anemia coincided with an elevated mean cell volume (MCV; Fig. 3D and Table 2), indicative of macrocytic anemia. Leukocytosis was also evident in p53+/R172HPML+/+ mice (Fig. 3E and Table 2). Together with the EMH described above, the constellation of macrocytic anemia with leukocytosis is suggestive of a myeloproliferative/myelodysplastic overlap syndrome according to current diagnostic criteria.29
Table 2. p53+/R172H mice manifested macrocytic anemia, but less frequently with PML depletion.
| mean values | C57BL.6 | PML−/− | p53+/R172H PML+/+ | p53+R172H PML+/− | p53+/R172HPML−/− | ||
|---|---|---|---|---|---|---|---|
| normal HGB | low HGB | normal HGB | low HGB | ||||
|
HGB (g/L) |
145 | 141.3 | 64.00 | 131.2 | 65.0 | 142.8 | 33.5 |
| p-value | ns 0.7355 | s < 0.0001 | ns 0.0490 | s < 0.0001 | ns 0.8304 | s < 0.0001 | |
| MCV (fL) | 51.11 | 49.90 | 59.41 | 49.28 | 64.28 | 50.56 | 62.01 |
| p-value | ns 0.3239 | s 0.0307 | ns 0.2110 | s 0.0027 | ns 0.7131 | s 0.0252 | |
| RBC | 9.078 | 8.775 | 4.281 | 8.106 | 3.583 | 8.628 | 2.195 |
| p-value | ns 0.4893 | s 0.0002 | s 0.0120 | s < 0.0001 | ns 0.4722 | s < 0.0001 | |
| WBCs (10 9 /L) | 3.317 | 4.138 | 9.250 | 7.276 | 5.700 | 6.139 | 8.215 |
| p-value | ns 0.3578 | s 0.0113 | ns 0.1583 | s 0.0394 | ns 0.3062 | ns 0.1419 | |
| No. mice | 9 | 4 | 15 | 17 | 5 | 18 | 10 |
Normal ranges: HGB 118–149 g/L; MCV 42.2–59.2 fL; RBC 7–10 × 1012/L; WBC 3–13 × 109/L ADVIA. *Significance (s) or no significance (ns) was determined using the unpaired t-test; p < 0.05,where all values were compared with C57BL.6.
Immunophenotyping of resected lymphomas and spleens from p53+/R172H mice identified B-cell lymphomas (Fig. 3F) dominating, in contrast to the predominant T-cell lymphomas in p53−/− mice.30 The identification of B-cell lymphomas is similarly consistent with their occurrence in the presence of other p53 mutations.2 These data are consistent with a single allele of mutant p53 driving B-cell lymphomagenesis, more profoundly in the presence of PML. It is interesting then that the loss of PML in p53+/R172H mice led to an increased abundance of T-cell lymphomas.
Mutant p53 accumulation was enhanced in the absence of PML
Immunoblotting of a range of tissues from male p53+/R172H PML+/+, p53+/R172H PML+/− and p53+/R172H PML−/− mice demonstrated that an increased accumulation of mutant p53 accompanied PML loss, both in mice that had developed lymphomas and sarcomas (Fig. 4A and B; quantified in Fig. S6). Further, levels of the key oncogenic stress response protein p19ARF, were identified to coincidently accumulate with increased levels of mutant p53, as PML diminished. Additional substantiation was provided by the examination of extra mice from each cohort, in which PML levels were verified. Interestingly, c-Myc, a known activator of p19ARF, was also most profoundly accumulated in the absence of PML, accompanying p53 mutation in lymphomas (with only very weak detection in sarcomas, Fig. S7). These data support the notion that PML loss promotes mutant p53 accumulation, which is vital for its “gain of function” capacity.

Figure 4. Western immunoblotting of a range of tissues from p53+/R172H mice stained for p53, p19ARF and HSP60 diagnosed with lymphomas (A) and sarcomas (B). Control (c) was p53+/+ irradiated splenocytes. c, Control; M, male.
PML loss and p53 mutation was identified in human leiomyosarcoma
PML and p53 staining of human sarcomas was undertaken to investigate whether the phenomena of PML loss and p53 mutation as identified in mice was a faithful indicator of genetic alterations in humans. PML depletion coincided with p53 mutation in a subset of leiomysarcomas (Table S3).
Discussion
Dysfunction of the tumor suppressor p53 network is a near universal hallmark of cancer31 that in at least 50% of human cancers is attributed to p53 mutations,32 which may also confer metastastic potential (as demonstrated in mouse models).2,3 A partial or complete loss of PML expression is also frequently observed in multiple types of cancer.16 In our study we examined for the combined influence of these common genetic alterations, and found that PML loss both reduced survival and profoundly altered the spectrum of tumors driven by the mouse equivalent of the human hotspot p53R175H mutation (in heterozygous p53+/R172H mice). Mice expressing PML predominantly exhibited lymphomas or altered hematopoiesis reminiscent of a myeloproliferative/myelodysplastic phenotype (as evidenced by EMH, Table 1B), hepatosplenomegaly (Fig. 3A and B, macrocytic anemia and leukocytosis, Table 2). Notably, p53 function is critical for restricting the numbers of proliferating hematopoietic stem cells (HSC) (ref. 33 and references within), and its absence leads to greater HSC proliferation.34,35 Strikingly, loss of PML resulted in a marked susceptibility to sarcomas and reduced survival. The more limited involvement of the hematopoietic compartment in the absence of PML may be explained, at least in part, by its role in quiescence maintenance of HSC and leukemic initiating cells (LIC; at least in some forms of leukemia).36 In the absence of PML, the leukemic stem cell pool becomes exhausted, which reduces the number of LIC and hence limits tumorigenic capacity (reviewed in ref. 37).
Unexpectedly, the effect of PML loss was gender-specific. Explicitly, PML absence from male mutant p53 mice resulted in reduced survival, associated with a high prevalence of soft tissue sarcomas; suggesting that in males, cooperation between these pathways drove a more aggressive disease. In this context, it is pertinent to note that significant PML loss in locally advanced and metastatic soft tissue human sarcomas corresponded with reduced time to tumor progression, duration of response and overall survival.38,39 Clearly it would be important to know the status of p53 in these tumors, where other studies have defined a ~30% incidence of p53 mutations in human soft tissue sarcomas.32,40 We have addressed this in a small study on selected sarcoma types and grades and identified a subset of leiomysarcomas with coincident PML loss and p53 mutation (Table S3). Interestingly, this rare cancer type was identified among our mice bearing mutant p53 and reduced PML; it will be fascinating to investigate this correlation on a larger population of human leiomysarcomas (and also other muscle sarcomas), for which survival data are available, to validate the prognostic value of the identified correlation. Overall, our results support a role for PML loss in the context of p53 mutation in sarcomas.
Intriguingly, PML loss had no influence on the survival of mutant p53 female mice in our study. Previous studies have also shown that PML loss did not accelerate breast cancer induced by the (MMTV)/neu transgene.12 Further, while PML loss accelerated PTEN-induced invasive colon and prostate carcinomas in males, evaluation in females was preempted by an overriding lethality that resulted from a female dominant autoimmune disease.17 Together with our findings, it is tempting to speculate the fascinating possibility that PML loss affects males more than females, at least in the context of certain oncogenic events. It would be interesting to re-evaluate previous studies for possible gender differences, to further clarify the role of PML in solid tumors. The basis for the gender effect of PML is not clear. However, clues may lie in the recent finding that coincident mutation of the PML-regulated circadian clock gene Period2 (Per2)41 and p53 mutation resulted in reduced survival of male mice but did not further reduce the diminished female survival.22
PML affects multiple apoptotic pathways implicated in tumor suppression (reviewed in ref. 42). Most pertinently, PML is a key partner of wt p53;8–10 however, its capacity to also interact with mutant p5320 led us to examine the effect of PML loss on the stability of mutant p53. Indeed, we found elevated levels of mutant p53 protein in tumors lacking either one or two PML alleles (Fig. 4). This is the first study to suggest that PML loss can induce mutant p53 accumulation. Previously, mutant p53R172H has been shown to accumulate through the germ line elimination of Mdm2 or through the deletion of p16INK4a5 (leading to elevated levels of p19ARF, able to sequester Mdm2 and promote the stabilization of p53: wt43 and mutant p535). We observed that mutant p53 accumulation in the absence of PML was invariably associated with elevated p19ARF expression, without engineered deletion of p16INK4a (Fig. S7). Further, an absence of PML was also frequently coincident with both p53 and Myc accumulation (Fig. S7). Myc levels are likely to have been elevated through mutant p53-driven myc transcription,44 compounded by the absence of PML-driven Myc destabilization.45 Pertinently, Myc elevation was demonstrated to be promoted by p53R172H in a Ras-induced mouse skin cancer model.46 At least in the context of wt p53, Myc activates p19ARF and protects p53 from Mdm2.47 Together with previous findings of the role of PML in the protection of p53 from Mdm2,8–10,48 these observations support the involvement of Myc/p19ARF/Mdm2 in the accumulation of mutant p53 driven by PML loss (Fig. 5), but suggest that additional pathways cannot be excluded.
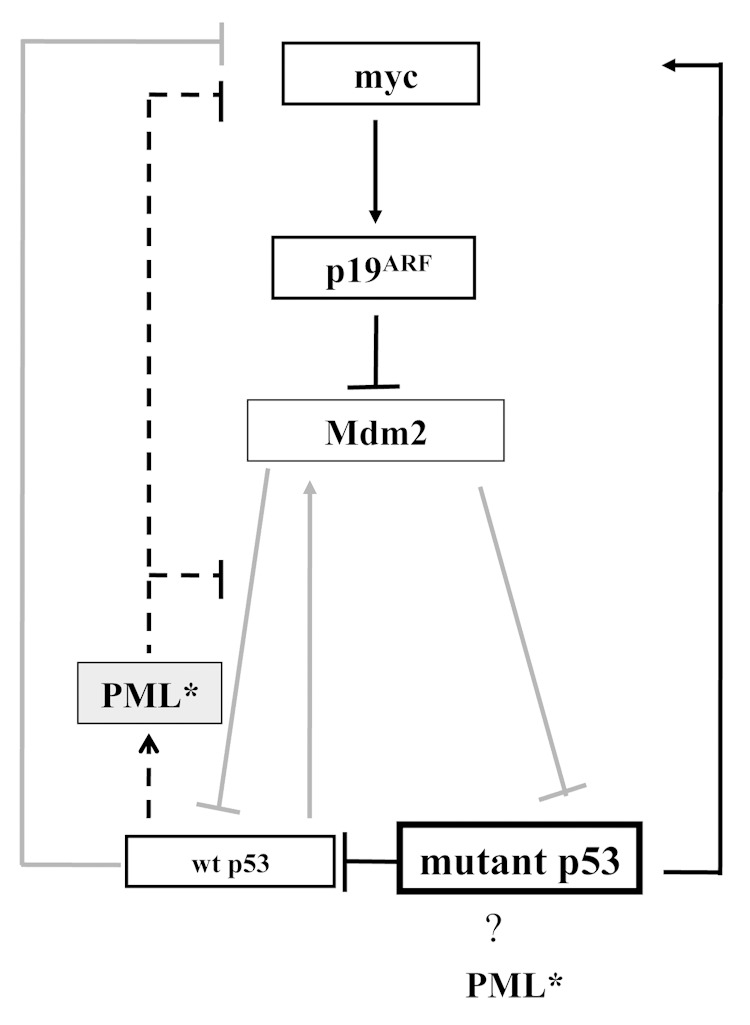
Figure 5. A postulated model mechanism of PML tumor protection in p53+/R172H mice. PML presence promotes wt p53 activity in response to stress provocation and preserves genomic integrity. In the absence of PML, wt p53 function is compromised through the loss of its co-activator and mutant p53 is accumulated and drives tumorigenesis. Dashed line indicates pathways dependent on PML presence. Grey pathways indicate pathways that are anticipated to be less active when PML is absent.
Soft tissue sarcomas are a diverse collection of malignancies that represent a disproportionate abundance (~15%) of cancers in the young, and metastatic disease is very aggressive with a 5-y survival rate of 10–30%.49 Our study reveals cooperation between PML loss and mutant p53, particularly in soft tissue sarcomas in males. These results parallel previous findings, which identified that high p53 levels (suggestive of mutant p53) and coincident low PML levels correlated with reduced patient survival in another malignancy: sporadic gall bladder cancer.50 Together, these data provide a rational basis for further exploration of mutant p53 and PML as disease markers in certain human hemopoietic malignancies and sarcomas and also predict exciting possibilities for combinatorial treatments of the new era of mutant p53 specific therapies together with PML enhancing drugs.51
Materials and Methods
Mouse husbandry and tumor analysis
Mice knocked-in for mutant p53R172H (bearing a G-to-A substitution at nucleotide 515 in a single p53 allele) were as previously described.3 PML-knockout mice (PML−/−) mice were also as previously described.19 Mutant mice were generated on an equivalently advanced C57BL.6 background (99.61% C57BL.6, achieved by backcrossing with C57BL.6 mice for eight generations, N8) and then intercrossed to generate the experimental cohorts (p53+/R172HPML+/+; p53+/R172HPML+/−; p53+/R172HPML−/−). Mice were aged until a tumor burden was evident, or until ethical endpoints were reached, then killed per institutional guidelines. The research was approved by the institute’s ethics committee. Necropsies were performed, and soft tissue organs were harvested and divided for protein analysis and microscopy. In preparation for microscopy, soft tissues were fixed in 10% neutral buffered formalin for 24 h. Skeletons were also fixed, then decalcified in hydrochloric acid/formic acid (8.5%:11%; 2 h). Fixed tissues were processed for paraffin embedding, sectioned and stained with hematoxylin and eosin. Histopathological analysis for each organ was performed by an onco-pathologist (CM). Tisssue sections were visualized using a BX-51 microscope (Olympus). Pictures were acquired using SPOT Version 4.7 software (Diagnostic Instruments).
Quantification of tumor burden and metastases
Histopathology of hematoxylin and eosin stained slides was performed on every soft tissue organ of the analyzed mice. Metastases were distinguished from primary tumors by histopathology. Mice with more than two tumor types were scored as containing multiple tumors. Decalcified bones were also analyzed where osteosarcoma was suspected.
The weight of each mouse was measured, and the weight of their livers and spleens were assessed as a percentage of total body weight.
Immunophenotyping
Mice from the p53+/R172H cohorts with enlarged spleens and nodes, suspected to have lymphoid malignancies were terminally resected. Single-cell suspensions of isolated lymphoid tissues were stained with the following antibodies: FITC-conjugated monoclonal anti-mouse Thy-1.2 (CD90.2; eBioscience); APC-conjugated monoclonal anti-mouse B220 (CD45R; eBioscience); FITC-conjugated anti-mouse CD4+ (L3T4; eBioscience); APC-conjugated monoclonal antibody anti-mouse CD8+ (Ly2; eBioscience). Data was collected using BD FACSCanto 2 flow cytometer and analyzed on FlowJo software. Immunophenotyping of lymphoid cells from selected animals enabled a lymphocyte spectrum analysis according to genotype.
Blood analysis
At death, cardiac puncture was performed and blood was immediately diluted 1:5 in heparin-sulfate (50 IU in 5 ml 0.9% NaCl, Pfizer) at collection and analyzed using the Advia blood analyzer for WBC and RBC.
Immunoblot analysis
Immunoblot analysis was performed essentially as described,52 where lysates of homogenized mouse tissues were loaded for equivalent protein (20–50 μg) and separated by SDS-PAGE. Electrophoresed proteins were transferred to nitrocellulose membranes (Biorad) and probed with antibodies prior to detection using the Odyssey® Imager (LI-COR). Immunodetection of mouse proteins was undertaken using a mouse monoclonal antibody to PML (clone 36.1–104) from Upstate Biotechnology, a rabbit polyclonal antibody to p53 (CM5) from Vector Laboratories, a rabbit polyclonal antibody to CDKN2A/p19Arf (ab80) from abcam and a rabbit polyclonal antibody to c-Myc (9402) from Cell Signaling. Secondary goat ant-rabbit (926–32,211) or goat anti-mouse (926–32,220) antibodies conjugated with infrared dyes (IRDye) were purchased from LI-COR. Densitometry analysis was performed using ImageJ software (NIH) as per the request of the ImageJ developers.
Immunohistochemistry
Human tissue sections (NY; covered by institute ethic approvals) were stained for PML51 and p53 (Santa Cruz rabbit polyclona antibody sc-6243, diluted 1:200). Staining was visualized using the chromogen diaminobenzidine together with hematoxylin as the nuclear counter stain.
Supplementary Material
Acknowledgments
The work was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia to Y.H. (NHMRC 509197); by the VESKI award and CASS Foundation. Y.H. is an NHMRC Senior Research Fellow.
Glossary
Abbreviations:
- PML
promyelocytic leukemia
- EMH
extramedullary hematopoiesis
- MCV
mean cell volume
- WBC
white blood cells
- RBC
red blood cells
- HGB
hemaglobin
- HSC
hematopoietic stem cells
- LIC
leukemic initiating cells
- IHC
immunohistochemistry
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authorship contributions
S.H. performed all the experimental work on the mice, with technical assistance from V.C.; S.H. designed the experiments and wrote the manuscript with Y.H.; pathology was assessed by C.M. and S.F.; hematological analysis was undertaken in consultation with J.S.; mutant p53 mice were provided by G.L., PML KO mice were provided by P.P.P.; human sarcoma samples were provided, stained for IHC and analyzed by M.C-M. D.M.B. and C.C-C.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24806
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/24805
References
- 1.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Donehower LA, Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer. 2009;9:831–41. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- 5.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 7.Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27:8284–95. doi: 10.1128/MCB.00050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP, Haupt Y. The promyelocytic leukemia protein protects p53 from Mdm2-mediated inhibition and degradation. J Biol Chem. 2003;278:33134–41. doi: 10.1074/jbc.M301264200. [DOI] [PubMed] [Google Scholar]
- 9.Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–72. doi: 10.1038/ncb1147. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Wu L, Maki CG. MDM2 and promyelocytic leukemia antagonize each other through their direct interaction with p53. J Biol Chem. 2003;278:49286–92. doi: 10.1074/jbc.M308302200. [DOI] [PubMed] [Google Scholar]
- 11.de Thé H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–84. doi: 10.1016/0092-8674(91)90113-D. [DOI] [PubMed] [Google Scholar]
- 12.Rego EM, Wang ZG, Peruzzi D, He LZ, Cordon-Cardo C, Pandolfi PP. Role of promyelocytic leukemia (PML) protein in tumor suppression. J Exp Med. 2001;193:521–9. doi: 10.1084/jem.193.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, et al. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–6. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- 14.Gottifredi V, Prives C. P53 and PML: new partners in tumor suppression. Trends Cell Biol. 2001;11:184–7. doi: 10.1016/S0962-8924(01)01983-3. [DOI] [PubMed] [Google Scholar]
- 15.Bellodi C, Kindle K, Bernassola F, Cossarizza A, Dinsdale D, Melino G, et al. A cytoplasmic PML mutant inhibits p53 function. Cell Cycle. 2006;5:2688–92. doi: 10.4161/cc.5.22.3504. [DOI] [PubMed] [Google Scholar]
- 16.Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst. 2004;96:269–79. doi: 10.1093/jnci/djh043. [DOI] [PubMed] [Google Scholar]
- 17.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–7. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scaglioni PP, Yung TM, Cai LF, Erdjument-Bromage H, Kaufman AJ, Singh B, et al. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell. 2006;126:269–83. doi: 10.1016/j.cell.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 19.Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, et al. Role of PML in cell growth and the retinoic acid pathway. Science. 1998;279:1547–51. doi: 10.1126/science.279.5356.1547. [DOI] [PubMed] [Google Scholar]
- 20.Haupt S, di Agostino S, Mizrahi I, Alsheich-Bartok O, Voorhoeve M, Damalas A, et al. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009;69:4818–26. doi: 10.1158/0008-5472.CAN-08-4010. [DOI] [PubMed] [Google Scholar]
- 21.Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185–95. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 2012;19:397–405. doi: 10.1038/cdd.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–80. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931–6. doi: 10.1016/S0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- 25.Chen X, Watkins R, Delot E, Reliene R, Schiestl RH, Burgoyne PS, et al. Sex difference in neural tube defects in p53-null mice is caused by differences in the complement of X not Y genes. Dev Neurobiol. 2008;68:265–73. doi: 10.1002/dneu.20581. [DOI] [PubMed] [Google Scholar]
- 26.Harutyunyan A, Klampfl T, Cazzola M, Kralovics R. p53 lesions in leukemic transformation. N Engl J Med. 2011;364:488–90. doi: 10.1056/NEJMc1012718. [DOI] [PubMed] [Google Scholar]
- 27.Peller S, Frenkel J, Lapidot T, Kahn J, Rahimi-Levene N, Yona R, et al. The onset of p53-dependent apoptosis plays a role in terminal differentiation of human normoblasts. Oncogene. 2003;22:4648–55. doi: 10.1038/sj.onc.1206541. [DOI] [PubMed] [Google Scholar]
- 28.Mellstedt H. Clinical signs and symptoms at diagnosis and its differential diagnosis. Ann Oncol. 2007;18(Suppl 1):i14–21. doi: 10.1093/annonc/mdl445. [DOI] [PubMed] [Google Scholar]
- 29.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press. Lyon: IARC Press, 2008. [Google Scholar]
- 30.Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA, Jr., Park SH, et al. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- 31.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 33.Abbas HA, Pant V, Lozano G. The ups and downs of p53 regulation in hematopoietic stem cells. Cell Cycle. 2011;10:3257–62. doi: 10.4161/cc.10.19.17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, et al. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–42. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol. 2003;31:521–7. doi: 10.1016/S0301-472X(03)00072-9. [DOI] [PubMed] [Google Scholar]
- 36.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Rich T, Watson CJ. PML: a tumor suppressor that regulates cell fate in mammary gland. Cell Cycle. 2009;8:2711–7. doi: 10.4161/cc.8.17.9462. [DOI] [PubMed] [Google Scholar]
- 38.Vincenzi B, Perrone G, Santini D, Grosso F, Silletta M, Frezza A, et al. PML down-regulation in soft tissue sarcomas. J Cell Physiol. 2010;224:644–8. doi: 10.1002/jcp.22161. [DOI] [PubMed] [Google Scholar]
- 39.Vincenzi B, Santini D, Schiavon G, Frezza AM, Silletta M, Crucitti P, et al. PML expression in soft tissue sarcoma: Prognostic and predictive value in alkylating agents/antracycline-based first line therapy. J Cell Physiol. 2012;227:1657–62. doi: 10.1002/jcp.22889. [DOI] [PubMed] [Google Scholar]
- 40.Das P, Kotilingam D, Korchin B, Liu J, Yu D, Lazar AJ, et al. High prevalence of p53 exon 4 mutations in soft tissue sarcoma. Cancer. 2007;109:2323–33. doi: 10.1002/cncr.22680. [DOI] [PubMed] [Google Scholar]
- 41.Miki T, Xu Z, Chen-Goodspeed M, Liu M, Van Oort-Jansen A, Rea MA, et al. PML regulates PER2 nuclear localization and circadian function. EMBO J. 2012;31:1427–39. doi: 10.1038/emboj.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernardi R, Papa A, Pandolfi PP. Regulation of apoptosis by PML and the PML-NBs. Oncogene. 2008;27:6299–312. doi: 10.1038/onc.2008.305. [DOI] [PubMed] [Google Scholar]
- 43.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 44.Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol. 1998;18:3735–43. doi: 10.1128/mcb.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buschbeck M, Uribesalgo I, Ledl A, Gutierrez A, Minucci S, Muller S, et al. PML4 induces differentiation by Myc destabilization. Oncogene. 2007;26:3415–22. doi: 10.1038/sj.onc.1210128. [DOI] [PubMed] [Google Scholar]
- 46.Torchia EC, Caulin C, Acin S, Terzian T, Kubick BJ, Box NF, et al. Myc, Aurora Kinase A, and mutant p53(R172H) co-operate in a mouse model of metastatic skin carcinoma. Oncogene. 2012;31:2680–90. doi: 10.1038/onc.2011.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene. 2008;27:3653–61. doi: 10.1038/sj.onc.1211036. [DOI] [PubMed] [Google Scholar]
- 49.Thompson PA, Chintagumpala M. Targeted therapy in bone and soft tissue sarcoma in children and adolescents. Curr Oncol Rep. 2012;14:197–205. doi: 10.1007/s11912-012-0223-2. [DOI] [PubMed] [Google Scholar]
- 50.Chang HJ, Yoo BC, Kim SW, Lee BL, Kim WH. Significance of PML and p53 protein as molecular prognostic markers of gallbladder carcinomas. Pathol Oncol Res. 2007;13:326–35. doi: 10.1007/BF02940312. [DOI] [PubMed] [Google Scholar]
- 51.Wolyniec K, Chan AL, Haupt S, Haupt Y. Restoring PML tumor suppression to combat cancer. Cell Cycle. 2012;11:3705–6. doi: 10.4161/cc.22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Louria-Hayon I, Alsheich-Bartok O, Levav-Cohen Y, Silberman I, Berger M, Grossman T, et al. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ. 2009;16:1156–66. doi: 10.1038/cdd.2009.31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.